

Abstract
Sepsis remains the leading cause of death in most intensive care units. Advances in understanding the immune response to sepsis provide the opportunity to develop more effective therapies. The immune response in sepsis can be characterized by a cytokine-mediated hyper-inflammatory phase, which most patients survive, and a subsequent immune-suppressive phase. Patients fail to eradicate invading pathogens and are susceptible to opportunistic organisms in the hypo-inflammatory phase. Many mechanisms are responsible for sepsis-induced immuno-suppression, including apoptotic depletion of immune cells, increased T regulatory and myeloid-derived suppressor cells, and cellular exhaustion. Currently in clinical trial for sepsis are granulocyte macrophage colony stimulating factor and interferon gamma, immune-therapeutic agents that boost patient immunity. Immuno-adjuvants with promise in clinically relevant animal models of sepsis include anti-programmed cell death-1 and interleukin-7. The future of immune therapy in sepsis will necessitate identification of the immunologic phase using clinical and laboratory parameters as well as biomarkers of innate and adaptive immunity.
Keywords: sepsis, immune therapy, cell exhaustion, immune suppression, adaptive immunity
Introduction
Sepsis is the major cause of death in most intensive care units in the United States with approximately a quarter of a million deaths annually. Although new treatment algorithms focusing on rapid administration of broad spectrum antibiotics and aggressive restoration of tissue oxygen delivery have led to decreases in mortality, the death rate is still 30%.1,2 Microbial virulence factors, the extent of bacterial tissue invasion, and patient co-morbidities interact to drive the host response that includes a wide array of manifestations including septic shock, adult respiratory distress syndrome (ARDS), multiple organ dysfunction syndrome (MODS), and systemic inflammatory response syndrome (SIRS).3,4 The heterogeneity of the disease encompasses a diverse interplay between immunological stimulation, systemic inflammation, and coagulopathy which vary in degree from patient to patient.5 This heterogeneity in patient response to sepsis is in part responsible for the over 30 failed clinical drug trials; beyond supportive care, there is presently no specific Food and Drug Administration-approved drug for the treatment of sepsis in humans.6 In this review we will focus on (1) the role of the adaptive immune system and detail its contribution to the host response in sepsis and (2) outline novel potential targets for individualized immune therapy in these patients.
Pro-Inflammatory Response: Cytokine Storm
Septic patients frequently present with fever, shock, and respiratory failure due to an uncontrolled pro-inflammatory response that has been termed SIRS.7 This initial immune recognition response is mediated by pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) originating from bacterial or fungal organisms and the host upon injury, respectively, that bind pattern recognition receptors expressed on innate immune cells.3,4,8 The activation of pattern recognition receptors results in the production of numerous pro-inflammatory molecules including TNF-α, IL-1β, IL-2, IL-6, IL-8, and IFN-γ and anti-inflammatory cytokines that induce a panoply of cellular responses and counter-responses. These responses include but are not limited to enhanced phagocytic activity, vascular endothelial injury with capillary leak, synthesis of acute phase proteins by the liver, chemotaxis of leukocytes to sites of infection/inflammation, and activation of the coagulation system.3,4,8,9
The coagulation system is closely linked to inflammation predominately through the innate immune response. In sepsis, inflammation is accompanied by coagulation activation, thrombin generation, and disseminated intravascular coagulation (DIC).10 The primary coagulation pathway activated in sepsis is the tissue factor pathway which results in upregulation on monocyte/macrophage membranes and damage to the endothelium.10 The intrinsic or contact factor pathway amplifies clotting in an autoactivation manner leading to widespread vasodilation and generation of bradykinin.10 Ultimately, the rapid consumption of coagulation factors leads to diffuse hemorrhaging in sepsis.10 Protease-activated receptors on activated endothelial cells, neutrophils, and monocytes bind to thrombin, factor Xa, and tissue factor:VIIa which increases the synthesis of pro-inflammatory mediators IL-6, IL-8, and adhesion molecules.10 This adhesion molecule expression on the vasculature recruits activated leukocytes, in particular activated neutrophils which produce lytic enzymes, reactive oxygen species, and nitrogen intermediates that contribute to microcirculatory and organ failure.10
Initially the pro-inflammatory response was believed to be the major cause of mortality in sepsis and was heavily targeted for therapeutic intervention.11 Clinical trials included, for example, TNF and IL-1β antagonists, toll-like receptor (TLR) blockers, platelet activating factor inhibitors (Xigris12-14), anti-coagulants, endotoxin antagonists, hemofiltration to remove soluble endotoxin and cytokines, and blocking super-antigens which ultimately showed no benefit or, in some cases, worsened outcome.11,15-17
Hypo-Inflammatory Response: Immuno-Paralysis
As patients survived the initial hyper-inflammatory, cytokine storm phase of sepsis, it became apparent that many septic patients developed a delayed and potentially prolonged counter-regulatory, anti-inflammatory state. This was initially referred to as a compensatory anti-inflammatory response syndrome (CARS).18-21 After considerable debate, a consensus developed that, sepsis can evolve into two phases: the first being hyper-inflammation (cytokine storm) and the second being hypo-inflammation (immune-paralysis)15,22 (Fig. 1). Although inflammation in sepsis is presented as a biphasic view in this review, genome-wide transcription profiling in human sepsis has indicated that mechanisms of pro- as well as anti-inflammatory mechanisms occur during variable times over the course of sepsis.23,24 However for the purpose of this review, the biphasic model explains the net effect on the inflammatory response, including both innate and adaptive immune function, where patients may cycle through each phase multiple times over the course of sepsis.15 This biphasic view maybe a simplistic explanation of a complex disease, yet provides a rational explanation for how the function of the immune system becomes altered during the course of sepsis. The intensity of the initial hyper-inflammatory phase varies depending upon a multitude of factors including the patient’s underlying physical state and co-morbidities, pathogen virulence factors, pathogen load, and genetic factors. Following this, perhaps in an effort to dampen systemic inflammation, subsequent immunosuppression may develop. As with the uncontrolled initial inflammation, this may overshoot and result in immune dysfunction that leads to increased host susceptibility to secondary bacterial infections (ventilator-associated pneumonia [VAP]),25 infections with typically avirulent or opportunistic organisms,26,27 reactivation of latent herpes viruses (cytomegalovirus [CMV]),28-30 increased risk of MODS,31 and loss of delayed type hypersensitivity response to common recall antigens.32 Research by many groups has shown that hypo-inflammation is due to a variety of immune defects including a dysfunctional adaptive immune response.15,33-40
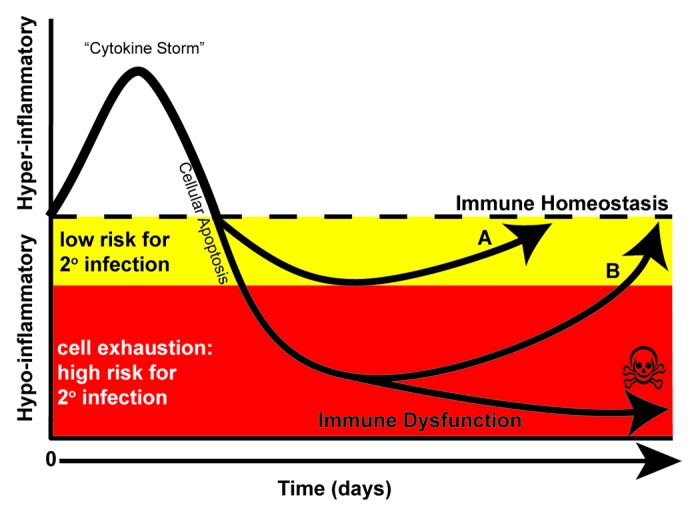
Figure 1. Immune response in sepsis. The immune response in sepsis is determined by many factors including co-morbidities (i.e., diabetes, heart disease, malignancy) as well as the pathogen virulence and size of the microbial inoculums. Although both pro- and anti-inflammatory processes are activated simultaneously during the onset of sepsis, during the first few days, a hyper-inflammatory response often dominates the clinical picture. The hyper-inflammatory phase has been termed a “cytokine storm” that is indicated by increased levels of TNF-α, IL-1β, and IL-6. A robust depletion of both innate and adaptive immune cells through apoptosis occurs to dampen the response. (A) At this stage, patients may undergo a controlled anti-inflammatory response enabling them to return to immune homeostasis. Alternatively, patients may undergo an uncontrolled anti-inflammatory response and enter a hypo-inflammatory phase yet survive (B) or succumb. Protracted time spent in this hypo-inflammatory phase may lead to cellular exhaustion; a cellular phenotype indicated by impaired function as well as increased PD-1 and decreased IL-7R expression on T lymphocytes. In this phase, patients fail to mount proper immune responses leading to viral re-activation and secondary infections, frequently caused by avirulent and opportunistic organisms and of ventilator-associated pneumonia.
The adaptive immune system consists of specialized cells that are antigen specific and critical for generating memory and recall responses to antigens. Antigen-specific T cells are necessary for driving specific responses against intracellular (T-helper 1 [Th1]) or extracellular pathogens (T-helper 2 [Th2]) by cell-to-cell contact mechanisms or through soluble mediators (cytokines). CD4 T cells are typically classified as helper T cells and control cells of the adaptive system. CD8 T cells are classified as cytotoxic (CTL) and kill targeted cells such as virally infected or tumor cells. Antibody production, termed humoral immunity, by B cells requires T-cell help. Antigen-specific antibody can neutralize toxins, fix complement, and coat the surface for phagocytic uptake of pathogens by monocytes/macrophages. Antigen-presenting cells, including monocyte/macrophages and dendritic cells, sample the environment and present antigens to T cells for initiation of immune responses or induction of tolerance of the adaptive immune response. The effects of sepsis on the adaptive immune response and the potential therapeutic advantage of targeting these components during the pathogenesis of sepsis will be the focus of the following sections.
