

Abstract
Streptococcus pyogenes (group A streptococcus, GAS) is responsible for a wide range of pathologies ranging from mild pharyngitis and impetigo to severe invasive soft tissue infections. Despite the continuing susceptibility of the bacterium to β-lactam antibiotics there has been an unexplained resurgence in the prevalence of invasive GAS infection over the past 30 years. Of particular importance was the emergence of a GAS-associated sepsis syndrome that is analogous to the systemic toxicosis associated with TSST-1 producing strains of Staphylococcus aureus. Despite being recognized for over 20 years, the etiology of GAS associated sepsis and the streptococcal toxic shock syndrome remains poorly understood. Here we review the virulence factors that contribute to the etiology of GAS associated sepsis with a particular focus on coagulation system interactions and the role of the superantigens in the development of streptococcal toxic shock syndrome.
Keywords: group A streptococcus, sepsis, toxic shock syndrome, superantigen, STSS
Streptococcal Sepsis, Septic Shock, and Toxic Shock Syndrome
Sepsis is a progressively injurious systemic immunopathology that is triggered in response to a range of bacterial and fungal pathogens. The features of sepsis and septic shock were first described by Jacobs and Bone and have since been refined several times, along with the published guidelines for management of the condition.1-4 The initial features of sepsis are mild and largely non-specific however, severe sepsis is characterized by impaired organ function and may be associated with coagulation defects such as disseminated intravascular coagulation.1,2 The onset of septic shock is associated with a profound drop in arterial blood pressure that is refractory to adequate volume resuscitation, and precedes eventual multisystem failure and death.1,2
Streptococcal toxic shock syndrome (STSS) is defined by a number of criteria that largely mirror those defining septic shock, coupled with evidence of an invasive GAS infection (Table 1).3,5 While staphylococcal toxic shock syndrome presents as a discrete entity in association with otherwise mild or even occult infections (e.g., menstrual-related TSS in tampon users), STSS is commonly associated with systemic GAS bacteremia or pathological soft tissue necrosis. The defining criteria of STSS include some specific features that are not associated with toxic shock per se, but instead indicate the presence of the underlying GAS pathology (Table 1). As there is considerable overlap between the criteria for septic shock and STSS, and it is common for patients to satisfy the conditions for both, herein the term STSS is used interchangeably with streptococcal septic shock.
Table 1. The diagnostic criteria for septic shock and streptococcal toxic shock syndrome.
| Septic shock | Streptococcal toxic shock syndrome |
|---|---|
| Presumed or confirmed infection | Isolation of GAS from normally sterile site |
| Plus at least ONE of the following | Plus at least TWO of the following |
| Renal dysfunction | Renal dysfunction |
| Respiratory distress | Respiratory distress |
| Hepatic dysfunction | Hepatic dysfunction |
| Hematological abnormalities | Coagulopathy |
| Altered mental status | Erythroderma ± desquamation |
| Unexplained metabolic acidosis | Soft tissue necrosis |
| Tachycardia | Pain |
| Tissue destruction | |
| Skin discoloration | |
| AND | AND |
| Hypotension that is refractory to adequate volume resuscitation | Hypotension that is refractory to adequate volume resuscitation |
The Epidemiology of Invasive GAS Disease and STSS
STSS reportedly complicates approximately 10–16% of invasive GAS infections; however, the true incidence of toxic shock may well be higher as ICU admission is recorded in around 20% of invasive GAS cases.6 STSS is associated with a case fatality rate of 35–45% which is almost twice as high as that reported for invasive GAS cases lacking a shock manifestation.6,7 There is no correlation between STSS and any specific invasive pathology, and the condition is frequently encountered in association with bacteremia, necrotizing fasciitis, pneumonia, and puerperal sepsis.8
GAS strains are routinely divided into serotypes based upon the variable antigenic properties of the major surface M protein. While STSS can be caused by a large number of different GAS M serotypes, the condition is particularly associated with M1 and M3 strains, which together account for approximately 50% of STSS cases in Europe, and over 30% of all invasive disease cases in the United States.6,7 The recent resurgence of serious streptococcal disease has coincided with the emergence of a highly virulent clone of serotype M1T1 GAS which is frequently recovered from invasive infections and STSS cases (discussed in detail below).9,10 M1T1 clones can be distinguished from related serotype M1 isolates by the presence of the phage-encoded virulence factors speA (encoding a superantigen) and sdaI (encoding a DNase) within the accessory genome.11 Furthermore, M1T1 isolates are associated with heightened production of the cytolytic toxins streptolysin O and NAD+ glycohydrolase, which have been shown to contribute to the epithelial inflammation during streptococcal sepsis (discussed below).12 The enhanced virulence that is associated with these genetic alterations, coupled with the ability of M1T1 to switch from a superficial to an invasive disease phenotype in vivo has facilitated global dissemination of this clone over the past 30 years.9,10
While the presence of a single superantigen gene (or superantigen gene repertoire) cannot be uniquely associated with the development of STSS, the phage encoded speA and speC genes have received particular attention with regard to invasive infection.8,13,14 A high rate of speA and/or speC gene carriage has frequently been reported among STSS-associated isolates compared with those recovered from superficial infections however, the relevance of this association (if any) is yet to be elucidated.8,13,15
The Control of Virulence (Cov) System and Invasive GAS Infection
The CovR/S system (also known as CsrR/S) is a two component transcriptional regulator that modulates expression of 10–15% of the GAS transcriptome.16-18 Mutation of the CovR/S system results in transcriptional upregulation of an aggressive repertoire of virulence associated genes, and thus triggers a phenotypic switch from a superficial to an invasive disease phenotype.12,19-21 Indeed such mutations have been shown to account for the prolific phenotypic switching of M1T1 GAS in vivo. Of particular relevance is the reported upregulation of the speA and speJ genes which contribute to the inflammatory pathogenesis of STSS through non-specific T-cell activation (discussed in detail below).11 CovR/S mutation also results in derepression of a multitude of virulence factors that facilitate resistance to opsonophagocytosis, including the hyaluronic acid capsule, streptococcal inhibitor of complement (Sic) and the chemokine protease SpyCEP.10 Such mutations may help to perpetuate the symptoms of streptococcal sepsis by facilitating persistence of GAS at the nidus of infection.
