

Background: The thermophilic DNA topoisomerase reverse gyrase induces DNA-positive supercoiling.
Results: The novel reverse gyrase, PcalRG, is presented.
Conclusion: PcalRG is the most efficient and robust reverse gyrase known, and the first inducing ATP-dependent unwinding of Holliday junctions and annealing of single-stranded oligonucleotides.
Significance: PcalRG shares structural and functional features with evolutionary conserved helicase-topoisomerase complexes involved in genome stability.
Keywords: DNA Enzymes, DNA Helicase, DNA Recombination, DNA Repair, DNA Topoisomerase, Holliday Junction, Thermophiles
Abstract
Reverse gyrase is a DNA topoisomerase specific for hyperthermophilic bacteria and archaea. It catalyzes the peculiar ATP-dependent DNA-positive supercoiling reaction and might be involved in the physiological adaptation to high growth temperature. Reverse gyrase comprises an N-terminal ATPase and a C-terminal topoisomerase domain, which cooperate in enzyme activity, but details of its mechanism of action are still not clear. We present here a functional characterization of PcalRG, a novel reverse gyrase from the archaeon Pyrobaculum calidifontis. PcalRG is the most robust and processive reverse gyrase known to date; it is active over a wide range of conditions, including temperature, ionic strength, and ATP concentration. Moreover, it holds a strong ATP-inhibited DNA cleavage activity. Most important, PcalRG is able to induce ATP-dependent unwinding of synthetic Holliday junctions and ATP-stimulated annealing of unconstrained single-stranded oligonucleotides. Combined DNA unwinding and annealing activities are typical of certain helicases, but until now were shown for no other reverse gyrase. Our results suggest for the first time that a reverse gyrase shares not only structural but also functional features with evolutionary conserved helicase-topoisomerase complexes involved in genome stability.
Introduction
Reverse gyrase (RG)4 is the only DNA topoisomerase able to catalyze positive supercoiling and is a hallmark of hyperthermophilic microorganisms (1–4). Because positive supercoiling is associated with increased resistance of DNA to thermal denaturation, and RG is the only hyperthermophile-specific protein, it was suggested to play a role in the adaptation of bacteria and archaea living at high temperature, although knock-out experiments in different strains gave contradictory results (5–6). Experiments in vivo suggest that, at least in the archaeon Sulfolobus solfataricus, RG is involved in maintenance of genome stability (7–10).
RG is a modular enzyme, as it comprises a C-terminal type IA topoisomerase domain and an N-terminal region containing sequence motifs typical of the SF2 helicase family, including an ATP-binding domain. For its intriguing features, RG has attracted considerable interest over the last few years, and several members of this family have been characterized with the aim of elucidating its mechanism, which is still not completely understood. Despite considerable primary sequence similarity (see supplemental Fig. S1), different RGs show both structural and functional differences. For instance, although all RGs are thermophilic enzymes, their optimal reaction temperatures range between 75 and 90 °C. Whereas an NTP is absolutely required for the positive supercoiling reaction, some RGs are able to relax DNA in the absence of nucleotide, whereas others are not. Differences in processivity and ionic strength preference are also seen (11–17).
The isolated C- and N-terminal domains of a few RGs can exist in stable form either naturally (e.g. the Nanoarchaeum equitans enzyme) or as a result of a deletion mutagenesis (e.g. Sulfolobus RG), retaining their own activities and restoring the positive supercoiling activity when combined. In both enzymes, the C-terminal domain stimulates the N-terminal ATPase activity (13, 18, 19). In contrast, in Thermotoga maritima RG, each domain was suggested to attenuate the individual activities of the other, although only the N-terminal domain of this enzyme was tested in isolation (20–22).
It was previously proposed that communication between the N-terminal and C-terminal domains is mediated by the so-called latch, a poorly conserved region of the N-terminal domain. Deletion of this region has different effects in different RGs: for example, in the Archaeoglobus fulgidus enzyme, the latch suppresses DNA relaxation in the absence of ATP, but is not essential for positive supercoiling (11, 12); in the Thermoanaerobacter tengcongensis RG the latch is required for positive DNA supercoiling, but not for DNA binding (17); finally, in the T. maritima enzyme it contributes to DNA binding and is required for positive supercoiling and ATP-dependent transient duplex separation (23, 24).
The molecular basis of these differences are currently not clear. Primary sequence alignments show regions highly conserved in all RGs, in particular in the topoisomerase domain, whereas considerable sequence and length variability is seen in the N-terminal domain (supplemental Fig. S1). The two RG three-dimensional structures available showed overall similarities, but significantly different folds in the N-terminal domain, in particular in the latch and the H1 sub-domain (25, 26). However, these structural data failed to provide straightforward explanation of the action mechanism as well as the functional differences among RGs.
Here, we present a biochemical characterization of the RG from the crenarchaeon Pyrobaculum calidifontis (PcalRG), which appears even more peculiar as compared with other RGs, as it shows: (i) high thermal activity and salt tolerance of its positive DNA supercoiling activity; (ii) the ability of inducing unwinding of synthetic Holliday junctions (HJs); (iii) single strand (ss) DNA annealing; and (iv) strong ssDNA cleavage activity. ATP has an essential modulatory role, not only in positive supercoiling, but also in all these activities: it stimulates ssDNA annealing, inhibits DNA cleavage, and is required for HJ unwinding. These features make PcalRG distinct from all other RGs studied so far and reveal additional properties of this class of unconventional enzymes.
EXPERIMENTAL PROCEDURES
Cloning of PcalRG
The PcalRG gene (GenBankTM database accession number CP000561, gene identifier number GI: 126459306) was amplified from P. calidifontis genomic DNA using oligonucleotides matching the 5′- and the 3′-terminal ends of the coding sequence with the addition of an NdeI restriction site at the 5′-end (sequences available upon request). The amplified gene product was ligated to TA cloning vector pTZ57R/T (Thermo Scientific); the recombinant plasmid, named Rg-pTZ57, was digested with NdeI and HindIII to liberate the PcalRG gene, which was ligated to pET-21a(+) (Novagen) digested with the same restriction enzymes. The resulting recombinant expression vector was named Rg-pET. The presence of PcalRG gene was confirmed by DNA sequencing using CEQ800 Beckman Coulter sequencing system. Multiple sequence alignments were performed by using ClustalW (27) and T-coffee (www.tcoffee.org) programs.
