

Background: Tight junctions play an essential role in the maintenance of endothelial barrier function.
Results: 15(S)-HETE stimulated ZO-1 phosphorylation at Thr-770/772 residues in PKCϵ-dependent MEK1-ERK1/2 activation disrupting endothelial tight junctions and barrier function.
Conclusion: PKCϵ by interrupting ZO-1 and occludin interactions plays a role in 15(S)-HETE-induced endothelial TJ disruption.
Significance: 12/15-Lipoxygenase appears to be a crucial player in the modulation of endothelial barrier permeability.
Keywords: Eicosanoid, Endothelial Cell, Lipoxygenase Pathway, Signal Transduction, Tight Junctions
Abstract
Disruption of tight junctions (TJs) perturbs endothelial barrier function and promotes inflammation. Previously, we have shown that 15(S)-hydroxyeicosatetraenoic acid (15(S)-HETE), the major 15-lipoxygenase 1 (15-LO1) metabolite of arachidonic acid, by stimulating zona occludens (ZO)-2 tyrosine phosphorylation and its dissociation from claudins 1/5, induces endothelial TJ disruption and its barrier dysfunction. Here, we have studied the role of serine/threonine phosphorylation of TJ proteins in 15(S)-HETE-induced endothelial TJ disruption and its barrier dysfunction. We found that 15(S)-HETE enhances ZO-1 phosphorylation at Thr-770/772 residues via PKCϵ-mediated MEK1-ERK1/2 activation, causing ZO-1 dissociation from occludin, disrupting endothelial TJs and its barrier function, and promoting monocyte transmigration; these effects were reversed by T770A/T772A mutations. In the arteries of WT mice ex vivo, 15(S)-HETE also induced ZO-1 phosphorylation and endothelial TJ disruption in a PKCϵ and MEK1-ERK1/2-dependent manner. In line with these observations, in WT mice high fat diet feeding induced 12/15-lipoxygenase (12/15-LO) expression in the endothelium and caused disruption of its TJs and barrier function. However, in 12/15-LO−/− mice, high fat diet feeding did not cause disruption of endothelial TJs and barrier function. These observations suggest that the 12/15-LO-12/15(S)-HETE axis, in addition to tyrosine phosphorylation of ZO-2, also stimulates threonine phosphorylation of ZO-1 in the mediation of endothelial TJ disruption and its barrier dysfunction.
Introduction
Atherosclerosis is one of the leading causes of death and disability in the world (1, 2). It is characterized by the presence of lipid-laden macrophages and smooth muscle cells as well as other inflammatory cells in the vessel wall (3, 4). The major risk factors of this disease are hypercholesterolemia, dyslipidemia, diabetes, obesity, and smoking, and all of these risk factors are associated with induced expression or enhanced activity of 12/15-lipoxygenase (12/15-LO),2 a murine ortholog of human 15-LO1 (5–9). Both human 15-LO1 and 15-LO2 metabolize arachidonic acid (AA) mainly to 15(S)-hydroperoxyeicosatetraenoic acid, which is subsequently reduced to 15(S)-HETE (10, 11). Furthermore, 15-HETE was found to be the predominant eicosanoid formed in the arteries of Watanabe heritable hyperlipidemic and cholesterol-fed rabbits (12, 13). Besides its role in the conversion of AA, 15-LO1 has been shown to oxidize low density lipoprotein (LDL), a crucial mediator of atherogenesis (14, 15). In addition, either inhibition of 15-LO1 or deficiency of 12/15-LO decreased lipid peroxidation and reduced the burden of atherogenesis (16, 17). Furthermore, overexpression of human 15-LO1 in the endothelium of LDL receptor-deficient mice exacerbated early atherosclerosis (18). Thus, there is a convincing body of evidence for the role of 15-LO1 in atherogenesis. However, besides its capacity in LDL oxidation, the other possible mechanisms by which 15-LO1 (12/15-LO in mice) could play a role in atherogenesis are still not clear.
The endothelium forms a continuous inner lining of the blood vessels and provides a selective nonthrombogenic permeability barrier between the vascular wall and blood (19, 20). Tight junctions (TJs) play a crucial role in the regulation of endothelial barrier function, and their disruption leads to increased paracellular permeability and endothelial dysfunction (21, 22). Endothelial dysfunction is generally regarded as the initial step in atherosclerotic plaque formation (23, 24). Because many risk factors for cardiovascular diseases increase the expression or activity of 12/15-LO and atherosclerotic but not healthy arteries produce 15-HETE, we asked the question whether this lipid molecule alters endothelial barrier function promoting transmigration of monocytes, thereby setting the stage for atherogenesis. In this context, it should be noted that studies from other laboratories have reported that deletion of the 12/15-LO gene reduces vascular permeability in a mouse model of acute lung injury (25). In addition, a recent report showed that 12(S)-HETE by NADPH oxidase-dependent reactive oxygen species production and VEGF receptor-2 tyrosine phosphorylation increases retinal endothelial cell permeability (26). Previously, we have demonstrated that 15(S)-HETE by stimulating ZO-2 tyrosine phosphorylation and its dissociation from claudins 1/5 disrupts endothelial TJs and its barrier function. We also showed that 15(S)-HETE elicits similar effects in mouse arteries ex vivo and deletion of 12/15-LO gene prevents high fat diet-induced disappearance of ZO-2 and claudins 1/5 from aortic endothelial TJs (27). In this study, we asked the question whether 15(S)-HETE also triggers serine/threonine phosphorylation of TJ proteins in the dissemblance of endothelial TJs causing increased vascular permeability. Here, we report that 15(S)-HETE stimulates intracellular TJ protein, zona occludens-1 (ZO-1) phosphorylation at Thr-770/772 residues in PKCϵ-mediated MEK1-ERK1/2 activation and that phosphorylation of these amino acid residues is required for its dissociation from the TJ transmembrane protein, occludin, causing endothelial TJ disruption and its barrier dysfunction, and thereby promoting monocyte transmigration. High fat diet feeding also caused ZO-1 dislocation from endothelial TJs leading to increased vascular permeability only in WT mice but not in 12/15-LO−/− mice.
MATERIALS AND METHODS
Reagents
5(S)-HETE, 12(S)-HETE, 15(R)-HETE, 15(S)-HETE, anti-15-LO1 antibodies, and PD098059 were purchased from Cayman Chemical Co. (Ann Arbor, MI). Growth factor-reduced Matrigel was obtained from BD Biosciences. Evans blue dye, trypan blue dye, fluorescein isothiocyanate/dextran, and thrombin were bought from Sigma. Anti-phospho-p44/42 and anti-phospho-p38 MAPK antibodies were obtained from Cell Signaling Technology (Beverly, MA). Anti-ERK2, anti-Jam-B, anti-p38 MAPK, and anti-GFP antibodies as well as PKCϵ inhibitor peptide were bought from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-phospho-Ser/Thr antibody was purchased from Abcam (Cambridge, MA). Anti-ZO-1, anti-ZO-2, anti-claudin-1, anti-claudin-5, anti-occludin, and anti-Jam-C antibodies, BCECF-AM, Hoechst 33342, goat anti-rabbit, goat anti-mouse, and goat anti-rat secondary antibodies conjugated with Alexa Fluor 568, Alexa Fluor 488, ProLong Gold Antifade reagent, pAd/CMV/V5-GW/lacZ vector, medium 200, low serum growth supplements, and gentamycin/amphotericin solution were from Invitrogen. Anti-Jam-A antibodies were bought from R & D Systems (Minneapolis, MN). Rat anti-mouse CD31 antibodies were obtained from BD Biosciences. All primers and oligonucleotides were synthesized by IDT (Coralville, IA).
Adenoviral Vectors
Construction of Ad-GFP, Ad-dnMEK1, Ax-PKC D/Nδ (Ad-dnPKCδ), and Ax-PKC D/Nϵ (Ad-dnPKCϵ) was described previously (28, 29). Ad-dnPKCζ was constructed by retrieving FLAG.PKCζ.KW from pCMV5.PKCζ.KW vector (30) by digestion with EcoRI and cloning it into the same sites of pENTR3C vector generating pENTR3C-PKCζ.KW. The pENTR3C-PKCζ.KW was subjected to recombination using pAd/CMVV5-DEST vector to generate pAd-dnPKCζ, which was linearized by digestion with PacI and transfected into HEK293 cells. To generate Ad-lacZ, pAd/CMV/V5-GW/lacZ was digested with PacI, gel-purified, and transfected to HEK293A cells. The adenovirus was purified by cesium chloride gradient ultracentrifugation as described previously (28).
Cell Culture
Human umbilical vein endothelial cells (HUVECs) were obtained from Invitrogen and cultured in medium 200 containing low serum growth supplements, 10 μg/ml gentamycin, and 0.25 μg/ml amphotericin B. THP1 cells were purchased from American Type Culture Collection (ATCC) (Manassas, VA) and grown in RPMI 1640 medium containing 50 μm 2-mercaptoethanol, 10% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Mouse pancreatic endothelial cells were obtained from ATCC and cultured in DMEM/F-12 medium supplemented with 10% FBS, 50 units/ml penicillin, and 50 μg/ml streptomycin. The human peripheral blood CD14+ monocytes were purchased from ReachBio LLC (Seattle, WA), suspended in RPMI 1640 medium containing 50 μm 2-mercaptoethanol, 100 IU/ml penicillin, and 100 μg/ml streptomycin, and used for transmigration assay. To collect peritoneal macrophages, WT mice were injected with 2 ml of 4% autoclaved thioglycollate, and 4 days later, the animals were anesthetized with ketamine and xylazine, and the peritoneal lavage was collected in RPMI 1640 medium. Cells were cultured at 3 × 105 cells/cm2 in RPMI 1640 medium containing 50 μm 2-mercaptoethanol, 10% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin. After 3 h, floating cells (mostly RBC) were removed by washing with cold PBS. Adherent cells (macrophages) were used for transmigration assay. Cultures were maintained at 37 °C in a humidified 95% air and 5% CO2 atmosphere. HUVECs between 6 and 10 passages were growth-arrested by incubating in medium 200 for 12 h and used to perform the experiments unless otherwise indicated.