Adaptive Immunity: Apoptosis of Immune Effector Cells
Programmed cell death, also called apoptosis, is one way in which the immune system maintains homeostasis by eliminating activated cells. Central to apoptosis are caspases which are cysteine proteases that degrade cellular proteins and NFκB, a transcription factor which will activate transcription of both pro-apoptotic and pro-survival genes. While the hyper-inflammatory response of sepsis requires NFκB for production of pro-inflammatory cytokines and IL-1β activation by caspase cleavage, both NFκB and caspases concurrently induce apoptosis of adaptive immune cells.8,41,42 Consistent with this, a concurrent apoptotic response has been shown to be present in sepsis in association with the pro-inflammatory response. Our group has shown both in mouse models of sepsis43,44 as well as most recently in patients with severe sepsis that there is a profound depletion of T, B, and dendritic cells.45-48 Within the first 24 h of sepsis diagnosis in humans, marked lymphopenia occurs which is due to recruitment of lymphocytes from the circulation to sites of inflammation/infection and to apoptotic depletion of CD4 and CD8 T cells in the blood.45 Postmortem analysis of spleens and lymph nodes from individuals who succumbed to sepsis confirmed the highly significant loss of CD4 and CD8 T cells.46 Memory CD8 T cells are highly susceptible to apoptosis in systemic inflammatory states such as septic shock and are depleted in sepsis.49 These results indicate that depletion of adaptive immune cells is a major pathologic component of sepsis with potential debilitating effects on host immunity.
Although the depletion of adaptive immune cells is recognized as an important part of the pathology of sepsis, the mechanisms responsible for this are not totally understood.50,51 In general apoptosis is divided into two mechanistic pathways, i.e., an extrinsic pathway that conveys a signal through a death receptor such as the tumor-necrosis family of receptors (i.e., CD95/CD95L, TNF/TNFR) or an intrinsic pathway that disrupts mitochondrial integrity resulting in release of cytochrome C.52,53 The activation of caspase 8, through death domains, and/or the activation of caspase 9, by the release of cytochrome C, results in the cleavage and activation of caspase 3 an effector caspase that induces extensive substrate cleavage of many critical proteins ultimately leading to cell death.52,53 These pathways of apoptosis have multiple points of crosstalk.54 Bid, a member of the Bcl-2 family of proteins, is cleaved by activated caspase-8, an effector of the extrinsic pathway, to form truncated Bid.54 This truncated Bid translocates to the mitochondrion where it binds Bax/Bak, members of the Bcl-2 family associated with the intrinsic pathway, to further induce apoptosis.54 Heat-shock proteins can either be constitutively expressed or rapidly induced in response to stress, like inflammation, yet modulate both the mitorchondrial and receptor mediated apoptotic pathways.54 Adaptor molecules like receptor interacting protein (RIP) or members of the TNF receptor-associated factor family (TRAF) activate transcription factor families associated with both survival and apoptosis including NFκB and mitogen-activated protein kinase (MAPK) pathways.54 To add to this complexity, a wide range of protein modifications and interactions form diverse yet interconnected signaling cascades that regulate signaling pathways such as PI3K-Akt/PKB, Ras-Raf-Mek-Erk MAPK, NFκB, and protein kinase C that are pro-survival while JNK/p38 stress MAPK or ROS/ceramide-mediate signaling which are pro-apoptotic.54
As noted earlier, CD4 and CD8 T cells are highly susceptible to sepsis-induced apoptosis. To test the premise that soluble factors induce lymphocyte apoptosis in sepsis, serum from patients with SIRS was added to cultures of lymphocytes from healthy volunteers. Apoptosis of CD4 T cells was observed when plasma from SIRS patients was added but apoptosis was not present when serum from healthy donors was used.55 Although this soluble factor has yet to be identified, it is likely that a multitude of soluble factors are involved in modulating lymphocyte apoptosis. Some gram-positive bacteria have a unique property, an exotoxin termed a superantigen that activates CD4 T cells through a tetrad signaling complex consisting of a correctly folded superantigen, the CD28 dimer interface, Vβ loop of the TCR, and a binding site outside the epitope presenting groove on MHC class II molecules.56 This interaction occurs independent of MHC-Class II molecules and results in rapid activation, secretion of pro-inflammatory cytokines and ultimately cell death.8 In polymicrobial sepsis the activation of the peroxisome proliferator activated receptor (PPARγ) leads to decreased IL-2 gene expression, a T-cell survival factor that induces expression of the potently anti-apoptotic protein Bcl-2.57 The massive release of TNF-α early in sepsis results in activation of additional pro-inflammatory cytokines and chemokines which act in conjunction with TNF-α to induce T-cell apoptosis.58 In mouse models of sepsis, the complement factor C5a binds to the receptor C5aR on thymocytes and induces apoptosis.59 Therefore, in sepsis a diversity of soluble factors are present that potentially regulate apoptosis and add to the heterogeneity of sepsis pathology.
Lymphocyte fate is determined by the balance of pro- vs anti-apoptotic mechanisms. In our postmortem study of sepsis, we documented decreased expression of CD28 on T lymphocytes on septic patients when compared with control patients.46 The co-stimulatory molecule CD28, when engaged by B7 molecules on antigen presenting cells, is an essential requirement for IL-2 production and Bcl-XL expression, both pro-survival factors of T cells.60 Antigen-presenting cells, including dendritic cells and monocytes/marcrophages, isolated from either the spleen or the lung of septic patients also had decreased expression of B7 molecules (B7-1/CD80 and B7-2/CD86) potentially limiting the ability of T cells to receive co-stimulation.46 Without the necessary co-stimulation through CD28/B7 when the T-cell receptor engages antigen, T cells will undergo a process termed death by neglect or become functionally unresponsive (anergic).
Our recent studies have also indicated that T cells upregulate surface receptors that inhibit T-cell functions. We identified increased expression of programmed death receptor-1 (PD-1), CTLA-4, and B and T lymphocyte attenuator (BTLA) on T cells when isolated from post-mortem spleen and lung as well as from blood 7 days after initial diagnosis with sepsis.45,46 Furthermore, we detected increased expression of the ligands for PD-1, programmed death ligand-1 and -2 (PD-L1 and PD-L2), on antigen-presenting cells isolated from the blood after 7 days of initial septic diagnosis and from lymphocytes isolated from postmortem spleen and lung.45,46 The expression of PD-L and herpesvirus entry mediator (HVEM), the ligand for BTLA, was observed on the epithelium and on macrophages of the lung in septic patients.45,46 The expression of PD-L on antigen presenting cells and endothelial cells has been associated with the induction of tolerance,61,62 providing a mechanism where the tissue regulates lymphocyte homeostasis. The engagement of PD-1, CTLA-4, or BTLA, all members of the CD28-superfamily of receptors, results in T-cell apoptosis or cellular unresponsiveness that aid in returning the host to immune homeostasis.63 PD-L and CTLA-4 as well as TNFR ligand family members such as Fas-Ligand (CD95L) are expressed on a specialized type of CD4 T cell that expresses the transcription factor FoxP3 and high surface levels of CD25 (IL-2Rα) termed regulatory T cells (Tregs).64 Regulatory T cells are capable of suppressing T-cell proliferation, cytokine secretion and induce T-cell apoptosis.65 These cells are increased in percentage in septic patients due to their resistance to apoptosis and a decrease in CD4+CD25− T cells35,36,45,66,67 provides yet another potential mechanism of immune dysfunction. The expression of these inhibitory receptors/ligands on T cells, antigen presenting cells and tissue presents an environment that is conducive to T-cell depletion and unresponsiveness in septic patients.
Adaptive Immunity: Cellular Hypo-Responsiveness
The anti-inflammatory cytokine IL-10 has been detected in serum of septic patients very early during their illness.68 A high ratio of IL-10 to TNF-α in septic patients correlated with mortality in patients with community acquired infection.68,69 The anti-inflammatory cytokine IL-10 is produced by Tregs and Th2 type cells and suppresses the Th1 (CD8 T cell) response further potentiating an anti-inflammatory environment.70,71 This suppressive environment results in a marked decrease in stimulated monocyte production of pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 (typically less than 10–20% that of healthy controls).72-76 This compensatory anti-inflammatory response syndrome, or immune-paralysis as it was originally referred to, is mediated by a predominance of a Th2 response, increased Tregs, apoptosis of lymphocytes and decreased MHC class II (HLA-DR) molecules on monocytes/macrophages.10,18 Our group reported that immune effector cells isolated from postmortem spleens with patients dying of sepsis and peripheral blood mononuclear cells isolated from blood of septic individuals 7 days after onset of sepsis have impaired secretion of both pro- and anti-inflammatory cytokines (TNF-α, IFN-γ, IL-6, IL-10).45,46 Furthermore, this cytokine secretion defect in septic patients was present after stimulation with lipopolysaccharide (a monocyte/macrophage activator) and after stimulation with anti-CD3/anti-CD28 (a T-cell activator) indicating that both the innate and adaptive immune systems are suppressed.45,46
Adaptive Immunity: Cellular Exhaustion
Both the postmortem and prospective blood study, previously discussed, demonstrated that T lymphocytes from septic patients expressed inhibitory receptors and that these T cells were unable to produce cytokines after ex vivo stimulation.45,46,77 In the postmortem study, T cells isolated from the spleen had increased expression of CD69, PD-1, and CD25 and decreased expression of the IL-7 receptor (IL-7R, CD127) and CD28 when compared with age-matched controls.46 T cells isolated from the lung had increased expression of PD-1 and BTLA indicating a degree of tissue specificity in sepsis.46 Furthermore, increased expression of inhibitory receptors, PD-1, BTLA, CTLA-4, T cell membrane protein-3 (TIM-3), lymphocyte-activation gene-3 (LAG-3), and decreased expression of the IL-7R on T cells isolated from the blood of septic patients occurs over the course of sepsis.45 This expression of inhibitory receptors and decreased cytokine secretion resemble a recently described phenotype observed in chronic viral infections like HIV and hepatitis C that is termed cellular exhaustion.
T-cell exhaustion is a step-wise progressive loss of T-cell functions in the presence of high antigen load that can result in T-cell deletion.78 Originally described in the lymphocytic choriomeningitis virus (LCMV) mouse model, T-cell exhaustion has also been documented in HIV, hepatitis C, hepatitis B, polyoma virus, adenovirus, Friend leukemia virus, and in malignancies.78 Exhausted T cells have high expression of CD69, CD43 (1B11), PD-1, TIM-3, and LAG-3, low expression levels of CD62L and CD127 (IL-7R), increased expression of the transcription factor BLIMP-1 (CD8 T cells) and decreased secretion of TNF-α, IL-2, IFN-γ, and Granzyme B.78 The engagement of PD-1 by its ligands on exhausted T cells results in IL-10 production, functional unresponsiveness and/or apoptosis.78 In sepsis, there is high antigen load due to chronic infection, T cells acquire expression of inhibitory receptors over time, as well as the expression of inhibitory ligands on antigen presenting cells and in a tissue-specific manner, provide a unique environment for T-cell exhaustion as an additional mechanism of immune suppression in sepsis.45,46 In support of this concept of sepsis-induced T-cell exhaustion, tissue expression of PD-L and HVEM the ligands for PD-1 and BTLA on T cells was present in the lungs of septic individuals but not in lungs of transplant recipients or age-matched non-septic control patients.46 These findings are particularly relevant given the high incidence of ventilator associated pneumonia which is the most common nosocomial infection in the intensive-care unit.79-81 The increased susceptibility of the lung to secondary infection in septic patients may be due in part to expression of inhibitory ligands on lung parenchyma resulting in local T-cell exhaustion.
