

Background: The FAK protein level decreases, but its mRNA level remains constant, during skeletal myogenesis, suggesting that an E3 ligase could induce FAK ubiquitination.
Results: The E3 ligase MG53 induces FAK ubiquitination and degradation.
Conclusion: MG53-mediated FAK ubiquitination and degradation is induced during myogenesis.
Significance: This work provides a molecular mechanism for the negative feedback regulation of skeletal myogenesis.
Keywords: Focal Adhesion Kinase, Myogenesis, Skeletal Muscle, Ubiquitin-conjugating Enzyme (Ubc), Ubiquitin-dependent Protease
Abstract
The striated muscle-specific mitsugumin 53 (MG53) is a novel E3 ligase that induces the ubiquitination of insulin receptor substrate 1 (IRS-1) during skeletal myogenesis, negatively regulating insulin-like growth factor and insulin signaling. Here we show that focal adhesion kinase (FAK) is the second target of MG53 during skeletal myogenesis. The FAK protein level gradually decreased, whereas its mRNA level was constant during myogenesis in C2C12 cells and MyoD-overexpressing mouse embryonic fibroblasts. The FAK protein was associated with the E2 enzyme UBE2H and the E3 enzyme MG53 in endogenous and exogenous immunoprecipitation experiments. FAK ubiquitination and degradation was induced by MG53 overexpression in myoblasts but abolished by MG53 or UBE2H knockdown in myotubes. Because RING-disrupted MG53 mutants (C14A and ΔR) did not induce FAK ubiquitination and degradation, the RING domain was determined to be required for MG53-induced FAK ubiquitination. Taken together, these data indicate that MG53 induces FAK ubiquitination with the aid of UBE2H during skeletal myogenesis.
Introduction
Focal adhesions are cellular compartments that anchor mammalian cells to the extracellular matrix. Focal adhesion complexes containing integrin, talin, vinculin, paxillin, focal adhesion kinase (FAK)2 and Src are necessary for transmitting signals from the extracellular environment to the cell interior for actin remodeling and gene activation. FAK is a non-receptor protein-tyrosine kinase (PTK) that functions as a scaffold protein during organismal disease and development (1–3). FAK is the first kinase to be identified as an integrin-regulated protein-tyrosine kinase, and its activity and phosphorylation status are regulated by integrin-dependent cell adhesion (4). FAK itself is regulated by its tyrosine phosphorylation state and intracellular distribution (5). FAK is also known to regulate skeletal myogenesis. For example, FAK regulates heterochromatin remodeling to modulate myogenin expression and skeletal myogenesis in C2C12 cells (6). In mouse primary myoblasts, FAK plays an essential role in skeletal myogenesis by regulating the expression of profusion genes, including caveolin 3 and the β1D integrin subunit (7).
Precise temporal and spatial control of protein synthesis, processing, and degradation plays a fundamental role in regulating skeletal muscle structure and function (8, 9). In fact, the turnover rates and steady-state concentrations of all cellular proteins are controlled by protein degradation (9). The most widely known degradation process is proteolysis via the ubiquitin proteasome system. Initially, free ubiquitin (Ub) is activated by the formation of a thiol ester linkage between the E1 ubiquitin-activating enzyme and the carboxyl terminus of ubiquitin. The activated ubiquitin is then transferred to one of several different E2 ubiquitin-conjugating enzymes. A specific E3 ubiquitin protein ligase interacts with the E2 to transfer the ubiquitin to its specific substrate. The polyubiquitinated substrate protein is then susceptible to degradation by the proteasome complex (10, 11).
It has been proposed that mitsugumin 53 (MG53), also known as tripartite motif-containing 72 (TRIM72), is a novel E3 ligase that induces ubiquitination and proteasomal degradation of insulin receptor substrate 1 (IRS-1) in skeletal muscle (12–14). MG53 is highly up-regulated during skeletal myogenesis because its promoter contains E-box- and myocyte enhancer factor-binding sites, which are binding sites for MyoD and myocyte enhancer factor, respectively (15). With the catalytic RING finger domain in its N terminus, MG53 induces IRS-1 ubiquitination with the aid of the E2 enzyme UBE2H, negatively regulating myogenesis and insulin signaling in C2C12 cells, mouse embryonic fibroblasts (MEFs), and mouse skeletal muscles (13). MG53 is also associated with dysferlin and caveolin 3 (Cav-3), forming membrane repair machinery after acute membrane damage in skeletal and cardiac muscles (16–18). Indeed, recent studies indicate that MG53 is a promising therapeutic target protein for muscular dystrophy, cardioprotection, and insulin resistance (13, 16, 18).
In this study, we provide evidence that FAK protein was down-regulated, but its mRNA level remained constant, during skeletal myogenesis in C2C12 cells and MyoD-overexpressing MEFs. In addition, FAK formed a complex with its E2 enzyme, UBE2H, and E3 enzyme, MG53. Such interactions promote FAK ubiquitination and proteasomal degradation. Overall, this study demonstrates that MG53 is necessary for FAK ubiquitination during skeletal myogenesis.