Cell Viability
HUVECs were allowed to grow to a confluent monolayer, rested for 12 h, and then treated with vehicle or 15(S)-HETE for 2 h. After treatments, cells were trypsinized, and trypan blue solution (0.4%) was added to the cell suspension (1:1) and incubated at RT for 10 min, and viable cells were counted on a hemocytometer. The cell viability was expressed as percentage of viable cells.
Western Blot Analysis
Western blot analysis was performed as described previously (28).
Flux Assay
HUVECs were seeded on the apical side of the polycarbonate membrane transwell insert with 0.4-μm pores, allowed to grow to full confluence for the formation of a monolayer, and growth-arrested for 6 h. FITC-conjugated dextran (∼70,000 Da) was added (100 μg/ml) to the basal chamber. 15(S)-HETE (0.1 μm) was added to both the apical and basal chambers, and after 2 h, 100 μl of the medium from each chamber was collected, and the fluorescence intensity was measured using SpectraMax microplate fluorometer (Molecular Devices). The flux was expressed as % dextran diffused/h/cm2.
Immunoprecipitation
After growth arresting, the cells were treated with and without 15(S)-HETE (0.1 μm) for a required time period and then lysed in lysis buffer containing 20 mm HEPES, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 1 mm Na3VO4, 50 mm NaF, 50 mm β-glycerophosphate, 10 mm PMSF, and 100 μg/ml aprotinin. To ensure complete rupture of cell membranes, cells were sonicated at a 40 MHz pulse for 40 s followed by centrifugation at 6000 rpm for 10 min. The cell extracts containing an equal amount of protein from control and each treatment were incubated with appropriate antibodies overnight at 4 °C followed by the addition of protein A/B-Sepharose CL4B beads for an additional 2 h with gentle rocking at 4 °C. The beads were collected by centrifugation at 1000 rpm for 1 min, washed in lysis buffer, and heated in Laemmli sample buffer for 10 min, and the released proteins were resolved on SDS-PAGE and immunoblotted with the desired antibodies.
Immunofluorescence
HUVECs were grown on cell culture grade glass coverslips to confluence, quiesced, and treated with vehicle or 15(S)-HETE (0.1 μm) for various time periods. After treatment, cells were washed with PBS, fixed with 3% paraformaldehyde for 10 min at 37 °C, permeabilized in TBS (10 mm Tris-HCl, pH 8.0, 150 mm NaCl) containing 3% BSA and 0.1% Triton X-100 for 10 min at room temperature, and blocked in 3% BSA in TBS overnight at 4 °C. Then the cells were incubated with mouse anti-ZO-1 antibodies in combination with rabbit anti-occludin antibodies followed by goat anti-mouse and goat anti-rabbit secondary antibodies conjugated with Alexa Fluor 568 and Alexa Fluor 488, respectively, counter-stained with Hoechst 33342 (1:3000 dilution in PBS) for 1 min at room temperature, and mounted onto glass slides with Prolong Gold antifade mounting medium. Fluorescence images of cells were captured using an inverted Zeiss fluorescence microscope (AxioObserver Z1) via a ×40 NA 0.6 objective and AxioCam MRm camera without any enhancements using the microscope operating and image analysis software AxioVision version 4.7.2 (Carl Zeiss Imaging Solutions GmbH). The TJ-localized ZO-1 levels were quantified using Nikon's NIS Elements AR 3.1 imaging software and expressed as relative fluorescence units.
Monocyte Transmigration
Monocyte endothelial transmigration was measured as described by Bardin et al. (31). BCECF-AM-labeled quiescent THP1 cells (1 × 105 cells/well) were seeded onto the HUVEC monolayer and treated with vehicle or 15(S)-HETE (0.1 μm) for 6 h. At the end of the treatment period, cells from the apical side were removed by a cotton swab; the membrane was cut, fixed, and mounted onto glass slides with Prolong Gold antifade mounting medium, and the THP1 cells transmigrated through the HUVEC monolayer to the dorsal side of the membrane were observed under a Zeiss inverted microscope (Zeiss AxioObserver Z1, type, plan-Apochromat; magnification, ×10/0.45 NA), and fluorescence images were captured with a Zeiss AxioCam MRm camera using the microscope operating and image analysis software AxioVision Version 4.7.2 (Carl Zeiss Imaging Solutions GmbH).
Site-directed Mutagenesis
The rZO1 mutants T770A, T772A, and T770A/T772A were generated by the QuikChange site-directed mutagenesis kit (Agilent Technologies, CA) using GFP-tagged rZO-1 containing vector (pEGFPZO-1, Addgene) and the following primers: T770A mutant, forward, 5′-ATC TCA TAA ACT TGC TAA AAA TAA TCA CCA TCT TTT TGC AAC TAC AAT TAA CTT AAA TTC-3′, and reverse, 5′-GAA TTT AAG TTG TAG TTG CAA AAG ATG GTG ATT ATT TTT AGC AAG TTT ATG AGA T-3′; T772A mutant, forward, 5′-CTA AAA ATA ATC ACC ATC TTT TTA CAA CTG CAA TTA ACT TAA ATT CAA TGA ATG ATG GT-3′, and reverse, 5′-ACC ATC ATT CAT TGA ATT TAA GTT AAT TGC AGT TGT AAA AAG ATG GTG ATT ATT TTT AG-3′; and T770A/772A mutant, forward, 5′-ACT TGC TAA AAA TAA TCA CCA TCT TTT TGC AAC TGC AAT TAA CTT AAA TTC AAT GAA TGA TGG-3′, and reverse, 5′-CCA TCA TTC ATT GAA TTT AAG TTA ATT GCA GTT GCA AAA AGA TGG TGA TTA TTT TTA GCA AGT-3′. The mutations were verified by DNA sequencing.
Mass Spectrometry
HUVEC cells at 60% confluence were quiesced for 24 h and then treated with 15(S)-HETE (0.1 μm) for 30 min. After washing with PBS, cells were lysed in the lysis buffer as described above under “Immunoprecipitation” and immunoprecipitated with anti-ZO-1 antibodies. The immunocomplexes were separated on a 0.1% SDS, 7% PAGE and visualized by Coomassie Brilliant Blue R-250 staining. The ZO-1 band was excised and subjected to in-gel digestion with trypsin. The resulting peptides were analyzed by LC-ESI-MS/MS to identify the phosphorylated amino acid residues at the Bioanalytical and Mass Spectrometry Facility at the University of Alabama at Birmingham School of Medicine.
Animals
WT (C57BL/6) and 12/15-LO−/− (B6129S2-Alox15tm1fun/J) mice were obtained from The Jackson Laboratory. Mice were bred and maintained according to the Institutional Animal Facility Guidelines. All the studies using animals were approved by the Animal Care and Use Committee of the University of Tennessee Health Science Center, Memphis, TN. The mice were fed with chow diet or high fat diet containing 21.2% fat, 0.2% cholesterol, 17.3% protein, and 48.5% carbohydrate (TD88137 from Harlan, Teklad, Madison, MI) for 3 months and used as needed.
Immunofluorescence Staining of Aortas
Aortas from WT and 12/15-LOX−/− mice were dissected out, cleaned from connective and fat tissue, and treated with vehicle, AA (2.5 μm), 15-(S)-HETE (0.5 μm), or 12(S)-HETE (0.5 μm) in the presence and absence of PD098059 (10 μm) or PKCϵ inhibitor peptide (20 μm) for 30 min. The aortas were then opened longitudinally, fixed with 3% paraformaldehyde for 30 min followed by 0.2% picric acid for 1 h, permeabilized in TBS (10 mm Tris-HCl, pH 8.0, 150 mm NaCl) containing 3% BSA and 0.2% Triton X-100 for 10 min, and blocked in 3% BSA for 1 h at room temperature. The aortas were then incubated with rabbit anti-ZO-1 antibodies followed by goat anti-rabbit secondary antibodies conjugated with Alexa Fluor 568 and mounted onto glass slides with Prolong Gold antifade mounting medium. In another set of experiments, aortas were isolated from both WT and 12/15-LO−/− mice that were on chow diet or high fat diet for 3 months, fixed, and stained for ZO-1 as described above. Fluorescence images of the luminal side of aortas were captured using an inverted Zeiss fluorescence microscope (AxioObserver Z1) via a ×40 NA/0.6 and AxioCam MRm camera using the microscope operating and image analysis software AxioVision version 4.7.2 (Carl Zeiss Imaging Solutions GmbH) without any enhancements. The TJ-localized ZO-1 levels were quantified using Nikon's NIS Elements AR 3.1 imaging software and expressed as relative fluorescence units.
Extravasation Assay
Vascular permeability was examined by visualizing the leakage of Evans blue dye into the vascular wall as described previously (32). WT and 12/15-LO−/− mice that were fed a chow diet or high fat diet were anesthetized by ketamine/xylazine, and Evans blue dye (0.1 ml of 1% Evans blue dye in phosphate-buffered saline) was injected into the inferior vena cava. After 30 min, the mice were perfused through left ventricle with 4% paraformaldehyde for 10 min and sacrificed with an overdose of ketamine/xylazine. Aortas were dissected out and cleaned, and pictures were taken using Nikon SLR camera model D7100.
Statistics
All the experiments were repeated three times, and the data are presented as mean ± S.D. The treatment effects were analyzed by one-way ANOVA followed by post hoc t test in Microsoft Excel. The p values <0.05 were considered statistically significant.