Expression of PcalRG Gene and Purification of the Recombinant Protein
Escherichia coli BL21 CodonPlus(DE3)-RIL cells transformed with Rg-pET were grown in 3 liters of ZY autoinduction medium (28) containing 100 μg/ml of ampicillin. Autoinduction systems enable regulated protein expression in E. coli without the need to monitor the culture or add inducer during cell growth. Cultures were incubated at 37 °C for 3–4 h until the A600 reached 1.5–2.0 followed by further overnight incubation at 17 °C. Cells were harvested by centrifugation, the cell pellet was suspended in Resuspension buffer (25 mm NaH2PO4, 25 mm Na2HPO4, 1.0 mm DTT, 0.1 mm EDTA, 0.1 m NaCl, and 10 mm MgCl2, pH 7.4) and subjected to three cycles of freeze-thawing followed by enzymatic digestion with lysozyme (1 mg/g wet cells) at room temperature for 1 h on shaking after adding 0.25% Triton X-100, Benzonase (25 units/g of cells), and protease inhibitors (PMSF 1.0 mm, leupeptin 20 μm and aprotinin 0.8 μm). Partially broken lysate was immediately lysed using three cycles of French Press. Lysate was centrifuged at 110,000 × g for 25 min at 4 °C, the supernatant was heated at 80 °C for 20 min and centrifuged again at 110,000 × g for 25 min at 4 °C. The resulting supernatant was decanted followed by addition of 0.8 m (NH4)2SO4 and 1.2 m NaCl and loaded onto a phenyl Sepharose 26/10 column (GE Healthcare) equilibrated with phenyl buffer (25 mm phosphate buffer, 1.2 m NaCl, 0.8 m (NH4)2SO4, 1.0 mm EDTA, 1.0 mm DTT, pH 7.4). Proteins were eluted with a linear gradient of (NH4)2SO4 (0.8 to 0 m), and positive fractions (which were eluted at 0 m (NH4)2SO4) were pooled and concentrated after addition of 40 mm Tris-HCl, pH 7.5, 0.05% Triton X-100, 0.5 mm DTT, and 0.5 mm EDTA and loaded onto a 10/300 GL Superdex S200 column (GE Healthcare) equilibrated with gel filtration buffer (40 mm Tris-HCl, 0.05% Triton X-100, 0.5 mm DTT, and 0.5 mm EDTA, pH 7.5). Throughout purification, fractions were analyzed by SDS-PAGE and Western blotting using the anti-TopR1 polyclonal antibody (10). Positive fractions were pooled, concentrated, and stored at −20 °C with the addition of 20% glycerol after positive supercoiling activity analysis by two-dimensional gel electrophoresis as described (29).
Production of the PcalRG-Y966F Mutant
Site-directed mutagenesis was performed using the GeneTailorTM Site-directed Mutagenesis System (Invitrogen) as previously reported (10); the mutant protein was expressed and purified as described above for the wild-type.
DNA Substrates
Oligonucleotides, either unmodified or with Cy5 or Cy3 modifications, were purchased from PRIMM (Milan, Italy). Substrates used in DNA binding and unwinding assays were prepared by annealing oligonucleotides in appropriate combinations: M-HJ, A1-A2-A3-A4; IM-HJ, A1-A2-A5-A6; Y-shaped Fork, A1-A2; 40 bp-double strand (ds), A2-A7; Flap, A1–21lead (30); dsFork, A1+A2 + 30Lag and 21Lead (39). The single strand (ss) 70 bp Lead and Lag oligonucleotides (39) were used for annealing assays. Annealing and purification of substrates were performed as described (30).
Positive Supercoiling Assay
Standard assays were performed as reported (8, 29) using plasmid pQE31 (Qiagen) as substrate, RG buffer (35 mm Tris-HCl, 0.1 mm EDTA, 30 mm MgCl2, 2.0 mm DTT, pH 7.0, and ATP as indicated) and the indicated temperatures and time spans. Appropriate controls were included in each experiment and, to minimize variations among different samples, a single mix with all common components was set up. After incubation, reactions were stopped by adding 2% SDS at room temperature and immediately loaded on 1.2% agarose gel run in the first dimension without intercalants and with ethidium bromide (0.01 μg/ml) in the second dimension. After electrophoresis, gels were stained with ethidium bromide (1 μg/ml), analyzed under UV light using a VersaDoc 4000™ apparatus and quantified with QuantityOne software (Bio-Rad). Briefly, the fluorescence intensity of each band was determined and used to calculate the amount of total DNA (i.e. unprocessed substrate plus all products) and the fraction of each different product (nicked, relaxed and positive) and unprocessed substrate (negative). Each assay was performed at least three times.
DNA Unwinding Assay
HJ, Y-shaped fork, or ds oligonucleotides (200 nm) were incubated in HJ buffer (2.5 mm Tris-HCl, 0.25 mm β-mercaptoethanol, 5.0 mm sodium acetate, 0.5 mm MgCl2, pH 8.0) at 55 °C for 5 min without or with different amounts of PcalRG. ATP (1.0 mm) was added where indicated. To minimize variations among samples within each experiment, a single mix with all common components was set up. Reactions were terminated by adding 2.5 μl of 5× Stop solution (0.5% SDS, 40 mm EDTA, 0.5 mg/ml proteinase K, 20% glycerol), immediately loaded on 8% polyacrylamide gel containing 0.1% SDS, and run in 0.5× Tris borate-EDTA buffer at 150 V/cm. Reaction substrates and products were analyzed as described previously (30) by gel imaging with a VersaDoc 4000™ using the preset laser excitation and emission setting for the Cy5 and Cy3 fluorophores (red led and filter at 695 nm for Cy5, green led and filter at 605 nm for Cy3). Each assay was performed at least three times. The relative percentage of each DNA species in reactions was determined by the Quantity One software and plotted by GraFit version 5.0.11 (Erithacus Software Limited).
Electrophoretic Mobility Shift Assay (EMSA)
DNA binding assays were performed using fluorescently labeled substrates and increasing PcalRG concentrations in binding buffer (20 mm Tris-HCl, 10% glycerol, 50 mm KCl, pH 8.0). After incubation for 10 min at 37 °C, samples were immediately loaded on non-denaturing 5% polyacrylamide gels in 0.5× Tris borate-EDTA buffer and run at 100 V at room temperature. Fluorescence was determined and quantified as described above.
Strand-pairing Assay
Reaction mixtures (10 μl) contained 1× HJ buffer and 40 nm of each oligonucleotide. PcalRG was added to the mixture, and samples were incubated for 10 min at appropriate temperature chosen to minimize spontaneous annealing. Reactions were stopped by the addition of 5× Stop solution, samples were subjected to gel electrophoresis as described for the DNA unwinding assay, and reaction substrates and products were analyzed and quantified by gel imaging as described above.