EXPERIMENTAL PROCEDURES
Cell Culture and Differentiation
The mouse myoblast cell line C2C12 and HEK 293 cells were obtained from the ATCC and cultured in growth medium containing DMEM supplemented with 2% penicillin/streptomycin and 10% FBS in a 5% CO2 incubator at 37 °C. Approximately 90–100% confluent C2C12 myoblasts were differentiated into myotubes by shifting to differentiation medium (DMEM supplemented with 2% penicillin/streptomycin and 2% horse serum). Every 48 h, the myotubes were fed with fresh differentiation medium. MEFs were prepared according to a method described previously (19). The differentiation of the MEFs was induced by adenoviral MyoD overexpression at a dosage of 5 × 109 viral particles/ml, and the cells were incubated with differentiation medium.
Antibodies
The anti-MG53 antibodies were generated as described previously. The anti-Cav-3 antibody was obtained from BD Transduction Laboratories. The anti-MyoD, UBE2H, GAPDH, HA, Myc, FLAG (rabbit polyclonal), and Ub antibodies were obtained from Santa Cruz Biotechnology. The anti-FLAG antibody (mouse monoclonal) was obtained from Sigma. The anti-FAK antibody was obtained from Millipore. The anti-pFAK Tyr-576/577 antibody was obtained from Cell Signaling Technology. The anti-MyHC antibody was obtained from the Developmental Studies Hybridoma Bank. The anti-Mgn antibody was obtained from BD Biosciences.
Adenovirus Preparation and Infection
Adenoviruses harboring MG53, C14A, and ΔR were produced according to a method described previously (12). Adenovirus containing MyoD was obtained from Cell Biolabs. To amplify the virus, viral stocks were reinfected into AD293 cells and purified by double cesium chloride-gradient ultracentrifugation. Cesium-banded virus stocks were dialyzed against 10 mm Tris (pH 8.0), 2 mm MgCl2, and 5% sucrose. The stocks were then aliquoted and stored at −80 °C until use. C2C12 myoblasts or MEFs were infected with adenovirus at a dosage of 5 × 109 viral particles/ml.
Plasmids and Transient Transfection
FAK, ubiquitin, MG53, and UBE2H cDNA constructs were generated by PCR and cloned into the pCMV-Tag2b and pCMV-3Tag4a vectors. Different MG53 and FAK mutant constructs were prepared as described previously (12, 20).
Plasmids for pRK5-HA-Ubiquitin-WT, pRK5-HA-Ubiquitin-K48, pRK5-HA-Ubiquitin-K48R, and pRK5-HA-Ubiquitin-K29R were purchased from Ted Dawson. DNA transfection was performed using Polyfect (Qiagen) or electroporation (Invitrogen) according to the protocol of the manufacturer.
RNA Interference
siRNA oligomers targeting MG53 (si-MG53) or UBE2H (si-UBE2H) and a scrambled oligomer (si-control) were obtained from Ambion. The target sequences of MG53 and UBE2H were 5′-AAGCACGCCUCAAGACACAGC-3′ and 5′-CUAUGAUCUUACCAAUAUAtt-3′, respectively. C2C12 myoblasts were transfected with si-control, si-MG53, or si-UBE2H by electroporation (Invitrogen) according to the protocol of the manufacturer.
Immunoblotting and Immunoprecipitation
Cells were collected and washed once with ice-cold PBS and lysed with lysis buffer (PBS containing 0.5% Nonidet P-40, protease inhibitor mixture, and phosphatase inhibitors (Roche)) for 30 min on ice. After microcentrifugation (14,000 rpm) for 10 min at 4 °C, the proteins in the whole cell lysate were separated by SDS-PAGE and transferred to a PVDF membrane. The membranes were blocked for 1 h at room temperature with 5% w/v nonfat dry milk in TBS containing 0.1% Tween 20 and subsequently allowed to react with a sequence of specific primary antibodies and HRP-conjugated secondary antibodies. The signals were visualized with ECL reagents using an automatic image analysis system (LAS 3000, Fuji Life Science). For immunoprecipitation, 1 mg of the whole cell lysate was incubated with 2 μg of the designated antibody overnight at 4 °C and subsequently incubated with 30 μl of protein A-agarose (Roche) on a rotator for 1 h at 4 °C. The immunoprecipitates were then analyzed by immunoblotting. To determine whether FAK phosphorylation was required for the formation of MG53, UBE2H, and FAK complex, the whole cell lysates were obtained from 3-day differentiated C2C12 myotubes and incubated with or without λ phosphatase (1000 units, Calbiochem) for 30 min at 30 °C. The reaction was stopped by the addition of 5 mm of sodium orthovanadate. Molecular interactions among MG53, UBE2H, and FAK were determined by immunoprecipitation.
