

Background: Effect of Ninjurin1 deletion in the experimental autoimmune encephalomyelitis (EAE) mice has not been examined.
Results: Ninjurin1 knock-out (KO) mice are resistance to EAE due to a defect of leukocyte recruitment into lesion sites.
Conclusion: Ninjurin1 is a potent target molecule for treating inflammatory diseases such as multiple sclerosis.
Significance: Our study proved contribution of Ninjurin1 in EAE pathogenesis in vivo and supports the importance of its targeting strategies.
Keywords: Autoimmune Diseases, Autoimmunity, Inflammation, Leukocyte, Multiple Sclerosis, EAE, Ninjurin1, Leukocyte Trafficking, Transendothelial Migration
Abstract
Ninjurin1 is a homotypic adhesion molecule that contributes to leukocyte trafficking in experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis. However, in vivo gene deficiency animal studies have not yet been done. Here, we constructed Ninjurin1 knock-out (KO) mice and investigated the role of Ninjurin1 on leukocyte trafficking under inflammation conditions such as EAE and endotoxin-induced uveitis. Ninjurin1 KO mice attenuated EAE susceptibility by reducing leukocyte recruitment into the injury regions of the spinal cord and showed less adhesion of leukocytes on inflamed retinal vessels in endotoxin-induced uveitis mice. Moreover, the administration of a custom-made antibody (Ab26–37) targeting the Ninjurin1 binding domain ameliorated the EAE symptoms, showing the contribution of its adhesion activity to leukocyte trafficking. In addition, we addressed the transendothelial migration (TEM) activity of bone marrow-derived macrophages and Raw264.7 cells according to the expression level of Ninjurin1. TEM activity was decreased in Ninjurin1 KO bone marrow-derived macrophages and siNinj1 Raw264.7 cells. Consistent with this, GFP-tagged mNinj1-overexpressing Raw264.7 cells increased their TEM activity. Taken together, we have clarified the contribution of Ninjurin1 to leukocyte trafficking in vivo and delineated its direct functions to TEM, emphasizing Ninjurin1 as a beneficial therapeutic target against inflammatory diseases such as multiple sclerosis.
Introduction
Immune responses mediated by leukocytes from blood have a great influence on the pathogenesis of several inflammatory central nervous system (CNS) diseases including multiple sclerosis, stroke, Alzheimer disease, Parkinson disease, trauma, and epilepsy (1). Multiple sclerosis is caused by autoimmune T cells coordinating with monocytes, dendritic cells, and B cells recruited from the blood; it is one of the most common diseases with neuromyelitis and encephalomyelitis (2). In particular, the remarkable accumulation of leukocytes in the plaque of a damaged region is a hallmark of multiple sclerosis and its animal model, EAE.4 Therefore, there have been active attempts to modulate leukocyte diapedesis in these CNS diseases (3).
The process of leukocyte recruitment from blood to the lesion is explained as sequential adhesive events, including the tethering, rolling, arresting, firming, crawling, and transmigration of leukocytes against the endothelium (4, 5). This response begins with the capture and adhesion of circulating leukocytes on the stimulated endothelium of venules. The diverse homotypic or heterotypic bindings of adhesion molecules on leukocytes and endothelial cells are responsible for the leukocyte adhesion cascade in a stage-restrictive or non-restrictive manner (6), for example, Selectin/PSGL-1 (7) for rolling, LFA-1/ICAM-1 (8) and VLA4/VACM1 (9) for arrest and crawling, and PECAM-1/PECAM-1(10), CD99/CD99 (11), LFA-1/ICAM-2 (12), and LFA-1/JAM-1 (13) for transmigration.