ATPase Assay
Samples were incubated in Activity Buffer (35 mm Tris-HCl, 5.0 mm MgCl2, pH 8.0) with appropriate concentrations of ATP (10 μm to 2.0 mm), and the appropriate amount of enzyme (10–40 ng/μl) was added. For kinetic analysis, each reaction was divided into aliquots and incubated at 85 °C for different time spans. The nucleotide hydrolysis was measured spectrophotometrically using the Malachite Green Phosphate Assay Kit (BioAssay System, Hayward, CA) as described (13). Each value was corrected for the spontaneous hydrolysis of ATP under the same conditions; values were plotted against time to obtain the initial velocity, v0 (in nmol Pi/min). The GraphPad Prism 5™ software (version 5.01) was used to plot v0 values against ATP concentrations by fitting the following substrate inhibition equation: v = Vmax × [S]/(Km × [S] × (1 + [S]/Ki)), allowing the determination of the steady-state kinetic constants. v0 values at each substrate concentration were used to calculate the enzyme specific activities, expressed as units/mg, where one unit was the amount of enzyme able to hydrolyze 1.0 μmol of ATP to ADP and inorganic phosphate in 1 min under the conditions indicated.
RESULTS
PcalRG Identification and Purification
P. calidifontis is an aerobic crenarchaeon with optimal growth temperature between 90 and 95 °C and pH 7.0 (31), encoding a single RG, we named PcalRG. Sequence comparison with bacterial and archaeal RGs shows all conserved motifs typical of the SF2 family helicases in the PcalRG N-terminal domain (including an ATP binding site), and overall high similarity in the C-terminal topoisomerase domain (supplemental Fig. S1). A single putative Zn finger, conserved in all RGs, is present at the N terminus (aa 11–20); the region corresponding to the latch is longer than in most of the other RGs (aa 432 to 536), as is the insert in the H1 domain (aa 242–290).
We have cloned the PcalRG gene in a vector for heterologous expression in E. coli and purified the recombinant untagged protein. The protein preparation showed reproducibly, besides a ∼130 kDa band corresponding to the full-length protein, a ∼70 kDa protein band corresponding to a truncated product, as shown by its cross-reactivity with an antibody directed against the S. solfataricus TopR1 RG (10). Such truncated fragments were present in variable amounts in independent enzyme preparations, and accumulated with prolonged storage in parallel with reduction of all enzyme activities (data not shown). We thus believe that the active form is the full-length enzyme, corresponding to the high molecular weight band, whereas faster band(s) are due to a proteolytic process that leads to enzyme inactivation. Propensity to fragmentation, both in vitro and in vivo, has been reported for other RGs (9, 10, 13, 32).
PcalRG Topoisomerase and ATPase Activities
The topoisomerase activity of the purified recombinant PcalRG was analyzed by two-dimensional agarose gel electrophoresis, which allows separation of positive and negative plasmid topoisomers (Fig. 1A). Such assay allows qualitative and quantitative analysis of topoisomerase activity, taking into account both the amount of plasmid topological isomers (topoisomers) produced and their Linking number (ΔLk), and gives information not only on the efficiency, but also on the distributive or processive character of the reaction. PcalRG showed the hallmark of all RGs, i.e. ATP-dependent positive supercoiling of covalently closed DNA plasmids (Fig. 1B). This activity required the presence of ATP and high temperature: whereas at 50 °C the enzyme showed only a weak DNA relaxation activity, both the amount and ΔLk of positive topoisomers increased with temperature; in reactions above 90 °C all products were highly positive, and a significant activity was seen even at 100 °C (Fig. 1, B and C). At all temperatures, relaxed and/or positive topoisomers were produced when a certain amount of the negative substrate was still present, thus suggesting that the enzyme is highly processive, i.e. it performs multiple supercoiling cycles before detaching from DNA and attacking a new substrate molecule. Processivity greatly increased with temperature, and above 90 °C highly positive and highly negative topoisomers coexisted (Fig. 1B). Positive supercoiling efficiency increased with increasing protein/DNA (P/D) ratio (data not shown). Time course experiments confirmed the processive character of PcalRG: at both 80 °C and 90 °C the enzyme produced positively supercoiled topoisomers since the first minute of reaction, when the negatively supercoiled DNA substrate was still present (data not shown).
FIGURE 1.

The PcalRG ATP-dependent positive supercoiling activity. A, diagram of two-dimensional gels showing the relative migration of plasmid topoisomers and nicked forms; −SC, +SC, highly negative and positive topoisomers, respectively; negative topoisomers with different degree of relaxation migrate on the left arm and topoisomers with different degree of positive supercoiling on the right arm of the arc. B, temperature dependence of the PcalRG positive supercoiling activity. Standard assays were set up in a final volume of 20 μl, incubated for 10 min at the indicated temperatures (°C), and subjected to two-dimensional agarose gel electrophoresis. All reactions contained 6.0 nm (300 ng) pQE31, 50 nm (150 ng) of PcalRG (P/D molar ratio 8/1), and ATP (1.0 mm). S50, S95, plasmid DNA was mock incubated at either 50 or 95 °C, respectively. Symbols are as in A. Each incubation condition was tested at least three times; representative gels are shown. C, quantification of the PcalRG positive supercoiling activity at different temperatures. The intensity of all bands in two-dimensional gels determined by densitometric analysis was used to calculate the amount of total DNA and the fraction of products (nicked and relaxed/positive) as well as unprocessed substrate (negative). Values are the mean of three independent experiments set as in B. Numbers on the x-axis indicate the incubation temperature (°C); the No protein control is plasmid DNA mock incubated at 95 °C.
In the absence of ATP, PcalRG showed weak Type I topoisomerase-like DNA relaxation activity (Fig. 2, A and B). Interestingly, this activity could only be seen between 60 and 80 °C, whereas at higher temperatures the plasmid was converted into nicked form. To confirm that the DNA nicking activity seen in Figs. 1B and 2A is due to PcalRG, we produced a site-directed mutant carrying a substitution of the putative catalytic tyrosine in the C-terminal domain (PcalRG-Y966F). The mutant was purified following the same procedure described for the wild-type enzyme. As expected, PcalRG-Y966F did not show topoisomerase or DNA nicking activity at either 50 or 80 °C (Fig. 2C), although it was proficient in DNA binding (data not shown). This result strongly suggests that the DNA nicking activity is indeed due to PcalRG and not a contaminant nuclease.
FIGURE 2.

A, temperature dependence of the PcalRG ATP-independent DNA relaxation activity. Reactions were set and analyzed as described in the legend to Fig. 1B with the exception that no ATP was included. Each incubation condition was tested at least three times; representative gels are shown. B, quantification of the PcalRG DNA relaxation activity at different temperatures. Values determined as described in the legend to Fig. 1C are the mean of three independent experiments set as described in A. Numbers on the x-axis indicate the incubation temperature (°C); the No protein control is plasmid DNA mock incubated at 95 °C. C, two-dimensional gel analysis of the PcalRG-Y966F mutant activity. Reactions were set and analyzed as described in the legend to Fig. 1B using the mutant protein instead of wild-type. The temperature of incubation is indicated.