Interestingly, the cysteine protease SpeB undergoes reciprocal regulation by CovR/S, yet is also implicated in the pathogenesis of necrotizing fasciitis and STSS.22 SpeB can augment inflammation through activation of the kallikrein-kinin system (discussed in detail below) and by cleaving interleukin 1β precursor to form biologically active IL-1β.23,24 While SpeB is therefore predicted to enhance the classical symptoms of shock, the precise role of the molecule during STSS remains unclear. Recently Ikebe et al. have reported that the frequency of CovR/S mutation is higher among strains recovered from STSS patients than those isolated from superficial infections although the significance of this finding remains the subject of some debate.25,26
GAS Interactions with the Coagulation System during Severe Sepsis
The pathophysiology of sepsis-associated coagulopathy
Blood coagulation (thrombogenesis) is an essential process that maintains the integrity of the circulatory system and provides innate protection against systemic infection through the isolation of invading pathogens.27,28 Thrombogenesis is initiated following vascular injury and involves a stepwise series of proteolytic reactions that culminate in the formation of a fibrinous clot.29 Vascular injury also facilitates adhesion and activation of circulating platelets which subsequently become incorporated into the growing clot.29 Streptococcal sepsis is often associated with aberrant thrombogenesis resulting in the consumption of clotting factors and the formation of circulating microthrombi.28,30 The pathological effect of this disseminated intravascular coagulation (DIC) is 2-fold. The formation of circulating microthrombi has been shown to result in venous thrombosis and infarction of the subcutaneous tissues in a murine model, while trafficking of microthrombi to the organs is thought to contribute to the pathogenesis of organ dysfunction.31-33 In addition, the depletion of platelets that results from microthrombus formation has been shown to impair normal clot formation resulting in severe secondary bleeding when vascular injury occurs.28,32
M1 protein interactions with fibrinogen
GAS has been shown to facilitate platelet aggregation through a series of stepwise, immune mediated reactions (Fig. 1). The initial interaction may be facilitated by a fibrinogen intermediate which simultaneously binds to the GAS M1 protein and the αIIbβ3 integrins present on the surface of platelets.34 Alternatively GAS may colocalize with circulating platelets at sites of vascular damage where the components of the subendothelial matrix have become exposed. Subendothelial collagen in particular provides a platform for the multimerization of circulating von Willebrand factor, which has been shown to facilitate platelet immobilization upon the vascular endothelium under sheer force conditions.35,36 Regardless of how the initial interaction occurs, the adherent platelets are subsequently activated by the anti-GAS IgG response which engages with platelet Fc receptors (FcγRII) and initiates clot formation.34 Soluble M1 protein that has been released from the bacterial cell surface is similarly capable of activating and aggregating platelets in the presence of anti-M1 IgG.37 M1 activated platelets may also interact with neutrophils and monocytes resulting in the activation of both cell types and the generation of more tissue factor.37
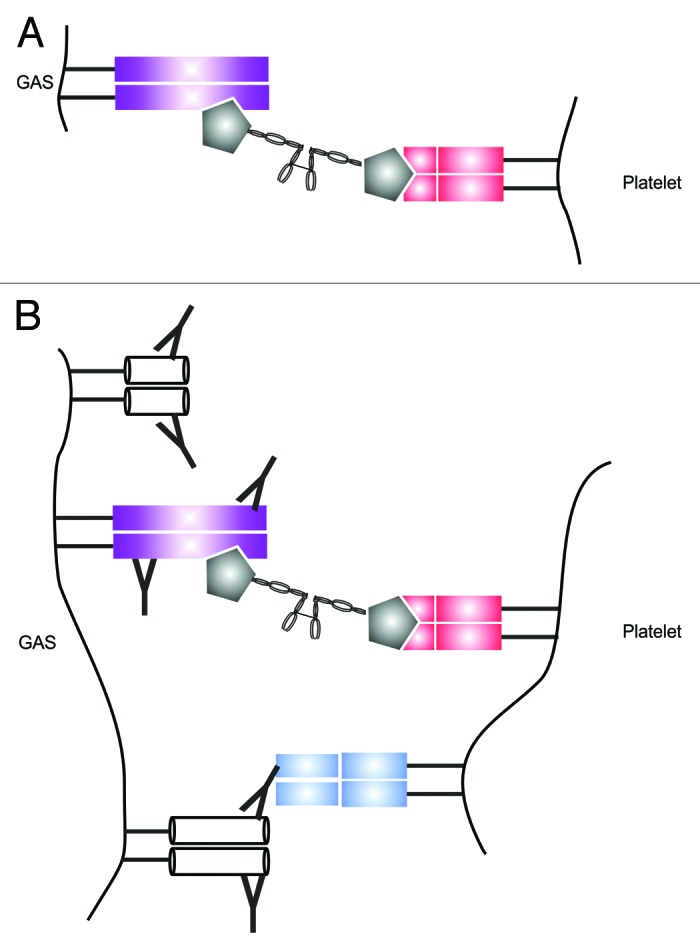
Figure 1. GAS-mediated platelet aggregation and activation. The initial cross linking interaction occurs via simultaneous binding of circulating fibrinogen (gray) by the M protein (purple) and the platelet αIIbβ3 integrin (red) (A). Platelet activation occurs when the Fc receptor (blue) comes into contact with surface associated IgG (B).
In addition to platelet activation, binding of fibrinogen to soluble M1 has been shown to result in the formation of large aggregates (M1/Fg complexes) that are capable of activating neutrophil β2 integrins.38,39 β2 integrin activation triggers a release of heparin binding protein, a soluble inflammatory mediator that has been shown to induce localized vascular leakage. The crystal structure of these M1/Fg complexes has been resolved and the irregular coiled coil structure of the M1 protein has been shown to cross link four fibrinogen molecules leading to the construction of a supramolecular network.38 Intravenous administration of M1 protein into mice induces colocalization of activated neutrophils and protein aggregates within the tissues of the lung, resulting in localized inflammation and pulmonary vascular leakage.39 M1/Fg complexes have been detected in necrotic tissue biopsies recovered from STSS patients supporting a role for these complexes in the induction of vascular leakage during human infection.39
GAS and the extrinsic pathway of coagulation
One of the most important early initiators of the coagulation cascade is tissue factor (TF). TF is produced by the subendothelial tissues and monocytes, and forms an active complex with the serine protease FVIIa on contact with the blood.29 The TF-driven (or extrinsic) coagulation pathway is known to be activated during sepsis, and several studies have reported increased plasma TF levels in patients manifesting trauma-associated SIRS and severe sepsis.40-43 While TF upregulation in GAS sepsis patients has never explicitly been examined, serotype M1 and M3 (but not serotype M6) GAS have been shown to elicit TF synthesis from freshly isolated human monocytes.44 A similar effect was achieved using purified soluble M protein indicating that this virulence factor may be partly responsible for GAS-mediated DIC.30 This hypothesis is supported by recent data demonstrating that purified soluble M protein stimulates the release of TF-rich microparticles from activated monocytes, and that these microparticles can be identified in the blood of septic patients.30,45 TF/factor VII complexes have also been shown to stimulate production of the proinflammatory cytokines IL-1β and TNF-α from macrophages and neutrophils via protease-activated receptor 1 activation.46
GAS and the intrinsic pathway of coagulation
In addition to the extrinsic arm of the coagulation cascade, GAS may also trigger coagulation via the intrinsic pathway through a specific interaction with the contact (or kallikrein–kinin) system. The contact pathway helps to stabilize the formed clot through activation of factor XII, and stimulates the generation of small proinflammatory peptides known as kinins from high molecular weight kininogen.28 Activation of the contact system at the GAS cell surface triggers the release of bradykinin, a potent vasodilator that induces vascular leakage, localized edema, hypotension, and pain.47 Bradykinin release results from the proteolytic processing of the soluble zymogen H-kininogen, which can bind to the GAS cell surface via the M protein. Subsequent recruitment of plasma kallikrein results in a massive release of active bradykinin and thus promotes localized inflammation and vasodilation.48,49 The secreted GAS cysteine protease SpeB has also been shown to promote cleavage of H-kininogen, suggesting a role for this protease during in vivo kinin activation and inflammation.23 Systemic cleavage of H-kininogen has been demonstrated in vivo using a murine model of streptococcal sepsis and ex vivo using inflamed tissue from invasive disease cases.50,51 Patients with severe GAS infections and STSS often demonstrate evidence of isolated activation of the intrinsic pathway of coagulation, as a surrogate of contact system activation.52 Taken together, these studies suggest that activation of the contact pathway at the GAS cell surface may be responsible for much of the abnormal vascular physiology observed during STSS.
Superantigen-Mediated Modulation of the Host Immune Response
The superantigens
The superantigens (SAgs) are a family of toxins that are capable of activating a large set of human T cells, resulting in a massive production of proinflammatory cytokines. Conventional antigens are processed into short fragments by antigen presenting cells prior to translocation to the antigen binding domain (ABD, also known as the epitope groove) of the MHC class II molecule.53 The amino acids that line the ABD are highly polymorphic and as a result each MHC class II isoform is only capable of presenting a finite number of antigens, and activating a very small subset circulating T cells (Fig. 2).

Figure 2. The different modes of SAg binding to MHC class II molecules (red) and the T-cell receptor (yellow). The mitogenicity of a conventional antigen (blue) is limited by its ability to cross link the hypervariable ABD with the TCR (A). SAgs are capable of activating many more T cells by binding to a specific repertoire of Vβ subsets. SAg presentation is facilitated by non-specific binding of the MHC α chain (e.g., SpeA in green) or β chain (e.g., SpeC in purple) (B) or by zinc-dependent β chain binding and engagement of the bound peptide antigen (C). Zinc-dependent β chain binding may also occur following dimerization of SpeC (D); however, the significance of this with regards T-cell activation is yet to be elucidated.
