

Background: The chemokine receptor CXCR3-B initiates inhibitory signals.
Results: CXCR3-B-mediated signal induced apoptosis and inhibited autophagy of breast cancer cells. It is associated with nuclear translocation of Bach-1, nuclear export of Nrf2, and down-regulation of HO-1.
Conclusion: A CXCR3-B-mediated signal promotes apoptosis of breast cancer cells.
Significance: Induction of CXCR3-B-mediated signaling can serve as a novel therapeutic approach for the treatment of breast cancer.
Keywords: Apoptosis, Chemokines, Heme Oxygenase, Nrf2, Nuclear Transport, Signal Transduction, Signaling
Abstract
Chemokines and their receptors play diverse roles in regulating cancer growth and progression. The receptor CXCR3 can have two splice variants with opposite functions. CXCR3-A promotes cell growth, whereas CXCR3-B mediates growth-inhibitory signals. However, the negative signals through CXCR3-B in cancer cells are not well characterized. In this study, we found that CXCR3-B-mediated signaling in MCF-7 and T47D breast cancer cells induced apoptotic cell death. Signals through CXCR3-B decreased the levels of the antiapoptotic proteins Bcl-2 and Bcl-xL and increased the expression of apoptotic cleaved poly(ADP-ribose) polymerase. Along with up-regulation in apoptosis, CXCR3-B signals were associated with a decrease in cellular autophagy with reduced levels of the autophagic markers Beclin-1 and LC3B. Notably, CXCR3-B down-regulated the expression of the cytoprotective and antiapoptotic molecule heme oxygenase-1 (HO-1) at the transcriptional level. There was an increased nuclear localization of Bach-1 and nuclear export of Nrf2, which are important negative and positive transcription factors, respectively, for HO-1 expression. We also observed that CXCR3-B promoted the activation of p38 MAPK and the inhibition of ERK-1/2. CXCR3-B could not induce cancer cell apoptosis at the optimal level when we either inhibited p38 activity or knocked down Bach-1. Further, CXCR3-B-induced apoptosis was down-regulated when we overexpressed HO-1. Together, our data suggest that CXCR3-B mediates a growth-inhibitory signal in breast cancer cells through the modulations of nuclear translocation of Bach-1 and Nrf2 and down-regulation of HO-1. We suggest that the induction of CXCR3-B-mediated signaling can serve as a novel therapeutic approach where the goal is to promote tumor cell apoptosis.
Introduction
Chemokines and their receptors play key roles in chemotaxis, primarily regulating the migration of leukocytes to the site of inflammation and secondary lymphoid organs (1, 2). However, recent studies clearly suggest that both chemokines and chemokine receptors are expressed in tumor cells and the cells of tumor microenvironment and that they play a critical role in tumor growth, progression, and metastasis (3–8).
CXCR3 belongs to the CXC chemokine receptor subfamily and is classically expressed by T cells and natural killer cells (9). CXCR3 promotes the regulation of leukocyte trafficking through interaction with its ligands CXCL9, CXCL10, and CXCL11 (10). Interestingly, CXCR3 is also expressed by tumor cells, including skin, ovary, renal, and breast cancer cells (5, 6, 8, 11, 12). It has been shown that an alternate splicing of CXCR3 generates at least two splice variants, CXCR3-A and CXCR3-B, with completely opposite functions (10, 11, 13). The ligands CXCL9, CXCL10, and CXCL11 bind to both the splice variants, whereas CXCL4 is a specific ligand for CXCR3-B. Upon activation, each of the splice variants mediates different intracellular signaling events and exhibits distinct biological functions (10, 13, 14). CXCR3-A signaling promotes cell proliferation and chemotaxis, whereas CXCR3-B mediates negative signals for growth inhibition. It has been demonstrated that cellular signaling via CXCR3-A results in increased calcium influx and that CXCR3-B mediates the increase in intracellular cyclic AMP levels. In addition, CXCR3-A- and CXCR3-B-mediated signaling can be associated with different G protein coupling.
Although the expression of CXCR3 is increased in different types of tumors, recent studies have revealed that the growth-promoting CXCR3-A is up-regulated and that the growth-inhibitory CXCR3-B is markedly down-regulated in cancer cells and tissues (15, 16). We have demonstrated previously that the activation of oncogenic ras promotes the down-regulation of CXCR3-B in human breast cancer cells, and we suggested that, in the absence/low presence of CXCR3-B, the overexpressed ligand CXCL10 can induce cancer cell proliferation, possibly through CXCR3-A (11). We have also shown that the overexpression of CXCR3-B in renal cancer cells can restrict tumor cell growth through the down-regulation of cytoprotective molecules (17). Gacci et al. (18) demonstrated that the expression of CXCR3-B is possibly correlated with tumor necrosis. However, the detailed mechanism(s) of CXCR3-B-mediated negative signals in cancer cells and how they are linked to the regulations of specific transcription factor(s) is not well defined.
The transcription factor Bach-1 functions as a repressor of the enhancers of stress-inducible genes, like heme oxygenase 1 (HO-1), by forming heterodimers with the small Maf proteins (19, 20). The expression of HO-1 can be tightly controlled by the positive regulator nuclear factor E2-related factor 2 (Nrf2) and the negative regulator Bach-1 (19, 21). The HO-1 gene has two important distal enhancer regions, E1 and E2, located upstream of the transcription start site (21, 22). The inducible enhancers of HO-1 carry multiple stress-responsive elements that are closely related to Maf recognition elements. The heterodimers of Nrf2 and small Maf proteins activate HO-1 through binding to Maf recognition elements. In contrast, the heterodimers of Bach1 and small Maf proteins (like MafK) repress transcription (20, 23). Depending on a specific type of signal(s), there is either an enhanced nuclear accumulation or nuclear exclusion of Bach1 and Nrf2 to regulate gene expression.
HO-1 is a cytoprotective enzyme that degrades heme into carbon monoxide (CO), biliverdin, and ferrous iron. It classically functions to maintain cellular homeostasis under stress conditions (24). The byproducts of heme degradation play a crucial role in reducing cellular inflammation and apoptosis and inducing cell proliferation and angiogenesis (24, 25). Despite its cytoprotective properties, recent evidence clearly suggests a critical role of HO-1 in promoting cancer (26, 29). HO-1 and its positive regulator, Nrf2, are overexpressed in different types of cancer and can play a significant role in the survival of tumor cells by regulating prosurvival and antiapoptotic pathways (27, 28).
In this study, we show that CXCR3-B mediates a growth-inhibitory signal in human breast cancer cells through the down-regulation of antiapoptotic HO-1. It is associated with decreased phosphorylation of ERK-1/2 and increased phosphorylation of p38 MAPK. In addition, CXCR3-B-induced signals promote increased nuclear translocation of Bach-1 and nuclear export of Nrf2.
EXPERIMENTAL PROCEDURES
Reagents
Gene-specific siRNAs for CXCR3-B along with control siRNA were purchased from Invitrogen. Bach-1 siRNA was purchased from Qiagen. Cells were transfected with siRNA using Lipofectamine 2000 (Invitrogen). The recombinant CXCL4 was purchased from R&D Systems. SB203580 was obtained from Calbiochem. Cobalt protoporphyrine was purchased from Frontier Scientific. The plasmid DNAs were transfected using Effectene transfection reagent (Qiagen).