It was previously reported that, when the Sulfolobus RG reaction is stopped with detergents and quickly chilled, a fraction of the enzyme molecules is trapped in covalent complexes with cleaved DNA molecules; nicked DNA can be released from these complexes upon RG digestion with proteinase K (33, 34). In our experiments, reactions were stopped with SDS but no protease, thus the nicked plasmid forms do not derive from covalent DNA-enzyme complexes, rather result from abortive cycles, in which the enzyme disengages from DNA after cleavage without completing the ligation reaction. When ATP was present, comparable amounts of nicked plasmid were produced at all temperatures (Fig. 1, B and C); in contrast, in the absence of ATP the amount of nicked plasmid increased with increasing temperature (Fig. 2, A and B), thus showing that concomitant high temperature and absence of the nucleotide favor interruption of the catalytic cycle. These results suggest that ATP is required for a correct coordination of DNA cleavage with ΔLk modification and ligation, in particular at high temperature.
PcalRG was able to induce positive supercoiling with ATP concentrations as low as 0.1 μm; the efficiency of the reaction increased with increasing nucleotide concentrations, with an optimum between 0.1 and 1.0 mm. At this latter nucleotide concentration, corresponding to a 1:1 ratio of Mg2+ to ATP, the efficiency of reaction decreased (Fig. 3A), raising the possibility that the nucleotide may be chelating the metal ion required for enzyme activity. PcalRG required divalent cations for catalysis: Mg2+, Ca2+, and, at much lesser extent, Co2+-supported positive supercoiling, whereas Mn2+, Zn2+, Ni2+, and Cu2+ were essentially ineffective (data not shown).
FIGURE 3.

ATP concentration dependence and ATPase activity of PcalRG. A, positive supercoiling activity at different of ATP concentrations. Reactions (P/D = 8/1) were set and analyzed as described in the legend to Fig. 1B with the indicated ATP concentrations; incubation was at 85 °C. B and C, kinetic analysis of the ATPase activity. Steady-state kinetic constants were determined using the Malachite Green kit (see “Experimental Procedures”). Reactions containing 10 ng/μl of PcalRG, 200 ng of pQE31, and varying ATP concentrations were incubated at 85 °C for different times (0, 1, 2, 5, 10 min). Specific activities are expressed as units/mg, where one unit is the amount of enzyme able to hydrolyze 1.0 μmol of ATP to ADP and inorganic phosphate in 1 min under the conditions indicated. Values are the mean ± S.E. of three independent experiments.
Fig. 3B reports a kinetic analysis of the ATP hydrolysis reaction at 85 °C. The kinetic constants (Fig. 3C) were in the same order of magnitude of those reported for other RGs (13, 19, 20). Surprisingly, however, steady state ATPase activity of PcalRG deviates from the Michaelis-Menten behavior, showing inhibition of the hydrolysis reaction at ATP concentrations higher than 300 μm. Such inhibition has not been reported for other RGs; for instance, the A. fulgidus RG ATPase activity shows hyperbolic dependence on ATP concentration up to 0.3 mm of ATP, and constant velocity from 0.3 to 1.0 mm (15). PcalRG was active over a wide range of NaCl concentrations: it catalyzed positive supercoiling from 0 to 1 m, and showed slight relaxation activity even at 1.2 m (Fig. 4).
FIGURE 4.
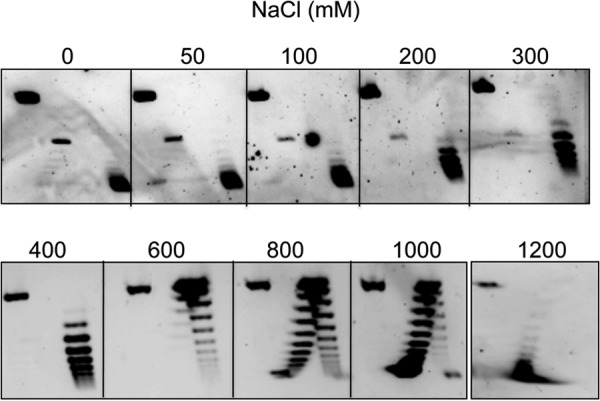
Effect of ionic strength on the PcalRG positive supercoiling activity. Reactions were set and analyzed as described in the legend to Fig. 1 with the exception that buffer contained the indicated NaCl concentration and incubation was at 85 °C.
PcalRG Unwinding and Annealing Activities on Synthetic Oligonucleotides
In line with previously characterized RGs, PcalRG bound ss, ds and mixed DNA oligonucleotides (Fig. 5, A and B), with a preference for ssDNA or substrates containing ss tails (such as Y-shaped fork or flap). Binding was not affected by the presence of Mg2+ or ATP (data not shown). Previously, S. solfataricus TopR1 was shown to induce ATP-independent destabilization of synthetic substrates resembling Y-shaped forks and Holliday junctions (HJ); high concentrations of the enzyme (P/D ratio >10) were required for this process, where substrate destabilization was due to non-enzymatic, TopR1 binding-induced distortion (30). We thus sought to test PcalRG by the same experimental approach, i.e. using synthetic Cy3 or Cy5 end-labeled oligonucleotides annealed in appropriate combinations to form either Y-shaped forks or HJs. We chose an incubation temperature of 55 °C as a compromise allowing relative substrate stability, but also enzyme activity; however, even at this temperature, the Y-shaped fork was quite unstable existing as a mixture of fork and ss species (Fig. 6, A and B, lanes 1). In the absence of ATP, PcalRG catalyzed efficient cleavage of the Cy3-labeled oligonucleotide, which was enzyme concentration-dependent over a wide range of P/D ratios (Fig. 6A), suggesting that in the absence of ATP the enzyme strongly cleaves not only supercoiled (Fig. 2) but also unconstrained substrates. Cleavage occurred on both fork and ssDNA, with slight preference for the former (Fig. 6A and data not shown). In the presence of ATP, cleavage was inhibited and no apparent activity by PcalRG on the Y-shaped fork was seen (Fig. 6B); however, we cannot rule out the possibility that, if substrate unwinding occurred, the ss oligonucleotides eventually produced were annealed back, either spontaneously or by the action of PcalRG (see below).
FIGURE 5.

A, EMSA analysis of PcalRG DNA binding activity. The indicated amounts (ng) of PcalRG were incubated in 10-μl reaction mixture for 10 min at 37 °C with the indicated fluorescently end labeled substrates (40 nm each); ss was the 40-nt A2 oligonucleotide (30). /: no protein. In each gel the free substrate is indicated. B, quantification of DNA binding activity: for each DNA ligand, the fraction of shifted DNA in each lane versus the amount of protein used was plotted. Values are the mean ± S.E. of three independent experiments.