In contrast, SAgs are capable of non-specifically cross linking loaded MHC class II molecules with the variable N-terminal domain of the T-cell receptor (TCR) β chain (denoted the Vβ region) without prior processing (Fig. 2).54,55 Subsequent ligation with the dimer interface of CD28 results in a massive release of proinflammatory cytokines (most notably IL-2, IFN-γ, and TNF-α) which in turn potentiates the acute shock and systemic vascular leakage that is associated with STSS.54,56-59
The majority of GAS SAgs are also capable of binding to MHC class II receptors via high affinity zinc binding (Table 2). Zinc binding is facilitated by the formation of a tetravalent metal complex that incorporates a conserved zinc atom located within the MHC class II β-domain.59,60 Subsequent interactions with the N-terminus of the bound peptide antigen facilitates promiscuous MHC II engagement. Zinc-dependent binding thus facilitates deposition of SAg molecules upon the antigen presenting cell surface, leading to further T-cell activation.59,61
Table 2. The Vβ specificities and binding preferences of the GAS SAgs.
| SAg | MHC II chain bound | Zinc binding | Vβ specificity |
|---|---|---|---|
| SpeA | α | Y | 2.1, 12.2, 14.1, 15.1 |
| SpeC | β | Y | 2.1, 3.2, 12.5, 15.1 |
| SpeG* | β | Y | 2.1, 4.1, 6.9, 9.1, 12.3 |
| SpeH | β | Y | 2.1, 7.3, 9.1, 23.1 |
| SpeI | β | Y | 6.9, 9.1, 18.1, 22 |
| SpeJ* | β | Y | 2.1 |
| SpeK/L | β | Y | 1.1, 5.1, 23.1 |
| SpeL/M | β | Y | 1.1, 5.1, 23.1 |
| SpeM | ? | Y | 1.1, 5.1, 23.1 |
| SSA | α | N | 1.1, 3, 15 |
| SMEZ-1* | β | Y | 2.1, 4.1, 7.3, 8.1 |
| SMEZ-2* | β | Y | 4.1, 8.1 |
*SMEZ and SpeG are chromosomally encoded and encoded ubiquitously within the GAS metagenome. SpeJ is also chromosomally encoded; however, speJ negative clones of GAS are extremely common. While SpeB and SpeF were initially identified as SAgs, they have since been reclassified as a cysteine protease and a DNase (DNaseB) respectively. Table adapted from reference 59.
In addition to these monovalent binding strategies, SpeA and SpeC are capable of forming monomeric dimers that are potentially capable of interacting with two MHC II molecules (and/or TCRs) simultaneously.62,63 While the physiological relevance of this dimerization remains the subject of some debate, the current evidence suggests that it is not essential for the mitogenic activity of either SAg.62-65
Each SAg is specific for a distinct repertoire of Vβ gene products, 20–30 of which exist within the human genome (Table 2) and as such a single SAg is capable of activating up to 20% of all circulating naive T cells.53,66 Cytokine production in response to secreted SAg is extremely rapid, as evidenced by animal models where gene transcription and systemic cytokine release can be detected within one hour of toxin exposure.67,68 While the benefits of SAg production remain incompletely understood, the ubiquitous presence of SAg genes within the GAS metagenome suggests that SAg-mediated T-cell activation imposes a significant selective advantage upon toxigenic GAS isolates.8,55,69
Superantigen-mediated TLR upregulation
Further to direct TCR binding, streptococcal SAgs may augment cytokine release via a number of additional pathways. It has been reported that SAg exposure can enhance the TLR response to gram-negative lipopolysaccharide, thus enhancing TNF cytokine responses during natural coinfection.70 This effect is particularly marked where there is hypoperfusion of the gastrointestinal tract that affects mucosal integrity. While such synergy has been hard to demonstrate in vivo, the effect has been replicated in vitro using primary human monocytes. In vitro priming of monocytes with physiological concentrations of streptococcal SAgs increases membrane expression of TLR4 via MHC class II ligation, and thus enhances the response to endotoxin and other TLR4 ligands.71 MHC class II recognition of streptococcal SAgs may therefore contributes to the pathophysiology of sepsis through upregulation of the proinflammatory cytokines TNF-α, IL-1β, and IL-6.71
In vitro SAg exposure also upregulates monocyte TLR2 expression, enhancing the potential for further synergistic interactions.72 TLR2 signaling in response to peptidoglycan and lipoteichoic acid results in activation of the transcription factors NFκB and AP1, stimulating the release of proinflammatory cytokines and a downstream induction of the adaptive immune response.73,74 TLR2 transcription and expression has been shown to be upregulated in neutrophils and monocytes from streptococcal and non-streptococcal sepsis patients compared with healthy controls.72,75,76 This suggests that TLR signaling may drive the immunopathological symptoms of sepsis prior to activation of the adaptive immune response.
Superantigen-mediated epithelial inflammation
The ability of SAgs to interact with epithelial cells has been demonstrated using a variety of cell lines however, most studies focus mainly on the staphylococcal superantigens.77-79 Stimulation of vaginal epithelial cells with SpeA reportedly triggers production of the proinflammatory cytokines IL-6, IL-8, and MIP-3α in vitro; however, the specific receptor binding interactions that elicit this effect remain uncharacterized.77 A concomitant production of streptolysin O, a hemolytic exotoxin that promotes cytolysis through the formation of transmembrane pores, is thought to enhance mucosal inflammation through localized tissue destruction and the exposure of further epithelial cells.77 This “outside-in” signaling may result in SAg mediated T-cell activation at the submucosa and could therefore facilitate early activation of proinflammatory cytokine cascade during streptococcal septic shock.
Experimental Studies of the Effect of the GAS Superantigens
Laboratory studies
In 1989, Lee and Schlievert reported that the administration of purified SpeA stimulated an STSS-like pathology in a rabbit model, providing early evidence that this family of toxins could elicit features of profound inflammation and shock.80 However, this study was conducted prior to the identification of several additional GAS SAgs, one of which (SMEZ) is a highly potent activator of rabbit Vβ T cells, and is a recognized contaminant of crudely purified SpeA preparations.81,82 Indeed targeted mutagenesis of the speA gene has little impact on the overall mitogenic activity of toxigenic GAS in vitro despite the presence of high SpeA concentrations (>500 ng/ml) within the tested culture supernatants.83 It should be noted that SAg production in vitro is markedly reduced compared with in vivo synthesis, and that this effect is particularly apparent when phage encoded SAgs are considered (potentially as a result of in vivo phage induction).84-86 Nevertheless, the targeted mutagenesis data suggests that a large amount of functional redundancy exists among the GAS SAgs, and that the differential Vβ specificities allow the repertoire of each isolate to act in concert to stimulate a potent T-cell mitogenesis.
The interpretation of experiments using bolus SAg administration is complicated by the rapid clearance of injected SAg toxins and the natural resistance of mice to bacterial SAgs. Rabbits, pigs, and non-human primates demonstrate greater SAg responsiveness than mice and are more amenable to SAg infusion; however, reagents to study such models are limited and present obvious ethical and cost implications. To circumvent these issues, humanized transgenic mouse models of streptococcal sepsis that express human MHC class II molecules have been used to demonstrate the impact of SAgs produced during GAS soft tissue infection.85,87 Using a high SpeA-producing scarlet fever isolate and an isogenic speA knockout mutant, it was possible to show that Vβ-specific T-cell expansion did indeed occur in response to SAg exposure, and that T-cell activation takes place in a number of tissues during invasive infection.83 Using a similar pair of isogenic GAS clones that differed in ability to produce SMEZ, a specific release of cytokines was attributed to SAg synthesis during sepsis, consistent with the widely held view that SAgs can and do trigger a cytokine storm.88 Importantly this also indicates that STSS can occur in the absence of phage-encoded SAgs suggesting that carriage of one or more chromosomal SAg genes is sufficient to stimulate cytokine release.69,89
SMEZ elicits detectable T-cell proliferation at sub-picomolar concentrations and exhibits a high degree of antigenic variation that does not affect the potency or Vβ specificity of the molecule.90 This pattern of antigenic variation suggests that SMEZ is uniquely important for survival of GAS during invasive pathogenesis, and that the need to escape antibody neutralization has driven the progressive variation within the smez locus, without affecting the mitogenicity of the molecule. Despite these observations, a highly conserved naturally occurring smez mutation has been shown to abrogate production of SMEZ by M3 GAS isolates, including those recovered from active STSS cases.91 Taken together these data suggest that a combination of GAS virulence factors that include, but are not limited to, the SAgs are required to stimulate the signs of STSS.