RT-PCR
Total RNA (1–2 μg) was isolated from C2C12 myoblasts and myotubes in addition to MEFs using TRIzol reagent (Ambion) and was reverse-transcribed into cDNA using Moloney murine leukemia virus reverse transcriptase (Invitrogen). Semiquantitative PCR was performed using Taq polymerase (Genemed) with the following primers: FAK, 5′-GTGGTCCTACCAGCACTTTTGGG-3′ and 5′-CGATCGCAGGTGACTGAGGCG-3′; MyHC, 5′-AGAAGGAGGAGGCAACTTCTG-3′ and 5′-ACATACTCATTGCCGACCTTG-3′;and GAPDH, 5′-CTGCACCACCAACTGCTTAGC-3′ and 5′-CTTCACCACCTTCTTGATGTC-3′.
FAK Ubiquitination
HEK 293 cells were cotransfected with FLAG-FAK and His-Ub along with Myc-UBE2H and HA-MG53, HA-C14A, or HA-ΔR. After 36 h of transfection, the cells were treated with MG132 (5 μm) for another 12 h and then harvested with lysis buffer. The lysates were immunoprecipitated with an anti-FLAG antibody, and the immunoprecipitates were immunoblotted with an anti-His antibody. Adenoviral MG53- or siRNA-treated C2C12 cells were treated with MG132 (5 μm) for 12 h and lysed with lysis buffer. Whole cell lysates were immunoprecipitated with an anti-FAK antibody. Endogenous FAK ubiquitination was detected by immunoblotting with an anti-ubiquitin antibody.
RESULTS
FAK Protein Is Down-regulated during Skeletal Myogenesis
There are conflicting data regarding the expression level of FAK during skeletal myogenesis. For example, the FAK expression level gradually decreases during myogenesis in primary mouse myoblast cultures but remains constant during C2C12 myogenesis (7, 21). To reconcile this difference, we re-evaluated the level of FAK expression during C2C12 myogenesis. Immunoblot analysis revealed a significant decline in the FAK protein expression level during C2C12 myogenesis (Fig. 1A). After day 3, the FAK protein level gradually decreased and reached a low level after 5 days. However, other myogenic marker proteins, such as myosin heavy chain (MyHC), myogenin (Mgn), and caveolin 3 (Cav-3), were gradually increased during myogenesis, indicating that skeletal myogenesis was well induced. Interestingly, the E2 enzyme UBE2H and the E3 enzyme MG53 were up-regulated during myogenesis. Although the FAK protein level was down-regulated, its mRNA level was nearly constant throughout C2C12 myogenesis (Fig. 1B). We also reconfirmed the decrease in FAK expression level during MyoD-driven myogenesis in MEFs. After adenoviral overexpression of MyoD, the MEFs were induced to differentiate into myotubes. As shown in Fig. 1C, the levels of myogenic marker proteins, such as MyHC, Mgn, and Cav-3, were increased greatly during myogenesis, indicating that MyoD overexpression led to myogenesis induction. On days 5 and 7, the FAK expression level was decreased greatly, whereas its mRNA level was constant, in the MyoD-induced myotubes of MEFs. In addition, UBE2H and MG53 were up-regulated during MyoD-induced myogenesis of MEFs. The data in Fig. 1, A–D, indicate that FAK is degraded during skeletal myogenesis in C2C12 cells and MyoD-overexpressing MEFs.
FIGURE 1.
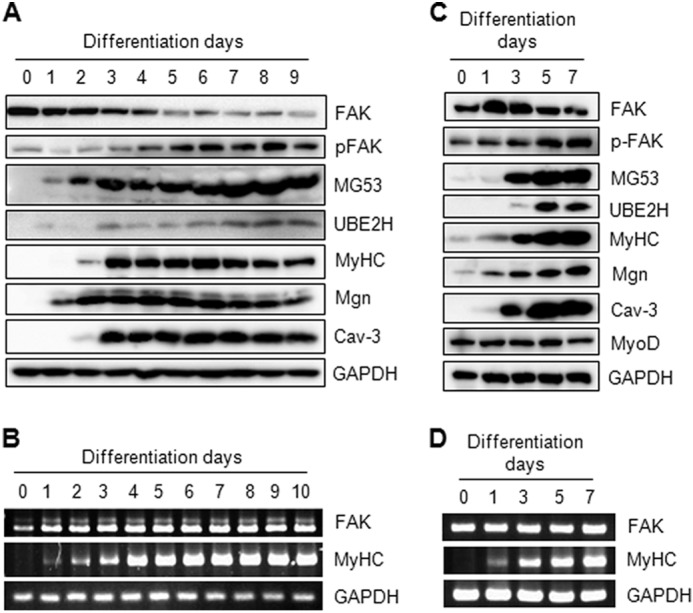
The protein expression level of FAK decreases during skeletal myogenesis. A and B, C2C12 myoblasts were differentiated into myotubes for the indicated days. The expression levels of FAK, phospho-FAK (p-FAK) at Tyr-576/577, MG53, UBE2H, MyHC, Mgn, and Cav-3 were determined by immunoblotting using GAPDH as a loading control (A). The mRNA levels of FAK and MyHC were determined by RT-PCR using GAPDH as a loading control (B). C and D, MEFs were infected with adenoviral MyoD for 12 h and differentiated into myotubes for the indicated number of days. The protein levels of FAK, p-FAK, MG53, UBE2H, MyHC, Mgn, Cav-3, and GAPDH were determined by immunoblotting (C), and the mRNA levels of FAK, MyHC, and GAPDH were determined by RT-PCR (D).