FIGURE 6.
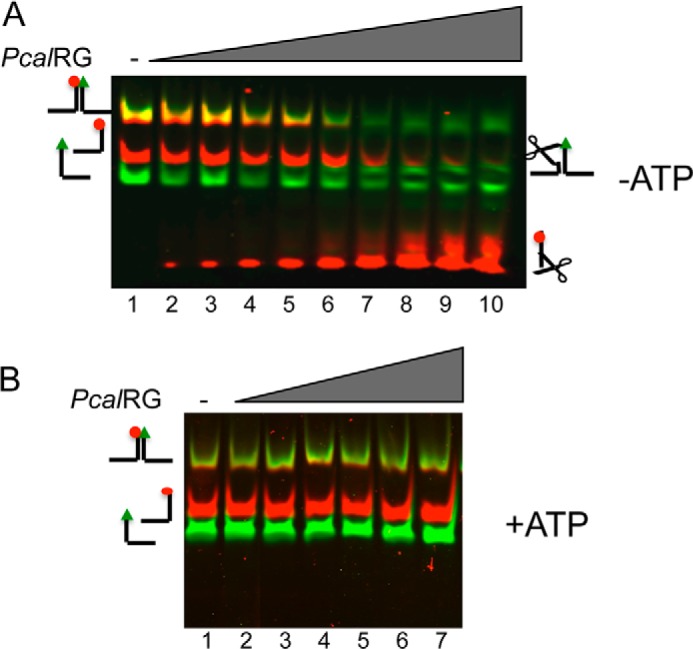
Activity of PcalRG on the Y-shaped fork. A, Cy3, Cy5-end-labeled Y-shaped fork was incubated at 55 °C for 10 min without (lane 1) or with increasing amounts of PcalRG in the absence of 1.0 mm ATP. The following P/D ratios were used: lane 2: 1/40; lane 3: 1/20; lane 4: 1/15; lane 5: 1/10; lane 6, 1/5; lane 7: 1/2.5; lane 8: 1/1; lane 9: 2/1; lane 10: 5/1. B, Y-shaped fork was incubated as in A without (lane 1) or with increasing amounts of PcalRG in the presence of 1.0 mm ATP. The following P/D ratios were used: lane 2: 1/15; lane 3: 1/10; lane 4: 1/5; lane 5: 1/2.5; lane 6: 1/1; lane 7: 2/1. Samples were run, and gel was scanned under the appropriate conditions (as described under “Experimental Procedures”) to follow the Cy3 and Cy5 fluorophores; the merged image of each gel is shown.
We then tested PcalRG activity on a synthetic 20 bp arm HJ, in which the 3′- and 5′-ends of one arm were labeled with the Cy3 and Cy5 fluorophores, respectively (IM-HJ, 30). Interestingly, PcalRG was able to process the IM-HJ following two completely different pathways, according to the presence or absence of ATP (Fig. 7). In the absence of nucleotide, the main activity was cleavage of the Cy3-labeled oligonucleotide at a preferential site; cleavage efficiency and parallel HJ decrease were enzyme concentration-dependent, starting from P/D = 2/1, and essentially no fork or ssDNA were produced (Fig. 7, A and C). In the presence of ATP cleavage was strongly inhibited, but in this case the enzyme catalyzed unwinding of the junction to Y-shaped forks. HJ unwinding required lower enzyme concentration as compared with DNA cleavage, as it was seen already at a P/D of 1/20; fork accumulation showed a moderate increase by increasing the enzyme concentration up to P/D of 1/1, above which it decreased; the opposite behavior was seen for the HJ (Fig. 7, A and B; see below). AMP could not substitute for ATP in the HJ unwinding, thus suggesting that nucleotide binding cannot support this reaction, but nucleotide hydrolysis is required (data not shown).
FIGURE 7.

Resolution of IM-HJ by PcalRG. A, IM-HJ was incubated at 55 °C for 10 min without (lane 1) or with increasing amounts of PcalRG in the absence or presence of 1 mm ATP as indicated. The following P/D ratios were used: lanes 2, 3: 1/20; lanes 4, 5: 1/10; lanes 6, 7: 1/5; lanes 8, 9: 1/2.5; lanes 10, 11: 1/1; lanes 12, 13: 2/1; lanes 14, 15: 5/1. Samples were run, and gel was scanned under the appropriate conditions (as described under “Experimental Procedures”) to follow the fluorophores shown on the left. Reaction substrate and products are shown on the right; scissors indicate cleavage products. B and C, quantification of the results shown in A. For clarity, only the fractions of HJs and Y-shaped forks versus protein concentration in each Cy5-scanned gel are plotted using GraFit version 5.0.11 (Erithacus Software Limited). In samples without ATP, the total amount of HJs +forks was reduced with increasing protein concentration due to the cleavage activity of the enzyme. To simplify the pattern, the quantification of the cleavage product is not shown. Values are the mean ± S.E. of three independent experiments.
We sought to confirm these results using a “mobile” HJ (M-HJ), identical to the IM-HJ, except for a 4-nucleotide homologous core allowing the molecule to shift between two conformations (30), a feature that usually facilitates HJ recognition and resolution by junction resolving enzymes. PcalRG showed ATP-dependent unwinding and ATP-independent cleavage on this substrate as well (Fig. 8). The cleavage activity was more efficient with the M-HJ than with the IM-HJ (Fig. 8, A and C), suggesting that it is affected by the higher substrate instability and thus, likely, by transient exposure of ss-regions. In contrast, unwinding efficiency was comparable with both substrates, suggesting that this reaction is less dependent on intrinsic substrate features. In addition, as seen with the IM-HJ, the efficiency of M-HJ unwinding increased moderately with the enzyme concentration, and the ratio between HJ and fork was inverted at the highest P/D (Figs. 8B and 7B).
FIGURE 8.

Resolution of M-HJ by PcalRG. A, experiment was set and analyzed as described in the legend to Fig. 6 with the exception that the M-HJ was used. The following P/D ratios were used: lanes 2, 3: 1/10; lanes 4, 5: 1/5; lanes 6, 7: 1/2.5; lanes 8, 9: 1/1; lanes 10, 11: 2/1; lanes 12, 13: 5/1. B and C, quantification of the results shown in A.