Clinical studies
Despite the advances made through the study of SAgs under experimental conditions, definitive clinical evidence of SAg induced T-cell proliferation is lacking. Most clinicians report that lymphocyte levels are too low to conduct such studies at the time of presentation; however, a few case studies reporting T-cell repertoire changes during STSS have been published.92,93 In addition several studies describe a sequential release of cytokines during STSS and invasive soft tissue infection that is consistent with SAg mediated T-cell activation.94,95 SAg production and immune recognition is also evidenced by a handful of studies that successfully detected circulating SAg and anti-SAg antibodies within the blood of STSS patients by bioassay and ELISA.94,96
Several clinical studies have suggested that the SAgs may suppress phagocyte recruitment during active disease and therefore promote survival of bacteria at the nidus of infection.97-99 However, the observed reduction in neutrophil recruitment during severe GAS infection most likely results from chemokine cleavage by SpyCEP and C5a peptidase.100,101 Furthermore, conflicting data recovered from experimental analysis has suggested that the SAgs may enhance inflammation and subsequent phagocyte recruitment when produced in isolation. These concomitant effects are thought to result from the rapid induction of endothelium-activating cytokines such as TNF, and CXC chemokines.83,102-104 Given that SAgs undergo a specific interaction with the components of the adaptive immune response, collateral interference with T-follicular helper cell function may be expected. Consequent interference with memory B cell activation may impact on clearance of GAS at the nidus of infection if the generation of de novo anti-GAS antibody is impaired.57,96,105 The current data therefore suggests that SAg production is strongly immunostimulatory, but with an as yet unclear impact on anti-GAS immunity and bacterial clearance.
Inherited and Acquired Host Susceptibilities to Streptococcal Toxic Shock Syndrome
The lack of association between a specific SAg profile and the provocation of STSS, coupled with the observation that highly toxigenic GAS clones can be isolated from a spectrum of disease manifestation suggests that the immunogenetics of the host may be partly responsible for the outcome of toxigenic GAS infection.106-108 The human MHC genes are highly polymorphic and three pairs of linked α and β chains (denoted HLA-DP, HLA-DQ, and HLA-DR) may give rise to four isoforms of MHC class II molecule. With the exception of the monomeric DRα locus, a multitude of different alleles has been described for each locus.53 Structural characterization of several SAg-MHC complexes has established that the structure of the SAg binding domain is highly variable between different MHC class II isoforms.109-111 Polymorphism within the DQα1 locus has a direct effect on SpeA binding, and alleles that facilitate a higher affinity interaction have been shown to trigger a more prolific T-cell response in vitro.108 MHC class II polymorphism has also been shown to influence the outcome of SAg mediated disease in vivo. Certain “protective” haplotypes have been shown to confer strong protection against STSS through the elicitation of a polarized, anti-inflammatory cytokine response.107,112 Interestingly, SAg cytokine responses in experimental rodents are doubled in females compared with males, underlining the importance of conducting such experiments in study groups controlled carefully for gender.113 Whether such sexual dimorphism influences the incidence and outcome of STSS is unclear from existing human epidemiological studies, and may be offset by the generally enhanced male predisposition to acquisition of bacterial infection. Pre-existing immunity to streptococcal virulence factors such as the SAgs and the M protein may also influence disease severity, notwithstanding prevention of superficial and invasive infection acquisition, providing a rationale for the use of pooled intravenous immunoglobulin as an adjunct therapy for STSS.57,114
A number of large epidemiological studies have attempted to identify additional preventable risk factors for severe streptococcal disease. However, as invasive GAS is rare (~3 cases per 100 000 in the UK) and STSS somewhat rarer, clear patterns are hard to identify and most risk factors relate to acquisition of infection rather than susceptibility to severe disease or death. One study of invasive GAS infection has reported that the risk of death was highest among those with a prior diagnosis of malignancy, those admitted from long-term residential care and those with alcoholism listed as a comorbidity.115 Mortality from GAS invasive infection was greatest during the winter months (December–April) when acquisition of invasive disease is also common, suggesting that the factors that influence immunity can also affect severity. Cases of invasive disease diagnosed in October were 80% less likely to have a fatal outcome than those in January, although surges in specific, highly virulent M types could explain this seasonal pattern.115
Concluding Remarks
The resurgence of invasive GAS infection over the past 30 years is a matter of great concern especially when one considers the rapid onset and high mortality rates associated with sterile site infections manifesting STSS. While SAg production is responsible for the most recognizable symptoms of toxic shock, there are many other facets that must be considered if novel treatment options are to be explored. Importantly the question of how and why the most highly virulent clones of GAS can produce such profoundly different disease pathologies between cases remains unanswered.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This manuscript was supported by UKCRC and Wellcome Trust (Centre for Infection Prevention & Management, M.R.). S.S. acknowledges the support of the NIHR Biomedical Research Centre at Imperial College.
Glossary
Abbreviations:
- GAS
group A streptococcus
- TSST-1
toxic shock syndrome toxin 1
- STSS
streptococcal toxic shock syndrome
- TLR
toll like receptor
- SAg
superantigen
- ABD
antigen binding domain
- TCR
T-cell receptor
- DIC
disseminated intravascular coagulation
- TF
tissue factor
References
- 1.Jacobs ER, Bone RC. Clinical indicators in sepsis and septic adult respiratory distress syndrome. Med Clin North Am. 1986;70:921–32. doi: 10.1016/s0025-7125(16)30932-4. [DOI] [PubMed] [Google Scholar]
- 2.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ, ACCP/SCCM Consensus Conference Committee Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. 1992. Chest. 2009;136(Suppl):e28. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 3.Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G, International Sepsis Definitions Conference 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Intensive Care Med. 2003;29:530–8. doi: 10.1007/s00134-003-1662-x. [DOI] [PubMed] [Google Scholar]
- 4.Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, Sevransky JE, Sprung CL, Douglas IS, Jaeschke R, et al. Surviving Sepsis Campaign Guidelines Committee including The Pediatric Subgroup Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med. 2013;39:165–228. doi: 10.1007/s00134-012-2769-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breiman RF, Davis JP, Facklam RR, et al. The Working Group on Severe Streptococcal Infections Defining the group A streptococcal toxic shock syndrome. Rationale and consensus definition. JAMA. 1993;269:390–1. doi: 10.1001/jama.1993.03500030088038. [DOI] [PubMed] [Google Scholar]