Ninjurin1 was originally identified as an adhesion molecule, ∼17 kDa by Araki et al. with two transmembrane domains (14). The 12 residues, from Pro26 to Asn37, on the N-terminal ectodomain of Ninjurin1 are critical for its homophilic binding (15). Our group previously reported that Ninjurin1 is preferentially expressed in myeloid cells and in the inflamed endothelium in the EAE rat brain and that its overexpression promotes the adhesion of leukocytes onto endothelial cell monolayers in vitro (16, 17). Consistent with our results, Ifergan et al. (18) demonstrated that Ninjurin1 is restricted to endothelial cells and myeloid cells, particularly to dendritic cells at lesions from human brains with multiple sclerosis. Furthermore, the functional blockage of Ninjurin1 decreases the transendothelial migration (TEM) of monocytes by blocking rolling and other adhesive steps on endothelial cells, whereas it attenuates the clinical symptoms of EAE mice by decreasing leukocyte infiltration (18). Recently, it has been reported that in highly migratory T cells activated in the lungs of EAE rats, Ninjurin1 is transiently up-regulated and participates in the intravascular crawling of T cells in the CNS vessels (19). These previous results suggest that Ninjurin1 is a beneficial candidate that targets the TEM of leukocytes including myeloid-lineage cells and T cells.
However, the in vivo role of Ninjurin1 in a gene-deficient animal model and its direct regulation via Ninjurin1 own expression pertaining to the processes of TEM should be explored. We herein clarified the in vivo relevance of Ninjurin1 using both KO mice and a blocking antibody generated by immunization with the homophilic binding domain as the specific antigen (Ab26–37). Ninjurin1 KO and Ab26–37-administrated mice exhibited protective effects against EAE by reducing leukocyte infiltration in the lesion site. In addition to the well known homophilic binding activity of Ninjurin1, we found that Ninjurin1 directly enhances TEM activity in a dose-dependent manner, which is shown in the Ninjurin1 KO bone marrow-derived macrophage (BMDM)s and Ninjurin1 siRNA or stable overexpressing Raw264.7 cells in vitro. Altogether, we determined the role of Ninjurin1 in vivo and discovered that TEM is regulated based on the amount of Ninjurin1 expression during the trafficking of immune cells under inflammatory conditions.
EXPERIMENTAL PROCEDURES
Animals
Ninjurin1 KO mice (C57BL/6J background) were backcrossed with C57BL/6 for at least seven generations. The breeding colony was established and maintained under pathogen-free conditions in the animal housing facility of the College of Pharmacy, Seoul National University, for the duration of the experiments under the rule of the Committee for Care and Use of Laboratory Animals at Seoul National University (SNU-101011-1). The primer sequences for genotyping are as follows: wild type (forward), 5′-GAG ATA GAG GGA GCA CGA CG-3′; Neo (forward), 5′-ACG CGT CAC CTT AAT ATG CG-3′; reverse primer, 5′-CGG GTT GTT GAG GTC ATA CTT G-3′.
EAE Induction and Clinical Scoring
Sex- and age (6–10 weeks)-matched C57BL/6 mice were immunized subcutaneously with an emulsion containing 100 μg of myelin oligodendrocyte glycoprotein-(35–55) (MOG35–55; Peptron Inc.) in complete Freund's adjuvant (CFA; Mycobacterium tuberculosis H37Ra, 4 mg/ml). Each mouse was injected with 300 ng of pertussis toxin intraperitoneally at 0 and 2 days after immunization. The mice were weighed and observed daily for clinical signs of EAE. The progression of EAE was graded according to the following scale: 0, no symptoms; 1, floppy tail; 2, mild paralysis of the hind limbs; 3, complete paralysis of one hind limbs and partial paralysis of another one; 4, complete paralysis of both hind limbs; 5, a moribund state or death.
Endotoxin-induced Uveitis (EIU) and Quantification of Retinal Adherent Leukocytes
Each mouse (male, 6–10 weeks) received a single intraperitoneal injection of lipopolysaccharide (LPS, Sigma) in PBS at a dose of 9 mg/kg body weight. Twenty-four hours after the LPS injection, the chest cavity was opened, and a 24-gauge syringe was introduced into the left ventricle under deep anesthesia. After injection of 5 ml PBS to remove the erythrocytes and non-adherent leukocytes, 5 ml of rhodamine-conjugated concanavalin A (5 mg/kg) (Vector Laboratories) was perfused. After the eyes were enucleated, the retinas were flat-mounted. The flat mounts were imaged with a fluorescence microscope, and the total number of concanavalin A-positive adherent leukocytes per retina was counted.