Cell Lines
Human breast cancer cell lines (MCF-7 and T47D) and primary mammary epithelial cells were obtained from the ATCC. Cancer cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (Invitrogen). Normal epithelial cells were grown in mammary epithelial cell basal medium supplemented with growth factors (ATCC).
Plasmids
The CXCR3-B overexpression plasmid (pcDNA3-CXCR3-B) was a gift from P. Romagnani (University of Florence, Italy) (13). The pCMV-HO-1 plasmid was used to overexpress human HO-1 (29). The human HO-1 promoter-luciferase plasmid was a gift from J. Alam (Alton Ochsner Medical Foundation, New Orleans, LA). The plasmid phHO4luc was constructed by cloning the human HO-1 promoter sequence (bp −4067 to +70 relative to the transcription start site) into the luciferase reporter gene vector pSKluc (27).
RNA Isolation and Real-time PCR
Total RNA was prepared using the RNeasy isolation kit (Qiagen), and cDNA was synthesized using a cloned avian myeloblastosis virus (AMV) first-strand synthesis kit (Invitrogen). To analyze mRNA expression, we performed real-time PCR using the Assay-on-Demand Gene Expression product (TaqMan, mammalian gene collection probes) according to the instructions of the manufacturer (Applied Biosystems). As an internal control, GAPDH mRNA was amplified and analyzed under identical conditions. Gene-specific primer-probe sets were obtained from Applied Biosystems. The Ct value (the cycle number at which emitted fluorescence exceeded an automatically determined threshold) for the gene of interest was corrected by the Ct value for GAPDH and expressed as ΔCt. The fold change of the mRNA amount was calculated as follows: (fold changes) = 2X (where X = ΔCt for the control group − ΔCt for the experimental group).
Luciferase Assays
Breast cancer cells (0.5 × 105) were cotransfected with the HO-1 promoter-luciferase plasmid and the CXCR3-B overexpression plasmid. The cells were then treated with CXCL4/vehicle alone as described. The cells were harvested 48 h after transfection, and luciferase activity was quantified using a standard assay kit (Promega) in a luminometer. As an internal control for transfection efficiency, the cells were cotransfected with the β-galactosidase gene under the control of the cytomegalovirus immediate early promoter, and β-galactosidase activity was measured by a standard assay system (Promega). Luciferase activity values were corrected for the transfection efficiency by calculating the ratio of luciferase units to β-galactosidase units and represented as relative luciferase counts.
Western Blot Analysis
Protein samples were run on SDS-polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Millipore Corp.). The membranes were incubated with anti-HO-1 (R&D Systems); anti-CXCR3-B (Proteintech); anti-Nrf2, anti-Bach-1, and anti-Sp1 (Santa Cruz Biotechnology); anti-p38, anti-phospho-p38, anti-ERK-1/2, anti-phospho-ERK-1/2, anti-Bcl-xL, anti-Bcl-2, and anti-poly(ADP-ribose) polymerase (Cell Signaling); or anti-Beclin-1, anti-LC3B, anti-β-Actin, and anti-GAPDH (Sigma-Aldrich) and subsequently incubated with peroxidase-linked secondary antibody. Reactive bands were detected using chemiluminescent substrate (Pierce).
Preparation of Nuclear Extracts
Nuclear extracts were prepared from the cells using a nuclear extraction kit (Active Motif). Following treatment, cells were collected in PBS in the presence of phosphatase inhibitors. The cells were resuspended in hypotonic buffer (supplied with the kit) and incubated on ice for 15 min. Cytoplasmic fractions were collected after adding detergent (supplied with the kit) and centrifuging the cells. The nuclear pellets were resuspended in complete lysis buffer and incubated on ice for 30 min. The nuclear fractions were collected by centrifuging the cells for 10 min.
Cell Proliferation Assay
Cell proliferation was measured by MTT2 cell proliferation assay (ATCC) following the protocol of the manufacturer. Briefly, cells were plated in 96-well plates. Following transfection/ligand treatment, 10 μl of MTT reagent was added to each well. When purple crystals of formazan became clearly visible under the microscope, 100 μl of detergent reagent was added, and the cells were incubated in the dark for 4 h. Absorbance was measured at 570 nm and corrected against blanks, which consisted of culture medium processed in the same way as above in the absence of cells. The reading at 570 nm is directly proportional to cell proliferation (number of viable cells).
Apoptosis Assay
Cellular apoptosis was measured by annexin V and propidium iodide staining using an allophycocyanin (APC) annexin V apoptosis detection kit (eBioscience) according to the protocol of the manufacturer. Briefly, 0.5 × 105 cells were seeded into each well of a 6-well plate and treated as described. For the assay, the cells were trypsinized, washed with PBS and 1× binding buffer, and then resuspended in the binding buffer at a concentration of 1–2 × 106 cells/ml. 5 μl of APC-conjugated annexin V was added to 100 μl of the cell suspension and incubated at room temperature for 15 min. The cells were washed and resuspended in 200 μl of binding buffer. 5 μl of propidium iodide was added to the cells, and the cells were analyzed by flow cytometry on a FACSCalibur.
Autophagy Assay
Cellular autophagy was monitored using a Cyto-ID autophagy detection kit (Enzo Life Sciences) following the protocol of the manufacturer. The 488-nm excitable Cyto-ID green autophagy detection reagent supplied with the kit becomes brightly fluorescent in vesicles produced during autophagy and, thus, serves as a convenient tool to detect autophagy at a cellular level. Following treatment, the cells were trypsinized, washed in PBS, and resuspended in 2000× dilution of the detection reagent. After 30 min of incubation at 37 °C, the cells were washed and analyzed by flow cytometry. Another group of Cyto-ID stained cells were mixed with Prolong Gold (Invitrogen) at a 1:1 ratio, allowed to settle for 5 min on a glass slide, and then sealed with a coverslip and visualized using a Zeiss epifluorescence microscope with appropriate filters. Nuclei were counterstained in blue with Hoechst 33342 dye.
Detection of Reactive Oxygen Species (ROS)
Cellular ROS were measured using a total ROS detection kit (Enzo Life Sciences). This kit is designed to directly monitor real-time ROS production in live cells using flow cytometry. The kit includes an oxidative stress detection reagent (green) as the major component. This non-fluorescent, cell-permeable, total ROS detection dye reacts directly with a wide range of reactive species, yielding a green fluorescent product indicative of cellular production of ROS.
Statistical Analysis
Statistical significance was determined by Student's t test. Differences with p < 0.05 were considered statistically significant.