- 6.Lamagni TL, Darenberg J, Luca-Harari B, Siljander T, Efstratiou A, Henriques-Normark B, Vuopio-Varkila J, Bouvet A, Creti R, Ekelund K, et al. Strep-EURO Study Group Epidemiology of severe Streptococcus pyogenes disease in Europe. J Clin Microbiol. 2008;46:2359–67. doi: 10.1128/JCM.00422-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Loughlin RE, Roberson A, Cieslak PR, Lynfield R, Gershman K, Craig A, Albanese BA, Farley MM, Barrett NL, Spina NL, et al. Active Bacterial Core Surveillance Team The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin Infect Dis. 2007;45:853–62. doi: 10.1086/521264. [DOI] [PubMed] [Google Scholar]
- 8.Luca-Harari B, Darenberg J, Neal S, Siljander T, Strakova L, Tanna A, Creti R, Ekelund K, Koliou M, Tassios PT, et al. Strep-EURO Study Group Clinical and microbiological characteristics of severe Streptococcus pyogenes disease in Europe. J Clin Microbiol. 2009;47:1155–65. doi: 10.1128/JCM.02155-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aziz RK, Kotb M. Rise and persistence of global M1T1 clone of Streptococcus pyogenes. Emerg Infect Dis. 2008;14:1511–7. doi: 10.3201/eid1410.071660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cole JN, Barnett TC, Nizet V, Walker MJ. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol. 2011;9:724–36. doi: 10.1038/nrmicro2648. [DOI] [PubMed] [Google Scholar]
- 11.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. Genome-wide analysis of group a streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2006;2:e5. doi: 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sumby P, Porcella SF, Madrigal AG, Barbian KD, Virtaneva K, Ricklefs SM, Sturdevant DE, Graham MR, Vuopio-Varkila J, Hoe NP, et al. Evolutionary origin and emergence of a highly successful clone of serotype M1 group a Streptococcus involved multiple horizontal gene transfer events. J Infect Dis. 2005;192:771–82. doi: 10.1086/432514. [DOI] [PubMed] [Google Scholar]
- 13.Rantala S, Vähäkuopus S, Siljander T, Vuopio J, Huhtala H, Vuento R, Syrjänen J. Streptococcus pyogenes bacteraemia, emm types and superantigen profiles. Eur J Clin Microbiol Infect Dis. 2012;31:859–65. doi: 10.1007/s10096-011-1385-9. [DOI] [PubMed] [Google Scholar]
- 14.Michaelsen TE, Andreasson IK, Langerud BK, Caugant DA. Similar superantigen gene profiles and superantigen activity in norwegian isolates of invasive and non-invasive group a streptococci. Scand J Immunol. 2011;74:423–9. doi: 10.1111/j.1365-3083.2011.02594.x. [DOI] [PubMed] [Google Scholar]
- 15.Maripuu L, Eriksson A, Norgren M. Superantigen gene profile diversity among clinical group A streptococcal isolates. FEMS Immunol Med Microbiol. 2008;54:236–44. doi: 10.1111/j.1574-695X.2008.00469.x. [DOI] [PubMed] [Google Scholar]
- 16.Graham MR, Smoot LM, Migliaccio CA, Virtaneva K, Sturdevant DE, Porcella SF, Federle MJ, Adams GJ, Scott JR, Musser JM. Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc Natl Acad Sci U S A. 2002;99:13855–60. doi: 10.1073/pnas.202353699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Treviño J, Perez N, Ramirez-Peña E, Liu Z, Shelburne SA, 3rd, Musser JM, Sumby P. CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect Immun. 2009;77:3141–9. doi: 10.1128/IAI.01560-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heath A, DiRita VJ, Barg NL, Engleberg NC. A two-component regulatory system, CsrR-CsrS, represses expression of three Streptococcus pyogenes virulence factors, hyaluronic acid capsule, streptolysin S, and pyrogenic exotoxin B. Infect Immun. 1999;67:5298–305. doi: 10.1128/iai.67.10.5298-5305.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turner CE, Kurupati P, Jones MD, Edwards RJ, Sriskandan S. Emerging role of the interleukin-8 cleaving enzyme SpyCEP in clinical Streptococcus pyogenes infection. J Infect Dis. 2009;200:555–63. doi: 10.1086/603541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurupati P, Turner CE, Tziona I, Lawrenson RA, Alam FM, Nohadani M, Stamp GW, Zinkernagel AS, Nizet V, Edwards RJ, et al. Chemokine-cleaving Streptococcus pyogenes protease SpyCEP is necessary and sufficient for bacterial dissemination within soft tissues and the respiratory tract. Mol Microbiol. 2010;76:1387–97. doi: 10.1111/j.1365-2958.2010.07065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levin JC, Wessels MR. Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol Microbiol. 1998;30:209–19. doi: 10.1046/j.1365-2958.1998.01057.x. [DOI] [PubMed] [Google Scholar]
- 22.Olsen RJ, Sitkiewicz I, Ayeras AA, Gonulal VE, Cantu C, Beres SB, Green NM, Lei B, Humbird T, Greaver J, et al. Decreased necrotizing fasciitis capacity caused by a single nucleotide mutation that alters a multiple gene virulence axis. Proc Natl Acad Sci U S A. 2010;107:888–93. doi: 10.1073/pnas.0911811107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herwald H, Collin M, Müller-Esterl W, Björck L. Streptococcal cysteine proteinase releases kinins: a virulence mechanism. J Exp Med. 1996;184:665–73. doi: 10.1084/jem.184.2.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kapur V, Majesky MW, Li LL, Black RA, Musser JM. Cleavage of interleukin 1 beta (IL-1 beta) precursor to produce active IL-1 beta by a conserved extracellular cysteine protease from Streptococcus pyogenes. Proc Natl Acad Sci U S A. 1993;90:7676–80. doi: 10.1073/pnas.90.16.7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maamary PG, Sanderson-Smith ML, Aziz RK, Hollands A, Cole JN, McKay FC, McArthur JD, Kirk JK, Cork AJ, Keefe RJ, et al. Parameters governing invasive disease propensity of non-M1 serotype group A streptococci. J Innate Immun. 2010;2:596–606. doi: 10.1159/000317640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ikebe T, Ato M, Matsumura T, Hasegawa H, Sata T, Kobayashi K, Watanabe H. Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog. 2010;6:e1000832. doi: 10.1371/journal.ppat.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Renné T. The procoagulant and proinflammatory plasma contact system. Semin Immunopathol. 2012;34:31–41. doi: 10.1007/s00281-011-0288-2. [DOI] [PubMed] [Google Scholar]
- 28.Shannon O, Herwald H, Oehmcke S. Modulation of the coagulation system during severe streptococcal disease. Curr Top Microbiol Immunol. 2013;368:189–205. doi: 10.1007/82_2012_283. [DOI] [PubMed] [Google Scholar]
- 29.McMichael M. New models of hemostasis. Top Companion Anim Med. 2012;27:40–5. doi: 10.1053/j.tcam.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 30.Påhlman LI, Malmström E, Mörgelin M, Herwald H. M protein from Streptococcus pyogenes induces tissue factor expression and pro-coagulant activity in human monocytes. Microbiology. 2007;153:2458–64. doi: 10.1099/mic.0.2006/003285-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ashbaugh CD, Warren HB, Carey VJ, Wessels MR. Molecular analysis of the role of the group A streptococcal cysteine protease, hyaluronic acid capsule, and M protein in a murine model of human invasive soft-tissue infection. J Clin Invest. 1998;102:550–60. doi: 10.1172/JCI3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gawaz M, Dickfeld T, Bogner C, Fateh-Moghadam S, Neumann FJ. Platelet function in septic multiple organ dysfunction syndrome. Intensive Care Med. 1997;23:379–85. doi: 10.1007/s001340050344. [DOI] [PubMed] [Google Scholar]