Antibodies
For a custom-made anti-moue Ninjurin1 antibody, rabbits were immunized with keyhole limpet hemocyanin-conjugated synthetic peptide-bearing mouse Ninjurin1 residues 26–37 (Ab26–37) and 1–15 (Ab1–15) following standard procedures (Peptron, Inc. and Abfrontier, Inc.); these anti-Ninjurin1 antibodies were purified in each case with antigen-specific affinity chromatography. The isotype rabbit IgG purified with a Protein A column (Upstate Biotechnology) from the serum of normal rabbits was used for the in vivo blocking experiment in the EAE model. Ab1–15 was used for immunostaining and Western blotting.
Cell Culture
Raw264.7 and MBEC4 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen) and maintained in an incubator with a humidified atmosphere of 95% O2 and 5% CO2 at 37 °C. For the BMDM culture, bone marrow cells were isolated from femurs and tibias and cultured in RPMI 1640 medium supplemented with 10% FBS, 1% penicillin, and streptomycin for 3 days and differentiated in RPMI 1640 containing 20 ng/ml macrophage-colony stimulating factor (Peprotech) for 3 days.
Construction of the Expression Vector and Stable Raw264.7 Cells
For the construction of stable Ninjurin1-overexpressed Raw264.7 cells, full-length cDNA corresponding to mouse Ninjurin1 (NM_013610) was inserted into the pEGFP vector (GFP-mNinj1). The constructed GFP-mNinj1 plasmid was transfected in Raw264.7 cells (Nucleofector, Amaxa) and maintained in complete DMEM with G418 (500 μg/ml). After several days the surviving colonies were selected. Purity and homogeneity (>90%) were determined by means of fluorescence microscope observations and Western blotting.
Cell Adhesion and TEM Assays
To measure the adhesion activity with Ab26–37 treatment, Raw264.7 cells activated by TNFα and IFNγ (10 ng/ml, 12 h, Peprotech) were labeled with CFSE (5 μm, 5 min) and preincubated with Ab26–37 for 1 h corresponding to each concentration. CFSE-labeled Raw264.7 cells were added to a MBEC4 monolayer activated with TNFα and IFNγ (10 ng/ml, 12 h) and incubated for 20 min. After washing three times with PBS, the adherent cells were imaged in at least six positions by microscopy, and the percentages of CFSE-labeled cells were quantified in each picture.
For the measurement of TEM activity with Ab26–37 treatment, MBEC4 was seeded in the upper chamber of each trans-well (6.5 mm diameter, 5 μm pore size; Costar), and the cells were grown to form a confluent monolayer. CFSE-labeled Raw264.7 cells preincubated with Ab26–37 for 1 h corresponding to each concentration were added to the upper chamber, and 10 ng/ml TNFα and IFNγ were used as a chemoattractant in the lower chamber. After incubation for 12 h, the trans-well was fixed with 4% paraformaldehyde, and its upper side was cleaned with cotton and then mounted. The migrated cells were imaged at six positions by means of microscopy and analyzed by counting the percentages of CFSE-labeled cells that had transmigrated. For TEM activity of BMDMs, siNinj1, or stable Raw264.7 cells, each cells were labeled with CFSE and added to MBEC4 monolayer containing with TNFα and IFNγ (10 ng/ml, 12 h) in the lower chamber as chemoattractants and incubated for 12 h. The migrated cells were quantified using microscopy.
RNAi Interference
For Ninjurin1 knockdown by RNAi interference, siRNAs (siNinj1) against mouse Ninjurin1 (NM_013610) were designed using design software and purchased from Invitrogen. Negative control siRNAs (siCont) were predesigned from Bioneer Inc. (Korea). Each sequence of the siRNAs is as follows: siCont, 5′-CCT ACG CCA CCA AUU UCG U dTdT-3′; siNinj1, 5′-ACC GGC CCA UCA AUG UAA ACC AUU A-3′. Each siRNA was transfected in Raw264.7 cells by Nucleofector (Amaxa) at a concentration of <200 pmol per sample. After 24 h, the Ninjurin1 knockdown efficiency was determined by Western blotting of anti-GFP and the custom-made anti-Ninjurin1 antibody (Ab1–15).