Immuno-Therapy: Targeting the Adaptive Immune System
Despite active research in understanding human sepsis, the number of registered human clinical trials is disappointing, with most observational in nature and few being interventional (Table 1). Many septic patients have a relatively short-lived hyper-inflammatory phase; therefore, the success of drugs targeting inflammation has only a narrow time frame to be effective. The keys to drugs targeting this hyper-inflammation are: drugs should be short lived, applied early and used only in patients with elevated levels of pro-inflammatory cytokines (i.e., TNF-α, IL-1β, or IL-6).3,4 Phase I–III clinical trials using drugs to reduce this pro-inflammatory cascade have had disappointing results in 28-day mortality rates in septic patients when compared with placebo-treated controls.14,17,82-90 Most septic patients residing in ICUs have survived the hyper-inflammatory stage of the disease yet have prolonged periods of hospitalization and recovery with the potential to become immune compromised and develop nosocomial infections and/or multiorgan dysfunction.34,91,92 Therefore, one of the keys to decreasing mortality in sepsis may be the development of therapies to augment host immune responses. Collectively, results of studies designed to block this hyper-inflammatory phase suggest partial benefit in early onset sepsis in highly-defined populations of septic patients yet are potentially detrimental to septic patients who have progressed into immune-paralysis.15,33
Table 1. Immune enhancing therapy: clinical trials in sepsis.
| Agent | Study | Outcomes | References |
|---|---|---|---|
| G-CSF | RCT in patients with pneumonia and severe sepsis | Increased WBC counts No reduction in mortality Well tolerated |
Root et al.103 |
| G-CSF | RCT in patients with multilobar pneumonia | Increased WBC counts Reduction in mortality (trend) Well tolerated |
Nelson et al.101 |
| GM-CSF | RCT in patients with sepsis or septic shock and sepsis induced immunesuppression | Increased HLA-DR expression Restored cytokine secretion in monocytes Improved patient outcomes |
Meisel et al.105 |
| GM-CSF, rIFN-γ (ongoing) | Effects of immunostimulation with GM-CSF or IFN-γ on immunoparalysis following human endotoxemia | Cytokine secretion by lymphocytes HLA-DR expression Monocyte/neutrophil function Lymphocyte gene expression Volunteer responses |
NCT01374711 |
| rIFN-γ | RCT in trauma patients | Increased HLA-DR expression Decreased severe infections (trend) |
Polk et al.106 |
| rIFN-γ | RCT in patients with burns | No improved patient outcomes | Wasserman et al.107 |
| rIFN-γ | RCT in trauma patients | Reduced infection related deaths | Dries et al.108 |
| rIFN-γ (ongoing) | Effects of interferon-gamma on sepsis-induced immunoparalysis | Cytokine secretion by lymphocytes HLA-DR and receptor expression (PD-1) Lymphocyte gene expression Reversibility of monocyte dysfunction Patient outcomes |
NCT01649921 |
Thus far, the focus of enhancing immune function in sepsis has been limited to trials for GM-CSF or G-CSF, a stimulator of the innate immune system, and IFN-γ, a stimulator of the adaptive immune system. Abbreviations: RCT, randomized controlled trial; GM-CSF, granulocyte-macrophage colony-stimulating factor; G-CSF, granulocyte colony-stimulating factor; rIFN-γ, recombinant interferon gamma; PD-1, programmed death receptor-1; WBC, white blood cell count; NCT, ClinicalTrials.gov identifier.
Even before the molecular pathophysiology of sepsis was thoroughly understood, investigators attempted to stimulate the innate and adaptive immune systems with IFN-γ, granulocyte macrophage colony stimulating factor (GM-CSF) or granulocyte colony stimulating factor G-CSF. In an attempt to reverse monocyte/macrophage dysfunction and increase IL-17, a cytokine necessary for the recruitment of neutrophils to the infected site, septic patients were treated with IFN-γ.93 Treatment with GM-CSF was also predicted to reverse the dysfunction of dendritic cells and macrophages/monocytes.94 G-CSF was utilized to increase numbers of polymorphonuclear leukocytes, in an effort to enhance pathogen clearance. Although a recent meta-analysis performed on GM-CSF and G-CSF studies conducted from 1998 to 2011 failed to show survival benefit,95 some studies did show limited efficacy on research endpoints.96-105 In summary, GM-CSF or G-CSF treatment increased neutrophil phagocytosis, resolved infections earlier, decreased secretion of toxic metabolites, increased expression of CD11b and HLA-DR on neutrophils/monocytes, and reversed immune-paralysis in selected septic patients.3,4 Two recent small phase 2 trials of GM-CSF that have targeted its use to septic patients with low monocyte HLA-DR expression have shown benefit. Mechanically-ventilated patients with sepsis that received GM-CSF had improved health evaluation scores, shortened time of mechanical ventilation, and an accompanied increased HLA-DR expression and in secretion of pro-inflammatory cytokines (IL-6 and TNF-α) ex vivo as compared with controls.105 At least 2 clinical trials of GM-CSF are currently enrolling patients in sepsis (NCT01374711 and NCT01653665, Table 2), so it will be highly interesting to observe these results when they become available.
Table 2. Clinical trials for the immune system in sepsis.
| Identifier | Title | Study type | Sponsor |
|---|---|---|---|
| NCT01600989 | Mitochondrial Function of Immune Cells in Sepsis (MitoSepsis) (2012) | Observational | University Hospital Inselspital |
| NCT01530932 | Immune Activation, Hypoxia and Vasoreaction in Sepsis of Pulmonary Vs. Abdominal Origin (2011) | Observational | University Hospital Mannheim |
| NCT00187824 | Regulation of Endocrine, Metabolic, Immune and Bioenergetic Responses in Sepsis (2005) | Observational | University College London Hospital |
| NCT01410526 | Assessment of Peritoneal Immune Response in Patients with Severe Intra-abdominal Sepsis Managed with Laparostomy and Vacuum Assisted Closure (VAC) (2011) | Observational | Aristotle University of Thessaloniki |
| NCT01472952 | System-level Monitoring of Immune Activation Concerning Susceptibility to Sepsis in Trauma Patients (2011) | Observational | University Hospital Mannheim |
| NCT01155674 | Innate Immune Functions of Immature Neutrophils (2010) | Observational | University Hospital Geneva |
| NCT01766414 | In Vivo Effects of C1-esteraase Inhibitor on the Innate Immune Response During Human Endotoxemia – VECTOR II (2013) | Interventional | Radboud University |
| NCT01649921 | The Effects of Interferon-gamma on Sepsis-induced Immunoparalysis (2012) | Interventional | Radboud University |
| NCT00294697 | Genetic Variation and Immune Response After Injury (2006) | Observational | National Institute of General Medical Sciences |
| NCT00638521 | Immune-cell Membrane Trafficking (2008) | Observational | University of Washington |
| NCT01099813 | Sepsis Pathophysiological and Organisational Timing (SPOT[Light]) (2010) | Observational | Intensive Care National Audit and Research Centre |
| NCT01756755 | Endotoxin Adsorber Hemoperfusion and Microcirculation (2012) | Interventional | National Taiwan University Hospital |
| NCT01275976 | Effect of C1-esterase Inhibitor on Systemic Inflammation in Trauma Patients with a Femur Fracture (CAESAR) (2011) | Interventional | UMC Utrecht |
| NCT01005589 | CD64 Meaurement in Neonatal Infection and Necrotising Enterocolitis (2009) | Observational | Newcastle-upon-Tyne Hospitals NHS Trust |
| NCT00527384 | Biomarker Analysis of Stress (2007) | Observational | National Institute of Environmental Health Sciences |
| NCT01397058 | Reactivation of CMV Infection in Immunocompetent Patients Under Severe Stress (RECYSTRESS) (2011) | Observational | University of Athens |
| NCT01374711 | Effects of Immunostimulation with GM-CSF or IFN-γ on Immunoparalysis Following Human Endotoxemia (2011) | Interventional | Radboud University |
| NCT01653665 | Does GM-CSF Restore Neutrophil Phagocytosis in Critical Illness? (2012) | Interventional | Newcastle-upon-Tyne Hospitals NHS Trust |
A search on ClinicalTrials.gov was performed using the search terms “Sepsis and Immune” (48 studies) or “Sepsis and Biomarkers” (74 studies) filtered by “open studies”. This represents a list of the current open and enrolling clinical trials for sepsis in regards to the immune system. Interestingly, most clinical trials in sepsis have been initiated within the past 10 years as indicated by the start date of the clinical trial in parenthesis following the trial title.
Thus far, the only attempt to modulate the adaptive immune response in a sepsis clinical trial was the administration of IFN-γ (Table 2). Treatment with IFN-γ in critical illness has yielded conflicting results. In two multi-center trials with patients of trauma and burns, treatment with IFN-γ had no effect when compared with placebo.106,107 However, in an additional trial in burn patients, IFN-γ treatment reduced infection-related deaths when compared with placebo.108 Interestingly, when IFN-γ was co-administered with GM-CSF to those septic patients whose mononcytes had decreased HLA-DR expression of less than 35% of normal, therapy raised HLA-DR co-expression up to ~50% and restored TNF-α secretion from ex vivo stimulated peripheral blood mononuclear cells.109 These results further confirm the importance of knowing the relative pro- and anti-inflammatory balance of the septic patients before administering immune therapies in sepsis. In our prospective study of septic patients, IFN-γ was determined to be the cytokine that was significantly decreased at the earliest time point following sepsis onset.45 Furthermore, peripheral blood mononuclear cells isolated from septic individuals that were rested overnight in fresh media recovered their ability to secrete IFN-γ in response to ex vivo stimulation.45 These data suggest depressed secretion of IFN-γ is an early indicator of hypo-inflammation and may be a reversible defect in lymphocytes; therefore, pathways that augment IFN-γ may be good potential therapeutic targets. Currently Radboud University in the Netherlands is conducting an IFN-γ trial in sepsis to study the effects on “sepsis-induced immunoparalysis”, which will be completed in December 2013 (ClinicalTrials.gov; NCT01649921).