- 33.Gustot T. Multiple organ failure in sepsis: prognosis and role of systemic inflammatory response. Curr Opin Crit Care. 2011;17:153–9. doi: 10.1097/MCC.0b013e328344b446. [DOI] [PubMed] [Google Scholar]
- 34.Sjöbring U, Ringdahl U, Ruggeri ZM. Induction of platelet thrombi by bacteria and antibodies. Blood. 2002;100:4470–7. doi: 10.1182/blood-2002-01-0069. [DOI] [PubMed] [Google Scholar]
- 35.Huizinga EG, Tsuji S, Romijn RA, Schiphorst ME, de Groot PG, Sixma JJ, Gros P. Structures of glycoprotein Ibalpha and its complex with von Willebrand factor A1 domain. Science. 2002;297:1176–9. doi: 10.1126/science.107355. [DOI] [PubMed] [Google Scholar]
- 36.Reininger AJ, Heijnen HF, Schumann H, Specht HM, Schramm W, Ruggeri ZM. Mechanism of platelet adhesion to von Willebrand factor and microparticle formation under high shear stress. Blood. 2006;107:3537–45. doi: 10.1182/blood-2005-02-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shannon O, Hertzén E, Norrby-Teglund A, Mörgelin M, Sjöbring U, Björck L. Severe streptococcal infection is associated with M protein-induced platelet activation and thrombus formation. Mol Microbiol. 2007;65:1147–57. doi: 10.1111/j.1365-2958.2007.05841.x. [DOI] [PubMed] [Google Scholar]
- 38.Macheboeuf P, Buffalo C, Fu CY, Zinkernagel AS, Cole JN, Johnson JE, Nizet V, Ghosh P. Streptococcal M1 protein constructs a pathological host fibrinogen network. Nature. 2011;472:64–8. doi: 10.1038/nature09967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herwald H, Cramer H, Mörgelin M, Russell W, Sollenberg U, Norrby-Teglund A, Flodgaard H, Lindbom L, Björck L. M protein, a classical bacterial virulence determinant, forms complexes with fibrinogen that induce vascular leakage. Cell. 2004;116:367–79. doi: 10.1016/S0092-8674(04)00057-1. [DOI] [PubMed] [Google Scholar]
- 40.Gando S, Nanzaki S, Sasaki S, Aoi K, Kemmotsu O. Activation of the extrinsic coagulation pathway in patients with severe sepsis and septic shock. Crit Care Med. 1998;26:2005–9. doi: 10.1097/00003246-199812000-00030. [DOI] [PubMed] [Google Scholar]
- 41.Gando S, Kameue T, Nanzaki S, Hayakawa T, Nakanishi Y. Participation of tissue factor and thrombin in posttraumatic systemic inflammatory syndrome. Crit Care Med. 1997;25:1820–6. doi: 10.1097/00003246-199711000-00019. [DOI] [PubMed] [Google Scholar]
- 42.Gando S, Nanzaki S, Morimoto Y, Ishitani T, Kemmotsu O. Tissue factor pathway inhibitor response does not correlate with tissue factor-induced disseminated intravascular coagulation and multiple organ dysfunction syndrome in trauma patients. Crit Care Med. 2001;29:262–6. doi: 10.1097/00003246-200102000-00006. [DOI] [PubMed] [Google Scholar]
- 43.Stief TW, Ijagha O, Weiste B, Herzum I, Renz H, Max M. Analysis of hemostasis alterations in sepsis. Blood Coagul Fibrinolysis. 2007;18:179–86. doi: 10.1097/MBC.0b013e328040bf9a. [DOI] [PubMed] [Google Scholar]
- 44.Bryant AE, Hayes-Schroer SM, Stevens DL. M type 1 and 3 group A streptococci stimulate tissue factor-mediated procoagulant activity in human monocytes and endothelial cells. Infect Immun. 2003;71:1903–10. doi: 10.1128/IAI.71.4.1903-1910.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oehmcke S, Mörgelin M, Malmström J, Linder A, Chew M, Thorlacius H, Herwald H. Stimulation of blood mononuclear cells with bacterial virulence factors leads to the release of pro-coagulant and pro-inflammatory microparticles. Cell Microbiol. 2012;14:107–19. doi: 10.1111/j.1462-5822.2011.01705.x. [DOI] [PubMed] [Google Scholar]
- 46.Egorina EM, Sovershaev MA, Hansen JB. The role of tissue factor in systemic inflammatory response syndrome. Blood Coagul Fibrinolysis. 2011;22:451–6. doi: 10.1097/MBC.0b013e328346ef3f. [DOI] [PubMed] [Google Scholar]
- 47.Leeb-Lundberg LM, Marceau F, Müller-Esterl W, Pettibone DJ, Zuraw BL. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57:27–77. doi: 10.1124/pr.57.1.2. [DOI] [PubMed] [Google Scholar]
- 48.Bengtson SH, Sandén C, Mörgelin M, Marx PF, Olin AI, Leeb-Lundberg LM, Meijers JC, Herwald H. Activation of TAFI on the surface of Streptococcus pyogenes evokes inflammatory reactions by modulating the kallikrein/kinin system. J Innate Immun. 2009;1:18–28. doi: 10.1159/000145543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ben Nasr A, Herwald H, Sjöbring U, Renné T, Müller-Esterl W, Björck L. Absorption of kininogen from human plasma by Streptococcus pyogenes is followed by the release of bradykinin. Biochem J. 1997;326:657–60. [PMC free article] [PubMed] [Google Scholar]
- 50.Sriskandan S, Kemball-Cook G, Moyes D, Canvin J, Tuddenham E, Cohen J. Contact activation in shock caused by invasive group A Streptococcus pyogenes. Crit Care Med. 2000;28:3684–91. doi: 10.1097/00003246-200011000-00025. [DOI] [PubMed] [Google Scholar]
- 51.Linder A, Johansson L, Thulin P, Hertzén E, Mörgelin M, Christensson B, Björck L, Norrby-Teglund A, Akesson P. Erysipelas caused by group A streptococcus activates the contact system and induces the release of heparin-binding protein. J Invest Dermatol. 2010;130:1365–72. doi: 10.1038/jid.2009.437. [DOI] [PubMed] [Google Scholar]
- 52.Sriskandan S, Cohen J. Kallikrein-kinin system activation in streptococcal toxic shock syndrome. Clin Infect Dis. 2000;30:961–2. doi: 10.1086/313827. [DOI] [PubMed] [Google Scholar]
- 53.Murphy K, Travers P, Walport M, Janeway C. Janeway's immunobiology. New York: Garland Science, 2008. [Google Scholar]
- 54.Proft T, Fraser JD. Bacterial superantigens. Clin Exp Immunol. 2003;133:299–306. doi: 10.1046/j.1365-2249.2003.02203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spaulding AR, Salgado-Pabón W, Kohler PL, Horswill AR, Leung DY, Schlievert PM. Staphylococcal and streptococcal superantigen exotoxins. Clin Microbiol Rev. 2013;26:422–47. doi: 10.1128/CMR.00104-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bisno AL, Stevens DL. Streptococcal infections of skin and soft tissues. N Engl J Med. 1996;334:240–5. doi: 10.1056/NEJM199601253340407. [DOI] [PubMed] [Google Scholar]
- 57.Shah SS, Hall M, Srivastava R, Subramony A, Levin JE. Intravenous immunoglobulin in children with streptococcal toxic shock syndrome. Clin Infect Dis. 2009;49:1369–76. doi: 10.1086/606048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arad G, Levy R, Nasie I, Hillman D, Rotfogel Z, Barash U, Supper E, Shpilka T, Minis A, Kaempfer R. Binding of superantigen toxins into the CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock. PLoS Biol. 2011;9:e1001149. doi: 10.1371/journal.pbio.1001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fraser JD, Proft T. The bacterial superantigen and superantigen-like proteins. Immunol Rev. 2008;225:226–43. doi: 10.1111/j.1600-065X.2008.00681.x. [DOI] [PubMed] [Google Scholar]
- 60.Li Y, Li H, Dimasi N, McCormick JK, Martin R, Schuck P, Schlievert PM, Mariuzza RA. Crystal structure of a superantigen bound to the high-affinity, zinc-dependent site on MHC class II. Immunity. 2001;14:93–104. doi: 10.1016/S1074-7613(01)00092-9. [DOI] [PubMed] [Google Scholar]