RESULTS
15(S)-HETE Increases Endothelial Barrier Permeability
To test the role of LOs in the regulation of endothelial barrier function, we have studied the effects of 5(S)-HETE, 12(S)-HETE, 15(S)-HETE, and 15(R)-HETE, the 5-, 12-, and 15-LO metabolites of AA, and the enantiomer of 15(S)-HETE, respectively, on HUVEC barrier permeability. As measured by fluorescein isothiocyanate (FITC)-labeled dextran flux assay, 5(S)-HETE, 12(S)-HETE, and 15(S)-HETE increased HUVEC barrier permeability compared with control, with 5(S)-HETE exhibiting lower effects over those of 12(S)-HETE or 15(S)-HETE (Fig. 1A). However, both 12(S)-HETE and 15(S)-HETE increased HUVEC permeability to almost the same level. 15(R)-HETE, the enantiomer of 15(S)-HETE, had little or no effect on HUVEC barrier permeability suggesting that the effects of HETEs on EC barrier permeability are stereospecific. To find whether the increase in EC barrier permeability has any influence on transmigration of inflammatory cells, we studied the effects of 5(S)-HETE, 12(S)-HETE, 15(S)-HETE, and 15(R)-HETE on THP1 cell trans-endothelialization through a HUVEC monolayer. Consistent with their effects on HUVEC barrier permeability, 5(S)-HETE, 12(S)-HETE, and 15(S)-HETE enhanced transmigration of THP1 cells through this EC monolayer as compared with vehicle control, and with 5(S)-HETE the effects were lower as compared with those of 12(S)-HETE or 15(S)-HETE (Fig. 1C). Between 12(S)-HETE and 15(S)-HETE, both exhibited similar effects on THP1 cell transmigration. 15(R)-HETE, which had little effect on HUVEC barrier permeability, also had little or no effect on THP1 cell transmigration. Because both 15-LO1 and 15-LO2, which are expressed in humans, convert AA mostly to 15(S)-HETE (10, 11), we focused the rest of the studies on this eicosanoid. The dose-response study showed that 15(S)-HETE increases both EC barrier permeability and THP1 cell transmigration almost to a maximum level at 100 nm (Fig. 1, B and D). To test whether the increased HUVEC barrier permeability by 15(S)-HETE was due to cell death, we examined its effects on HUVEC survival. As determined by trypan blue dye exclusion assay, no differences were found in HUVEC survival between control and 15(S)-HETE treatment, suggesting that the increases in 15(S)-HETE-induced HUVEC barrier permeability were not due to cell death (Fig. 1E). To explore the pathophysiological significance of the effects of 15(S)-HETE on HUVEC barrier permeability and THP1 cell transmigration, we also tested its effects on transmigration of primary human and mouse macrophages through EC monolayers. 15(S)-HETE induced transmigration of both human and mouse primary macrophages through their respective EC monolayers (Fig. 1F). To find the potency of 15(S)-HETE in inducing EC barrier permeability, we compared its effects with that of thrombin on HUVEC barrier permeability. 15(S)-HETE was found as potent as thrombin in the induction of HUVEC barrier permeability (Fig. 1G). These observations clearly indicate that 15(S)-HETE increases EC barrier permeability facilitating transmigration of monocytes. Based on these findings, we next focused on the elucidation of mechanisms underlying 15(S)-HETE-induced HUVEC barrier permeability.
FIGURE 1.
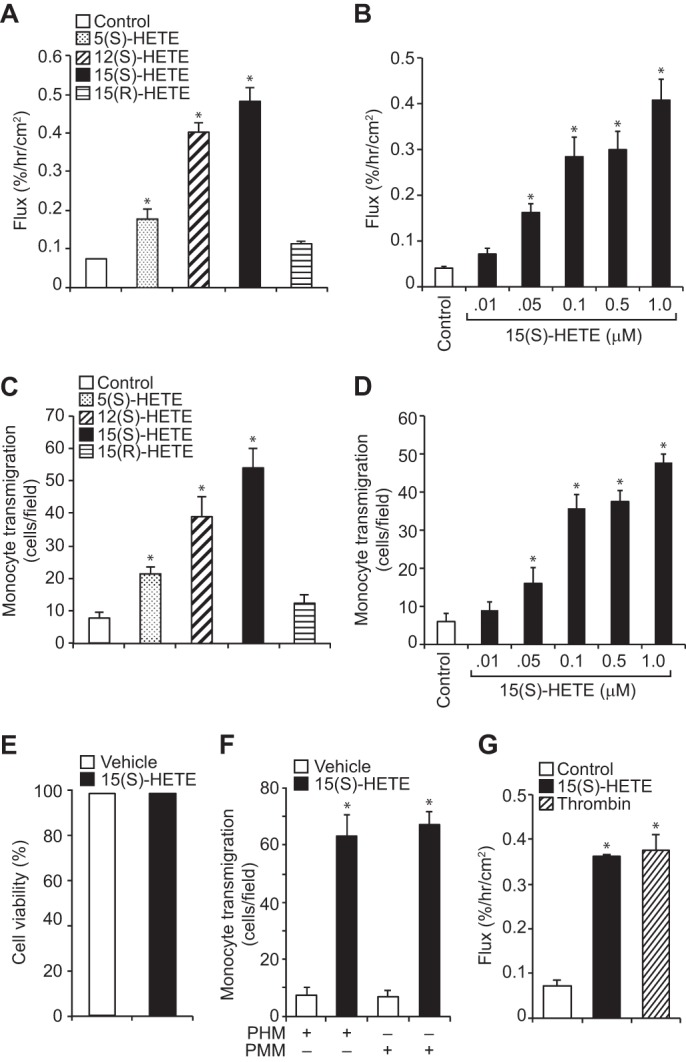
Effects of HETEs on EC barrier permeability and THP1 cell transmigration. A and B, quiescent HUVEC monolayer was treated with vehicle, 0.1 μm of the indicated HETE, or the indicated doses of 15(S)-HETE for 2 h, and dextran flux was measured. C and D, BCECF-AM-labeled THP1 cells were seeded onto a quiescent HUVEC monolayer and treated with vehicle, 0.1 μm of the indicated HETE, or the indicated doses of 15(S)-HETE for 8 h, and THP1 cell transmigration was measured. E, HUVEC viability in response to vehicle or 0.1 μm 15(S)-HETE treatment for 2 h was measured by trypan blue dye exclusion assay as described under “Materials and Methods.” F, primary human and mouse macrophage transmigration through their respective EC monolayer in response to vehicle or 0.1 μm 15(S)-HETE treatment was measured as described in C. G, dextran flux through HUVEC monolayer in response to vehicle, 15(S)-HETE (0.1 μm), or thrombin (0.1 unit/ml) treatment was measured as described in A. The values are means ± S.D. of three experiments. *, p < 0.05 versus vehicle control. PHM, primary human monocytes; PMM, primary mouse monocytes.
15(S)-HETE Promotes EC Barrier Permeability by Disruption of TJs
TJs play an important role in the regulation of endothelial barrier function (21, 22). Therefore, to understand the mechanisms by which 15(S)-HETE induces EC barrier permeability, we tested its effects on endothelial TJs. 15(S)-HETE had no major effect on the steady-state levels of TJ proteins, claudin-1, claudin-5, junctional adhesion molecule-A (Jam-A), Jam-B, Jam-C, occludin, ZO-1, and ZO-2, for at least 2 h, a time point before which maximal increases in HUVEC barrier permeability occurred by 15(S)-HETE (Fig. 2A). Therefore, we hypothesized that 15(S)-HETE might be disrupting the TJs by stimulating the post-translational modifications of TJ proteins causing their dissociation from the multimeric protein complexes. In line with this view, we found that 15(S)-HETE induced the Ser/Thr phosphorylation of claudin-1, claudin-5, occludin, ZO-1, and ZO-2 in a time-dependent manner with maximum effects at 10 and 30 min (Fig. 2B). 15(S)-HETE did not affect the Ser/Thr phosphorylation of Jam-A, Jam-B, or Jam-C. Among the TJ proteins whose phosphorylation was increased more than 2-fold by 15(S)-HETE, the time course of ZO-1 phosphorylation was found to be quite acute compared with others, as it peaked at 10 min and returned to basal levels by 60 min, a time course that correlates well with the transient increase in HUVEC barrier permeability. Because 5(S)-HETE, 12(S)-HETE, and 15(S)-HETE induced HUVEC barrier permeability, although differentially, we next asked the question whether these HETEs stimulate ZO-1 phosphorylation in correlation to their capacities on the induction of HUVEC barrier permeability. Consistent with their effects on HUVEC barrier permeability, all three HETEs stimulated ZO-1 phosphorylation; the effects by both 12(S)-HETE and 15(S)-HETE were 5-fold higher than control and 3-fold higher than the effect of 5(S)-HETE (Fig. 2C). 15(R)-HETE, the enantiomer of 15(S)-HETE, which does not have much effect on HUVEC permeability, also failed to stimulate ZO-1 phosphorylation significantly (Fig. 2C). These findings point to a possible role of ZO-1 phosphorylation in the induction of HUVEC barrier permeability by HETEs. Therefore, to understand the role of Ser/Thr phosphorylation of TJ proteins in the regulation of TJ integrity, we have focused on ZO-1. Because 15(S)-HETE also induced the Ser/Thr phosphorylation of other TJ proteins, namely claudin-5, occludin, and ZO-2 more robustly, we have examined their interactions with ZO-1 by co-immunoprecipitation (co-IP) assays. Co-IP experiments showed that ZO-1 exists in complex with claudin-5, occludin, and ZO-2 in a confluent HUVEC monolayer and upon treatment with 15(S)-HETE, it dissociated from these protein complexes with a robust and acute dissociation from occludin (Fig. 2D). Reassociation of ZO-1 with occludin was observed at 60 min and thereafter. Double immunofluorescence staining of a HUVEC monolayer treated with and without 15(S)-HETE for various time periods for ZO-1 and occludin showed that the dissociation of ZO-1 from occludin in the TJs in response to 15(S)-HETE occurs as acutely as 10 min, reaches maximum at 30 min, and starts reassociation at 60 min (Fig. 2E). Therefore, based on the co-IP and co-localization experiments, it appears that 15(S)-HETE disrupts endothelial TJs in a transient manner.