The PcalRG concentration effect in the ATP-dependent reaction with both HJs could be explained assuming that, whereas at lower protein concentration the preferred reaction in the presence of ATP is unwinding, higher protein concentrations either inhibit unwinding or switch the reaction from unwinding to annealing. This observation led us to test the potential annealing activity by PcalRG of the four ss oligonucleotides forming the IM-HJ. When incubated at 55 °C these oligonucleotides annealed spontaneously forming Y-shaped forks (Fig. 9A, lane 1); in the absence of ATP, the enzyme showed again strong cleavage activity (Fig. 9A, lanes 2–4); in contrast, in the presence of the nucleotide, cleavage was inhibited and formation of IM-HJ was stimulated (Fig. 9A, lane 7). Annealing was dependent on PcalRG concentration and occurred in the presence of either 1 mm ATP or AMP, thus suggesting that nucleotide hydrolysis is not required for the reaction (Fig. 9B).
FIGURE 9.

PcalRG-promoted DNA annealing. A, annealing of the HJ components. The oligonucleotides A1, A2, A5, and A6 (50 nm each) were incubated at 55 °C for 10 min without (lane 1) or with increasing amounts of PcalRG and 1.0 mm ATP where indicated, at the following P/D: lanes 2, 5: 1/1; lanes 3, 6: 2/1; lanes 4,7: 5/1. B, quantification of the HJ components annealing in the presence of 1.0 mm ATP or 1 mm AMP (last sample). The fraction of HJ versus protein concentration is plotted. Values are the mean ± S.E. of three independent experiments. The plot of samples without nucleotide is not shown because the cleavage activity prevented proper quantification. C, annealing of fully complementary oligonucleotides at high temperature. 70-Lead and 70-Lag oligonucleotides (40 nm each), were incubated at 85 °C for 10 min without (lanes 1 and 6) or with increasing amounts of PcalRG and 1.0 mm ATP where indicated, at the following P/D: lanes 2, 7: 0.6/1; lanes 3, 8: 1.25/1; lanes 4, 9: 2.5/1; lanes 5, 10: 5/1. The gel was scanned under the appropriate conditions to detect the Cy5 fluorophore. D, quantification of the 70-Lead-70-Lag oligonucleotides annealing in the presence of 1.0 mm ATP. The fraction of dsDNA versus protein concentration is plotted. Values are the mean ± S.E. of three independent experiments. The plot of samples without ATP is not shown because the cleavage activity prevented proper quantification of dsDNA.
ssDNA annealing was also tested with two 70-base fully complementary oligonucleotides, chosen because the duplex was relatively stable at 85 °C: again, the enzyme cleaved the substrate in the absence of ATP, and ATP inhibited cleavage while significantly stimulated the annealing reaction (Fig. 9, C and D). In helicase assays with 1 mm ATP at 55 °C PcalRG showed no activity on a 70-bp ds oligonucleotide (data not shown); however, as for the Y-shaped fork, we cannot rule out that unwinding occurs, but annealing is the prevailing reaction with this substrate. Taken together, these experiments show that PcalRG induces ATP-dependent unwinding of the HJ and ATP-stimulated annealing of ssDNA.
DISCUSSION
We have shown that PcalRG is a very active RG, as a relatively low P/D ratio is sufficient to obtain positive supercoiling. It is very processive, as it produces highly positively supercoiled products directly from the negatively supercoiled substrate, and it is active over a wide range of temperatures (up to 100 °C), ionic strength (0–1.2 m NaCl), and ATP concentrations (0.1–10,000 μm). In addition, it has a strong DNA cleavage activity in the absence of nucleotides, which is inhibited by ATP. These features make PcalRG a very robust and extreme enzyme if compared with well-characterized RGs. For instance, of the two Sulfolobus RGs, TopR2 was shown to be a very processive enzyme, with comparable positive supercoiling efficiency from 60 °C up to its temperature maximum of 75 °C (16). In contrast, TopR1 was found optimally active at 90 °C, typically distributive, and the supercoiling level of its products was modulated by temperature. Although it is difficult to compare different enzymes under different experimental conditions, PcalRG was more similar to TopR1 in terms of temperature dependence and thermoactivity, whereas its processivity was comparable to that of TopR2. The ability of PcalRG to work at very high salt concentration is unique among not only RGs, but DNA topoisomerases in general, because binding to DNA is very sensitive to ionic strength; as for RGs, they are only active at low-medium ionic strength (10–300 mm NaCl or KCl; 13, 16, 17), with the exception of TopR2, which was active up to 600 mm NaCl (16).
Another peculiarity of PcalRG is the substrate inhibition of the ATPase reaction. ATP inhibits the hydrolysis reaction at concentrations proficient for the positive supercoiling, a result not reported previously for any RG (13, 19, 20). Whether substrate inhibition is a biologically relevant regulatory mechanism is at moment not clear; in principle, one can speculate that substrate inhibition, decreasing the rate of the nucleotide cycle, might favor the coordination of ATP hydrolysis with all steps of the positive supercoiling reaction.
Besides allowing positive supercoiling, ATP has impact on the overall PcalRG behavior. In the absence of the nucleotide the enzyme showed strong cleavage activity of supercoiled as well as unconstrained substrates; by producing a site-directed mutant in the catalytic tyrosine of PcalRG we showed that the nicking activity is the result of interrupted enzyme cycles. Cleaved products accumulation was strongly reduced in the presence of ATP, suggesting that either cleavage is suppressed or ligation is stimulated when the nucleotide is present, as previously suggested for the S. shibatae RG (33, 34). All topoisomerases hold potential hazard for genomes, because the covalent enzyme-DNA complexes can be frozen by topoisomerase inhibitors or DNA lesions (35); our results suggest that the PcalRG temperature-stimulated nicking activity, if uncontrolled, might be an additional threat for hyperthermophile genomes.
The possible role of RGs in DNA protection/repair was supported by previous results showing that TopR1 destabilizes synthetic HJs by an ATP independent mechanism (30). We have also found an ATP-independent activity of PcalRG on HJs; however, whereas TopR1 produced mainly Y-shaped forks and ssDNA, and only slight amounts of cleaved products, PcalRG produced essentially cleaved products. This difference might be due to the efficient PcalRG cleavage activity of forks and ssDNA (Fig. 6A). It would be interesting to test other RGs to establish whether the ATP-independent HJ destabilizing activity might be a general feature of these enzymes.
PcalRG has two unique abilities: ATP hydrolysis-dependent unwinding of HJs and annealing of ssDNA. Various bacterial and archaeal RGs were previously tested for processive ATP-dependent DNA helicase activity, but none showed this ability (13, 18, 19). Recently, Ganguly et al. (24) showed that the full-length T. maritima RG can transiently unwind short DNA duplexes in the presence of the non-hydrolysable ATP analog ADP·BeFx, but not ATP. This result was explained suggesting that the enzyme is able to unwind DNA locally, but is not a processive helicase (24). Although PcalRG-induced unwinding of HJ requires ATP hydrolysis, our result is not necessarily in contrast with that reported by Ganguly et al. (24), since we use completely different experimental systems.