- 61.Fernández MM, Guan R, Swaminathan CP, Malchiodi EL, Mariuzza RA. Crystal structure of staphylococcal enterotoxin I (SEI) in complex with a human major histocompatibility complex class II molecule. J Biol Chem. 2006;281:25356–64. doi: 10.1074/jbc.M603969200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baker MD, Gendlina I, Collins CM, Acharya KR. Crystal structure of a dimeric form of streptococcal pyrogenic exotoxin A (SpeA1) Protein Sci. 2004;13:2285–90. doi: 10.1110/ps.04826804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roussel A, Anderson BF, Baker HM, Fraser JD, Baker EN. Crystal structure of the streptococcal superantigen SPE-C: dimerization and zinc binding suggest a novel mode of interaction with MHC class II molecules. Nat Struct Biol. 1997;4:635–43. doi: 10.1038/nsb0897-635. [DOI] [PubMed] [Google Scholar]
- 64.Tripp TJ, McCormick JK, Webb JM, Schlievert PM. The zinc-dependent major histocompatibility complex class II binding site of streptococcal pyrogenic exotoxin C is critical for maximal superantigen function and toxic activity. Infect Immun. 2003;71:1548–50. doi: 10.1128/IAI.71.3.1548-1550.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Swietnicki W, Barnie AM, Dyas BK, Ulrich RG. Zinc binding and dimerization of Streptococcus pyogenes pyrogenic exotoxin C are not essential for T-cell stimulation. J Biol Chem. 2003;278:9885–95. doi: 10.1074/jbc.M206957200. [DOI] [PubMed] [Google Scholar]
- 66.Cunningham MW. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev. 2000;13:470–511. doi: 10.1128/CMR.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Faulkner L, Cooper A, Fantino C, Altmann DM, Sriskandan S. The mechanism of superantigen-mediated toxic shock: not a simple Th1 cytokine storm. J Immunol. 2005;175:6870–7. doi: 10.4049/jimmunol.175.10.6870. [DOI] [PubMed] [Google Scholar]
- 68.Litton MJ, Sander B, Murphy E, O’Garra A, Abrams JS. Early expression of cytokines in lymph nodes after treatment in vivo with Staphylococcus enterotoxin B. J Immunol Methods. 1994;175:47–58. doi: 10.1016/0022-1759(94)90330-1. [DOI] [PubMed] [Google Scholar]
- 69.Commons R, Rogers S, Gooding T, Danchin M, Carapetis J, Robins-Browne R, Curtis N. Superantigen genes in group A streptococcal isolates and their relationship with emm types. J Med Microbiol. 2008;57:1238–46. doi: 10.1099/jmm.0.2008/001156-0. [DOI] [PubMed] [Google Scholar]
- 70.Kim YB, Watson DW. A purified group A streptococcal pyrogenic exotoxin. Physiochemical and biological properties including the enhancement of susceptibility to endotoxin lethal shock. J Exp Med. 1970;131:611–22. doi: 10.1084/jem.131.3.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hopkins PA, Fraser JD, Pridmore AC, Russell HH, Read RC, Sriskandan S. Superantigen recognition by HLA class II on monocytes up-regulates toll-like receptor 4 and enhances proinflammatory responses to endotoxin. Blood. 2005;105:3655–62. doi: 10.1182/blood-2004-07-2523. [DOI] [PubMed] [Google Scholar]
- 72.Hopkins PA, Pridmore AC, Ellmerich S, Fraser JD, Russell HH, Read RC, Sriskandan S. Increased surface toll-like receptor 2 expression in superantigen shock. Crit Care Med. 2008;36:1267–76. doi: 10.1097/CCM.0b013e31816a0a78. [DOI] [PubMed] [Google Scholar]
- 73.Gao H, Leaver SK, Burke-Gaffney A, Finney SJ. Severe sepsis and Toll-like receptors. Semin Immunopathol. 2008;30:29–40. doi: 10.1007/s00281-007-0101-4. [DOI] [PubMed] [Google Scholar]
- 74.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 75.Härter L, Mica L, Stocker R, Trentz O, Keel M. Increased expression of toll-like receptor-2 and -4 on leukocytes from patients with sepsis. Shock. 2004;22:403–9. doi: 10.1097/01.shk.0000142256.23382.5d. [DOI] [PubMed] [Google Scholar]
- 76.Armstrong L, Medford AR, Hunter KJ, Uppington KM, Millar AB. Differential expression of Toll-like receptor (TLR)-2 and TLR-4 on monocytes in human sepsis. Clin Exp Immunol. 2004;136:312–9. doi: 10.1111/j.1365-2249.2004.02433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brosnahan AJ, Schlievert PM. Gram-positive bacterial superantigen outside-in signaling causes toxic shock syndrome. FEBS J. 2011;278:4649–67. doi: 10.4049/jimmunol.0803283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brosnahan AJ, Schaefers MM, Amundson WH, Mantz MJ, Squier CA, Peterson ML, Schlievert PM. Novel toxic shock syndrome toxin-1 amino acids required for biological activity. Biochemistry. 2008;47:12995–3003. doi: 10.1021/bi801468w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peterson ML, Ault K, Kremer MJ, Klingelhutz AJ, Davis CC, Squier CA, Schlievert PM. The innate immune system is activated by stimulation of vaginal epithelial cells with Staphylococcus aureus and toxic shock syndrome toxin 1. Infect Immun. 2005;73:2164–74. doi: 10.1128/IAI.73.4.2164-2174.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee PK, Schlievert PM. Quantification and toxicity of group A streptococcal pyrogenic exotoxins in an animal model of toxic shock syndrome-like illness. J Clin Microbiol. 1989;27:1890–2. doi: 10.1128/jcm.27.8.1890-1892.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Unnikrishnan M, Altmann DM, Proft T, Wahid F, Cohen J, Fraser JD, Sriskandan S. The bacterial superantigen streptococcal mitogenic exotoxin Z is the major immunoactive agent of Streptococcus pyogenes. J Immunol. 2002;169:2561–9. doi: 10.1111/j.1574-6968.2000.tb09187.x. [DOI] [PubMed] [Google Scholar]
- 82.Kamezawa Y, Nakahara T, Nakano S, Abe Y, Nozaki-Renard J, Isono T. Streptococcal mitogenic exotoxin Z, a novel acidic superantigenic toxin produced by a T1 strain of Streptococcus pyogenes. Infect Immun. 1997;65:3828–33. doi: 10.1128/iai.65.9.3828-3833.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sriskandan S, Unnikrishnan M, Krausz T, Cohen J. Molecular analysis of the role of streptococcal pyrogenic Exotoxin A (SPEA) in invasive soft-tissue infection resulting from Streptococcus pyogenes. Mol Microbiol. 1999;33:778–90. doi: 10.1046/j.1365-2958.1999.01525.x. [DOI] [PubMed] [Google Scholar]
- 84.Broudy TB, Pancholi V, Fischetti VA. The in vitro interaction of Streptococcus pyogenes with human pharyngeal cells induces a phage-encoded extracellular DNase. Infect Immun. 2002;70:2805–11. doi: 10.1128/IAI.70.6.2805-2811.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Unnikrishnan M, Cohen J, Sriskandan S. Complementation of a speA negative Streptococcus pyogenes with speA: effects on virulence and production of streptococcal pyrogenic exotoxin A. Microb Pathog. 2001;31:109–14. doi: 10.1006/mpat.2001.0453. [DOI] [PubMed] [Google Scholar]
- 86.Sriskandan S, Moyes D, Buttery LK, Krausz T, Evans TJ, Polak J, Cohen J. Streptococcal pyrogenic exotoxin A release, distribution, and role in a murine model of fasciitis and multiorgan failure due to Streptococcus pyogenes. J Infect Dis. 1996;173:1399–407. doi: 10.1093/infdis/173.6.1399. [DOI] [PubMed] [Google Scholar]
- 87.Sriskandan S, Unnikrishnan M, Krausz T, Dewchand H, Van Noorden S, Cohen J, Altmann DM. Enhanced susceptibility to superantigen-associated streptococcal sepsis in human leukocyte antigen-DQ transgenic mice. J Infect Dis. 2001;184:166–73. doi: 10.1086/322018. [DOI] [PubMed] [Google Scholar]
- 88.Müller-Alouf H, Proft T, Zollner TM, Gerlach D, Champagne E, Desreumaux P, Fitting C, Geoffroy-Fauvet C, Alouf JE, Cavaillon JM. Pyrogenicity and cytokine-inducing properties of Streptococcus pyogenes superantigens: comparative study of streptococcal mitogenic exotoxin Z and pyrogenic exotoxin A. Infect Immun. 2001;69:4141–5. doi: 10.1128/IAI.69.6.4141-4145.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lintges M, van der Linden M, Hilgers RD, Arlt S, Al-Lahham A, Reinert RR, Plücken S, Rink L. Superantigen genes are more important than the emm type for the invasiveness of group A Streptococcus infection. J Infect Dis. 2010;202:20–8. doi: 10.1086/653082. [DOI] [PubMed] [Google Scholar]