FAK Interacts with MG53 and UBE2H
MG53 regulates skeletal myogenesis and insulin signaling via its E3 ubiquitin ligase activity, which leads to IRS-1 ubiquitination in C2C12 cells, MEFs, and mouse skeletal muscle (13, 14). Indeed, levels of MG53 and the E2 enzyme UBE2H were increased during skeletal myogenesis in C2C12 cells and MyoD-overexpressing MEFs, as shown in Fig. 1, A and C. These observations prompted us to investigate the involvement of MG53 in FAK ubiquitination and degradation in skeletal muscle cells. To explore this hypothesis, we first examined whether there was a molecular interaction between FAK and MG53. Evidence for this molecular interaction was revealed by reciprocal endogenous immunoprecipitation in 3-day differentiated C2C12 myotubes (Fig. 2, A and B). Because UBE2H is an E2 partner for MG53 (13), we also confirmed whether the FAK complex contained UBE2H and MG53. As shown in Fig. 2C, UBE2H interacted with FAK and MG53 in 3-day differentiated C2C12 myotubes.
FIGURE 2.

FAK interacts with MG53 and UBE2H. A–C, the molecular association of FAK with MG53 and UBE2H was determined by endogenous immunoprecipitation with anti-FAK, MG53, and UBE2H antibodies in 3-day differentiated C2C12 myotubes. WCL, whole cell lysates. D–F, the molecular interaction of MG53 and UBE2H was independent of the phosphorylation status of FAK. Whole cell lysates were obtained from 3-day differentiated C2C12 myotubes and incubated with or without 1000 units of λ phosphatase for 30 min at 30 °C. The reaction was stopped by adding 5 mm sodium orthovanadate, and the reactants were analyzed by immunoprecipitation.
Next, we tested whether FAK phosphorylation was required for the complex formation of MG53, UBE2H, and FAK after treating the whole cell lysates of C2C12 myotubes with or without λ phosphatase. As shown in Fig. 2, D–E, the phosphorylated FAK disappeared after the treatment of λ phosphatase. However, the reciprocal immunoprecipitation showed that molecular interactions among MG53, UBE2H, and FAK were not prevented by the addition of λ phosphatase (Fig. 2, D–E). These data indicate that the complex formation of MG53, UBE2H, and FAK is not dependent on FAK phosphorylation.
The molecular interaction between FAK and MG53 was further confirmed by exogenous immunoprecipitation in HEK 293 cells overexpressing both HA-FAK and Myc-MG53 (Fig. 3A). To identify the MG53-interacting FAK domain, we overexpressed different FAK deletion mutants with MG53 (Fig. 3B, top panel) and monitored their molecular interaction by coimmunoprecipitation. The immunoprecipitation assays in HEK 293 cells showed that His-FAK interacted with Myc-MG53 through its 4.1 protein/ezrin/radixin/moesin (FERM) and kinase domains (Fig. 3C). We next determined the FAK-interacting MG53 domain by immunoprecipitation in cells overexpressing FAK and different deletion mutants of MG53 (Fig. 3B, bottom panel). As shown in Fig. 3D, the B-box of MG53 might be a FAK-interacting domain because Myc-MG53 deletion mutants lacking a B-box (CC-SPRY (coiled-coil domain and spla and ryanodine receptor domain) and SPla and RYanodine receptor domain) did not interact with His-FAK.
FIGURE 3.

The B-box of MG53 is a binding domain for FAK. A, different combinations of Myc-MG53 (0.5 μg) and HA-FAK (1.5 μg) were cotransfected into HEK 293 cells for 36 h. The cells were further treated with MG132 (5 μm) for 12 h, and then the molecular association of MG53 with FAK was monitored by immunoprecipitation (IP) with anti-HA and anti-Myc antibodies. WCL, whole cell lysate. B, FAK and MG53 deletion constructs. F, full-length; FERM, 4.1 protein/ezrin/radixin/moesin; FAT, focal adhesion targeting domain; FRNK, FAK-related non-kinase domain; CC, coiled-coil domain; SPRY, SPla and RYanodine receptor domain; RBCC, RING, B-box, and coiled-coil domains. C and D, HEK 293 cells were cotransfected with Myc-MG53 (0.5 μg) and various His-FAK mutants (1.5 μg) or with His-FAK (1.5 μg) and various Myc-MG53 mutants (0.5 μg) for 48 h and subsequently treated with MG132 (5 μm) for 12 h. The molecular association of MG53 with FAK was determined by immunoprecipitation (IP). EV, empty vector.
MG53 Induces FAK Degradation
To confirm MG53-induced FAK degradation, we next monitored the expression level of FAK protein after transient expression of different combinations of HA-FAK and Myc-MG53 in HEK 293 cells. The FAK protein level was decreased drastically by overexpressing MG53 and was fully restored when the proteasome inhibitor MG132 was added (Fig. 4, A and B). MG53-induced FAK degradation occurred in a MG53 concentration-dependent manner in the absence of MG132 (Fig. 4C). These results suggest that MG53-induced FAK degradation is dependent on the proteasome pathway.