RESULTS
Induction of CXCR3-B-medited Signaling Promotes Apoptosis and Inhibits Autophagy of Breast Cancer Cells
We observed that the expression of CXCR3-B is much lower in breast cancer cells compared with normal breast epithelial cells (Fig. 1A, top left panel). To examine the effect of CXCR3-B-mediated signaling in regulating the cell death pathways of human breast cancer cells (MCF-7 and T47D), we made use of a specific CXCR3-B overexpression plasmid. We first confirmed that the transfection of breast cancer cells with the CXCR3-B plasmid significantly overexpressed the gene at both the mRNA and protein level (Fig. 1A, top right and bottom panels). We then checked the effect of CXCR3-B-induced signal on the apoptotic regulation of breast cancer cells. Cells were transfected with either the CXCR3-B plasmid or an empty vector and then treated with either the CXCR3-B-specific ligand CXCL4 or vehicle alone. Following treatment, the cells were stained with annexin V and propidium iodide and analyzed by flow cytometry to check the apoptotic index. As shown in Fig. 1B (top panels), the transfection with CXCR3-B in MCF-7 cells increased cellular apoptosis compared with vector-transfected cells. The percentage of total apoptotic cells (early and late) increased from 2.01% (1.53 + 0.48%) to 16.64% (4.76 + 11.88%). We suggest that CXCR3-B interacts in an autocrine manner with CXCR3-binding ligands that are overexpressed in cancer cells (11, 16) and, thus, generates an apoptotic signal. Next, we treated the CXCR3-B-transfected cells with its specific ligand, CXCL4, using the same concentration (250 ng/ml) as in our previous study (11). As shown in Fig. 1B, CXCL4 treatment markedly increased cellular apoptosis compared with vector-transfected and vehicle-treated cells. The percentage of total apoptotic cells (early and late) increased from 2.75% (1.76 + 0.99%) to 24.16% (9.17 + 14.99%). We found a similar result in T47D cells (data not shown). Similar results were obtained with the lower concentrations (50–100 ng/ml) of CXCL4 (supplemental Fig. 1A). The induction of CXCR3-B-mediated signaling in normal breast epithelial cells also induced apoptosis (supplemental Fig. 1B). In addition, we observed that CXCR3-B-mediated signaling significantly inhibited breast cancer cell proliferation compared with vector-transfected cells, as observed by MTT assay (Fig. 1C).
FIGURE 1.

CXCR3-B induces apoptosis and inhibits autophagy of breast cancer cells. A, the mRNA expression of CXCR3-B in normal breast epithelial cells and MCF-7 cells was analyzed by real-time PCR (top left panel). MCF-7 cells were transfected with either the CXCR3-B overexpression plasmid (0.5 μg) or the empty vector. Cells were harvested after 48 h, and the expression of CXCR3-B mRNA, CXCR3-B, and total CXCR3 (A and B) protein were analyzed by real-time PCR (top right panel), Western blot analysis (bottom left panel), and FACS analysis (bottom right panel), respectively. The lower Ct value reflects overexpression of the gene. B, MCF-7 cells were transfected with either the CXCR3-B overexpression plasmid (0.5 μg) or the empty vector and then treated with either CXCL4 (250 ng/ml) or vehicle alone. After 48 h of plasmid transfection, the apoptotic index of cells was determined by annexin V (APC) and propidium iodide staining as described under “Experimental Procedures.” C, MCF-7 cells were transfected with either the CXCR3-B overexpression plasmid (0.5 μg) or the empty vector and then treated with CXCL4 (250 ng/ml). After 72 h, cell proliferation was measured by MTT assay. D, cell lysate from B was used to measure the expression levels of poly(ADP-ribose) polymerase (PARP), Bcl-2, Bcl-xL, and β-actin (internal control) by Western blot analysis. E, MCF-7 cells were transfected with either the CXCR3-B overexpression plasmid (0.5 μg) or the empty vector and then treated with CXCL4 (250 ng/ml). After 48 h, cellular autophagy was measured by flow cytometry by staining the cells with Cyto-ID green autophagy detection reagent (left panel). The morphologies of these Cyto-ID-stained cells were analyzed under a fluorescence microscope (center panel). Cell lysates were used to check the expression of the autophagic markers Beclin-1, LC3B (I and II), and β-actin by Western blot analysis (right panel). A and C, the columns represent the mean ± S.D. of triplicate readings of two samples. *, p < 0.05. B, D, and E, data are representative of three independent experiments.
Next, we evaluated how CXCR3-B can modulate the expression of apoptotic and antiapoptotic molecules in breast cancer cells. MCF-7 cells were transfected with the CXCR3-B plasmid/empty vector and then treated with either CXCL4 or vehicle alone. As shown in Fig. 1D, the CXCR3-B-mediated signal (both in the absence or presence of CXCL4) increased the expression of apoptotic cleaved poly(ADP-ribose) polymerase. CXCR3-B also decreased the expression of antiapoptotic Bcl-2 and Bcl-xL compared with vector-transfected cells.
It has been shown that the process of autophagy (type II cell death) can be induced to promote the survival of breast tumor cells under hypoxic or nutrient-deprived condition (30, 31). Here we tested whether a CXCR3-B-mediated signal can alter autophagy in breast cancer cells. The cells were transfected with either the CXCR3-B plasmid or an empty vector and then treated with CXCL4. Following treatment, the cells were stained with Cyto-ID green autophagy detection reagent and analyzed by flow cytometry. As shown in Fig. 1E (left panel), in both MCF-7 and T47D cells (data not shown), the CXCR3-B-CXCL4-mediated signal markedly inhibited cellular autophagy, as observed by the left-hand shift of the histogram. We analyzed the morphology of these cells under a fluorescence microscope. We found that the overexpression of CXCR3-B is associated with a decrease in the intensity of green autophagic vesicles representing the punctate structures (Fig. 1E, center panel). We also confirmed by Western blot analysis that CXCR3-B down-regulated the expression of the autophagic proteins Beclin-1 and LC3B-II (Fig. 1E, right panel). Together, our findings clearly suggest that CXCR3-B can mediate a novel growth-inhibitory signal in breast cancer cells to promote apoptosis and down-regulate autophagy.
Knockdown of CXCR3-B Promotes the Survival and Proliferation of Breast Cancer Cells
As mentioned earlier, CXCR3-B is expressed at moderate to low levels in breast cancer cells. We utilized CXCR3-B-specific siRNA to knock down the gene and confirm the role of CXCR3-B in regulating the apoptosis and proliferation of breast cancer cells. We first checked that the siRNA significantly knocked down CXCR3-B at both the mRNA and protein levels (Fig. 2A). As shown in Fig. 2B, the knockdown of CXCR3-B markedly decreased cellular apoptosis compared with control cells. The total apoptotic cells (early and late) decreased from 3.29% (1.05 + 2.24%) to 1.41% (0.38 + 1.03%). We also found that CXCR3-B knockdown increased breast cancer cell proliferation compared with control cells (Fig. 2C).
FIGURE 2.

Knockdown of CXCR3-B is associated with the survival and proliferation of breast cancer cells. MCF-7 cells were transfected with either CXCR3-B siRNA (50 nm) or control siRNA for 72 h. A, cells were harvested, and the expression of CXCR3-B mRNA and protein was analyzed by real-time PCR (left panel) and Western blot analysis (right panel). B, the apoptotic index of siRNA-transfected cells was determined by annexin V (APC) and propidium iodide staining. C, cell proliferation of siRNA-transfected cells was measured by MTT assay. A and C, the columns represent the mean ± S.D. of triplicate readings of two different samples. *, p < 0.05. B, results are representative of three independent experiments.