- 90.Proft T, Moffatt SL, Weller KD, Paterson A, Martin D, Fraser JD. The streptococcal superantigen SMEZ exhibits wide allelic variation, mosaic structure, and significant antigenic variation. J Exp Med. 2000;191:1765–76. doi: 10.1084/jem.191.10.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Turner CE, Sommerlad M, McGregor K, Davies FJ, Pichon B, Chong DL, Farzaneh L, Holden MT, Spratt BG, Efstratiou A, et al. Superantigenic activity of emm3 Streptococcus pyogenes is abrogated by a conserved, naturally occurring smeZ mutation. PLoS One. 2012;7:e46376. doi: 10.1371/journal.pone.0046376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Michie C, Scott A, Cheesbrough J, Beverley P, Pasvol G. Streptococcal toxic shock-like syndrome: evidence of superantigen activity and its effects on T lymphocyte subsets in vivo. Clin Exp Immunol. 1994;98:140–4. doi: 10.1111/j.1365-2249.1994.tb06620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Watanabe-Ohnishi R, Low DE, McGeer A, Stevens DL, Schlievert PM, Newton D, Schwartz B, Kreiswirth B, Kotb M, Ontario Streptococcal Study Project Selective depletion of V β-bearing T cells in patients with severe invasive group A streptococcal infections and streptococcal toxic shock syndrome. J Infect Dis. 1995;171:74–84. doi: 10.1093/infdis/171.1.74. [DOI] [PubMed] [Google Scholar]
- 94.Sriskandan S, Moyes D, Cohen J. Detection of circulating bacterial superantigen and lymphotoxin-alpha in patients with streptococcal toxic-shock syndrome. Lancet. 1996;348:1315–6. doi: 10.1016/S0140-6736(05)65800-X. [DOI] [PubMed] [Google Scholar]
- 95.Norrby-Teglund A, Thulin P, Gan BS, Kotb M, McGeer A, Andersson J, Low DE. Evidence for superantigen involvement in severe group a streptococcal tissue infections. J Infect Dis. 2001;184:853–60. doi: 10.1086/323443. [DOI] [PubMed] [Google Scholar]
- 96.Proft T, Sriskandan S, Yang L, Fraser JD. Superantigens and streptococcal toxic shock syndrome. Emerg Infect Dis. 2003;9:1211–8. doi: 10.3201/eid0910.030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fast DJ, Schlievert PM, Nelson RD. Nonpurulent response to toxic shock syndrome toxin 1-producing Staphylococcus aureus. Relationship to toxin-stimulated production of tumor necrosis factor. J Immunol. 1988;140:949–53. [PubMed] [Google Scholar]
- 98.Cockerill FR, 3rd, Thompson RL, Musser JM, Schlievert PM, Talbot J, Holley KE, Harmsen WS, Ilstrup DM, Kohner PC, Kim MH, et al. Southeastern Minnesota Streptococcal Working Group Molecular, serological, and clinical features of 16 consecutive cases of invasive streptococcal disease. Clin Infect Dis. 1998;26:1448–58. doi: 10.1086/516376. [DOI] [PubMed] [Google Scholar]
- 99.Davis JP, Chesney PJ, Wand PJ, LaVenture M. Toxic-shock syndrome: epidemiologic features, recurrence, risk factors, and prevention. N Engl J Med. 1980;303:1429–35. doi: 10.1056/NEJM198012183032501. [DOI] [PubMed] [Google Scholar]
- 100.Ji YD, McLandsborough L, Kondagunta A, Cleary PP. C5a peptidase alters clearance and trafficking of group A streptococci by infected mice. Infect Immun. 1996;64:503–10. doi: 10.1128/iai.64.2.503-510.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zinkernagel AS, Timmer AM, Pence MA, Locke JB, Buchanan JT, Turner CE, Mishalian I, Sriskandan S, Hanski E, Nizet V. The IL-8 protease SpyCEP/ScpC of group A Streptococcus promotes resistance to neutrophil killing. Cell Host Microbe. 2008;4:170–8. doi: 10.1016/j.chom.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Neumann B, Engelhardt B, Wagner H, Holzmann B. Induction of acute inflammatory lung injury by staphylococcal enterotoxin B. J Immunol. 1997;158:1862–71. [PubMed] [Google Scholar]
- 103.Diener K, Tessier P, Fraser J, Köntgen F, McColl SR. Induction of acute inflammation in vivo by staphylococcal superantigens I: Leukocyte recruitment occurs independently of T lymphocytes and major histocompatibility complex Class II molecules. Lab Invest. 1998;78:647–56. [PubMed] [Google Scholar]
- 104.Tessier PA, Naccache PH, Diener KR, Gladue RP, Neote KS, Clark-Lewis I, McColl SR. Induction of acute inflammation in vivo by staphylococcal superantigens. II. Critical role for chemokines, ICAM-1, and TNF-alpha. J Immunol. 1998;161:1204–11. [PubMed] [Google Scholar]
- 105.Sriskandan S, Ferguson M, Elliot V, Faulkner L, Cohen J. Human intravenous immunoglobulin for experimental streptococcal toxic shock: bacterial clearance and modulation of inflammation. J Antimicrob Chemother. 2006;58:117–24. doi: 10.1093/jac/dkl173. [DOI] [PubMed] [Google Scholar]
- 106.Norrby-Teglund A, Nepom GT, Kotb M. Differential presentation of group A streptococcal superantigens by HLA class II DQ and DR alleles. Eur J Immunol. 2002;32:2570–7. doi: 10.1002/1521-4141(200209)32:9<2570::AID-IMMU2570>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 107.Kotb M, Norrby-Teglund A, McGeer A, El-Sherbini H, Dorak MT, Khurshid A, Green K, Peeples J, Wade J, Thomson G, et al. An immunogenetic and molecular basis for differences in outcomes of invasive group A streptococcal infections. Nat Med. 2002;8:1398–404. doi: 10.1038/nm1202-800. [DOI] [PubMed] [Google Scholar]
- 108.Llewelyn M, Sriskandan S, Peakman M, Ambrozak DR, Douek DC, Kwok WW, Cohen J, Altmann DM. HLA class II polymorphisms determine responses to bacterial superantigens. J Immunol. 2004;172:1719–26. doi: 10.4049/jimmunol.172.3.1719. [DOI] [PubMed] [Google Scholar]
- 109.Llewelyn M. Human leukocyte antigen class II haplotypes that protect against or predispose to streptococcal toxic shock. Clin Infect Dis. 2005;41(Suppl 7):S445–8. doi: 10.1086/431986. [DOI] [PubMed] [Google Scholar]
- 110.Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Chi YI, Stauffacher C, Strominger JL, Wiley DC. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature. 1994;368:711–8. doi: 10.1038/368711a0. [DOI] [PubMed] [Google Scholar]
- 111.Sundberg E, Jardetzky TS. Structural basis for HLA-DQ binding by the streptococcal superantigen SSA. Nat Struct Biol. 1999;6:123–9. doi: 10.1038/5809. [DOI] [PubMed] [Google Scholar]
- 112.Nooh MM, Nookala S, Kansal R, Kotb M. Individual genetic variations directly effect polarization of cytokine responses to superantigens associated with streptococcal sepsis: implications for customized patient care. J Immunol. 2011;186:3156–63. doi: 10.4049/jimmunol.1002057. [DOI] [PubMed] [Google Scholar]
- 113.Faulkner L, Altmann DM, Ellmerich S, Huhtaniemi I, Stamp G, Sriskandan S. Sexual dimorphism in superantigen shock involves elevated TNF-alpha and TNF-alpha induced hepatic apoptosis. Am J Respir Crit Care Med. 2007;176:473–82. doi: 10.1164/rccm.200611-1712OC. [DOI] [PubMed] [Google Scholar]
- 114.Norrby-Teglund A, Kaul R, Low DE, McGeer A, Newton DW, Andersson J, Andersson U, Kotb M. Plasma from patients with severe invasive group A streptococcal infections treated with normal polyspecific IgG inhibits streptococcal superantigen-induced T cell proliferation and cytokine production. J Immunol. 1996;156:3057–64. [PubMed] [Google Scholar]
- 115.Lamagni TL, Neal S, Keshishian C, Powell D, Potz N, Pebody R, George R, Duckworth G, Vuopio-Varkila J, Efstratiou A. Predictors of death after severe Streptococcus pyogenes infection. Emerg Infect Dis. 2009;15:1304–7. doi: 10.3201/eid1508.090264. [DOI] [PubMed] [Google Scholar]