Immunoblotting
Tissues and cells were extracted in a cell lysis buffer containing 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 μg/ml leupeptin, and a protease inhibitor mixture. Lysates were immunoprecipitated or immunoblotted with antibodies of Ninjurin1 (Ab1–15), inducible nitric-oxide synthase (BD Biosciences), actin (Sigma), and green fluorescent protein (GFP) (Abcam).
Immunofluorescence Staining and Microscopy
Tissues or cells were incubated with the following antibodies against F4/80 (Serotec), CD45 (BD Biosciences), CD3 (R&D Systems), and MOG (R&D Systems). After incubation with the primary antibodies overnight at 4 °C, tissues or cells were visualized with Alexa 488-conjugated IgG or Alexa 546-conjugated IgG (Molecular Probes) as secondary antibodies. Nucleus-staining was performed with Hoechst 33342 (Molecular Probes). Both Axiovert M200 and LSM 700 microscopes (Carl Zeiss) were used for immunofluorescence imaging.
Accelerating Rotarod Test
Motor coordination and balance were assessed by an accelerating rotarod (Columbus Institute, Columbus, OH). The rotating rod was grooved to improve the grip. The experimental procedure consisted of two phases: constant-speed training and accelerating-speed testing. For the constant-speed training, mice were placed on the rod and allowed to adjust for 1 min at a fixed 2.0 rpm. If a mouse fell before the 1-min cutoff, the process was restarted. For the accelerating-speed test, the rotarod was accelerated (acceleration = 17 rpm/min), and the latency to fall from the rod was recorded. This procedure was repeated three times, and the results were analyzed.
Data Analysis and Statistics
All data are presented as the means ± S.E. and expressed as relative percentages and fundamental units. Statistical significance was calculated using an unpaired two-tailed Student t test for single comparisons and an analysis of variance test for multiple comparisons. *, p < 0.05 was considered statistically significant.
RESULTS
Construction of Ninjurin1 KO Mice
To determine the role of Ninjurin1 during CNS inflammation in vivo, Ninjurin1 KO mice were generated by removing exon 1 from among a total of 4 exons for Ninjurin1 located on chromosome 13 by homologous recombination (Fig. 1A) and were backcrossed with the C57BL/6 strain for at least seven generations. Ninjurin1 heterozygous (Hetero), homozygous (KO), and congenic wild-type (WT) mice were evaluated by genotyping via a genomic polymerase chain reaction (PCR) (Fig. 1B).
FIGURE 1.
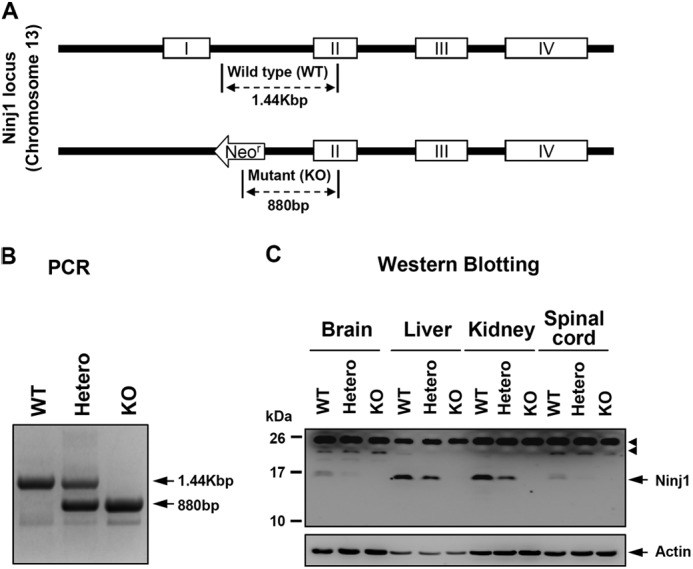
Construction of Ninjurin1 KO mice. A, schematic diagram of Ninjurin1 KO mice (upper). Targeted disruption of the Ninjurin1 gene was carried out by replacement of exon 1 among the 4 exons located on chromosome 13. B, the WT, Hetero, and homozygotes (KO) mice were identified by their PCR amplicons (WT = 1.44 Kbp, KO = 880 bp, and Hetero = 1.44 Kbp and 880 bp). C, one-dimensional SDS-PAGE of various tissues (brain, liver, kidney, and spinal cord) from WT and mutant mice was performed and blotted with Ab1–15 antibody and anti-actin antibody. Actin was used as a loading control, and the arrowhead indicates nonspecific band.