In addition to GM-CSF, G-CSF, and IFN-γ, other novel immune-adjuvant therapies may be effective in augmenting the adaptive immune system and restoring immunity. The profound apoptosis-induced depletion of lymphocytes in sepsis is one such attractive therapeutic target. The pro-survival cytokines IL-7 and IL-15 have had promising results in sepsis models in preventing lymphocyte apoptosis and restoring adaptive immunity. In a mouse model of sepsis, IL-15 treatment increased lymphocyte survival, decreased apoptosis of natural killer cells, dendritic cells, and T cells, and increased IFN-γ secretion.110 The administration of IL-15 may have additional benefits of increasing natural killer and dendritic cell survival111,112 whereas IL-7 is thought to be a more T cell targeted cytokine.113
Currently, the most promising cytokine in restoring T-cell function in a variety of disease states is IL-7. IL-7 is a pluripotent cytokine of the immune system that affects both T and B cells and induces proliferation of naïve and memory T cells.113,114 Administration of IL-7 by investigators at the National Cancer Institute to cancer patients led to a doubling of CD4 and CD8 T cells, did not expand regulatory T cells, and led to a corresponding increase in spleen and lymphnodes by roughly 50%.115-117 Recombinant IL-7 was also effective in inducing a doubling of circulatory CD4 and CD8 T cells in patients with lymphopenia due to HIV.118 Several groups including ours have shown that IL-7 treatment of mouse models of sepsis results in increased lymphocyte numbers, restored delayed type hypersensitivity responses, decreased lymphocyte apoptosis, reversed the impaired IFN-γ secretion of lymphocytes and improved survival.93,119,120 Furthermore, work by Venet et al. showed that IL-7 treatment of isolated lymphocytes from septic patients restored T-cell proliferation, IFN-γ secretion, STAT5 phosphorylation (an important downstream signaling molecule of the IL-7R), and Bcl-2 expression close to that of healthy controls.121 Treatment of HIV and cancer patients with hIL-7 has indicated IL-7 is well tolerated with less side effects then IL-2, another pro-survival cytokine for T cells, and has diverse effects including: increases in T-cell survival, restores function in exhausted T cells a phenotype identified in sepsis,77 increases expression of adhesion molecules and therefore trafficking to sites of infection, and increases T-cell receptor diversity thereby increasing pathogen recognition.118,122-124 Due to the diverse effects of IL-7 on the adaptive immune system and its good safety clinical profile, IL-7 has been consistently ranked as one of the top therapeutic molecules by the NCI.125 Therefore, we believe clinical trials of IL-7 in sepsis should be initiated.
Common to most septic patients is increased expression of PD-1 on T cells over the progression from hyper-inflammation to hypo-inflammation. As noted earlier, signaling through PD-1 inhibits T-cell proliferation, induces IL-10 secretion, induces apoptosis and anergy, and inhibits cytotoxicity of CD8 T cells.126,127 Four independent groups have shown that disruption of the PD-1/PD-L axis either by genetic deletion or by pharmacologic manipulation improved survival in bacterial and fungal murine sepsis.128-131 In both our prospective and post-mortem studies of septic patients, PD-1 expression was increased on CD4 and CD8 T cells while PD-L expression was increased on antigen presenting cells as well as on the tissue of the spleen and lung45,46 indicating this PD-1/PD-L axis is present and may be dysregulated in human sepsis. Furthermore, PD-1 overexpression on T cells from septic patients correlated with decreased T-cell proliferation, increased secondary infections and mortality.132 In oncology, anti-PD-1 and anti-PD-L antibody therapy has been used successfully to treat various tumors in 20–25% of treated patients.133,134 These data indicate that blocking the PD-1/PD-L axis is a promising target for restoring immune function in human sepsis.
As T-cell exhaustion is defined by the expression of multiple inhibitory receptors, not just PD-1 expression, the development of blocking antibodies to these additional receptors may also be promising in sepsis.77 For example the BTLA/HVEM axis also a negative regulator of T-cell responses135,136 is also present in human sepsis. Interestingly, T cells express BTLA while tissue of the lung, a common site of secondary nosocomial infections including ventilator-associated pneumonia, expresses HVEM.45,46 BTLA deficient mice have increased survival in a mouse model of sepsis,137 providing another immunologic target for ventilator associated pneumonia and sepsis. Additional receptors associated with T-cell exhaustion such as TIM-3, LAG-3, and CTLA-4 may also provide therapeutic targets in restoring T-cell functions. Cellular exhaustion is a characteristic not only of chronic viral infections but of cancer biology due to the presence of high antigen load.138 In mouse models of tumor formation, anti-TIM-3 therapy reduced tumor burden and increased IFN-γ secretion and cytotoxic ability of tumor-specific CD8 T cells.139,140 In addition to TIM-3, LAG-3 therapy has shown promising results in treating human cancers by restoring anti-tumor responses of T cells.141 With our rapidly expanding knowledge of T-cell exhaustion and the role of these inhibitory receptors in lymphocyte function, the potential for biologics that modulate the adaptive immune system and restore lymphocyte function in sepsis offers great promise (Fig. 2).

Figure 2. Pathways of immune dysfunction and targets for immune enhancing therapy in sepsis. In the initial pro-inflammatory response of sepsis, both the adaptive and innate immune systems are rapidly activated. This activation of monocytes, dendritic cells (DC), and macrophages (MAC), as well as CD4 helper and CD8 cytotoxic T cells results in the release of pro-inflammatory cytokines (TNF, IL-6, IL-1β) and chemokines. This pro-inflammatory response normally results in cellular activation and clearance of the primary pathogen (~~, pathogen). In the instance of a healthy individual, the immune system maintains homeostasis by employing counter inflammatory mechanisms such as regulatory T cells (Tregs), apoptosis, production of cytokines, expression of inhibitory receptors and myeloid-derived suppressor cells (MDSC) concurrently during inflammation. However, in some septic patients these normal homeostatic counter inflammatory mechanisms remain elevated such as expression of inhibitory receptors including: programmed death receptor -1 (PD-1), programmed death ligand (PD-L), B and T lymphocyte attenuator (BTLA), and herpesvirus entry mediator (HVEM) as well as the production of the immune modulating cytokine IL-10. Immune dysfunction occurs as activated innate and adaptive immune cells undergo rapid apoptosis while in the presence of increased suppressor cell populations like Tregs or MDSC. The primary infection fails to be cleared and may progress into immune suppression. Prolonged immune suppression and persistent antigen may result in T-cell exhaustion indicated by a T cell’s increased expression of PD-1 and decreased expression of the IL-7R as well as a functional impairment that includes failure to proliferate, secrete cytokines, and kill target cells. Potential targets for immune-therapy are indicted in the dotted GREEN line. Potential therapeutic targets include using blocking antibodies such as anti-IL-10 to decrease Treg function; anti-PD-1 and anti-PD-L to reverse the induction of T-cell exhaustion; and anti-HVEM or anti-BTLA to block tissue suppression of immune cells. IL-7 or IL-15 may be effective in blocking apoptosis and reversing cell exhaustion; GM-CSF to stimulate APC function by increasing recruitment and HLA-DR expression; and IFN-γ to increase PMN recruitment and function.
In the not too distant future, one can imagine a way to genetically manipulate T cells for the treatment of sepsis. Genetically modified T cells are currently in trial for the treatment of herpes viral infections in transplant patients (ClinicalTrials.gov; NCT01646645 and NCT01535885). These T cells have been manipulated to be specific against viral specific antigens.142 In sepsis, a modified T cell resistant to apoptosis and polyclonal for a variety of pathogens, including bacterial, fungal and viral, could be transfused during immune dysfunction to restore patient immunity. Preliminary evidence in mouse models of sepsis using genetically modified T cells that overexpress Bcl-2143-145 or Akt146 have resulted in increased T cell and mouse survival. Modified T cells incorporating a suicide gene would enable only the modified T cells to be destroyed once the infection is cleared leaving the patient’s own immune system intact.147,148 This would eliminate a potential concern of autoimmunity from long-term antibody therapy.
Final Thoughts: Immune-Enhancing Therapy
Many trials using immune-modulatory agents have yielded discouraging results in human clinical trials for sepsis.3,4 One reason for this failure is that animal studies may not always correlate with the human condition. As the majority of sepsis models use genetically similar young mice, confounding problems that are often present in clinical sepsis like age, genetic diversity, underlying co-morbidities, various sites of infections, susceptibility to virulence factors, and management protocols including nutrition and antibiotic usage are not accounted for.3,4,149,150 This heterogeneity of human sepsis is in part what makes performing clinical trials and their success very difficult to achieve. Recently, genetic profiling of inflammatory diseases in humans and mice have indicated that mouse gene transcriptional changes occurring in sepsis do not correlate with the human condition.151 These data indicate the need to undertake more human translational approaches to better our understanding of the underlying molecular pathophysiology of sepsis (Table 1).
To monitor sepsis in humans, clinical trials are currently underway to elucidate biomarkers that can identify patients who have increased or decreased inflammation (ClinicalTrials.gov, Table 1). Importantly, the kinetics of expression of the targeted receptors (i.e., PD-1/PD-L, HVEM/BTLA) as well as patient’s immune status needs to be fully understood so that therapy can be employed effectively. For example, inhibition of IL-10 in patients with increased inflammation dramatically increased pro-inflammatory cytokines with detrimental effects.152 Clinical trials that monitored HLA-DR expression as an indicator of immune function had moderate success in improving outcomes in sepsis.105,109 Therefore, a combination of flow cytometry for lymphocyte expression of surface receptors (i.e., HLA-DR, PD-1), functional assays for cytokine secretion (IFN-γ) as well as presence or absence of nosocomial infections (ventilator pneumonia, Candida spp.) and reactivation of latent viruses (i.e., herpes viruses) may help to guide patient immune-therapy.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Supported in part by National Institutes of Health (NIH) grant GM09839 to J.M.G. and R.S.H. and Institute of Clinical and Translational Sciences (ICTS) award CTSA601 to J.S.B.