FIGURE 2.

15(S)-HETE disrupts endothelial TJs by Ser/Thr phosphorylation of TJ proteins. A, quiescent HUVEC monolayer was treated with vehicle or 0.1 μm 15(S)-HETE for the indicated time periods, and cell extracts were prepared and analyzed by Western blot (WB) for the indicated TJ proteins using their respective antibodies followed by normalization to β-tubulin. B–D, equal amounts of proteins from control and the indicated time periods of 0.1 μm 15(S)-HETE-treated HUVECs or control and 30 min of the indicated HETE (0.1 μm)-treated HUVECs were immunoprecipitated (IP) with anti-Ser(P)/Thr or anti-ZO-1 antibodies, and the immunocomplexes were analyzed for the indicated TJ proteins by WB. The cell extracts in C were also analyzed by WB for total ZO-1 levels. E, HUVEC monolayer was treated with vehicle or 0.1 μm 15(S)-HETE for the indicated time periods, fixed, and probed with mouse anti-ZO-1 antibodies in combination with rabbit anti-occludin antibodies followed by Alexa Fluor 568-conjugated goat anti-mouse and Alexa Fluor 488-conjugated goat anti-rabbit secondary antibodies. Fluorescence images were captured using an inverted Zeiss fluorescence microscope (AxioObserver Z1) via a ×40 NA 0.6 objective, and AxioCam MRm camera without any enhancements. The TJ ZO-1 levels were calculated using NIS Elements AR 3.1 imaging software (Nikon, Tokyo, Japan). The experiments were repeated three times, and the values are expressed as means ± S.D. *, p < 0.05 versus vehicle control.
Inhibition of ZO-1 Phosphorylation Prevents TJ Disruption
To identify the Ser/Thr residues of ZO-1 phosphorylated by and involved in 15(S)-HETE-induced TJ disruption causing enhanced trans-endothelialization of monocytes, we performed mass spectrometry. Mass spectrometric analysis revealed that 15(S)-HETE induces phosphorylation of Thr-770 and Thr-772 of ZO-1 (Fig. 3A). Both Thr-770/772 are located in the guanylate kinase (GUK) homology region domain of the PDZ-SH3-GUK core motif as shown in blue in Fig. 3B (33). To understand the influence of phosphorylation of these amino acid residues of ZO-1 on its dissociation from occludin in the TJs in response to 15(S)-HETE, we mutated these amino acid residues alone or in combination to Ala in GFP-tagged rZO-1 expression vector and transfected HUVECs with the vector containing either WT or mutant rZO-1, treated with and without 15(S)-HETE for 30 min, and rZO-1 phosphorylation was measured. 15(S)-HETE increased WT rZO-1 phosphorylation by about 5-fold compared with control. However, mutation of either Thr-770 or Thr-772 to Ala alone or in combination suppressed 15(S)-HETE-induced rZO-1 phosphorylation, with complete blockade by the double mutant (Fig. 3C). Furthermore, consistent with their phosphorylation pattern, expression of rZO-1 with or without mutations formed a complex with endogenous occludin in a HUVEC monolayer. In response to 15(S)-HETE, rZO-1 without mutations was found to be dissociated from occludin, whereas the ones with mutations remained in the complex with occludin (Fig. 3D). To test if the Thr-770/772 phosphorylation of ZO-1 is also essential for its interactions with other TJ proteins such as Jam-A, we examined the interactions of WT and mutant rZO-1 with Jam-A. Co-immunoprecipitation assay revealed that rZO-1 regardless of its phosphorylation state at Thr-770/772 was found to be associated with Jam-A, suggesting that the state of ZO-1 phosphorylation at Thr-770/772 had no influence on its association with Jam-A. Immunofluorescence staining for endogenous ZO-1 and rZO-1 in the TJs also showed that although endogenous or rZO-1 was dissociated from TJs causing TJ disruption, the mutant rZO-1 remained in the TJs and prevented their dislocation from the TJs in response to 15(S)-HETE (Fig. 3E). Next, the effect of these mutants in 15(S)-HETE-induced HUVEC barrier permeability and THP1 cell transmigration was examined. As shown in Fig. 3, F and G, rZO-1 mutants attenuated 15(S)-HETE-induced increases in HUVEC permeability as well as THP1 cell transmigration, with a robust blockade by the double mutant.
FIGURE 3.

Mutation of ZO-1 T770/T772 to Ala prevents 15(S)-HETE-induced endothelial TJ disruption. A, characterization of phosphorylated amino acid residues of ZO-1 was carried out by LC-CID-MS/MS, which indicated double phosphorylation status on amino acids Thr-770 and Thr-772. A full MS/MS scan is shown in the upper panel with a zoomed-in region for a better visualization of the assigned −b and −y ions for the 4+ charge state parent ion at 762.9 m/z. There X corresponding score was 4.4 with a peptide probability of 96%, and there were 27 matching fragment ions, which were generally related to the most abundant ions in the spectra. In the lower panel, less abundant fragments, including water and ammonia losses on 2+ charge state daughter ions, are shown. Although the Thr-772 assignment was fair with an A-score of 12.3 and a localization score of 96%, there was some ambiguity between Thr-770 and Thr-772 with a slightly higher localization score for Thr-770 at 71%. There were no neutral loss ions, which is common on higher charge state peptides as is observed here, which made these assignments much more straightforward. B, structure of ZO-1 from Nomme et al. (33) pointing out in blue the Thr-770/772 residues located in the GUK domain. C and D. HUVECs that were transiently transfected with empty vector (EV) or GFP-tagged recombinant ZO-1 (rZO-1) expression vector, with and without mutations T770A, T772A, or T770A/T772A and grown to confluence, were quiesced and treated with vehicle or 0.1 μm 15(S)-HETE for 30 min; cell extracts were prepared and immunoprecipitated (IP) with anti-GFP antibodies, and the immunocomplexes were analyzed by WB with the antibodies of the indicated proteins. IB, immunoblot. E, all the conditions were the same as in C except that after quiescence and with the indicated treatments, the HUVEC monolayer was stained for endogenous ZO-1 using mouse anti-ZO-1 antibodies followed by developing with Alexa Fluor 568-conjugated goat anti-mouse secondary antibodies. The rZO-1 tagged with GFP was visualized by green fluorescence. Fluorescence images were captured, and TJ ZO-1 levels were measured as described in Fig. 2E. F and G, all the conditions were the same as in C except that after quiescence, the HUVEC monolayer was subjected to vehicle or 15(S)-HETE (0.1 μm)-induced dextran flux (F) or THP1 cell transmigration (G) assays. The bar graphs represent means ± S.D. values of three experiments. *, p < 0.05 versus vehicle control; **, p < 0.05 versus EV + 15(S)-HETE or rZO-1 + 15(S)-HETE. eZO-1, endogenous ZO-1; rZO-1, recombinant ZO-1.
ERK1/2 Mediate 15(S)-HETE-induced ZO-1 Phosphorylation
Previous studies from other laboratories have reported that MAPKs might play a role in the disruption of TJs via mediating phosphorylation of TJ proteins in response to external cues in epithelial cells (34, 35). Therefore, to investigate the upstream mechanisms of ZO-1 phosphorylation by 15(S)-HETE, we tested the role of MAPKs. 15(S)-HETE enhanced phosphorylation of ERK1/2 but not p38 MAPK in a time-dependent manner in HUVECs (Fig. 4A). Based on this observation, we next tested the role of ERK1/2 in 15(S)-HETE-induced ZO-1 phosphorylation. PD098059, a pharmacological inhibitor of MEK1 (36), an immediate upstream regulator of ERK1/2, attenuated 15(S)-HETE-induced ZO-1 phosphorylation (Fig. 4B). To confirm these results, we also used a dominant negative mutant approach. Adenovirus-mediated expression of dnMEK1 (37) also blocked 15(S)-HETE-induced ZO-1 phosphorylation (Fig. 4C). In line with these observations, both PD098059 and dnMEK1 blocked 15(S)-HETE-induced dissociation of ZO-1 from occludin (Fig. 4, D and E). In addition, inhibition of ERK1/2 by either pharmacological or the dominant negative mutant approach attenuated 15(S)-HETE-induced HUVEC TJ disruption, barrier permeability, and THP1 cell transmigration (Fig. 4, F–K).
FIGURE 4.

ERK1/2 mediates 15(S)-HETE-induced endothelial TJ disruption. A, HUVEC extracts of the control and the indicated treatments were analyzed by WB for pERK1/2 and pp38 MAPK levels followed by normalization to their total levels. B and C, HUVEC extracts of the control and the indicated treatments were immunoprecipitated (IP) with anti-Ser(P)/Thr antibodies, and the immunocomplexes were analyzed by WB for ZO-1 levels. Cell extracts were also analyzed by WB for total ZO-1 levels as well as for overexpression of dnMEK1 and GFP. IB, immunoblot. D and E, all the conditions were the same as in B or C except that the cell extracts were immunoprecipitated with anti-ZO-1 antibodies, and the immunocomplexes were analyzed by WB for occludin followed by normalization to ZO-1. The same cell extracts were also analyzed by WB for overexpression of dnMEK1 and GFP. F and G, all the conditions were the same as in B or C except that after quiescence, the HUVEC monolayers were treated with vehicle or 15(S)-HETE (0.1 μm) for 30 min and stained for ZO-1 and occludin as described in Fig. 2E legend. Fluorescence images were captured, and TJ-localized ZO-1 levels were quantified as described in Fig. 2E legend. H–K, all the conditions were the same as in B or C except that the HUVEC monolayers were subjected to vehicle or 15(S)-HETE (0.1 μm)-induced dextran flux (H and I) or THP1 cell transmigration (J and K) assays. The bar graphs represent means ± S.D. values of three experiments. *, p < 0.05 versus vehicle control or Ad-GFP; **, p < 0.05 versus 15(S)-HETE or Ad-GFP + 15(S)-HETE.