FIGURE 4.

The RING domain is required for MG53-induced FAK degradation. A and B, FAK was degraded by MG53. A, HEK 293 cells were cotransfected with HA-FAK (1.5 μg) and Myc-MG53 (0.5 μg) for 24 h and subsequently treated with or without MG132 (5 μm) for 12 h. The expression levels of FAK and MG53 were determined by immunoblotting. B, FAK protein levels were quantified by densitometry. This experiment was repeated three times (Student's t test; *, p < 0.01). C, FAK is degraded by MG53 in a concentration-dependent manner. HEK 293 cells were cotransfected with HA-FAK (1.5 μg) and increasing amounts of Myc-MG53 (0, 0.005, 0.01, 0.1, and 0.3 μg). In the presence or absence of MG132 for 12 h, the expression levels of FAK and MG53 were determined by immunoblotting. D and E, FAK is not degraded by RING-disrupted MG53 (C14A and ΔR). HEK 293 cells were cotransfected with His-FAK (1.5 μg) and HA-C14A or HA-ΔR (0.5 μg). In the absence of MG132, the expression levels of FAK and MG53 were determined by immunoblotting.
We demonstrated previously that the RING domain of MG53 is required for its E3 ligase activity because RING-disrupted MG53 mutants (C14A and ΔR) did not degrade the IRS-1 protein (13). Thus, we tested the requirement of the RING domain for MG53-induced FAK degradation after overexpressing His-FAK together with HA-C14A or HA-ΔR in HEK 293 cells. As shown in Fig. 4, D and E, C14A and ΔR did not degrade FAK, indicating that the RING domain is necessary for MG53-induced FAK degradation.
The RING Domain of MG53 Is Required for FAK Ubiquitination
Next, we tested MG53-induced FAK ubiquitination in HEK 293 cells after the transient expression of HA-FAK, Myc-MG53, and His-Ub in the presence of MG132. As shown in Fig. 5A, FAK ubiquitination was gradually induced with increasing amounts of MG53, suggesting that MG53 induced FAK ubiquitination in a concentration-dependent manner. Because Lys-48 in ubiquitin is critical for protein ubiquitination, we tested MG53-induced FAK ubiquitination after overexpressing HA-ubiquitin WT, Lys-48, K48R, or K29R along with Myc-MG53 and His-FAK in HEK 293 cells. In the Lys-48 mutant, all lysines except for amino acid position 48 are substituted for arginines. As shown in Fig. 5B, only the K48R ubiquitin mutant abolished FAK ubiquitination, indicating that the Lys-48 is a critical lysine for MG53-induced FAK ubiquitination.
FIGURE 5.

FAK ubiquitination requires the RING domain of MG53. A, MG53 induces FAK ubiquitination in a concentration-dependent manner. HEK 293 cells were cotransfected with HA-FAK (1.5 μg) and His-Ub (0.5 μg) along with increasing amounts of Myc-MG53 (0, 0.005, 0.01, 0.05, 0.1, 0.2, and 0.5 μg) for 24 h. After treatment with MG132 (5 μm) for 12 h, FAK ubiquitination was determined by immunoprecipitation (IP) with an anti-HA antibody. WCL, whole cell lysate. B, Lys-48 in ubiquitin is critical for FAK ubiquitination. HEK 293 cells were cotransfected with Myc-MG53 (0.5 μg) and His-FAK (1.5 μg) along with ubiquitin WT, Lys-48, K48R, or K29R (0.5 μg for each) for 24 h. All lysines, except for amino acid position 48, were substituted for arginines in the ubiquitin Lys-48 mutant. After MG132 treatment, FAK ubiquitination was determined by immunoprecipitation. EV, empty vector. C and D, the RING domain is required for MG53-induced FAK ubiquitination. HEK 293 cells were cotransfected with FLAG-FAK (1.5 μg) and His-Ub (0.5 μg) along with HA-MG53, HA-C14A, or HA-ΔR (0.5 μg) for 24 h in the indicated combinations. Twelve hours after MG132 treatment, FAK ubiquitination was determined by immunoprecipitation (C). C2C12 myoblasts were transfected with adenoviral LacZ, MG53, C14A, or ΔR for 24 h. Twelve hours after MG132 treatment, FAK ubiquitination was determined by immunoprecipitation (D). E, MG53 knockdown abolishes FAK ubiquitination. C2C12 myoblasts were treated with si-control or si-MG53 (200 nm) for 24 h and then further differentiated into myotubes for 3 days. After MG132 treatment for 12 h, FAK ubiquitination was determined by immunoprecipitation.