The second activity not seen before for any RGs is annealing of unconstrained substrates; however two previously reported observations might suggest this is a property shared by (all) other RGs. First, the A. fulgidus RG was shown to induce ATP-stimulated renaturation of ss circles yielding positively supercoiled products, thus suggesting that renaturation is a step of the supercoiling reaction (36); in addition, the experiment by Ganguly et al. (24) showing transient DNA unwinding by the T. maritima RG does imply an annealing step.
Taken together, our results suggest that in the ATP-dependent HJ resolution reaction PcalRG might use both its unwinding and annealing activities: the former induces the production of Y-shaped fork intermediates, which might be eventually unwound to ss products or annealed back; a dynamic equilibrium is established among HJ, Y-shaped forks and ss DNA at each enzyme concentration, with lower P/D ratio promoting unwinding and higher P/D driving reaction toward annealing. Combined unwinding/branch migration and annealing activities are shared by eukaryotic and archaeal helicases (37–41). The discovery that PcalRG holds such activities enabling the HJ resolution by a true ATP-dependent mechanism, at least in vitro, might support previous results suggesting the involvement of RG in recombination and/or repair (7–10).
The role of ATP in the PcalRG unwinding and annealing reactions is currently not understood, but it is clear that the nucleotide modulates all enzyme activities, preventing cleavage (or release of cleaved DNA strands) and stimulating base pairing or destabilization, depending on the enzyme concentration. Whether the nucleotide truly inhibits cleavage, or helps coordination of the cleavage and religation activities is currently not clear.
Our results might also shed some light on the molecular mechanism of RG positive supercoiling reaction, whose details are still partially obscure. An old model based on a presumptive ATP hydrolysis-driven helicase activity proposed that dsDNA unwinding by the N-terminal domain generates ssDNA regions, facilitating topoisomerase binding and thus strand passage. In a topologically closed molecule, such process would generate positive supercoils in front of the enzyme and negative supercoils behind, which could be relaxed by the topoisomerase domain leading to a net accumulation of positive supercoils (42). However, this model was till now challenged by the failure to show processive helicase activity for several RGs (13, 18, 19, 24). The finding that PcalRG holds processive duplex destabilization and annealing (rewinding) activities, at least with the peculiar HJ structure, might now support this model. Indeed, it is possible that the synthetic HJ is a good model substrate for the RG unwinding/rewinding activity and might recapitulate in vitro what happens in vivo with the genomic DNA of hyperthermophiles.
This work was supported in part by Ministero dell'Istruzione, Università e Ricerca (FIRB-Merit RBNE08YFN3 and FIRB-Futuro in Ricerca RBFR12OO1G_002 “Nematic”) and Ministero degli Affari Esteri (L.401/1990).

This article contains supplemental Fig. S1.
- RG
- reverse gyrase
- Pcal
- Pyrobaculum calidifontis
- HJ
- Holliday junctions
- ss
- single-stranded
- ds
- double-stranded.
REFERENCES
- 1. Brochier-Armanet C., Forterre P. (2007) Widespread distribution of archaeal reverse gyrase in thermophilic bacteria suggests a complex history of vertical inheritance and lateral gene transfers. Archaea 2, 83–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nadal M. (2007) Reverse gyrase: an insight into the role of DNA-topoisomerases. Biochimie 89, 447–455 [DOI] [PubMed] [Google Scholar]
- 3. Perugino G., Valenti A., D'amaro A., Rossi M., Ciaramella M. (2009) Reverse gyrase and genome stability in hyperthermophilic organisms. Biochem. Soc. Trans. 37, 69–73 [DOI] [PubMed] [Google Scholar]
- 4. Heine M., Chandra S. B. (2009) The linkage between reverse gyrase and hyperthermophiles: a review of their invariable association. J. Microbiol. 47, 229–234 [DOI] [PubMed] [Google Scholar]
- 5. Atomi H., Matsumi R., Imanaka T. (2004) Reverse gyrase is not a prerequisite for hyperthermophilic life. J. Bacteriol. 186, 4829–4833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang C., Tian B., Li S., Ao X., Dalgaard K., Gökce S., Liang Y., She Q. (2013) Genetic manipulation in Sulfolobus islandicus and functional analysis of DNA repair genes. Biochem. Soc. Trans. 41, 405–410 [DOI] [PubMed] [Google Scholar]
- 7. Napoli A., Valenti A., Salerno V., Nadal M., Garnier F., Rossi M., Ciaramella M. (2004) Reverse gyrase recruitment to DNA after UV light irradiation in Sulfolobus solfataricus. J. Biol. Chem. 279, 33192–33198 [DOI] [PubMed] [Google Scholar]
- 8. Napoli A., Valenti A., Salerno V., Nadal M., Garnier F., Rossi M., Ciaramella M. (2005) Functional interaction of reverse gyrase with single-strand binding protein of the archaeon Sulfolobus. Nucleic Acids Res. 33, 564–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Valenti A., Napoli A., Ferrara M. C., Nadal M., Rossi M., Ciaramella M. (2006) Selective degradation of reverse gyrase and DNA fragmentation induced by alkylating agent in the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 34, 2098–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Valenti A., Perugino G., Nohmi T., Rossi M., Ciaramella M. (2009) Inhibition of translesion DNA polymerase by archaeal reverse gyrase. Nucleic Acids Res. 37, 4287–4295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rodriguez A. C. (2002) Studies of a positive supercoiling machine. Nucleotide hydrolysis and a multifunctional “latch” in the mechanism of reverse gyrase. J. Biol. Chem., 277, 29865–29873 [DOI] [PubMed] [Google Scholar]
- 12. Rodríguez A. C. (2003) Investigating the role of the latch in the positive supercoiling mechanism of reverse gyrase. Biochemistry 42, 5993–6004 [DOI] [PubMed] [Google Scholar]
- 13. Valenti A., Perugino G., D'Amaro A., Cacace A., Napoli A., Rossi M., Ciaramella M. (2008) Dissection of reverse gyrase activities: insight into the evolution of a thermostable molecular machine. Nucleic Acids Res. 36, 4587–4597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsieh T. S., Capp C. (2005) Nucleotide- and stoichiometry-dependent DNA supercoiling by reverse gyrase. J. Biol. Chem. 280, 20467–20475 [DOI] [PubMed] [Google Scholar]