Glossary
Abbreviations:
- MDSC
myeloid derived suppressor cells
- APC
antigen presenting cells
- Th1
T lymphocyte type 1
- Th2
T lymphocyte type 2
- Treg
regulatory T cell
- SIRS
systemic inflammatory response syndrome
- VAP
ventilator-associated pneumonia
- PD-1
programmed death receptor 1
- PD-L
programmed death ligand
- BTLA
B and T lymphocyte attenuator
- HVEM
herpesvirus entry mediator
- MODS
multi-organ dysfunction syndrome
- ARDS
adult respiratory distress syndrome
- PAMPs
pathogen-associated molecular patterns
- DAMPs
danger-associated molecular patterns
- PRRs
pattern recognition receptors
- TLR
toll-like receptor
- CTL
cytotoxic T lymphocyte
- DC
dendritic cell
- CARS
compensatory anti-inflammatory response syndrome
- PBMCs
peripheral blood mononuclear cells
- LCMV
lymphocytic choriomeningitis virus
- ICU
intensive care unit
- G-CSF
granulocyte colony stimulating factor
- GM-CSF
granulocyte macrophage colony stimulating factor
- NCI
National Cancer Institute
- TIM-3
T cell membrane protein-3
- LAG-3
lymphocyte-activation gene-3
References
- 1.Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, Reinhart K, Angus DC, Brun-Buisson C, Beale R, et al. International Surviving Sepsis Campaign Guidelines Committee. American Association of Critical-Care Nurses. American College of Chest Physicians. American College of Emergency Physicians. Canadian Critical Care Society. European Society of Clinical Microbiology and Infectious Diseases. European Society of Intensive Care Medicine. European Respiratory Society. International Sepsis Forum. Japanese Association for Acute Medicine. Japanese Society of Intensive Care Medicine. Society of Critical Care Medicine. Society of Hospital Medicine. Surgical Infection Society. World Federation of Societies of Intensive and Critical Care Medicine Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36:296–327. doi: 10.1097/01.CCM.0000298158.12101.41. [DOI] [PubMed] [Google Scholar]
- 2.Balk RA. Optimum treatment of severe sepsis and septic shock: evidence in support of the recommendations. Dis Mon. 2004;50:168–213. doi: 10.1016/j.disamonth.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Christaki E, Anyfanti P, Opal SM. Immunomodulatory therapy for sepsis: an update. Expert Rev Anti Infect Ther. 2011;9:1013–33. doi: 10.1586/eri.11.122. [DOI] [PubMed] [Google Scholar]
- 4.Giamarellos-Bourboulis EJ, Raftogiannis M. The immune response to severe bacterial infections: consequences for therapy. Expert Rev Anti Infect Ther. 2012;10:369–80. doi: 10.1586/eri.12.2. [DOI] [PubMed] [Google Scholar]
- 5.van der Poll T, Opal SM. Host-pathogen interactions in sepsis. Lancet Infect Dis. 2008;8:32–43. doi: 10.1016/S1473-3099(07)70265-7. [DOI] [PubMed] [Google Scholar]
- 6.Ward PA, Bosmann M. A historical perspective on sepsis. Am J Pathol. 2012;181:2–7. doi: 10.1016/j.ajpath.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies MG, Hagen PO. Systemic inflammatory response syndrome. Br J Surg. 1997;84:920–35. doi: 10.1002/bjs.1800840707. [DOI] [PubMed] [Google Scholar]
- 8.Cinel I, Opal SM. Molecular biology of inflammation and sepsis: a primer. Crit Care Med. 2009;37:291–304. doi: 10.1097/CCM.0b013e31819267fb. [DOI] [PubMed] [Google Scholar]
- 9.Casey LC. Immunologic response to infection and its role in septic shock. Crit Care Clin. 2000;16:193–213. doi: 10.1016/S0749-0704(05)70107-X. [DOI] [PubMed] [Google Scholar]
- 10.Opal SM. Immunologic alterations and the pathogenesis of organ failure in the ICU. Semin Respir Crit Care Med. 2011;32:569–80. doi: 10.1055/s-0031-1287865. [DOI] [PubMed] [Google Scholar]
- 11.Angus DC. The search for effective therapy for sepsis: back to the drawing board? JAMA. 2011;306:2614–5. doi: 10.1001/jama.2011.1853. [DOI] [PubMed] [Google Scholar]
- 12.Abraham E, Laterre PF, Garg R, Levy H, Talwar D, Trzaskoma BL, François B, Guy JS, Brückmann M, Rea-Neto A, et al. Administration of Drotrecogin Alfa (Activated) in Early Stage Severe Sepsis (ADDRESS) Study Group Drotrecogin alfa (activated) for adults with severe sepsis and a low risk of death. N Engl J Med. 2005;353:1332–41. doi: 10.1056/NEJMoa050935. [DOI] [PubMed] [Google Scholar]
- 13.Nadel S, Goldstein B, Williams MD, Dalton H, Peters M, Macias WL, Abd-Allah SA, Levy H, Angle R, Wang D, et al. REsearching severe Sepsis and Organ dysfunction in children: a gLobal perspective (RESOLVE) study group Drotrecogin alfa (activated) in children with severe sepsis: a multicentre phase III randomised controlled trial. Lancet. 2007;369:836–43. doi: 10.1016/S0140-6736(07)60411-5. [DOI] [PubMed] [Google Scholar]
- 14.Opal SM, LaRosa SP. Recombinant human activated protein C as a therapy for severe sepsis: lessons learned? Am J Respir Crit Care Med. 2013;187:1041–3. doi: 10.1164/rccm.201303-0505ED. [DOI] [PubMed] [Google Scholar]
- 15.Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013;13:260–8. doi: 10.1016/S1473-3099(13)70001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen J, Opal S, Calandra T. Sepsis studies need new direction. Lancet Infect Dis. 2012;12:503–5. doi: 10.1016/S1473-3099(12)70136-6. [DOI] [PubMed] [Google Scholar]
- 17.Vincent JL, Sun Q, Dubois MJ. Clinical trials of immunomodulatory therapies in severe sepsis and septic shock. Clin Infect Dis. 2002;34:1084–93. doi: 10.1086/339549. [DOI] [PubMed] [Google Scholar]
- 18.Frazier WJ, Hall MW. Immunoparalysis and adverse outcomes from critical illness. Pediatr Clin North Am. 2008;55:647–68, xi. doi: 10.1016/j.pcl.2008.02.009. [xi.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–50. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 20.Munford RS, Pugin J. Normal responses to injury prevent systemic inflammation and can be immunosuppressive. Am J Respir Crit Care Med. 2001;163:316–21. doi: 10.1164/ajrccm.163.2.2007102. [DOI] [PubMed] [Google Scholar]
- 21.Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24:1125–8. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Ma S. The cytokine storm and factors determining the sequence and severity of organ dysfunction in multiple organ dysfunction syndrome. Am J Emerg Med. 2008;26:711–5. doi: 10.1016/j.ajem.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 23.Cavaillon JM, Annane D. Compartmentalization of the inflammatory response in sepsis and SIRS. J Endotoxin Res. 2006;12:151–70. doi: 10.1179/096805106X102246. [DOI] [PubMed] [Google Scholar]
- 24.Tang BM, Huang SJ, McLean AS. Genome-wide transcription profiling of human sepsis: a systematic review. Crit Care. 2010;14:R237. doi: 10.1186/cc9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kollef KE, Schramm GE, Wills AR, Reichley RM, Micek ST, Kollef MH. Predictors of 30-day mortality and hospital costs in patients with ventilator-associated pneumonia attributed to potentially antibiotic-resistant gram-negative bacteria. Chest. 2008;134:281–7. doi: 10.1378/chest.08-1116. [DOI] [PubMed] [Google Scholar]
- 26.Otto GP, Sossdorf M, Claus RA, Rödel J, Menge K, Reinhart K, Bauer M, Riedemann NC. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care. 2011;15:R183. doi: 10.1186/cc10332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monneret G, Venet F, Kullberg BJ, Netea MG. ICU-acquired immunosuppression and the risk for secondary fungal infections. Med Mycol. 2011;49(Suppl 1):S17–23. doi: 10.3109/13693786.2010.509744. [DOI] [PubMed] [Google Scholar]
- 28.Heininger A, Haeberle H, Fischer I, Beck R, Riessen R, Rohde F, Meisner C, Jahn G, Koenigsrainer A, Unertl K, et al. Cytomegalovirus reactivation and associated outcome of critically ill patients with severe sepsis. Crit Care. 2011;15:R77. doi: 10.1186/cc10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ziemann M, Sedemund-Adib B, Reiland P, Schmucker P, Hennig H. Increased mortality in long-term intensive care patients with active cytomegalovirus infection. Crit Care Med. 2008;36:3145–50. doi: 10.1097/CCM.0b013e31818f3fc4. [DOI] [PubMed] [Google Scholar]
- 30.Kalil AC, Florescu DF. Prevalence and mortality associated with cytomegalovirus infection in nonimmunosuppressed patients in the intensive care unit. Crit Care Med. 2009;37:2350–8. doi: 10.1097/CCM.0b013e3181a3aa43. [DOI] [PubMed] [Google Scholar]
- 31.Osuchowski MF, Welch K, Yang H, Siddiqui J, Remick DG. Chronic sepsis mortality characterized by an individualized inflammatory response. J Immunol. 2007;179:623–30. doi: 10.4049/jimmunol.179.1.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meakins JL, Pietsch JB, Bubenick O, Kelly R, Rode H, Gordon J, MacLean LD. Delayed hypersensitivity: indicator of acquired failure of host defenses in sepsis and trauma. Ann Surg. 1977;186:241–50. doi: 10.1097/00000658-197709000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. The sepsis seesaw: tilting toward immunosuppression. Nat Med. 2009;15:496–7. doi: 10.1038/nm0509-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hotchkiss RS, Opal S. Immunotherapy for sepsis--a new approach against an ancient foe. N Engl J Med. 2010;363:87–9. doi: 10.1056/NEJMcibr1004371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venet F, Chung CS, Monneret G, Huang X, Horner B, Garber M, Ayala A. Regulatory T cell populations in sepsis and trauma. J Leukoc Biol. 2008;83:523–35. doi: 10.1189/jlb.0607371. [DOI] [PubMed] [Google Scholar]