CXCR3-B-mediated Signaling Down-regulates the Expression of Antiapoptotic HO-1
We have demonstrated that HO-1 can promote survival of cancer cells through the inhibition of apoptosis (29). Here, we examined whether CXCR3-B can regulate HO-1 expression in breast cancer cells. MCF-7 cells were transfected with either the CXCR3-B plasmid or an empty vector and then treated with CXCL4/vehicle alone. We found that the CXCR3-B-mediated signal (both in the absence or presence of CXCL4) significantly down-regulated HO-1 expression at both the mRNA and protein levels compared with control cells, as observed by real-time PCR and Western blot analysis (Fig. 3, A and B). We also confirmed that siRNA-mediated knockdown of CXCR3-B increased HO-1 protein expression (Fig. 3C). Finally, by using the HO-1 promoter-luciferase plasmid construct, we observed that the CXCR3-B-mediated signal down-regulated HO-1 promoter activity, as observed by luciferase assay (Fig. 3D).
FIGURE 3.

CXCR3-B down-regulates the expression of HO-1. A and B, MCF-7 cells were transfected with either the CXCR3-B overexpression plasmid (0. 5 μg) or an empty vector and then treated with either CXCL4 (250 ng/ml) or vehicle alone. After 48 h of plasmid transfection, cells were lysed, and the expression of HO-1 mRNA and protein was measured by real-time PCR (A) and Western blot analysis (B). The expression of β-actin was measured as an internal control. C, MCF-7 cells were transfected with either CXCR3-B siRNA (50 nm or 25 nm) or control siRNA. After 72 h of siRNA transfection, cells were lysed, and HO-1 protein expression was measured by Western blot analysis. D, MCF-7 cells were cotransfected with the HO-1 promoter-luciferase plasmid (0.5 μg) and either the CXCR3-B plasmid (0.5 μg) or an empty vector. The cells were then treated with either CXCL4 (250 ng/ml) or vehicle alone. Following 48 h of transfection, cells were harvested, and HO-1 promoter activity was measured by luciferase assay. The percent decrease in luciferase activity was calculated from luciferase counts of each group of cells compared with that of cells transfected with the empty vector and treated with vehicle alone. A and C, the columns represent the mean ± S.D. of triplicate readings of two different samples. *, p < 0.05 compared with respective controls. B and D, data are representative of three independent experiments.
CXCR3-B Regulates the Expression of Bach-1 and Nrf2
The transcription of HO-1 is tightly controlled by the negative regulator Bach-1 and the positive regulator Nrf2 (21, 32). Because we found that the CXCR3-B-mediated signal regulates HO-1 expression, we wished to study whether CXCR3-B can modulate the expression of Bach-1 and Nrf2. We evaluated whether CXCR3-B can mediate any change in nuclear versus cytoplasmic localization of Bach-1 and Nrf2. To this end, breast cancer cells were transfected with either the CXCR3-B plasmid or an empty vector, and then nuclear and cytoplasmic fractions were isolated. The expression of Bach-1 and Nrf2 was measured by Western blot analysis. As shown in Fig-4A (left panel), the overexpression of CXCR3-B markedly increased Bach-1 and decreased Nrf2 expression in the nuclear fraction compared with the vector-transfected control. On the other hand, CXCR3-B overexpression decreased Bach-1 and increased Nrf2 expression in the cytoplasmic fraction (Fig. 4A, right panel). We also confirmed our findings through the knockdown of CXCR3-B using siRNA. As shown in Fig. 4B (left panel), the knockdown of CXCR3-B decreased Bach-1 and increased Nrf2 expression in the nuclear fractions compared with controls. In contrast, CXCR3-B knockdown increased Bach-1 and decreased Nrf2 expression in the cytoplasmic fractions (Fig. 4B, right panel). We found that there was no significant change in the expression of Bach-1 and Nrf2 total protein following CXCR3-B overexpression (data not shown). Together, our findings suggest that the CXCR3-B-mediated signal can induce nuclear translocation of Bach-1 and nuclear export of Nrf2, which can be associated with the down-regulation of HO-1 transcription.
FIGURE 4.
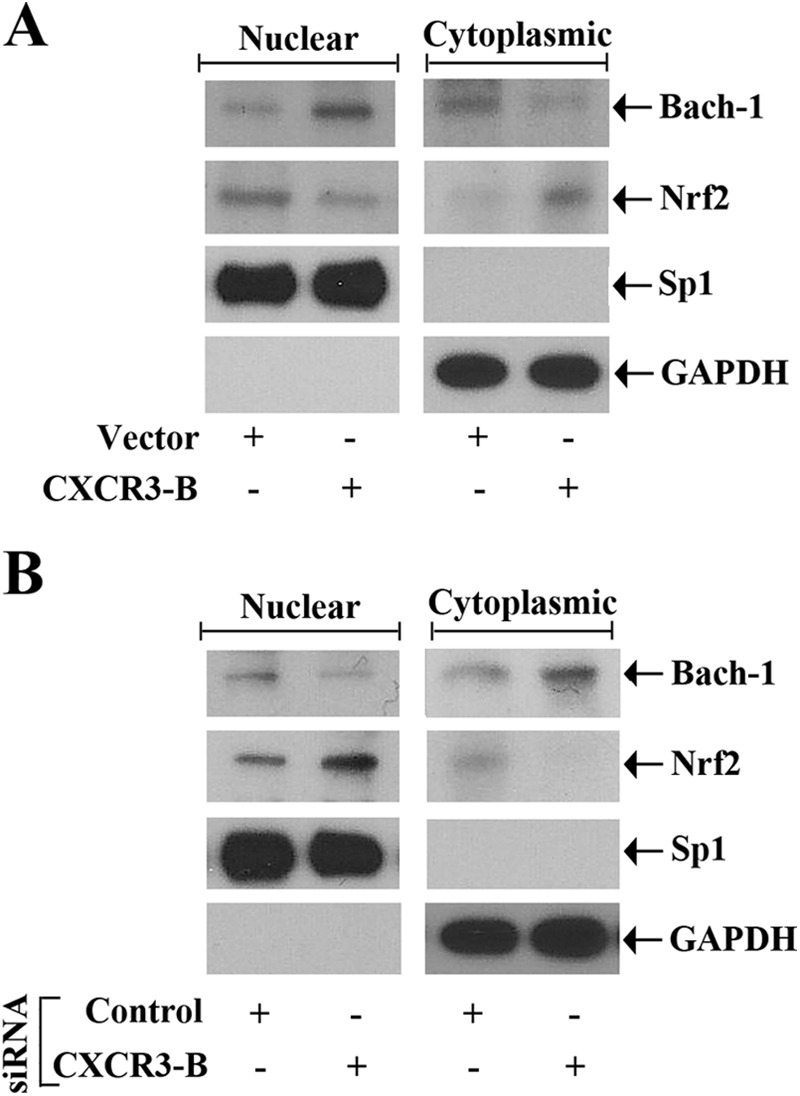
CXCR3-B-mediated signaling modulates Bach-1 and Nrf-2 nuclear localization. A, MCF-7 cells were transfected with either the CXCR3-B overexpression plasmid (0. 5 μg) or the empty vector. B, MCF-7 cells were transfected with either CXCR3-B siRNA (50 nm) or control siRNA. Following 48 h of plasmid transfection (A) and 72 h of siRNA transfection (B), nuclear and cytoplasmic fractions were isolated from these cells, and a Western blot analysis was performed to quantitate the expression of Bach-1 and Nrf2. The purities of nuclear and cytoplasmic fractions were evaluated by measuring the expression of Sp1 and GAPDH, respectively. Results shown are representative of three independent experiments.