PKCϵ Acts Upstream to ERK1/2 in the Mediation of ZO-1 Phosphorylation
It has been previously reported that PKCs play a predominant role in the regulation of TJ function (38–40). Hence, we wanted to test whether 15(S)-HETE activates any PKCs and, if so, their role in ERK1/2-mediated ZO-1 phosphorylation. The time course experiment revealed that 15(S)-HETE enhances phosphorylation of PKCδ, PKCϵ, and PKCζ in a time-dependent manner with maximum effects at 5–10 min (Fig. 5A). It had no major effect on PKCα/βII and PKCγ phosphorylation. Therefore, based on these observations, we next tested the role of PKCδ, PKCϵ, and PKCζ in ERK1/2 activation. Adenovirus-mediated expression of dnPKCϵ but not dnPKCδ or dnPKCζ blocked 15(S)-HETE-induced ERK1/2 activation, suggesting that PKCϵ acts upstream to ERK1/2 (Fig. 5B). To confirm this conclusion, we also tested the effect of blockade of MEK1 on PKCϵ activation. Inhibition of MEK1 by either a pharmacological or dominant negative mutant approach had no effect on 15(S)-HETE-induced PKCϵ phosphorylation, indicating that PKCϵ acts upstream of MEK1 in the phosphorylation of ERK1/2 (Fig. 5, C and D). We next tested the role of PKCϵ on 15(S)-HETE-induced ZO-1 phosphorylation. As shown in Fig. 5E, adenovirus-mediated expression of dnPKCϵ efficiently blocked the Ser/Thr phosphorylation of ZO-1. Similarly, dnPKCϵ prevented 15(S)-HETE-induced dissociation of ZO-1 from occludin and disruption of TJs (Fig. 5, F and G). 15(S)-HETE-induced HUVEC barrier permeability as well as THP1 cell transmigration were also suppressed by dnPKCϵ (Fig. 5, H and I).
FIGURE 5.

PKCϵ mediates 15(S)-HETE-induced endothelial TJ disruption. A, HUVEC extracts of the control and the indicated treatments were analyzed by WB or immunoprecipitation (IP) followed by WB for the indicated phospho-PKC and total PKC levels. B, HUVECs that were transduced with adenoviral vectors of the indicated dnPKC or GFP at 40 M.O.I. and quiesced were treated with vehicle or 0.1 μm 15(S)-HETE for 10 min; cell extracts were prepared and analyzed for pERK1/2 levels. The blot was reprobed sequentially with the antibodies of the indicated proteins for normalization or to show the overexpression of the indicated dnPKC or GFP. C, cell extracts of HUVECs that were treated with vehicle or 0.1 μm 15(S)-HETE in the presence and absence of MEK1 inhibitor PD098059 (10 μm) for 30 min were analyzed for phospho and total PKCϵ levels as described in Fig. 2B. IB, immunoblot. D, all the conditions were the same as in C except that cells were transduced with the indicated adenoviral vector (40 M.O.I.) and quiesced before subjecting to the indicated treatments and analyzing for phospho-PKCϵ and total PKCϵ levels. The same cell extracts were also analyzed by WB for overexpression of dnMEK1 or GFP. E and F, HUVECs that were transduced with the indicated adenoviral vector (40 M.O.I.) and quiesced were treated with vehicle or 0.1 μm 15(S)-HETE for 30 min; cell extracts were prepared and immunoprecipitated with anti-Ser(P)/Thr or anti-ZO-1 antibodies, and the immunocomplexes were analyzed by WB for ZO-1 and occludin levels. The same cell extracts were also analyzed for ZO-1, PKCϵ, or GFP levels for normalization or to show the overexpression of dnPKCϵ or GFP. The blot in F was reprobed for ZO-1. G, all the conditions were the same as in E except that after quiescence, the HUVEC monolayer was treated with vehicle or 0.1 μm 15(S)-HETE for 30 min and immunostained for ZO-1 and occludin as described in Fig. 2E. Fluorescence images were captured, and TJ-localized ZO-1 levels were quantified as described in Fig. 2E. H and I, all the conditions were the same as in E except that the HUVEC monolayer after quiescence was subjected to 15(S)-HETE (0.1 μm)-induced dextran flux (H) or THP1 cell transmigration (I) assays. The bar graphs represent means ± S.D. values of three experiments. *, p < 0.05 versus vehicle control or Ad-GFP; **, p < 0.05 versus 15(S)-HETE or Ad-GFP + 15(S)-HETE.
12/15-LO-12/15(S)-HETE Mediates High Fat Diet-induced Aortic Endothelial TJ Disruption
To better understand the pathophysiological relevance of the above observations, we have extended these studies to intact mouse arteries. Exposure of intact arteries from WT mice to 15(S)-HETE or 12(S)-HETE ex vivo caused increased phosphorylation of ZO-1, and this effect was negated by inhibitors of either PKCϵ or MEK1 (Fig. 6, A and B). Consistent with this observation, exposure of intact arteries from WT mice to 15(S)-HETE or 12(S)-HETE ex vivo also caused dislocation/disappearance of ZO-1 from endothelial TJs in PKCϵ and MEK1-dependent manner (Fig. 6, C and D). To confirm these findings, we also tested the effects of AA, the substrate of 12/15-LO, on endothelial TJ disruption using aortas from WT and 12/15-LO−/− mice. Exposure of aortas from WT but not 12/15-LO−/− mice to AA led to the dislocation/disappearance of ZO-1 from TJs (Fig. 6E). However, exposure of aortas from either WT or 12/15-LO−/− mice to 15(S)-HETE or 12(S)-HETE caused dislocation/disappearance of ZO-1 from TJs (Fig. 6E), suggesting that AA conversion through 12/15-LO is essential for its effects on TJ disruption. To substantiate these observations, WT and 12/15-LO−/− mice were fed a chow diet (CD) or high fat diet (HFD) for 3 months, and 12/15-LO expression in the aorta and aortic endothelial TJs was examined. First, compared with CD, HFD feeding induced 12/15-LO expression in the endothelium as determined by co-immunofluorescence staining for 12/15-LO and CD31 levels (Fig. 7A). Second, in line with its effect on 12/15-LO expression, HFD induced aortic endothelial TJ disruption only in WT but not 12/15-LO−/− mice (Fig. 7B). Consistent with these observations, in response to HFD feeding, aortas from WT mice showed increased permeability as determined by extravasation of Evans blue dye as compared with aortas from 12/15-LO−/− mice (Fig. 7C).
FIGURE 6.

15(S)-HETE and 12(S)-HETE stimulate Ser/Thr phosphorylation of ZO-1 and its dislocation from aortic endothelial TJs ex vivo. A and B, aortas from WT mice were incubated with vehicle, 15(S)-HETE (0.5 μm), or 12(S)-HETE (0.5 μm) in the presence and absence of PKCϵ inhibitor peptide (PKCϵI) (20 μm) or PD098059 (10 μm) for 30 min; tissue extracts were prepared and analyzed for phospho and total ZO-1 levels as described in Fig. 2B. IP, immunoprecipitation; IB, immunoblot. C and D, aortas from WT mice were incubated with vehicle, 15(S)-HETE (0.5 μm), or 12(S)-HETE (0.5 μm) in the presence and absence of PKCϵI (20 μm) or PD098059 (10 μm) for 30 min, opened longitudinally, fixed, permeabilized, blocked, and incubated with rabbit anti-ZO-1 antibodies followed by development with Alexa Fluor 568-conjugated goat anti-rabbit secondary antibodies. E, aortas from WT or 12-LO−/− mice were incubated with vehicle, AA (2.5 μm), 15(S)-HETE (0.5 μm), or 12(S)-HETE (0.5 μm) for 30 min and processed for ZO-1 staining as described in C. The immunofluorescence images in C–E were captured, and TJ-localized ZO-1 levels were quantified as described in Fig. 2E. The white arrows point out TJ disruption. The bar graphs represent means ± S.D. values of three experiments or six animals. *, p < 0.05 versus vehicle control; **, p < 0.05 versus AA (WT), 15(S)-HETE or 12(S)-HETE.
FIGURE 7.

12/15-LO mediates HFD-induced ZO-1 dislocation from endothelial TJs leading to increased vascular permeability. A, aortas from WT mice that were either on CD or HFD for 3 months were dissected out and fixed in OCT compound; cross-sections were made and co-immunostained for 15-LO1 and CD31 using sheep anti-rabbit 15-LO1 antibodies and rat anti-mouse CD31 antibodies followed by development with Alexa Fluor 568-conjugated donkey anti-sheep antibodies and Alexa Fluor 488-conjugated goat anti-rat antibodies; and the fluorescence images were captured as described in Fig. 2E. B, aortas from WT and 12/15-LO−/− mice that were kept on CD or HFD for 3 months were dissected out, opened longitudinally, fixed, permeabilized, blocked and incubated with rabbit anti-ZO-1 antibodies followed by Alexa Fluor 568-conjugated goat anti-rabbit secondary antibodies. Fluorescence images were captured, and TJ-localized ZO-1 levels were quantified as described in Fig. 2E. The white arrows point out TJ disruption. C, all the conditions were the same as in B except that animals after feeding with CD or HFD for 3 months were anesthetized, and 0.1 ml of 1% Evans blue dye was injected into inferior vena cava, and after 30 min the blood vessels were perfused with PBS through the left ventricle and fixed with 4% paraformaldehyde. The arteries were isolated and cleaned from fat tissue, and pictures were taken using Nikon SLR camera (model D7100). Six animals were used in each group, and the bar graphs represent means ± S.D. *, p < 0.05 versus CD; **, p < 0.05 versus WT (HFD).