- 36.Venet F, Chung CS, Kherouf H, Geeraert A, Malcus C, Poitevin F, Bohé J, Lepape A, Ayala A, Monneret G. Increased circulating regulatory T cells (CD4(+)CD25 (+)CD127 (-)) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med. 2009;35:678–86. doi: 10.1007/s00134-008-1337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leng FY, Liu JL, Liu ZJ, Yin JY, Qu HP. Increased proportion of CD4(+)CD25(+)Foxp3(+) regulatory T cells during the early-stage sepsis in ICU patients. J Microbiol Immunol Infect. 2012 doi: 10.1016/j.jmii.2012.06.012. Forthcoming. [DOI] [PubMed] [Google Scholar]
- 38.Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, O’Malley KA, Wynn JL, Antonenko S, Al-Quran SZ, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. 2007;204:1463–74. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Venet F, Lepape A, Monneret G. Clinical review: flow cytometry perspectives in the ICU - from diagnosis of infection to monitoring of injury-induced immune dysfunctions. Crit Care. 2011;15:231. doi: 10.1186/cc10333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferguson NR, Galley HF, Webster NR. T helper cell subset ratios in patients with severe sepsis. Intensive Care Med. 1999;25:106–9. doi: 10.1007/s001340050795. [DOI] [PubMed] [Google Scholar]
- 41.Senftleben U, Karin M. The IKK/NF-kappaB pathway. Crit Care Med. 2002;30(Supp):S18–26. doi: 10.1097/00003246-200201001-00003. [DOI] [PubMed] [Google Scholar]
- 42.Weighardt H, Holzmann B. Role of Toll-like receptor responses for sepsis pathogenesis. Immunobiology. 2007;212:715–22. doi: 10.1016/j.imbio.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 43.Unsinger J, Kazama H, McDonough JS, Griffith TS, Hotchkiss RS, Ferguson TA. Sepsis-induced apoptosis leads to active suppression of delayed-type hypersensitivity by CD8+ regulatory T cells through a TRAIL-dependent mechanism. J Immunol. 2010;184:6766–72. doi: 10.4049/jimmunol.0904054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Unsinger J, McDonough JS, Shultz LD, Ferguson TA, Hotchkiss RS. Sepsis-induced human lymphocyte apoptosis and cytokine production in “humanized” mice. J Leukoc Biol. 2009;86:219–27. doi: 10.1189/jlb.1008615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boomer JS, Shuherk-Shaffer J, Hotchkiss RS, Green JM. A prospective analysis of lymphocyte phenotype and function over the course of acute sepsis. Crit Care. 2012;16:R112. doi: 10.1186/cc11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD, 2nd, Kreisel D, Krupnick AS, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306:2594–605. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–51. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 48.Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE, Jr., Hui JJ, Chang KC, Osborne DF, Freeman BD, Cobb JP, Buchman TG, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol. 2001;166:6952–63. doi: 10.4049/jimmunol.166.11.6952. [DOI] [PubMed] [Google Scholar]
- 49.Kasten KR, Tschöp J, Goetzman HS, England LG, Dattilo JR, Cave CM, Seitz AP, Hildeman DA, Caldwell CC. T-cell activation differentially mediates the host response to sepsis. Shock. 2010;34:377–83. doi: 10.1097/SHK.0b013e3181dc0845. [DOI] [PubMed] [Google Scholar]
- 50.Ayala A, Perl M, Venet F, Lomas-Neira J, Swan R, Chung CS. Apoptosis in sepsis: mechanisms, clinical impact and potential therapeutic targets. Curr Pharm Des. 2008;14:1853–9. doi: 10.2174/138161208784980617. [DOI] [PubMed] [Google Scholar]
- 51.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–83. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30:3667–83. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chang KC, Unsinger J, Davis CG, Schwulst SJ, Muenzer JT, Strasser A, Hotchkiss RS. Multiple triggers of cell death in sepsis: death receptor and mitochondrial-mediated apoptosis. FASEB J. 2007;21:708–19. doi: 10.1096/fj.06-6805com. [DOI] [PubMed] [Google Scholar]
- 54.Klener P, Jr., Andera L, Klener P, Necas E, Zivný J. Cell death signalling pathways in the pathogenesis and therapy of haematologic malignancies: overview of therapeutic approaches. Folia Biol (Praha) 2006;52:119–36. [PubMed] [Google Scholar]
- 55.Vaki I, Kranidioti H, Karagianni V, Spyridaki A, Kotsaki A, Routsi C, Giamarellos-Bourboulis EJ. An early circulating factor in severe sepsis modulates apoptosis of monocytes and lymphocytes. J Leukoc Biol. 2011;89:343–9. doi: 10.1189/jlb.0410232. [DOI] [PubMed] [Google Scholar]
- 56.Arad G, Levy R, Nasie I, Hillman D, Rotfogel Z, Barash U, Supper E, Shpilka T, Minis A, Kaempfer R. Binding of superantigen toxins into the CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock. PLoS Biol. 2011;9:e1001149. doi: 10.1371/journal.pbio.1001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmidt MV, Paulus P, Kuhn AM, Weigert A, Morbitzer V, Zacharowski K, Kempf VA, Brüne B, von Knethen A. Peroxisome proliferator-activated receptor γ-induced T cell apoptosis reduces survival during polymicrobial sepsis. Am J Respir Crit Care Med. 2011;184:64–74. doi: 10.1164/rccm.201010-1585OC. [DOI] [PubMed] [Google Scholar]
- 58.He KL, Ting AT. A20 inhibits tumor necrosis factor (TNF) alpha-induced apoptosis by disrupting recruitment of TRADD and RIP to the TNF receptor 1 complex in Jurkat T cells. Mol Cell Biol. 2002;22:6034–45. doi: 10.1128/MCB.22.17.6034-6045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Riedemann NC, Guo RF, Laudes IJ, Keller K, Sarma VJ, Padgaonkar V, Zetoune FS, Ward PA. C5a receptor and thymocyte apoptosis in sepsis. FASEB J. 2002;16:887–8. doi: 10.1096/fj.02-0033fje. [DOI] [PubMed] [Google Scholar]
- 60.Boomer JS, Green JM. An enigmatic tail of CD28 signaling. Cold Spring Harb Perspect Biol. 2010;2:a002436. doi: 10.1101/cshperspect.a002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shiao SL, McNiff JM, Pober JS. Memory T cells and their costimulators in human allograft injury. J Immunol. 2005;175:4886–96. doi: 10.4049/jimmunol.175.8.4886. [DOI] [PubMed] [Google Scholar]
- 62.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–15. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 63.Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116–26. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 64.Kitazawa Y, Fujino M, Wang Q, Kimura H, Azuma M, Kubo M, Abe R, Li XK. Involvement of the programmed death-1/programmed death-1 ligand pathway in CD4+CD25+ regulatory T-cell activity to suppress alloimmune responses. Transplantation. 2007;83:774–82. doi: 10.1097/01.tp.0000256293.90270.e8. [DOI] [PubMed] [Google Scholar]
- 65.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kessel A, Bamberger E, Masalha M, Toubi E. The role of T regulatory cells in human sepsis. J Autoimmun. 2009;32:211–5. doi: 10.1016/j.jaut.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 67.Venet F, Pachot A, Debard AL, Bohe J, Bienvenu J, Lepape A, Powell WS, Monneret G. Human CD4+CD25+ regulatory T lymphocytes inhibit lipopolysaccharide-induced monocyte survival through a Fas/Fas ligand-dependent mechanism. J Immunol. 2006;177:6540–7. doi: 10.4049/jimmunol.177.9.6540. [DOI] [PubMed] [Google Scholar]
- 68.van Dissel JT, van Langevelde P, Westendorp RG, Kwappenberg K, Frölich M. Anti-inflammatory cytokine profile and mortality in febrile patients. Lancet. 1998;351:950–3. doi: 10.1016/S0140-6736(05)60606-X. [DOI] [PubMed] [Google Scholar]
- 69.Gogos CA, Drosou E, Bassaris HP, Skoutelis A. Pro- versus anti-inflammatory cytokine profile in patients with severe sepsis: a marker for prognosis and future therapeutic options. J Infect Dis. 2000;181:176–80. doi: 10.1086/315214. [DOI] [PubMed] [Google Scholar]
- 70.Jiang H, Chess L. Regulation of immune responses by T cells. N Engl J Med. 2006;354:1166–76. doi: 10.1056/NEJMra055446. [DOI] [PubMed] [Google Scholar]
- 71.Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. 2010;140:845–58. doi: 10.1016/j.cell.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 72.Ertel W, Kremer JP, Kenney J, Steckholzer U, Jarrar D, Trentz O, Schildberg FW. Downregulation of proinflammatory cytokine release in whole blood from septic patients. Blood. 1995;85:1341–7. [PubMed] [Google Scholar]
- 73.Munoz C, Carlet J, Fitting C, Misset B, Blériot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest. 1991;88:1747–54. doi: 10.1172/JCI115493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rigato O, Salomao R. Impaired production of interferon-gamma and tumor necrosis factor-alpha but not of interleukin 10 in whole blood of patients with sepsis. Shock. 2003;19:113–6. doi: 10.1097/00024382-200302000-00004. [DOI] [PubMed] [Google Scholar]
- 75.Sinistro A, Almerighi C, Ciaprini C, Natoli S, Sussarello E, Di Fino S, Calò-Carducci F, Rocchi G, Bergamini A. Downregulation of CD40 ligand response in monocytes from sepsis patients. Clin Vaccine Immunol. 2008;15:1851–8. doi: 10.1128/CVI.00184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weighardt H, Heidecke CD, Emmanuilidis K, Maier S, Bartels H, Siewert JR, Holzmann B. Sepsis after major visceral surgery is associated with sustained and interferon-gamma-resistant defects of monocyte cytokine production. Surgery. 2000;127:309–15. doi: 10.1067/msy.2000.104118. [DOI] [PubMed] [Google Scholar]