- 15. Jungblut S. P., Klostermeier D. (2007) Adenosine 5′-O-(3-thio)triphosphate (ATPγS) promotes positive supercoiling of DNA by T. maritima reverse gyrase. J. Mol. Biol. 371, 197–209 [DOI] [PubMed] [Google Scholar]
- 16. Bizard A., Garnier F., Nadal M. (2011) TopR2, the second reverse gyrase of Sulfolobus solfataricus, exhibits unusual properties. J. Mol. Biol. 408, 839–849 [DOI] [PubMed] [Google Scholar]
- 17. Li J., Liu J., Zhou J., Xiang H. (2011) Functional evaluation of four putative DNA-binding regions in Thermoanaerobacter tengcongensis reverse gyrase. Extremophiles 15, 281–291 [DOI] [PubMed] [Google Scholar]
- 18. Déclais A. C., Marsault J., Confalonieri F., de La Tour C. B., Duguet M. (2000) Reverse gyrase, the two domains intimately cooperate to promote positive supercoiling. J. Biol. Chem. 275, 19498–194504 [DOI] [PubMed] [Google Scholar]
- 19. Capp C., Qian Y., Sage H., Huber H., Hsieh T. S. (2010) Separate and combined biochemical activities of the subunits of a naturally split reverse gyrase. J. Biol. Chem. 285, 39637–39645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. del Toro Duany Y., Jungblut S. P., Schmidt A. S., Klostermeier D. (2008) The reverse gyrase helicase-like domain is a nucleotide-dependent switch that is attenuated by the topoisomerase domain. Nucleic Acids Res. 36, 5882–5895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. del Toro Duany Y., Klostermeier D., Rudolph M. G. (2011) The conformational flexibility of the helicase-like domain from Thermotoga maritima reverse gyrase is restricted by the topoisomerase domain. Biochemistry 50, 5816–5823 [DOI] [PubMed] [Google Scholar]
- 22. del Toro Duany Y., Klostermeier D. (2011) Nucleotide-driven conformational changes in the reverse gyrase helicase-like domain couple the nucleotide cycle to DNA processing. Phys. Chem. Chem. Phys. 13, 10009–10019 [DOI] [PubMed] [Google Scholar]
- 23. Ganguly A., Del Toro Duany Y., Rudolph M. G., Klostermeier D. (2011) The latch modulates nucleotide and DNA binding to the helicase-like domain of Thermotoga maritima reverse gyrase and is required for positive DNA supercoiling. Nucleic Acids Res. 39, 1789–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ganguly A., del Toro Duany Y., Klostermeier D. (2013) Reverse gyrase transiently unwinds double-stranded DNA in an ATP-dependent reaction. J. Mol. Biol. 425, 32–40 [DOI] [PubMed] [Google Scholar]
- 25. Rodríguez A. C., Stock D. (2002) Crystal structure of reverse gyrase: insights into the positive supercoiling of DNA. EMBO J. 21, 418–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rudolph M. G., del Toro Duany Y., Jungblut S. P., Ganguly A., Klostermeier D. (2013) Crystal structures of Thermotoga maritima reverse gyrase: inferences for the mechanism of positive DNA supercoiling. Nucleic Acids Res. 41, 1058–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Larkin M. A. (2007) Clustal W and Clustal X version 2.0. Bioinformatics, 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 28. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expression Purification 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 29. Napoli A., Zivanovic Y., Bocs C., Buhler C., Rossi M., Forterre P., Ciaramella M. (2002) DNA bending, compaction and negative supercoiling by the architectural protein Sso7d of Sulfolobus solfataricus. Nucleic Acids Res. 30, 2656–2662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Valenti A., Perugino G., Varriale A., D'Auria S., Rossi M., Ciaramella M. (2010) The archaeal topoisomerase reverse gyrase is a helix-destabilizing protein that unwinds four-way DNA junctions. J. Biol. Chem. 285, 36532–36541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Amo T., Paje M. L., Inagaki A., Ezaki S., Atomi H., Imanaka T. (2002) Pyrobaculum calidifontis sp. nov., a novel hyperthermophilic archaeon that grows in atmospheric air. Archaea 1, 113–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nadal M., Couderc E., Duguet M., Jaxel C. (1994) Purification and characterization of reverse gyrase from Sulfolobus shibatae. Its proteolytic product appears as an ATP-independent topoisomerase. J. Biol. Chem. 269, 5255–5263 [PubMed] [Google Scholar]
- 33. Jaxel C., Duguet M., Nadal M. (1999) Analysis of DNA cleavage by reverse gyrase from Sulfolobus shibatae B12. Eur. J. Biochem. 260, 103–111 [DOI] [PubMed] [Google Scholar]
- 34. Jaxel C., Nadal M., Mirambeau G., Forterre P., Takahashi M., Duguet M. (1989) Reverse gyrase binding to DNA alters the double helix structure and produces single-strand cleavage in the absence of ATP. EMBO J. 8, 3135–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Champoux J. J., (2001) DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 70, 369–413 [DOI] [PubMed] [Google Scholar]
- 36. Hsieh T. S., Plank J. L. (2006) Reverse gyrase functions as a DNA renaturase: annealing of complementary single-stranded circles and positive supercoiling of a bubble substrate. J. Biol. Chem. 281, 5640–5647 [DOI] [PubMed] [Google Scholar]
- 37. Li Z., Lu S., Hou G., Ma X., Sheng D., Ni J., Shen Y. (2008) Hjm/Hel308A DNA helicase from Sulfolobus tokodaii promotes replication fork regression and interacts with Hjc endonuclease in vitro. J. Bacteriol. 190, 3006–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Woodman I. L., Bolt E. L. (2009) Molecular biology of Hel308 helicase in archaea. Biochem. Soc. Trans. 37, 74–78 [DOI] [PubMed] [Google Scholar]
- 39. Valenti A., De Felice M., Perugino G., Bizard A., Nadal M., Rossi M., Ciaramella M. (2012) Synergic and opposing activities of thermophilic RecQ-like helicase and topoisomerase 3 proteins in Holliday junction processing and replication fork stabilization. J. Biol. Chem. 287, 30282–30295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rezazadeh S. (2012) On BLM helicase in recombination-mediated telomere maintenance. Mol. Biol. Rep. 39, 4527–4543 [DOI] [PubMed] [Google Scholar]
- 41. Larsen N. B., Hickson I. D. (2013) RecQ Helicases: Conserved Guardians of Genomic Integrity. Adv. Exp. Med. Biol. 767, 161–184 [DOI] [PubMed] [Google Scholar]
- 42. Jaxel C., Bouthier de la Tour C., Duguet M., Nadal M. (1996) Reverse gyrase gene from Sulfolobus shibatae B12: gene structure, transcription unit and comparative sequence analysis of the two domains. Nucleic Acids Res. 24, 4668–4675 [DOI] [PMC free article] [PubMed] [Google Scholar]