DISCUSSION
Endothelial TJs form strand-like structures between two adjacent cells and regulate the paracellular permeability of the endothelium to ions and macromolecules (22). Endothelial TJs are composed of transmembrane proteins (occludin, claudins, and junction adhesion molecules) and intracellular proteins (zona occludens 1–3 (ZO-1–3), protein incorporated later into tight junctions (PILT), junction-enriched and associated protein (JEAP), and multi-PDZ domain protein 1 (MUPP-1)) (21, 41, 42). Disruption in the interactions between these proteins perturbs the TJs affecting the permeability of the endothelium (22). Although the steady-state levels of transmembrane and intracellular TJ components are critical in the formation of TJ complexes, many studies have also reported that post-translational modifications, such as phosphorylation, play a role in the regulation of TJs (35, 38, 40, 42, 43). In this aspect, a role for both Ser/Thr and Tyr phosphorylation of TJ proteins in the formation and/or disruption of TJs has been reported (40, 44–46). The ZO-1, which was the first component of the TJ complexes identified, belongs to a family of multidomain proteins known as the membrane-associated guanylate kinases and consists of three PDZ domains, an Src homology 3 (SH3) domain, and a proline-rich region (47). Because of the presence of multiple protein-binding motifs, it possesses the capacity to interact with several proteins and organize them as multiprotein complexes. Many reports showed that ZO-1 forms heterodimers with claudins, occludin, and ZO-2 (22, 41, 47). It was also reported that ZO-1 interacts with several signaling proteins (48). However, the mechanisms of these interactions and their relevance to TJ assembly are poorly understood. A convincing body of evidence suggests that lipoxygenases, particularly 15-LO1, play a crucial role in the pathogenesis of various vascular diseases (12–18). Interestingly, 15-HETE, the major 15-LO product of AA, was also found to be the major metabolite of AA converted by the atherosclerotic arteries (12, 13). Similarly, we have reported that overexpression of 15-LO1 or 15-LO2 exacerbates restenosis in response to injury (49, 50). In addition, the previous studies by us as well as others have reported the presence of 15-LO or its inhibitor, nordihydroguaiaretic acid-sensitive production of 15(S)-HETE and 13(S)-hydroxyoctadecadienoic acid in response to incubation with AA and linoleic acid, respectively, in both vascular smooth muscle cells and endothelial cells (28, 51–54). Thus, all these reports may lend support to the role of 15-LO-15(S)-HETE axis in vascular diseases. Some studies also showed a role for cyclooxygenase in the production of 15(S)-HETE in vascular smooth muscle cells (55). Regardless of its source, how 15(S)-HETE is involved in the pathogenesis of these vascular diseases is not clear. Perturbation in TJs leads to endothelial dysfunction, which is considered to be hallmark in the initiation of atherosclerosis and other vascular diseases (19, 20). So, we hypothesized that 15(S)-HETE may cause endothelial dysfunction by altering TJs. Our findings reveal that 15(S)-HETE causes TJ disruption and facilitates transmigration of macrophages through the endothelium. Toward understanding the mechanisms, we observed that 15(S)-HETE induces Ser/Thr phosphorylation of several TJ proteins, among which the most striking is ZO-1. From mass spectrometric analysis, we found two Thr residues of ZO-1, namely Thr-770/772, to be phosphorylated by 15(S)-HETE, and mutation of these amino acid residues to Ala protected TJ disruption to a significant level, and this effect is dependent on PKCϵ-mediated activation of MEK1-ERK1/2 signaling.
Previous reports showed that ZO-1-associated kinase, a serine/threonine kinase, binds to the SH3 domain of ZO-1 and phosphorylates it in the carboxyl terminus (56). In addition, a pharmacological inhibitor of PKC was shown to inhibit ZO-1 phosphorylation and TJ assembly, suggesting that PKC-mediated phosphorylation of ZO-1 promotes TJ formation (57). In contrast, inhibition of MEK1 was shown to attenuate ZO-1 phosphorylation and enhance TJ assembly, indicating that the Ser/Thr phosphorylation of ZO-1 disassembles TJs (35). Some studies also indicated that phosphorylation of ZO-1 may not always correlate with barrier function (58). However, none of these studies identified specific Ser/Thr phosphorylation sites and therefore led to correlative conclusions. However, our results indicate that PKCϵ-dependent ERK1/2 activation is required for ZO-1 Thr-770/772 phosphorylation, and these events lead to its disassembly from TJs, affecting endothelial barrier permeability. Recently, it was shown that ZO-1 and occludin complex plays an important role in TJ assembly and barrier permeability (59). In this aspect, it is interesting to note that 15(S)-HETE-induced phosphorylation of ZO-1 at Thr-770/772 leads to its dissociation from occludin affecting TJ integrity and barrier function. These findings also reveal that the phosphorylation state of ZO-1 at Thr-770/772 determines its capacity to form a complex with occludin. The structural studies demonstrated that the GUK region in the ZO-1 is essential for its interactions with other TJ proteins such as occludin in forming TJs (60). As Thr-770/772 residues are located in the GUK region and phosphorylation of these amino acid residues prevents its interactions with occludin, it is likely that these amino acid residues play an important role in the interaction of ZO-1 with other proteins in the assembly of TJs. Because AA, the substrate of 12/15-LO, could affect the aortic endothelial TJ integrity of only WT but not 12/15-LO−/− mice, and 15(S)-HETE or 12(S)-HETE, the 15-LO1 and 12/15-LO metabolites of AA, respectively, disrupt aortic endothelial TJs from either mouse suggest that 15-LO1 (12/15-LO in mice) plays an important role in the modulation of TJ disassembly and barrier dysfunction. Furthermore, the findings that high fat diet feeding induces 12/15-LO expression in the endothelium of WT mice and disrupts aortic endothelial TJs only in WT but not 12/15-LO−/− mice reinforce the role of 12/15-LO in the endothelial TJ dysfunction.
A large body of evidence suggests that 12/15-LO plays a critical role in atherogenesis (12–18). Similarly, EC dysfunction was thought to be an early event in the pathogenesis of atherosclerosis (19, 20). Because 12/15-LO and its AA metabolites, 15(S)-HETE and 12(S)-HETE, appear to play a role in the disassembly of endothelial TJs and barrier dysfunction, it is possible that 12/15-LO via affecting TJ integrity and thereby endothelial barrier function is involved in the pathogenesis of atherosclerosis. In this context, it is worth to point out that aortic endothelial TJs in 12/15-LO−/− mice appear to be more intact than those in WT mice and in response to HFD feeding retained their integrity and prevented vascular leakiness. This enhanced vascular leakiness, which can be attributed to 12/15-LO, could be a causative factor in atherogenesis in response to risk factors such as HFD and infections (61). A large body of literature points out a link between oxidative stress and vascular diseases (62). A recent study showed that 12(S)-HETE increases retinal endothelial barrier permeability by NADPH oxidase-dependent reactive oxygen species production leading to phosphorylation and activation of VEGF receptor-2 (26). We have previously shown that 15(S)-HETE by activating nonreceptor tyrosine kinases, Src and Pyk2, stimulates ZO-2 tyrosine phosphorylation and its dissociation from claudins 1/5 and thereby disrupts aortic endothelial TJs in response to HFD feeding (27). We have also shown that 15(S)-HETE induces reactive oxygen species production via xanthine oxidase and an NADPH oxidase-dependent manner in macrophages (63). Furthermore, our studies have demonstrated that the macrophages from apoE−/−:12/15-LO−/− mice exhibited lower capacity in the production of oxidants as compared with apoE−/− mice in response to HFD feeding (63). In view of these findings and the present observations, it may be suggested that 15-LO1/15(S)-HETE axis (12/15-LO/12(S)-HETE axis in mice), via triggering both tyrosine and serine/threonine phosphorylation of various endothelial TJ proteins and thereby disrupting endothelial TJs, increasing vascular permeability, and enhancing vascular inflammation, might be playing a contributing role in atherogenesis in response to cardiovascular risk factors such as HFD. It is interesting to note that inhibition of either ZO-2 tyrosine phosphorylation or ZO-1 threonine phosphorylation completely blocked 15(S)-HETE-induced increases in endothelial permeability and monocyte transmigration. These observations may imply that both of these events are integrating in the mediation of 15(S)-HETE's effects on endothelial permeability and monocyte transmigration, in which case inhibition of either signaling event may affect the endothelial responses of 15(S)-HETE to the same level. A convincing body of evidence also suggests that polyunsaturated fatty acids such as AA and linoleic acid and their lipoxygenase metabolites, HETEs and hydroxyoctadecadienoic acids, respectively, are ligands for peroxisome proliferator-activated receptors, which upon activation in complex with retinoid X receptor bind to peroxisome proliferator response elements and regulate the target gene expression (64). In addition, some studies have provided clues for the involvement of G protein-coupled receptors in the mediation of cellular responses by 12(S)-HETE and 15-HETE (65, 66). Because 15(S)-HETE-induced endothelial TJ disruption appears to be dependent on transient phosphorylation of TJ proteins but not their steady-state levels, it is unlikely that peroxisome proliferator-activated receptors are involved in the mediation of these effects. However, although there is no putative receptor identified for HETEs thus far, based on the previous observations from other laboratories (65, 66) it is quite possible that 15(S)-HETE-induced endothelial TJ disruption may depend on activation of cell surface receptors such as G protein-coupled receptors. In any case, future studies are required to address whether 15(S)-HETE-induced TJ disruption in HUVECs in vitro and in arteries ex vivo/in vivo depends on activation of G protein-coupled receptors or some other receptors.
In brief, in addition to its role in tyrosine phosphorylation of ZO-2 and its dissociation from claudins 1/5 (27), 15(S)-HETE, as shown in Fig. 8, can also mediate the serine/threonine phosphorylation of other TJ proteins such as ZO-1 and its dissociation from occludin in the disruption of endothelial TJs and its barrier function.
FIGURE 8.

Schematic diagram showing the mechanism(s) of 12/15-LO-12/15(S)-HETE axis on the disruption of endothelial tight junctions and barrier function.
Acknowledgment
We thank Dr. Alex Toker for providing us the pCMV5F.PKCζ.KW plasmid.