The CXCR3-B-mediated Signal Activates p38 MAPK and Inhibits ERK-1/2 Phosphorylation
We and others have shown that the activation of Nrf2 and Bach-1 can be regulated by the members of the MAPK family, which can promote both the positive and negative signal (19, 21, 27, 32). Because we observed that the CXCR3-B-mediated signal regulates the expression of Nrf2 and Bach-1, we wished to check whether CXCR3-B can modulate the activation of ERK-1/2 and p38 MAPK. The breast cancer cells were transfected with either the CXCR3-B plasmid or an empty vector, and the cells were treated with CXCL4. Following treatment, the phosphorylation statuses of p38 and ERK-1/2 were measured by Western blot analysis. As shown in Fig. 5A, the CXCR3-B-mediated signal increased the phosphorylation of p38 MAPK and decreased the phosphorylation of ERK-1/2. There was no change in the expression of total p38 and total ERK-1/2. We also confirmed our findings by using CXCR3-B siRNA. As shown in Fig. 5B, the knockdown of CXCR3-B markedly decreased p38 phosphorylation and increased ERK-1/2 phosphorylation. In addition, we found that, when we blocked p38 activity by treating the cells with a specific pharmacological inhibitor (SB203580), CXCR3-B-induced nuclear translocation of Bach-1 was markedly down-regulated (Fig. 5C). However, there was no significant change (data not shown) in CXCR3-B-mediated nuclear export of Nrf2 following p38 inhibition. Thus, we suggest that the CXCR3-B-mediated negative signal may involve the activation of p38 and inactivation of ERK-1/2.
FIGURE 5.

CXCR3-B-mediated signaling activates p38 MAPK and inhibits ERK1/2 phosphorylation. A, MCF-7 cells were transfected with either the CXCR3-B overexpression plasmid (0. 5 μg) or the empty vector and then treated with CXCL4 (250 ng/ml). B, MCF-7 cells were transfected with either CXCR3-B siRNA (50 nm) or control siRNA. Following 48 h of plasmid transfection (A) and 72 h of siRNA transfection (B), cells were lysed, and a Western blot analysis was performed to measure the expression levels of phospho-p38, phopsho-ERK1/2 (p-ERK), total p38, total ERK1/2, and β-actin. C and D, MCF-7 cells were transfected with either the CXCR3-B overexpression plasmid (0.5 μg) or the empty vector and then treated with either SB203580 (25 μm) or vehicle alone for 48 h. C, nuclear and cytoplasmic fractions were isolated from these cells, and a Western blot analysis was performed to quantitate the expression of Bach-1. D, the apoptotic index of the cells was determined by annexin V (APC) and propidium iodide staining. A–D, results are representative of three independent experiments.
Next, we examined whether the inhibition of p38 MAPK can decrease the apoptotic effect of CXCR3-B. MCF-7 cells were transfected with the CXCR3-B plasmid/empty vector and then treated with either the p38 inhibitor SB203580 or vehicle alone. Following treatment, the cells were stained with annexin V and propidium iodide and analyzed by flow cytometry to check the apoptotic index. As shown in Fig. 5D, CXCR3-B markedly increased cellular apoptosis in vehicle-treated cells. The total apoptotic cells (early and late) increased from 2.78% (0.99 + 1.79%) to 22.03% (7.93 + 14.1%). However, CXCR3-B could not induce apoptosis up to a similar level in cells treated with p38 inhibitor. The total of apoptotic cells increased from 2.79% to only 13.88% (5.28 + 8.60%). Thus, the activation of p38 MAPK can play an important role in mediating CXCR3-B-induced growth-inhibitory signaling in breast cancer cells.
Knockdown of Bach-1 Down-regulates the Growth-inhibitory Functions of CXCR3-B
Our findings indicate that CXCR3-B can induce the nuclear translocation of Bach-1, which is known to negatively regulate HO-1. Here, we tested the effects of Bach-1 knockdown on growth-inhibitory functions of CXCR3-B. We first confirmed that the transfection of breast cancer cells with Bach-1 siRNA significantly decreased the expression of Bach-1 and increased HO-1 (Fig. 6A). To study the role of Bach-1 on CXCR3-B-mediated breast cancer cell proliferation and apoptosis, we first transfected MCF-7 cells with either Bach-1 siRNA or control siRNA. Following siRNA transfection, the cells were transfected with either CXCR3-B or an empty vector. Cells were then subjected to cell proliferation and apoptosis assays as described before. We found that CXCR3-B decreased cancer cell proliferation in control cells. However, following knockdown of Bach-1, CXCR3-B could not down-regulate cell proliferation up to a similar level as that observed in control cells (Fig. 6B). Similarly, we observed that CXCR3-B markedly increased cellular apoptosis in control cells. The total apoptotic cells (early and late) increased from 9.19% (4.06 + 5.13%) to 21.3% (10.3 + 11.0%) (Fig. 6C). However, following knockdown of Bach-1, it could not promote apoptosis up to a similar level as that observed in control cells. The total apoptotic cells increased from 9.19% to only 11.0% (4.22 + 6.78%). Thus, our observations suggest that Bach-1 is one of the critical mediators for CXCR3-B-induced growth-inhibitory functions.
FIGURE 6.

Inhibition of Bach-1 down-regulates the growth-inhibitory functions of CXCR3-B. A, MCF-7 cells were transfected with either Bach-1 siRNA (50 nm) or control siRNA. After 72 h of transfection, the mRNA expression of Bach-1 and HO-1 was checked by real-time PCR. B and C, MCF-7 cells were first transfected with either Bach-1 siRNA (50 nm) or control siRNA. Following 24 h of siRNA transfection, cells were transfected with either the CXCR3-B overexpression plasmid (0.5 μg) or an empty vector. After 48 h of plasmid transfection, either an MTT assay was performed to measure cell proliferation (B), or the apoptotic index of the cells was determined by annexin V (APC) and propidium iodide staining (C). A and B, the columns represent the mean ± S.D. of triplicate readings from two different samples. *, p < 0.05. C, results are representative of three independent experiments.
Overexpression of HO-1 Down-regulates the Growth-inhibitory Functions of CXCR3-B
As discussed, Bach-1 negatively regulates antiapoptotic HO-1 (19), and we found that the CXCR3-B-mediated signal activates Bach-1 and inhibits HO-1. Here, we evaluated how the growth-inhibitory functions of CXCR3-B can be altered if we overexpress HO-1 in cancer cells. We transfected breast cancer cells with the CXCR3-B plasmid in the absence or presence of the HO-1 overexpression plasmid. As shown in Fig. 7A, CXCR3-B increased the apoptosis of breast cancer cells. The total apoptotic cells (early and late) increased from 1.56% (0.54 + 1.02%) to 15.29% (3.09 + 12.2%). However, in the presence of overexpressed HO-1, CXCR3-B could not induce apoptosis up to a similar level as that observed in control cells. The total apoptotic cells increased from 1.56% to 10.83% (1.71 + 9.12%). The overexpression of HO-1 was confirmed by Western blot analysis (Fig. 7A, right panel). As an alternate approach, we also treated the cells with cobalt protoporphyrine, which is a known inducer of HO-1. We found that, following cobalt protoporphyrine treatment, there was also a down-regulation of CXCR3-B-mediated apoptosis of breast cancer cells (Fig. 7B). We also observed that, in the presence of overexpressed HO-1, CXCR3-B could not decrease cancer cell proliferation up to a similar level as that observed in control cells (Fig. 7C).