We also tested whether the RING domain of MG53 is required for MG53-induced FAK ubiquitination in HEK 293 cells after the overexpression of FLAG-FAK and His-Ub with HA-C14A or HA-ΔR. MG53 induced FAK ubiquitination, whereas C14A and ΔR did not, in the presence of MG132 (Fig. 5C), indicating that the RING domain of MG53 is required for FAK ubiquitination. We also demonstrated the requirement of the RING domain for MG53-induced FAK ubiquitination in C2C12 myoblasts after adenoviral overexpression of MG53, C14A, and ΔR (Fig. 5D). Furthermore, FAK ubiquitination was completely abrogated in C2C12 myotubes after MG53 knockdown (Fig. 5E). These findings suggest that MG53 could directly participate in FAK ubiquitination via its RING domain, which has ubiquitin ligase activity.
The E2 Enzyme UBE2H Is Essential for MG53-induced FAK Ubiquitination
We demonstrated previously that UBE2H is an E2 partner of MG53 for IRS-1 ubiquitination (13). Thus, we tested the involvement of UBE2H in MG53-induced FAK ubiquitination after UBE2H knockdown in C2C12 myotubes. As shown in Fig. 6, A and B, the expression level of FAK protein was increased 3-fold after UBE2H knockdown. Furthermore, FAK ubiquitination was abrogated after UBE2H knockdown in C2C12 myotubes (Fig. 6C). Next, we monitored the increase in FAK ubiquitination after overexpressing FLAG-FAK, HA-MG53, and His-Ub along with Myc-UBE2H in HEK 293 cells. MG53-induced FAK ubiquitination was enhanced by overexpressing UBE2H (Fig. 6D). Taken together, these data indicate that the UBE2H and MG53 complexes are required for FAK ubiquitination and degradation.
FIGURE 6.

UBE2H is required for MG53-induced FAK ubiquitination. A and B, C2C12 myoblasts were treated with si-control (si-con) or si-UBE2H (100 nm) for 24 h and then differentiated into myotubes for 3 days. The expression levels of FAK, UBE2H, MyHC, Cav-3, and GAPDH were determined by immunoblotting (A). The expression of FAK was also quantified by densitometry. These experiments were repeated three times (Student's t test; *, p < 0.01) (B). C, UBE2H knockdown C2C12 myotubes were treated with MG132 (5 μm) for 12 h, and FAK ubiquitination was determined by endogenous immunoprecipitation (IP). WCL, whole cell lysate. D, UBE2H overexpression increases MG53-induced FAK ubiquitination. HEK 293 cells were cotransfected with different combinations of FLAG-FAK (1.5 μg), His-Ub (0.5 μg), HA-MG53 (0.5 μg), and Myc-UBE2H (0.5 μg) for 24 h. After MG132 treatment for 12 h, FAK ubiquitination was determined by immunoprecipitation with an anti-FLAG antibody and immunoblotting with an anti-His antibody.
DISCUSSION
FAK has an essential role in skeletal myogenesis. For example, knockdown or overexpression of the dominant-negative focal adhesion targeting domain of FAK prevents cell fusion in mouse primary myoblasts (7). Early in C2C12 myogenesis, FAK is phosphorylated, activated, and, subsequently, translocated into the nucleus. In the nucleus, the activated FAK interacts with methyl CpG-binding domain protein 2 (MBD2), sequestering histone deacetylase complex 1 (HDAC1) from MBD2. Subsequently, the free HDAC1 regulates heterochromatin remodeling at the methyl CpG site in the Mgn promoter, triggering Mgn expression and muscle differentiation (6). As myotubes matured, the FAK protein level decreased, beginning as early as 3 days in C2C12 cells and MyoD-overexpressing MEFs (Fig. 1, A and C) and as early as 2 days in mouse primary myoblasts (7). Thus, the decreased FAK protein level might be necessary for negative feedback regulation of skeletal myogenesis, preventing excess Mgn transcription and myogenesis.
Proteasomal activity is increased during skeletal myogenesis in primary myoblast cultures (22). In this study, we demonstrated that the expression levels of the E2 enzyme UBE2H and the E3 enzyme MG53 were increased during skeletal myogenesis in C2C12 cells and MyoD-overexpressing MEFs (Fig. 1, A and C). We also demonstrated MG53-induced FAK ubiquitination. First, FAK was determined to be associated with MG53 and UBE2H by reciprocal endogenous and exogenous immunoprecipitation (Figs. 2 and 3). Second, MG53-induced FAK ubiquitination and degradation was demonstrated by MG53 and UBE2H overexpression in HEK 293 cells and C2C12 myoblasts and MG53 or UBE2H knockdown in C2C12 myotubes (Figs. 4–6). Third, we showed that the RING domain was required for MG53-induced FAK ubiquitination after overexpressing RING-disrupted MG53 mutants (C14A and ΔR) in HEK 293 cells and C2C12 myoblasts (Figs. 4 and 5). Taken together, these data indicate that FAK is ubiquitinated and degraded by the E2 enzyme UBE2H and the E3 enzyme MG53, which are up-regulated during skeletal myogenesis.