This work was supported, in whole or in part, by National Institutes of Health Grant HL074860 from NHLBI (to G. N. R.).
- 12/15-LO
- 12/15-lipoxygenase
- TJ
- tight junction
- ZO
- zona occludens
- HUVEC
- human umbilical vein endothelial cell
- HFD
- high fat diet
- CD
- chow diet
- AA
- arachidonic acid
- BCECF-AM
- 2′,7′-bis(carboxyethyl)-5,6-carboxyfluorescein
- 15(S)-HETE
- 15(S)-hydroxyeicosatetraenoic acid
- 15-LO1
- 15-lipoxygenase 1
- WB
- Western blot
- rZO-1
- recombinant ZO-1
- M.O.I.
- multiplicity of infection
- SH3
- Src homology 3
- GUK
- guanylate kinase
- EC
- endothelial cell.
REFERENCES
- 1. Hansson G. K., Hermansson A. (2011) The immune system in atherosclerosis. Nat. Immunol. 12, 204–212 [DOI] [PubMed] [Google Scholar]
- 2. Galkina E., Ley K. (2009) Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 27, 165–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ross R. (1999) Atherosclerosis–an inflammatory disease. N. Engl. J. Med. 340, 115–126 [DOI] [PubMed] [Google Scholar]
- 4. Stary H. C., Chandler A. B., Dinsmore R. E., Fuster V., Glagov S., Insull W., Jr., Rosenfeld M. E., Schwartz C. J., Wagner W. D., Wissler R. W. (1995) A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 92, 1355–1374 [DOI] [PubMed] [Google Scholar]
- 5. Bailey J. M., Makheja A. N., Lee R., Simon T. H. (1995) Systemic activation of 15-lipoxygenase in heart, lung, and vascular tissues by hypercholesterolemia: relationship to lipoprotein oxidation and atherogenesis. Atherosclerosis 113, 247–258 [DOI] [PubMed] [Google Scholar]
- 6. Natarajan R., Gerrity R. G., Gu J. L., Lanting L., Thomas L., Nadler J. L. (2002) Role of 12-lipoxygenase and oxidant stress in hyperglycaemia-induced acceleration of atherosclerosis in a diabetic pig model. Diabetologia 45, 125–133 [DOI] [PubMed] [Google Scholar]
- 7. Hatley M. E., Srinivasan S., Reilly K. B., Bolick D. T., Hedrick C. C. (2003) Increased production of 12/15 lipoxygenase eicosanoids accelerates monocyte/endothelial interactions in diabetic db/db mice. J. Biol. Chem. 278, 25369–25375 [DOI] [PubMed] [Google Scholar]
- 8. Chakrabarti S. K., Cole B. K., Wen Y., Keller S. R., Nadler J. L. (2009) 12/15-Lipoxygenase products induce inflammation and impair insulin signaling in 3T3-L1 adipocytes. Obesity 17, 1657–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoshida H., Sasaki K., Hirowatari Y., Kurosawa H., Sato N., Furutani N., Tada N. (2004) Increased serum iron may contribute to enhanced oxidation of low density lipoprotein in smokers in part through changes in lipoxygenase and catalase. Clin. Chim. Acta 345, 161–170 [DOI] [PubMed] [Google Scholar]
- 10. Kühn H., O'Donnell V. B. (2006) Inflammation and immune regulation by 12/15-lipoxygenases. Prog. Lipid Res. 45, 334–356 [DOI] [PubMed] [Google Scholar]
- 11. Chang M. S., Schneider C., Roberts R. L., Shappell S. B., Haselton F. R., Boeglin W. E., Brash A. R. (2005) Detection and subcellular localization of two 15S-lipoxygenases in human cornea. Invest. Ophthalmol. Vis. Sci. 46, 849–856 [DOI] [PubMed] [Google Scholar]
- 12. Henriksson P., Hamberg M., Diczfalusy U. (1985) Formation of 15-HETE as a major hydroxyeicosatetraenoic acid in the atherosclerotic vessel wall. Biochim. Biophys. Acta 834, 272–274 [DOI] [PubMed] [Google Scholar]
- 13. Simon T. C., Makheja A. N., Bailey J. M. (1989) Formation of 15-hydroxyeicosatetraenoic acid (15-HETE) as the predominant eicosanoid in aortas from Watanabe Heritable hyperlipidemic and cholesterol-fed rabbits. Atherosclerosis 75, 31–38 [DOI] [PubMed] [Google Scholar]
- 14. Cathcart M. K., McNally A. K., Chisolm G. M. (1991) Lipoxygenase-mediated transformation of human low density lipoprotein to an oxidized and cytotoxic complex. J. Lipid Res. 32, 63–70 [PubMed] [Google Scholar]
- 15. Ylä-Herttuala S., Luoma J., Viita H., Hiltunen T., Sisto T., Nikkari T. (1995) Transfer of 15-lipoxygenase gene into rabbit iliac arteries results in the appearance of oxidation-specific lipid-protein adducts characteristic of oxidized low density lipoprotein. J. Clin. Invest. 95, 2692–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sendobry S. M., Cornicelli J. A., Welch K., Bocan T., Tait B., Trivedi B. K., Colbry N., Dyer R. D., Feinmark S. J., Daugherty A. (1997) Attenuation of diet-induced atherosclerosis in rabbits with a highly selective 15-lipoxygenase inhibitor lacking significant antioxidant properties. Br. J. Pharmacol. 120, 1199–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cyrus T., Praticò D., Zhao L., Witztum J. L., Rader D. J., Rokach J., FitzGerald G. A., Funk C. D. (2001) Absence of 12/15-lipoxygenase expression decreases lipid peroxidation and atherogenesis in apolipoprotein E-deficient mice. Circulation 103, 2277–2282 [DOI] [PubMed] [Google Scholar]
- 18. Harats D., Shaish A., George J., Mulkins M., Kurihara H., Levkovitz H., Sigal E. (2000) Overexpression of 15-lipoxygenase in vascular endothelium accelerates early atherosclerosis in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 20, 2100–2105 [DOI] [PubMed] [Google Scholar]
- 19. Cines D. B., Pollak E. S., Buck C. A., Loscalzo J., Zimmerman G. A., McEver R. P., Pober J. S., Wick T. M., Konkle B. A., Schwartz B. S., Barnathan E. S., McCrae K. R., Hug B. A., Schmidt A. M., Stern D. M. (1998) Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 91, 3527–3561 [PubMed] [Google Scholar]
- 20. Pober J. S., Sessa W. C. (2007) Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 7, 803–815 [DOI] [PubMed] [Google Scholar]
- 21. Dejana E. (2004) Endothelial cell-cell junctions: happy together. Nat. Rev. Mol. Cell Biol. 5, 261–270 [DOI] [PubMed] [Google Scholar]
- 22. Bazzoni G., Dejana E. (2004) Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol. Rev. 84, 869–901 [DOI] [PubMed] [Google Scholar]
- 23. Ross R., Glomset J. A. (1973) Atherosclerosis and the arterial smooth muscle cell: Proliferation of smooth muscle is a key event in the genesis of the lesions of atherosclerosis. Science 180, 1332–1339 [DOI] [PubMed] [Google Scholar]
- 24. Ross R., Harker L. (1976) Hyperlipidemia and atherosclerosis. Science 193, 1094–1100 [DOI] [PubMed] [Google Scholar]
- 25. Zarbock A., Distasi M. R., Smith E., Sanders J. M., Kronke G., Harry B. L., von Vietinghoff S., Buscher K., Nadler J. L., Ley K. (2009) Improved survival and reduced vascular permeability by eliminating or blocking 12/15-lipoxygenase in mouse models of acute lung injury (ALI). J. Immunol. 183, 4715–4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Othman A., Ahmad S., Megyerdi S., Mussell R., Choksi K., Maddipati K. R., Elmarakby A., Rizk N., Al-Shabrawey M. (2013) 12/15-Lipoxygenase-derived lipid metabolites induce retinal endothelial cell barrier dysfunction: contribution of NADPH oxidase. PLoS One 8, e57254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kundumani-Sridharan V., Dyukova E., Hansen D. E., 3rd, Rao G. N. (2013) 12/15-Lipoxygenase mediates high fat diet-induced endothelial tight junction disruption and monocyte transmigration: a new role for 15(S)-hydroxyeicosatetraenoic acid in endothelial cell dysfunction. J. Biol. Chem. 288, 15830–15842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bajpai A. K., Blaskova E., Pakala S. B., Zhao T., Glasgow W. C., Penn J. S., Johnson D. A., Rao G. N. (2007) 15(S)-HETE production in human retinal microvascular endothelial cells by hypoxia: Novel role for MEK1 in 15(S)-HETE induced angiogenesis. Invest. Ophthalmol. Vis. Sci. 48, 4930–4938 [DOI] [PubMed] [Google Scholar]
- 29. Matsumura M., Tanaka N., Kuroki T., Ichihashi M., Ohba M. (2003) The η isoform of protein kinase C inhibits UV-induced activation of caspase-3 in normal human keratinocytes. Biochem. Biophys. Res. Commun. 303, 350–356 [DOI] [PubMed] [Google Scholar]
- 30. Chou M. M., Hou W., Johnson J., Graham L. K., Lee M. H., Chen C. S., Newton A. C., Schaffhausen B. S., Toker A. (1998) Regulation of protein kinase Cζ by PI 3-kinase and PDK-1. Curr. Biol. 8, 1069–1077 [DOI] [PubMed] [Google Scholar]
- 31. Bardin N., Blot-Chabaud M., Despoix N., Kebir A., Harhouri K., Arsanto J. P., Espinosa L., Perrin P., Robert S., Vely F., Sabatier F., Le Bivic A., Kaplanski G., Sampol J., Dignat-George F. (2009) CD146 and its soluble form regulate monocyte transendothelial migration. Arterioscler. Thromb. Vasc. Biol. 29, 746–753 [DOI] [PubMed] [Google Scholar]
- 32. Phinikaridou A., Andia M. E., Protti A., Indermuehle A., Shah A., Smith A., Warley A., Botnar R. M. (2012) Noninvasive magnetic resonance imaging evaluation of endothelial permeability in murine atherosclerosis using an albumin-binding contrast agent. Circulation 126, 707–719 [DOI] [PubMed] [Google Scholar]