FIGURE 7.

Overexpression of HO-1 down-regulates the growth-inhibitory functions of CXCR3-B. A, C, and D, MCF-7 cells were cotransfected with different combinations of CXCR3-B plasmid (0. 5 μg) or the HO-1 overexpression plasmid (0.5 μg). Control cells were transfected with respective empty vectors. B, MCF-7 cells were transfected with the CXCR3-B plasmid (0.5 μg) or an empty vector and then treated with Cobalt protoporphyrine (CoPP) (50 μm) or vehicle alone. After 48 h of transfection/treatment, the following assays were performed. A and B, the apoptotic index of the cells was determined by annexin V (APC) and propidium iodide staining. The overexpression of HO-1 following plasmid transfection was measured by Western blot analysis (A, right panel). C, cell proliferation was determined by MTT assay. D, cells were stained with oxidative stress detection reagent and analyzed for cellular ROS by flow cytometry. The mean fluorescence intensity is represented as bar graphs. A and B, results are representative of three independent experiments. C and D, the columns represent the mean ± S.D. of triplicate readings of two independent experiments. *, p < 0.05.
It is known that the generation of increased ROS can promote cellular apoptosis (33). Here, we checked whether CXCR3-B can modulate the level of ROS in breast cancer cells and whether it can be altered in the presence of HO-1. We measured the cellular ROS using a total ROS detection kit. As shown in Fig. 7D, CXCR3-B increased cellular ROS compared with control cells. However, in the presence of overexpressed HO-1, CXCR3-B could not increase cellular ROS up to a similar level. Together, our observations clearly suggest that CXCR3-B can mediate a novel growth-inhibitory signal, which involves modulation in the expression of antiapoptotic HO-1.
DISCUSSION
The mechanism(s) by which the chemokine receptor CXCR3-B can mediate novel growth-inhibitory signals in cancer cells is not well defined. In this study, we show that CXCR3-B-mediated signaling in human breast cancer cells promotes cellular apoptosis through the down-regulation of antiapoptotic HO-1. It is associated with an increased nuclear translocation of Bach-1 and nuclear export of Nrf2, which are critical positive and negative regulators of HO-1 transcription. We also demonstrate that CXCR3-B mediates the inactivation of ERK-1/2 and promotes increased phosphorylation of p38 MAPK.
The chemokine receptor CXCR3 is expressed on immune cells and also in different types of cancer cells, including renal and breast cancer cells (5, 6, 8, 11, 12). Classically, the tumor infiltration of CXCR3-positive immune cells can promote antitumor immunity. However, it has now been established that CXCR3 and its ligands may play important roles in the development and progression of human tumors by facilitating the migration and proliferation of malignant cells (3, 8, 12). Interestingly, as discussed earlier, the two splice variants of CXCR3 (CXCR3-A and CXCR3-B) can have completely opposite functions in terms of regulating tumor progression versus tumor inhibition (11, 16). It has been shown that CXCR3-B can mediate novel negative signal(s) for growth inhibition (13). There are some reports regarding CXCR3-B-mediated antitumorigenic functions. However, the nature of CXCR3-B-mediated signals in cancer cells has not yet been thoroughly explored. Wu et al. (15) have shown that the aberrant expression of CXCR3-A and down-regulation of CXCR3-B may promote the metastasis of prostate tumors by stimulating cell migration and invasion. We have demonstrated previously that the expression of CXCR3-B is markedly down-regulated in tumor tissues (16) and that the activation of the Ras-Raf pathway can inhibit the expression of CXCR3-B in breast cancer cells (11). The down-regulation CXCR3-B may also be associated with a shift in the splice variant. However, we found that, following either the overexpression or the knockdown of CXCR3-B in breast cancer cells, there was no significant change (data not shown) in the expression of CXCR3-A. We suggest that the down-regulation of CXCR3-B-mediated negative signals may become advantageous for the survival of tumors. In absence/low presence of CXCR3-B, the CXCR3-binding ligands (like CXCL10) may promote a rapid growth of cancer cells, possibly through CXCR3-A.
The apoptotic mechanism(s) of tumor cells often become(s) defective and favors tumor growth. We have demonstrated recently that the antiapoptotic HO-1 is up-regulated in tumor cells and can play a major role in cancer cell survival (27, 29). Here we found that the activation of CXCR3-B-mediated signaling promotes apoptosis and inhibits proliferation of breast cancer cells and that the expression of HO-1 is significantly down-regulated following CXCR3-B stimulation. Recent studies clearly suggest that, along with apoptosis, the process of autophagy can also play a crucial role in regulating tumor growth (30, 31). Under nutrient-deficient condition, autophagy can promote tumor growth through the recycling of essential nutrients. We have observed that the CXCR3-B-mediated signal down-regulates autophagy in breast cancer cells. It has been established that there can be a reverse correlation in the regulation of apoptosis and autophagy (34), which is clearly supportive to our findings. In addition, it has been demonstrated that autophagy can delay apoptotic death in MCF-7 cells (35). Thus, CXCR3-B can promote breast cancer cell death through the regulation of both apoptosis and autophagy, where apoptosis is induced and autophagy is inhibited.
The transcription of HO-1 can be tightly controlled by both positive and negative regulators (19, 21, 32). Here, we found that the CXCR3-B-mediated signal promotes the nuclear translocation of Bach-1, which is a negative regulator of HO-1. It is also associated with nuclear export of Nrf2, a positive regulator of HO-1 expression. Thus, it appears that CXCR3-B inhibits HO-1 transcription through the modulation of nuclear Bach-1 and Nrf2. We have demonstrated recently that the activation of ERK-1/2 in cancer cells plays a major role in the nuclear translocation of Nrf2 (27). In addition, it has been suggested that ERK-1/2 is required to inactivate Bach-1 (20). Our data suggest that CXCR3-B may promote nuclear translocation of Bach-1 and nuclear export of Nrf2 through the inactivation of ERK-1/2. We also observed that the CXCR3-B-induced signal in breast cancer cells promotes increased phosphorylation of p38 MAPK. In support of our finding, Petrai et al. (36) demonstrated that the activation of p38 can mediate the angiostatic effects of CXCR3-B. Also, it has been shown that p38 can promote apoptosis through the activation of Bad and inhibition of ERK-1/2 (37). Further, cells lacking p38α have been shown to be more resistant to apoptosis induced by different stimuli (38), and this is associated with increased nuclear accumulation of Nrf2 and its binding to the HO-1 promoter (39). In this study, we observed that, when the breast cancer cells were pretreated with a p38 MAPK pharmacological inhibitor (SB203580), CXCR3-B-induced cellular apoptosis is much reduced compared with control cells. We suggest that CXCR3-B may regulate a critical balance for the activation statuses of ERK-1/2 and p38 MAPK to mediate cancer cell apoptosis through the modulation of HO-1 expression. In support of our findings, Deng et al. (33) have shown that the treatment of MCF-7 cells with rotenone, an inhibitor of the mitochondrial electron transport chain, can cause the activation of p38 MAPK and the inactivation of ERK-1/2.