It is well known that FAK is also ubiquitinated and degraded by other E3 ligases, such as suppressor of cytokine signaling proteins and Casitas B-lineage lymphoma (Cbl) (23–25). Suppressor of cytokine signaling protein-induced FAK ubiquitination inhibits FAK-dependent signaling events, such as cell motility on fibronectin (23). Cbl-induced FAK ubiquitination leads to anoikis in myocytes and prevents integrin-mediated T-cell adhesion (24, 25). However, there have been no reports of E3 ligases involved in FAK ubiquitination during skeletal myogenesis. This study is the first report demonstrating that FAK is ubiquitinated and degraded through the MG53-dependent proteasomal pathway during skeletal myogenesis. Although it has been characterized as a putative E3 ligase, MG53 has a limited number of protein substrates. IRS-1, the first protein identified as a target of MG53, is also degraded by suppressor of cytokine signaling and Cbl (13, 26, 27), suggesting that IRS-1 and FAK may share additional common E3 ligases. Indeed, MG53 is another common E3 ligase for the ubiquitination of IRS-1 and FAK.
Although FAK and IRS-1 share a common E2 ligase (UBE2H) and E3 ligase (MG53) during skeletal myogenesis, their MG53-interacting domains are different. For example, among the different domains of MG53, the coiled-coil domain is utilized for IRS-1 interaction, and the B-box domain is utilized for FAK interaction. Although the FAK expression level decreased during myogenesis of C2C12 cells and MyoD-overexpressing MEFs, phosphorylation of FAK at Tyr-576/577 was increased gradually (Fig. 1, A and C). These findings suggest that FAK ubiquitination might require its phosphorylation because many proteins are ubiquitinated and degraded in a phosphorylation-dependent process (28). However, the molecular interaction between MG53 and FAK was not prevented in the presence of λ phosphatase (Fig. 2, D–F), indicating that MG53-induced FAK ubiquitination is not dependent on the phosphorylation of FAK. We also observed previously that MG53-IRS-1 interaction is not altered after IGF stimulation in C2C12 myotubes. With all these data, we can conclude that the molecular association of MG53 to IRS-1 or FAK is independent of the phosphorylation status of substrate proteins.
This work was supported by National Research Foundation Grants 2011-0030158 and 2011-0017562 (to Y. G. K.). This work was also partially supported by a Korea University grant (to Y. G. K.).
- FAK
- focal adhesion kinase
- Ub
- ubiquitin
- MEF
- mouse embryonic fibroblast.
REFERENCES
- 1. Bisht B., Dey C. S. (2008) Focal adhesion kinase contributes to insulin-induced actin reorganization into a mesh harboring glucose transporter-4 in insulin resistant skeletal muscle cells. BMC Cell Biol. 9, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Flück M., Ziemiecki A., Billeter R., Müntener M. (2002) Fibre-type specific concentration of focal adhesion kinase at the sarcolemma. Influence of fibre innervation and regeneration. J. Exp. Biol. 205, 2337–2348 [DOI] [PubMed] [Google Scholar]
- 3. Franchini K. G. (2012) Focal adhesion kinase. The basis of local hypertrophic signaling domains. J. Mol. Cell. Cardiol. 52, 485–492 [DOI] [PubMed] [Google Scholar]
- 4. Shen Y., Schaller M. D. (1999) Focal adhesion targeting. The critical determinant of FAK regulation and substrate phosphorylation. Mol. Biol. Cell 10, 2507–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mao H., Li F., Ruchalski K., Mosser D. D., Schwartz J. H., Wang Y., Borkan S. C. (2003) Hsp72 inhibits focal adhesion kinase degradation in ATP-depleted renal epithelial cells. J. Biol. Chem. 278, 18214–18220 [DOI] [PubMed] [Google Scholar]
- 6. Luo S. W., Zhang C., Zhang B., Kim C. H., Qiu Y. Z., Du Q. S., Mei L., Xiong W. C. (2009) Regulation of heterochromatin remodelling and myogenin expression during muscle differentiation by FAK interaction with MBD2. EMBO J. 28, 2568–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Quach N. L., Biressi S., Reichardt L. F., Keller C., Rando T. A. (2009) Focal adhesion kinase signaling regulates the expression of caveolin 3 and β1 integrin, genes essential for normal myoblast fusion. Mol. Biol. Cell 20, 3422–3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim J., Löwe T., Hoppe T. (2008) Protein quality control gets muscle into shape. Trends Cell Biol. 18, 264–272 [DOI] [PubMed] [Google Scholar]
- 9. Lundin V. F., Leroux M. R., Stirling P. C. (2010) Quality control of cytoskeletal proteins and human disease. Trends Biochem. Sci. 35, 288–297 [DOI] [PubMed] [Google Scholar]