- 77.Monneret G, Venet F. A rapidly progressing lymphocyte exhaustion after severe sepsis. Crit Care. 2012;16:140. doi: 10.1186/cc11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology. 2010;129:474–81. doi: 10.1111/j.1365-2567.2010.03255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xiao H, Siddiqui J, Remick DG. Mechanisms of mortality in early and late sepsis. Infect Immun. 2006;74:5227–35. doi: 10.1128/IAI.01220-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bonten MJ, Froon AH, Gaillard CA, Greve JW, de Leeuw PW, Drent M, Stobberingh EE, Buurman WA. The systemic inflammatory response in the development of ventilator-associated pneumonia. Am J Respir Crit Care Med. 1997;156:1105–13. doi: 10.1164/ajrccm.156.4.9610002. [DOI] [PubMed] [Google Scholar]
- 81.Pelekanou A, Tsangaris I, Kotsaki A, Karagianni V, Giamarellou H, Armaganidis A, Giamarellos-Bourboulis EJ. Decrease of CD4-lymphocytes and apoptosis of CD14-monocytes are characteristic alterations in sepsis caused by ventilator-associated pneumonia: results from an observational study. Crit Care. 2009;13:R172. doi: 10.1186/cc8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Abraham E, Laterre PF, Garbino J, Pingleton S, Butler T, Dugernier T, Margolis B, Kudsk K, Zimmerli W, Anderson P, et al. Lenercept Study Group Lenercept (p55 tumor necrosis factor receptor fusion protein) in severe sepsis and early septic shock: a randomized, double-blind, placebo-controlled, multicenter phase III trial with 1,342 patients. Crit Care Med. 2001;29:503–10. doi: 10.1097/00003246-200103000-00006. [DOI] [PubMed] [Google Scholar]
- 83.Fisher CJ, Jr., Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, Iberti TJ, Rackow EC, Shapiro MJ, Greenman RL, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. 1994;271:1836–43. doi: 10.1001/jama.1994.03510470040032. [DOI] [PubMed] [Google Scholar]
- 84.Opal SM, Fisher CJ, Jr., Dhainaut JF, Vincent JL, Brase R, Lowry SF, Sadoff JC, Slotman GJ, Levy H, Balk RA, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit Care Med. 1997;25:1115–24. doi: 10.1097/00003246-199707000-00010. [DOI] [PubMed] [Google Scholar]
- 85.Panacek EA, Marshall JC, Albertson TE, Johnson DH, Johnson S, MacArthur RD, Miller M, Barchuk WT, Fischkoff S, Kaul M, et al. Monoclonal Anti-TNF: a Randomized Controlled Sepsis Study Investigators Efficacy and safety of the monoclonal anti-tumor necrosis factor antibody F(ab’)2 fragment afelimomab in patients with severe sepsis and elevated interleukin-6 levels. Crit Care Med. 2004;32:2173–82. doi: 10.1097/01.ccm.0000145229.59014.6c. [DOI] [PubMed] [Google Scholar]
- 86.Rice TW, Wheeler AP, Morris PE, Paz HL, Russell JA, Edens TR, Bernard GR. Safety and efficacy of affinity-purified, anti-tumor necrosis factor-alpha, ovine fab for injection (CytoFab) in severe sepsis. Crit Care Med. 2006;34:2271–81. doi: 10.1097/01.CCM.0000230385.82679.34. [DOI] [PubMed] [Google Scholar]
- 87.Bellomo R, Tetta C, Ronco C. Coupled plasma filtration adsorption. Intensive Care Med. 2003;29:1222–8. doi: 10.1007/s00134-003-1796-x. [DOI] [PubMed] [Google Scholar]
- 88.Humes HD, Weitzel WF, Bartlett RH, Swaniker FC, Paganini EP, Luderer JR, Sobota J. Initial clinical results of the bioartificial kidney containing human cells in ICU patients with acute renal failure. Kidney Int. 2004;66:1578–88. doi: 10.1111/j.1523-1755.2004.00923.x. [DOI] [PubMed] [Google Scholar]
- 89.Tumlin J, Wali R, Williams W, Murray P, Tolwani AJ, Vinnikova AK, Szerlip HM, Ye J, Paganini EP, Dworkin L, et al. Efficacy and safety of renal tubule cell therapy for acute renal failure. J Am Soc Nephrol. 2008;19:1034–40. doi: 10.1681/ASN.2007080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, Wittebole X, Dugernier T, Perrotin D, Tidswell M, et al. ACCESS Study Group Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309:1154–62. doi: 10.1001/jama.2013.2194. [DOI] [PubMed] [Google Scholar]
- 91.Balk RA. Pathogenesis and management of multiple organ dysfunction or failure in severe sepsis and septic shock. Crit Care Clin. 2000;16:337–52, vii. doi: 10.1016/S0749-0704(05)70113-5. [vii.] [DOI] [PubMed] [Google Scholar]
- 92.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 93.Kasten KR, Prakash PS, Unsinger J, Goetzman HS, England LG, Cave CM, Seitz AP, Mazuski CN, Zhou TT, Morre M, et al. Interleukin-7 (IL-7) treatment accelerates neutrophil recruitment through gamma delta T-cell IL-17 production in a murine model of sepsis. Infect Immun. 2010;78:4714–22. doi: 10.1128/IAI.00456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Flohé SB, Agrawal H, Flohé S, Rani M, Bangen JM, Schade FU. Diversity of interferon gamma and granulocyte-macrophage colony-stimulating factor in restoring immune dysfunction of dendritic cells and macrophages during polymicrobial sepsis. Mol Med. 2008;14:247–56. doi: 10.2119/2007-00120.Flohe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bo L, Wang F, Zhu J, Li J, Deng X. Granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) for sepsis: a meta-analysis. Crit Care. 2011;15:R58. doi: 10.1186/cc10031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cheng AC, Limmathurotsakul D, Chierakul W, Getchalarat N, Wuthiekanun V, Stephens DP, Day NP, White NJ, Chaowagul W, Currie BJ, et al. A randomized controlled trial of granulocyte colony-stimulating factor for the treatment of severe sepsis due to melioidosis in Thailand. Clin Infect Dis. 2007;45:308–14. doi: 10.1086/519261. [DOI] [PubMed] [Google Scholar]
- 97.Hall MW, Knatz NL, Vetterly C, Tomarello S, Wewers MD, Volk HD, Carcillo JA. Immunoparalysis and nosocomial infection in children with multiple organ dysfunction syndrome. Intensive Care Med. 2011;37:525–32. doi: 10.1007/s00134-010-2088-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rosenbloom AJ, Linden PK, Dorrance A, Penkosky N, Cohen-Melamed MH, Pinsky MR. Effect of granulocyte-monocyte colony-stimulating factor therapy on leukocyte function and clearance of serious infection in nonneutropenic patients. Chest. 2005;127:2139–50. doi: 10.1378/chest.127.6.2139. [DOI] [PubMed] [Google Scholar]
- 99.Schefold JC, Zeden JP, Pschowski R, Hammoud B, Fotopoulou C, Hasper D, Fusch G, Von Haehling S, Volk HD, Meisel C, et al. Treatment with granulocyte-macrophage colony-stimulating factor is associated with reduced indoleamine 2,3-dioxygenase activity and kynurenine pathway catabolites in patients with severe sepsis and septic shock. Scand J Infect Dis. 2010;42:164–71. doi: 10.3109/00365540903405768. [DOI] [PubMed] [Google Scholar]
- 100.Stephens DP, Thomas JH, Higgins A, Bailey M, Anstey NM, Currie BJ, Cheng AC. Randomized, double-blind, placebo-controlled trial of granulocyte colony-stimulating factor in patients with septic shock. Crit Care Med. 2008;36:448–54. doi: 10.1097/01.CCM.0B013E318161E480. [DOI] [PubMed] [Google Scholar]
- 101.Nelson S, Belknap SM, Carlson RW, Dale D, DeBoisblanc B, Farkas S, Fotheringham N, Ho H, Marrie T, Movahhed H, et al. CAP Study Group A randomized controlled trial of filgrastim as an adjunct to antibiotics for treatment of hospitalized patients with community-acquired pneumonia. J Infect Dis. 1998;178:1075–80. doi: 10.1086/515694. [DOI] [PubMed] [Google Scholar]
- 102.Nelson S, Heyder AM, Stone J, Bergeron MG, Daugherty S, Peterson G, Fotheringham N, Welch W, Milwee S, Root R. A randomized controlled trial of filgrastim for the treatment of hospitalized patients with multilobar pneumonia. J Infect Dis. 2000;182:970–3. doi: 10.1086/315775. [DOI] [PubMed] [Google Scholar]
- 103.Root RK, Lodato RF, Patrick W, Cade JF, Fotheringham N, Milwee S, Vincent JL, Torres A, Rello J, Nelson S, Pneumonia Sepsis Study Group Multicenter, double-blind, placebo-controlled study of the use of filgrastim in patients hospitalized with pneumonia and severe sepsis. Crit Care Med. 2003;31:367–73. doi: 10.1097/01.CCM.0000048629.32625.5D. [DOI] [PubMed] [Google Scholar]
- 104.Weiss M, Gross-Weege W, Schneider M, Neidhardt H, Liebert S, Mirow N, Wernet P. Enhancement of neutrophil function by in vivo filgrastim treatment for prophylaxis of sepsis in surgical intensive care patients. J Crit Care. 1995;10:21–6. doi: 10.1016/0883-9441(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 105.Meisel C, Schefold JC, Pschowski R, Baumann T, Hetzger K, Gregor J, Weber-Carstens S, Hasper D, Keh D, Zuckermann H, et al. Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial. Am J Respir Crit Care Med. 2009;180:640–8. doi: 10.1164/rccm.200903-0363OC. [DOI] [PubMed] [Google Scholar]
- 106.Polk HC, Jr., Cheadle WG, Livingston DH, Rodriguez JL, Starko KM, Izu AE, Jaffe HS, Sonnenfeld G. A randomized prospective clinical trial to determine the efficacy of interferon-gamma in severely injured patients. Am J Surg. 1992;163:191–6. doi: 10.1016/0002-9610(92)90099-D. [DOI] [PubMed] [Google Scholar]
- 107.Wasserman D, Ioannovich JD, Hinzmann RD, Deichsel G, Steinmann GG, The Severe Burns Study Group Interferon-gamma in the prevention of severe burn-related infections: a European phase III multicenter trial. Crit Care Med. 1998;26:434–9. doi: 10.1097/00003246-199803000-00010. [DOI] [PubMed] [Google Scholar]
- 108.Dries DJ, Jurkovich GJ, Maier RV, Clemmer TP, Struve SN, Weigelt JA, Stanford GG, Herr DL, Champion HR, Lewis FR, et al. Effect of interferon gamma on infection-related death in patients with severe injuries. A randomized, double-blind, placebo-controlled trial. Arch Surg. 1994;129:1031–41, discussion 1042. doi: 10.1001/archsurg.1994.01420340045008. [DOI] [PubMed] [Google Scholar]