In conclusion, we suggest that that CXCR3-B can mediate novel signal(s) to promote growth inhibition and apoptosis of cancer cells. It modulates the activation of p38 MAPK and ERK-1/2 and is associated with increased nuclear translocation of Bach-1 and nuclear export of Nrf2, which leads to the decreased expression of antiapoptotic HO-1 as represented in the schematic in Fig. 8. Together, the induction of CXCR3-B-mediated signaling can serve as a novel therapeutic approach for the treatment of cancer where the goal is to promote tumor cell apoptosis.
FIGURE 8.
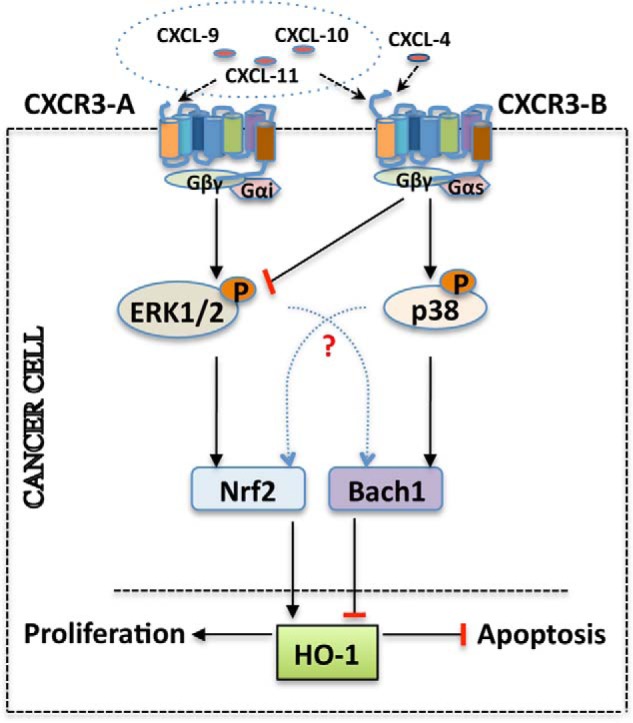
Schematic representing CXCR3-B-mediated growth-inhibitory pathways in breast cancer cells. The induction of CXCR3-B through its ligands promotes the activation of p38 MAPK and inhibition of ERK-1/2, and it is associated with an increased nuclear localization of Bach-1 and nuclear export of Nrf2. The modulation of Bach-1/Nrf2 nuclear localization through CXCR3-B-mediated signaling down-regulates HO-1, and these events can promote increased apoptosis and reduced proliferation of breast cancer cells.
Acknowledgments
We thank Dr. Arun T. Pores-Fernando and Evelyn Flynn for technical assistance, and also Dr. Pallavi Banerjee for helpful suggestions.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 CA131145 and R21 CA172946 (to S. P.).

This article contains supplemental Fig. 1.
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- ROS
- reactive oxygen species.
REFERENCES
- 1. Zlotnik A., Yoshie O. (2000) Chemokines. A new classification system and their role in immunity. Immunity 12, 121–127 [DOI] [PubMed] [Google Scholar]
- 2. Balkwill F., Mantovani A. (2001) Inflammation and cancer. Back to Virchow? Lancet 357, 539–545 [DOI] [PubMed] [Google Scholar]
- 3. Vandercappellen J., Van Damme J., Struyf S. (2008) The role of CXC chemokines and their receptors in cancer. Cancer Lett. 267, 226–244 [DOI] [PubMed] [Google Scholar]
- 4. Zipin-Roitman A., Meshel T., Sagi-Assif O., Shalmon B., Avivi C., Pfeffer R. M., Witz I. P., Ben-Baruch A. (2007) CXCL10 promotes invasion-related properties in human colorectal carcinoma cells. Cancer Res. 67, 3396–3405 [DOI] [PubMed] [Google Scholar]
- 5. Walser T. C., Rifat S., Ma X., Kundu N., Ward C., Goloubeva O., Johnson M. G., Medina J. C., Collins T. L., Fulton A. M. (2006) Antagonism of CXCR3 inhibits lung metastasis in a murine model of metastatic breast cancer. Cancer Res. 66, 7701–7707 [DOI] [PubMed] [Google Scholar]
- 6. Suyama T., Furuya M., Nishiyama M., Kasuya Y., Kimura S., Ichikawa T., Ueda T., Nikaido T., Ito H., Ishikura H. (2005) Up-regulation of the interferon γ (IFN-γ)-inducible chemokines IFN-inducible T-cell α chemoattractant and monokine induced by IFN-γ and of their receptor CXC receptor 3 in human renal cell carcinoma. Cancer 103, 258–267 [DOI] [PubMed] [Google Scholar]
- 7. Burns J. M., Summers B. C., Wang Y., Melikian A., Berahovich R., Miao Z., Penfold M. E., Sunshine M. J., Littman D. R., Kuo C. J., Wei K., McMaster B. E., Wright K., Howard M. C., Schall T. J. (2006) A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 203, 2201–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ma X., Norsworthy K., Kundu N., Rodgers W. H., Gimotty P. A., Goloubeva O., Lipsky M., Li Y., Holt D., Fulton A. (2009) CXCR3 expression is associated with poor survival in breast cancer and promotes metastasis in a murine model. Mol. Cancer Ther. 8, 490–498 [DOI] [PubMed] [Google Scholar]
- 9. Proudfoot A. E. (2002) Chemokine receptors. Multifaceted therapeutic targets. Nat. Rev. Immunol. 2, 106–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Romagnani P., Lasagni L., Annunziato F., Serio M., Romagnani S. (2004) CXC chemokines. The regulatory link between inflammation and angiogenesis. Trends Immunol. 25, 201–209 [DOI] [PubMed] [Google Scholar]
- 11. Datta D., Flaxenburg J. A., Laxmanan S., Geehan C., Grimm M., Waaga-Gasser A. M., Briscoe D. M., Pal S. (2006) Ras-induced modulation of CXCL10 and its receptor splice variant CXCR3-B in MDA-MB-435 and MCF-7 cells. Relevance for the development of human breast cancer. Cancer Res. 66, 9509–9518 [DOI] [PubMed] [Google Scholar]
- 12. Kawada K., Hosogi H., Sonoshita M., Sakashita H., Manabe T., Shimahara Y., Sakai Y., Takabayashi A., Oshima M., Taketo M. M. (2007) Chemokine receptor CXCR3 promotes colon cancer metastasis to lymph nodes. Oncogene 26, 4679–4688 [DOI] [PubMed] [Google Scholar]
- 13. Lasagni L., Francalanci M., Annunziato F., Lazzeri E., Giannini S., Cosmi L., Sagrinati C., Mazzinghi B., Orlando C., Maggi E., Marra F., Romagnani S., Serio M., Romagnani P. (2003) An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J. Exp. Med. 197, 1537–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kelsen S. G., Aksoy M. O., Yang Y., Shahabuddin S., Litvin J., Safadi F., Rogers T. J. (2004) The chemokine receptor CXCR3 and its splice variant are expressed in human airway epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 287, L584–591 [DOI] [PubMed] [Google Scholar]