- 33. Nomme J., Fanning A. S., Caffrey M., Lye M. F., Anderson J. M., Lavie A. (2011) The Src homology 3 domain is required for junctional adhesion molecule binding to the third PDZ domain of the scaffolding protein ZO-1. J. Biol. Chem. 286, 43352–43360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Petecchia L., Sabatini F., Usai C., Caci E., Varesio L., Rossi G. A. (2012) Cytokines induce tight junction disassembly in airway cells via an EGFR-dependent MAPK/ERK1/2-pathway. Lab. Invest. 92, 1140–1148 [DOI] [PubMed] [Google Scholar]
- 35. Chen Yh, Lu Q., Schneeberger E. E., Goodenough D. A. (2000) Restoration of tight junction structure and barrier function by down-regulation of the mitogen-activated protein kinase pathway in ras-transformed Madin-Darby canine kidney cells. Mol. Biol. Cell 11, 849–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alessi D. R., Cuenda A., Cohen P., Dudley D. T., Saltiel A. R. (1995) PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 270, 27489–27494 [DOI] [PubMed] [Google Scholar]
- 37. Zhao T., Wang D., Cheranov S. Y., Karpurapu M., Chava K. R., Kundumani-Sridharan V., Johnson D. A., Penn J. S., Rao G. N. (2009) A novel role for activating transcription factor-2 in 15(S)-hydroxyeicosatetraenoic acid-induced angiogenesis. J. Lipid Res. 50, 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Iden S., Misselwitz S., Peddibhotla S. S., Tuncay H., Rehder D., Gerke V., Robenek H., Suzuki A., Ebnet K. (2012) aPKC phosphorylates JAM-A at Ser285 to promote cell contact maturation and tight junction formation. J. Cell Biol. 196, 623–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chamorro D., Alarcón L., Ponce A., Tapia R., González-Aguilar H., Robles-Flores M., Mejía-Castillo T., Segovia J., Bandala Y., Juaristi E., González-Mariscal L. (2009) Phosphorylation of zona occludens-2 by protein kinase Cϵ regulates its nuclear exportation. Mol. Biol. Cell 20, 4120–4129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim J. H., Kim J. H., Jun H. O., Yu Y. S., Kim K. W. (2010) Inhibition of protein kinase Cδ attenuates blood-retinal barrier breakdown in diabetic retinopathy. Am. J. Pathol. 176, 1517–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morita K., Sasaki H., Furuse M., Tsukita S. (1999) Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. Cell Biol. 147, 185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Itoh M., Furuse M., Morita K., Kubota K., Saitou M., Tsukita S. (1999) Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J. Cell Biol. 147, 1351–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Elias B. C., Suzuki T., Seth A., Giorgianni F., Kale G., Shen L., Turner J. R., Naren A., Desiderio D. M., Rao R. (2009) Phosphorylation of Tyr-398 and Tyr-402 in occludin prevents its interaction with ZO-1 and destabilizes its assembly at the tight junctions. J. Biol. Chem. 284, 1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Collins N. T., Cummins P. M., Colgan O. C., Ferguson G., Birney Y. A., Murphy R. P., Meade G., Cahill P. A. (2006) Cyclic strain-mediated regulation of vascular endothelial occludin and ZO-1: influence on intercellular tight junction assembly and function. Arterioscler. Thromb. Vasc. Biol. 26, 62–68 [DOI] [PubMed] [Google Scholar]
- 45. Sonobe Y., Takeuchi H., Kataoka K., Li H., Jin S., Mimuro M., Hashizume Y., Sano Y., Kanda T., Mizuno T., Suzumura A. (2009) Interleukin-25 expressed by brain capillary endothelial cells maintains blood-brain barrier function in a protein kinase Cϵ-dependent manner. J. Biol. Chem. 284, 31834–31842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Murakami T., Felinski E. A., Antonetti D. A. (2009) Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J. Biol. Chem. 284, 21036–21046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Itoh M., Nagafuchi A., Yonemura S., Kitani-Yasuda T., Tsukita S. (1993) The 220-kD protein colocalizing with cadherins in non-epithelial cells is identical to ZO-1, a tight junction-associated protein in epithelial cells: cDNA cloning and immunoelectron microscopy. J. Cell Biol. 121, 491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Matter K., Balda M. S. (2003) Signalling to and from tight junctions. Nat. Rev. Mol. Cell Biol. 4, 225–236 [DOI] [PubMed] [Google Scholar]
- 49. Potula H. S., Wang D., Quyen D. V., Singh N. K., Kundumani-Sridharan V., Karpurapu M., Park E. A., Glasgow W. C., Rao G. N. (2009) Src-dependent STAT-3-mediated expression of monocyte chemoattractant protein-1 is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. J. Biol. Chem. 284, 31142–31155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chava K. R., Karpurapu M., Wang D., Bhanoori M., Kundumani-Sridharan V., Zhang Q., Ichiki T., Glasgow W. C., Rao G. N. (2009) CREB-mediated IL-6 expression is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 29, 809–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rao G. N., Baas A. S., Glasgow W. C., Eling T. E., Runge M. S., Alexander R. W. (1994) Activation of mitogen-activated protein kinases by arachidonic acid and its metabolites in vascular smooth muscle cells. J. Biol. Chem. 269, 32586–32591 [PubMed] [Google Scholar]
- 52. Liu Y., Ma C., Zhang Q., Yu L., Ma J., Zhang L., Hao X., Cao F., Wang L., Zhu D. (2012) The key role of transforming growth factor-β receptor I and 15-lipoxygenase in hypoxia-induced proliferation of pulmonary artery smooth muscle cells. Int. J. Biochem. Cell Biol. 44, 1184–1202 [DOI] [PubMed] [Google Scholar]
- 53. Li J., Rao J., Liu Y., Cao Y., Zhang Y., Zhang Q., Zhu D. (2013) 15-Lipoxygenase promotes chronic hypoxia-induced pulmonary artery inflammation via positive interaction with nuclear factor-κB. Arterioscler. Thromb. Vasc. Biol. 33, 971–979 [DOI] [PubMed] [Google Scholar]
- 54. Nishio E., Watanabe Y. (1997) The regulation of mitogenesis and apoptosis in response to the persistent stimulation of α1-adrenoceptors: a possible role of 15-lipoxygenase. Br. J. Pharmacol. 122, 1516–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bailey J. M., Bryant R. W., Whiting J., Salata K. (1983) Characterization of 11-HETE and 15-HETE, together with prostacyclin, as major products of the cyclooxygenase pathway in cultured rat aorta smooth muscle cells. J. Lipid Res. 24, 1419–1428 [PubMed] [Google Scholar]
- 56. Balda M. S., Anderson J. M., Matter K. (1996) The SH3 domain of the tight junction protein ZO-1 binds to a serine protein kinase that phosphorylates a region C-terminal to this domain. FEBS Lett. 399, 326–332 [DOI] [PubMed] [Google Scholar]
- 57. Stuart R. O., Nigam S. K. (1995) Regulated assembly of tight junctions by protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 92, 6072–6076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stevenson B. R., Anderson J. M., Braun I. D., Mooseker M. S. (1989) Phosphorylation of the tight-junction protein ZO-1 in two strains of Madin-Darby canine kidney cells which differ in transepithelial resistance. Biochem. J. 263, 597–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tash B. R., Bewley M. C., Russo M., Keil J. M., Griffin K. A., Sundstrom J. M., Antonetti D. A., Tian F., Flanagan J. M. (2012) The occludin and ZO-1 complex, defined by small angle X-ray scattering and NMR, has implications for modulating tight junction permeability. Proc. Natl. Acad. Sci. U.S.A. 109, 10855–10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Müller S. L., Portwich M., Schmidt A., Utepbergenov D. I., Huber O., Blasig I. E., Krause G. (2005) The tight junction protein occludin and the adherens junction protein α-catenin share a common interaction mechanism with ZO-1. J. Biol. Chem. 280, 3747–3756 [DOI] [PubMed] [Google Scholar]
- 61. Chen S., Shimada K., Zhang W., Huang G., Crother T. R., Arditi M. (2010) IL-17A is proatherogenic in high fat diet-induced and Chlamydia pneumoniae infection-accelerated atherosclerosis in mice. J. Immunol. 185, 5619–5627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Leopold J. A., Loscalzo J. (2009) Oxidative risk for atherothrombotic cardiovascular disease. Free Radic. Biol. Med. 47, 1673–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kotla S., Singh N. K., Heckle M. R., Tigyi G. J., Rao G. N. (2013) The transcription factor CREB enhances interleukin-17A production and inflammation in a mouse model of atherosclerosis. Sci. Signal. 6, ra83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Huang J. T., Welch J. S., Ricote M., Binder C. J., Willson T. M., Kelly C., Witztum J. L., Funk C. D., Conrad D., Glass C. K. (1999) Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature 400, 378–382 [DOI] [PubMed] [Google Scholar]
- 65. Guo Y., Zhang W., Giroux C., Cai Y., Ekambaram P., Dilly A. K., Hsu A., Zhou S., Maddipati K. R., Liu J., Joshi S., Tucker S. C., Lee M. J., Honn K. V. (2011) Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J. Biol. Chem. 286, 33832–33840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vonakis B. M., Vanderhoek J. Y. (1992) 15-Hydroxyeicosatetraenoic acid (15-HETE) receptors. Involvement in the 15-HETE-induced stimulation of the cryptic 5-lipoxygenase in PT-18 mast/basophil cells. J. Biol. Chem. 267, 23625–23631 [PubMed] [Google Scholar]