- 109.Döcke WD, Randow F, Syrbe U, Krausch D, Asadullah K, Reinke P, Volk HD, Kox W. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med. 1997;3:678–81. doi: 10.1038/nm0697-678. [DOI] [PubMed] [Google Scholar]
- 110.Inoue S, Unsinger J, Davis CG, Muenzer JT, Ferguson TA, Chang K, Osborne DF, Clark AT, Coopersmith CM, McDunn JE, et al. IL-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J Immunol. 2010;184:1401–9. doi: 10.4049/jimmunol.0902307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fehniger TA, Caligiuri MA. Interleukin 15: biology and relevance to human disease. Blood. 2001;97:14–32. doi: 10.1182/blood.V97.1.14. [DOI] [PubMed] [Google Scholar]
- 112.Steel JC, Waldmann TA, Morris JC. Interleukin-15 biology and its therapeutic implications in cancer. Trends Pharmacol Sci. 2012;33:35–41. doi: 10.1016/j.tips.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol. 2006;24:657–79. doi: 10.1146/annurev.immunol.24.021605.090727. [DOI] [PubMed] [Google Scholar]
- 114.Fry TJ, Mackall CL. The many faces of IL-7: from lymphopoiesis to peripheral T cell maintenance. J Immunol. 2005;174:6571–6. doi: 10.4049/jimmunol.174.11.6571. [DOI] [PubMed] [Google Scholar]
- 115.Rosenberg SA, Sportès C, Ahmadzadeh M, Fry TJ, Ngo LT, Schwarz SL, Stetler-Stevenson M, Morton KE, Mavroukakis SA, Morre M, et al. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. J Immunother. 2006;29:313–9. doi: 10.1097/01.cji.0000210386.55951.c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sportès C, Babb RR, Krumlauf MC, Hakim FT, Steinberg SM, Chow CK, Brown MR, Fleisher TA, Noel P, Maric I, et al. Phase I study of recombinant human interleukin-7 administration in subjects with refractory malignancy. Clin Cancer Res. 2010;16:727–35. doi: 10.1158/1078-0432.CCR-09-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lundström W, Fewkes NM, Mackall CL. IL-7 in human health and disease. Semin Immunol. 2012;24:218–24. doi: 10.1016/j.smim.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lévy Y, Sereti I, Tambussi G, Routy JP, Lelièvre JD, Delfraissy JF, Molina JM, Fischl M, Goujard C, Rodriguez B, et al. Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: results of a phase I/IIa randomized, placebo-controlled, multicenter study. Clin Infect Dis. 2012;55:291–300. doi: 10.1093/cid/cis383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Unsinger J, Burnham CA, McDonough J, Morre M, Prakash PS, Caldwell CC, Dunne WM, Jr., Hotchkiss RS. Interleukin-7 ameliorates immune dysfunction and improves survival in a 2-hit model of fungal sepsis. J Infect Dis. 2012;206:606–16. doi: 10.1093/infdis/jis383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Unsinger J, McGlynn M, Kasten KR, Hoekzema AS, Watanabe E, Muenzer JT, McDonough JS, Tschoep J, Ferguson TA, McDunn JE, et al. IL-7 promotes T cell viability, trafficking, and functionality and improves survival in sepsis. J Immunol. 2010;184:3768–79. doi: 10.4049/jimmunol.0903151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Venet F, Foray AP, Villars-Méchin A, Malcus C, Poitevin-Later F, Lepape A, Monneret G. IL-7 restores lymphocyte functions in septic patients. J Immunol. 2012;189:5073–81. doi: 10.4049/jimmunol.1202062. [DOI] [PubMed] [Google Scholar]
- 122.Sportès C, Hakim FT, Memon SA, Zhang H, Chua KS, Brown MR, Fleisher TA, Krumlauf MC, Babb RR, Chow CK, et al. Administration of rhIL-7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J Exp Med. 2008;205:1701–14. doi: 10.1084/jem.20071681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Morre M, Beq S. Interleukin-7 and immune reconstitution in cancer patients: a new paradigm for dramatically increasing overall survival. Target Oncol. 2012;7:55–68. doi: 10.1007/s11523-012-0210-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sereti I, Dunham RM, Spritzler J, Aga E, Proschan MA, Medvik K, Battaglia CA, Landay AL, Pahwa S, Fischl MA, et al. ACTG 5214 Study Team IL-7 administration drives T cell-cycle entry and expansion in HIV-1 infection. Blood. 2009;113:6304–14. doi: 10.1182/blood-2008-10-186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunol Rev. 2008;222:357–68. doi: 10.1111/j.1600-065X.2008.00604.x. [DOI] [PubMed] [Google Scholar]
- 126.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–45. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 128.Lázár-Molnár E, Gácser A, Freeman GJ, Almo SC, Nathenson SG, Nosanchuk JD. The PD-1/PD-L costimulatory pathway critically affects host resistance to the pathogenic fungus Histoplasma capsulatum. Proc Natl Acad Sci U S A. 2008;105:2658–63. doi: 10.1073/pnas.0711918105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, Hotchkiss RS. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol. 2010;88:233–40. doi: 10.1189/jlb.0110037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang Y, Zhou Y, Lou J, Li J, Bo L, Zhu K, Wan X, Deng X, Cai Z. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care. 2010;14:R220. doi: 10.1186/cc9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, Swan R, Kherouf H, Monneret G, Chung CS, et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U S A. 2009;106:6303–8. doi: 10.1073/pnas.0809422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Guignant C, Lepape A, Huang X, Kherouf H, Denis L, Poitevin F, Malcus C, Chéron A, Allaouchiche B, Gueyffier F, et al. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit Care. 2011;15:R99. doi: 10.1186/cc10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4:670–9. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- 136.Compaan DM, Gonzalez LC, Tom I, Loyet KM, Eaton D, Hymowitz SG. Attenuating lymphocyte activity: the crystal structure of the BTLA-HVEM complex. J Biol Chem. 2005;280:39553–61. doi: 10.1074/jbc.M507629200. [DOI] [PubMed] [Google Scholar]
- 137.Shubin NJ, Chung CS, Heffernan DS, Irwin LR, Monaghan SF, Ayala A. BTLA expression contributes to septic morbidity and mortality by inducing innate inflammatory cell dysfunction. J Leukoc Biol. 2012;92:593–603. doi: 10.1189/jlb.1211641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jin HT, Jeong YH, Park HJ, Ha SJ. Mechanism of T cell exhaustion in a chronic environment. BMB Rep. 2011;44:217–31. doi: 10.5483/BMBRep.2011.44.4.217. [DOI] [PubMed] [Google Scholar]
- 139.Ngiow SF, Teng MW, Smyth MJ. Prospects for TIM3-Targeted Antitumor Immunotherapy. Cancer Res. 2011;71:6567–71. doi: 10.1158/0008-5472.CAN-11-1487. [DOI] [PubMed] [Google Scholar]
- 140.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71:3540–51. doi: 10.1158/0008-5472.CAN-11-0096. [DOI] [PubMed] [Google Scholar]
- 141.Sierro S, Romero P, Speiser DE. The CD4-like molecule LAG-3, biology and therapeutic applications. Expert Opin Ther Targets. 2011;15:91–101. doi: 10.1517/14712598.2011.540563. [DOI] [PubMed] [Google Scholar]
- 142.Savoldo B, Huls MH, Liu Z, Okamura T, Volk HD, Reinke P, Sabat R, Babel N, Jones JF, Webster-Cyriaque J, et al. Autologous Epstein-Barr virus (EBV)-specific cytotoxic T cells for the treatment of persistent active EBV infection. Blood. 2002;100:4059–66. doi: 10.1182/blood-2002-01-0039. [DOI] [PubMed] [Google Scholar]
- 143.Wagner TH, Drewry AM, Macmillan S, Dunne WM, Chang KC, Karl IE, Hotchkiss RS, Cobb JP. Surviving sepsis: bcl-2 overexpression modulates splenocyte transcriptional responses in vivo. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1751–9. doi: 10.1152/ajpregu.00656.2006. [DOI] [PubMed] [Google Scholar]
- 144.Coopersmith CM, Chang KC, Swanson PE, Tinsley KW, Stromberg PE, Buchman TG, Karl IE, Hotchkiss RS. Overexpression of Bcl-2 in the intestinal epithelium improves survival in septic mice. Crit Care Med. 2002;30:195–201. doi: 10.1097/00003246-200201000-00028. [DOI] [PubMed] [Google Scholar]
- 145.Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, Korsmeyer SJ, Karl IE. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci U S A. 1999;96:14541–6. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bommhardt U, Chang KC, Swanson PE, Wagner TH, Tinsley KW, Karl IE, Hotchkiss RS. Akt decreases lymphocyte apoptosis and improves survival in sepsis. J Immunol. 2004;172:7583–91. doi: 10.4049/jimmunol.172.12.7583. [DOI] [PubMed] [Google Scholar]
- 147.June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest. 2007;117:1466–76. doi: 10.1172/JCI32446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.June CH. Principles of adoptive T cell cancer therapy. J Clin Invest. 2007;117:1204–12. doi: 10.1172/JCI31446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dyson A, Singer M. Animal models of sepsis: why does preclinical efficacy fail to translate to the clinical setting? Crit Care Med. 2009;37(Suppl):S30–7. doi: 10.1097/CCM.0b013e3181922bd3. [DOI] [PubMed] [Google Scholar]
- 150.Esmon CT. Why do animal models (sometimes) fail to mimic human sepsis? Crit Care Med. 2004;32(Suppl):S219–22. doi: 10.1097/01.CCM.0000127036.27343.48. [DOI] [PubMed] [Google Scholar]
- 151.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. Inflammation and Host Response to Injury, Large Scale Collaborative Research Program Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110:3507–12. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Standiford TJ, Strieter RM, Lukacs NW, Kunkel SL. Neutralization of IL-10 increases lethality in endotoxemia. Cooperative effects of macrophage inflammatory protein-2 and tumor necrosis factor. J Immunol. 1995;155:2222–9. [PubMed] [Google Scholar]