- 15. Wu Q., Dhir R., Wells A. (2012) Altered CXCR3 isoform expression regulates prostate cancer cell migration and invasion. Mol. Cancer 11, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Datta D., Contreras A. G., Grimm M., Waaga-Gasser A. M., Briscoe D. M., Pal S. (2008) Calcineurin inhibitors modulate CXCR3 splice variant expression and mediate renal cancer progression. J. Am. Soc. Nephrol. 19, 2437–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Datta D., Banerjee P., Gasser M., Waaga-Gasser A. M., Pal S. (2010) CXCR3-B can mediate growth-inhibitory signals in human renal cancer cells by down-regulating the expression of heme oxygenase-1. J. Biol. Chem. 285, 36842–36848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gacci M., Serni S., Lapini A., Vittori G., Alessandrini M., Nesi G., Palli D., Carini M. (2009) CXCR3-B expression correlates with tumor necrosis extension in renal cell carcinoma. J. Urol. 181, 843–848 [DOI] [PubMed] [Google Scholar]
- 19. Shan Y., Lambrecht R. W., Ghaziani T., Donohue S. E., Bonkovsky H. L. (2004) Role of Bach-1 in regulation of heme oxygenase-1 in human liver cells. Insights from studies with small interfering RNAS. J. Biol. Chem. 279, 51769–51774 [DOI] [PubMed] [Google Scholar]
- 20. Suzuki H., Tashiro S., Hira S., Sun J., Yamazaki C., Zenke Y., Ikeda-Saito M., Yoshida M., Igarashi K. (2004) Heme regulates gene expression by triggering Crm1-dependent nuclear export of Bach1. EMBO J. 23, 2544–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alam J., Wicks C., Stewart D., Gong P., Touchard C., Otterbein S., Choi A. M., Burow M. E., Tou J. (2000) Mechanism of heme oxygenase-1 gene activation by cadmium in MCF-7 mammary epithelial cells. Role of p38 kinase and Nrf2 transcription factor. J. Biol. Chem. 275, 27694–27702 [DOI] [PubMed] [Google Scholar]
- 22. Ryter S. W., Choi A. M. (2002) Heme oxygenase-1. Molecular mechanisms of gene expression in oxygen-related stress. Antioxid. Redox Signal. 4, 625–632 [DOI] [PubMed] [Google Scholar]
- 23. Sun J., Brand M., Zenke Y., Tashiro S., Groudine M., Igarashi K. (2004) Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc. Natl. Acad. Sci. U.S.A. 101, 1461–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Agarwal A., Nick H. S. (2000) Renal response to tissue injury. Lessons from heme oxygenase-1 gene ablation and expression. J. Am. Soc. Nephrol. 11, 965–973 [DOI] [PubMed] [Google Scholar]
- 25. Nath K. A. (2006) Heme oxygenase-1. A provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 70, 432–443 [DOI] [PubMed] [Google Scholar]
- 26. Jozkowicz A., Was H., Dulak J. (2007) Heme oxygenase-1 in tumors. Is it a false friend? Antioxid. Redox Signal. 9, 2099–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Banerjee P., Basu A., Datta D., Gasser M., Waaga-Gasser A. M., Pal S. (2011) The heme oxygenase-1 protein is overexpressed in human renal cancer cells following activation of the Ras-Raf-ERK pathway and mediates anti-apoptotic signal. J. Biol. Chem. 286, 33580–33590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim T. H., Hur E. G., Kang S. J., Kim J. A., Thapa D., Lee Y. M., Ku S. K., Jung Y., Kwak M. K. (2011) NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Cancer Res. 71, 2260–2275 [DOI] [PubMed] [Google Scholar]
- 29. Banerjee P., Basu A., Wegiel B., Otterbein L. E., Mizumura K., Gasser M., Waaga-Gasser A. M., Choi A. M., Pal S. (2012) Heme oxygenase-1 promotes survival of renal cancer cells through modulation of apoptosis- and autophagy-regulating molecules. J. Biol. Chem. 287, 32113–32123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jain K., Paranandi K. S., Sridharan S., Basu A. (2013) Autophagy in breast cancer and its implications for therapy. Am. J. Cancer Res. 3, 251–265 [PMC free article] [PubMed] [Google Scholar]
- 31. Kimmelman A. C. (2011) The dynamic nature of autophagy in cancer. Genes Dev. 25, 1999–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suzuki H., Tashiro S., Sun J., Doi H., Satomi S., Igarashi K. (2003) Cadmium induces nuclear export of Bach1, a transcriptional repressor of heme oxygenase-1 gene. J. Biol. Chem. 278, 49246–49253 [DOI] [PubMed] [Google Scholar]
- 33. Deng Y. T., Huang H. C., Lin J. K. (2010) Rotenone induces apoptosis in MCF-7 human breast cancer cell-mediated ROS through JNK and p38 signaling. Mol. Carcinog. 49, 141–151 [DOI] [PubMed] [Google Scholar]
- 34. Xu K., Xu P., Yao J. F., Zhang Y. G., Hou W. K., Lu S. M. (2013) Reduced apoptosis correlates with enhanced autophagy in synovial tissues of rheumatoid arthritis. Inflamm. Res. 62, 229–237 [DOI] [PubMed] [Google Scholar]
- 35. Abedin M. J., Wang D., McDonnell M. A., Lehmann U., Kelekar A. (2007) Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 14, 500–510 [DOI] [PubMed] [Google Scholar]
- 36. Petrai I., Rombouts K., Lasagni L., Annunziato F., Cosmi L., Romanelli R. G., Sagrinati C., Mazzinghi B., Pinzani M., Romagnani S., Romagnani P., Marra F. (2008) Activation of p38(MAPK) mediates the angiostatic effect of the chemokine receptor CXCR3-B. Int. J. Biochem. Cell Biol. 40, 1764–1774 [DOI] [PubMed] [Google Scholar]
- 37. Grethe S., Pörn-Ares M. I. (2006) p38 MAPK regulates phosphorylation of Bad via PP2A-dependent suppression of the MEK1/2-ERK1/2 survival pathway in TNF-α induced endothelial apoptosis. Cell Signal. 18, 531–540 [DOI] [PubMed] [Google Scholar]
- 38. Porras A., Zuluaga S., Black E., Valladares A., Alvarez A. M., Ambrosino C., Benito M., Nebreda A. R. (2004) P38 α mitogen-activated protein kinase sensitizes cells to apoptosis induced by different stimuli. Mol. Biol. Cell 15, 922–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Naidu S., Vijayan V., Santoso S., Kietzmann T., Immenschuh S. (2009) Inhibition and genetic deficiency of p38 MAPK up-regulates heme oxygenase-1 gene expression via Nrf2. J. Immunol. 182, 7048–7057 [DOI] [PubMed] [Google Scholar]