- 10. Fang S., Weissman A. M. (2004) A field guide to ubiquitylation. Cell. Mol. Life Sci. 61, 1546–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen C., Seth A. K., Aplin A. E. (2006) Genetic and expression aberrations of E3 ubiquitin ligases in human breast cancer. Mol. Cancer Res. 4, 695–707 [DOI] [PubMed] [Google Scholar]
- 12. Lee C. S., Yi J. S., Jung S. Y., Kim B. W., Lee N. R., Choo H. J., Jang S. Y., Han J., Chi S. G., Park M., Lee J. H., Ko Y. G. (2010) TRIM72 negatively regulates myogenesis via targeting insulin receptor substrate-1. Cell Death Differ. 17, 1254–1265 [DOI] [PubMed] [Google Scholar]
- 13. Yi J. S., Park J. S., Ham Y. M., Nguyen N., Lee N. R., Hong J., Kim B. W., Lee H., Lee C. S., Jeong B. C., Song H. K., Cho H., Kim Y. K., Lee J. S., Park K. S., Shin H., Choi I., Lee S. H., Park W. J., Park S. Y., Choi C. S., Lin P., Karunasiri M., Tan T., Duann P., Zhu H., Ma J., Ko Y. G. (2013) MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat. Commun. 4, 2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Song R., Peng W., Zhang Y., Lv F., Wu H. K., Guo J., Cao Y., Pi Y., Zhang X., Jin L., Zhang M., Jiang P., Liu F., Meng S., Zhang X., Jiang P., Cao C. M., Xiao R. P. (2013) Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature 494, 375–379 [DOI] [PubMed] [Google Scholar]
- 15. Jung S. Y., Ko Y. G. (2010) TRIM72, a novel negative feedback regulator of myogenesis, is transcriptionally activated by the synergism of MyoD (or myogenin) and MEF2. Biochem. Biophys. Res. Commun. 396, 238–245 [DOI] [PubMed] [Google Scholar]
- 16. Alloush J., Weisleder N. (2013) TRIM proteins in therapeutic membrane repair of muscular dystrophy. JAMA Neurol. 70, 928–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cai C., Masumiya H., Weisleder N., Matsuda N., Nishi M., Hwang M., Ko J. K., Lin P., Thornton A., Zhao X., Pan Z., Komazaki S., Brotto M., Takeshima H., Ma J. (2009) MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 11, 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hausenloy D. J., Yellon D. M. (2010) Cell membrane repair as a mechanism for ischemic preconditioning? Circulation 121, 2547–2549 [DOI] [PubMed] [Google Scholar]
- 19. Shim E. H., Kim J. I., Bang E. S., Heo J. S., Lee J. S., Kim E. Y., Lee J. E., Park W. Y., Kim S. H., Kim H. S., Smithies O., Jang J. J., Jin D. I., Seo J. S. (2002) Targeted disruption of hsp70.1 sensitizes to osmotic stress. EMBO Rep. 3, 857–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ahn S., Kim H. J., Chi S. G., Park H. (2012) XIAP reverses various functional activities of FRNK in endothelial cells. Biochem. Biophys. Res. Commun. 419, 419–424 [DOI] [PubMed] [Google Scholar]
- 21. Clemente C. F., Corat M. A., Saad S. T., Franchini K. G. (2005) Differentiation of C2C12 myoblasts is critically regulated by FAK signaling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R862–870 [DOI] [PubMed] [Google Scholar]
- 22. Gardrat F., Montel V., Raymond J., Azanza J. L. (1997) Proteasome and myogenesis. Mol. Biolo. Rep. 24, 77–81 [DOI] [PubMed] [Google Scholar]
- 23. Liu E., Côté J. F., Vuori K. (2003) Negative regulation of FAK signaling by SOCS proteins. EMBO J. 22, 5036–5046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rafiq K., Guo J., Vlasenko L., Guo X., Kolpakov M. A., Sanjay A., Houser S. R., Sabri A. (2012) c-Cbl ubiquitin ligase regulates focal adhesion protein turnover and myofibril degeneration induced by neutrophil protease cathepsin G. J. Biol. Chem. 287, 5327–5339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sekine Y., Tsuji S., Ikeda O., Sugiyma K., Oritani K., Shimoda K., Muromoto R., Ohbayashi N., Yoshimura A., Matsuda T. (2007) Signal-transducing adaptor protein-2 regulates integrin-mediated T cell adhesion through protein degradation of focal adhesion kinase. J. Immunol. 179, 2397–2407 [DOI] [PubMed] [Google Scholar]
- 26. Kawaguchi T., Yoshida T., Harada M., Hisamoto T., Nagao Y., Ide T., Taniguchi E., Kumemura H., Hanada S., Maeyama M., Baba S., Koga H., Kumashiro R., Ueno T., Ogata H., Yoshimura A., Sata M. (2004) Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am. J. Pathol. 165, 1499–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakao R., Hirasaka K., Goto J., Ishidoh K., Yamada C., Ohno A., Okumura Y., Nonaka I., Yasutomo K., Baldwin K. M., Kominami E., Higashibata A., Nagano K., Tanaka K., Yasui N., Mills E. M., Takeda S., Nikawa T. (2009) Ubiquitin ligase Cbl-b is a negative regulator for insulin-like growth factor 1 signaling during muscle atrophy caused by unloading. Mol. Cell. Biol. 29, 4798–4811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Swaney D. L., Beltrao P., Starita L., Guo A., Rush J., Fields S., Krogan N. J., Villén J. (2013) Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 10, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]