Next, we raised an antibody (Ab1–15) to detect Ninjurin1 expression, which was immunized with the peptide from Met1 to Arg15 of the mouse Ninjurin1 as an antigen (20). Western blotting with Ab1–15 antibody revealed the complete disappearance of the expected band in the Ninjurin1 KO mice, clearly demonstrating the well controlled Ninjurin1 expression on the protein level as well as the specificity of the Ab1–15 antibody to the mouse Ninjurin1 (Fig. 1C). Notably, the level of Ninjurin1 expression is heterogeneous in various tissues and is gene dosage-dependent when WT and mutant mice were compared (Fig. 1C).
By analyzing the phenotypical characteristics, some of the Ninjurin1 KO mice showed developmental retardation and dysfunctions such as ataxia and hydrocephalus (data not shown), whereas the others had normal phenotypes. In this study we used Ninjurin1 KO mice with a normal appearance and a hemogram similar to that of WT mice (Table 1).
TABLE 1.
Hemogram of WT or Ninjurin1 KO mice
Peripheral blood was collected from the retro-orbital sinus of 10-week-old mice (WT; n = 8, KO; n = 6). Cell counts were performed using an automated cell counter with veterinary parameters and reagents. Significantly different between WT and Ninjurin1 KO mice (p < 0.05). WBC, white blood cells.
| Variable | WT |
KO |
p Value | ||
|---|---|---|---|---|---|
| Mean | S.E. | Mean | S.E. | ||
| 103/μl | |||||
| WBC | 7.10 | 0.15 | 7.84 | 0.22 | 0.30 |
| Neutrophils | 0.95 | 0.03 | 1.22 | 0.05 | 0.07 |
| Lymphocytes | 5.87 | 0.12 | 6.29 | 0.17 | 0.44 |
| Monocytes | 0.21 | 0.01 | 0.28 | 0.01 | 0.15 |
| Eosinophils | 0.05 | 0.005 | 0.05 | 0.003 | 0.71 |
| Basophils | 0.02 | 0.002 | 0.02 | 0.001 | 0.40 |
| Erythrocytes 106/μl | 8.91 | 0.04 | 8.88 | 0.03 | 0.85 |
| Platelets | 563.9 | 21.47 | 563.0 | 12.35 | 0.99 |
Ninjurin1 Mutant Mice Attenuate the Susceptibility of MOG35–55-immunized EAE in a Gene Dosage-dependent Manner
Initially, the susceptibilities to EAE were compared between WT and Ninjurin1 KO mice for a 30-day period after (MOG)35–55 immunization. Interestingly, the Ninjurin1 KO mice showed significantly lower EAE clinical scores from 17 to 22 days after immunization compared with the WT mice (Fig. 2A) with a difference in their peak scores and without a difference in the incidence and onset time of the disease (Fig. 2B). Furthermore, when we compare the EAE susceptibility between the Ninjurin1 Hetero and KO mice, the difference is likely to be distinguishable despite the loss of statistical significance in terms of the clinical score for EAE (Fig. 2C). Ninjurin1 KO mice had reduced mean and peak values, but a similar onset time for EAE compared with the Hetero group (Fig. 2D). Nevertheless, the difference in the EAE severity between the Hetero and KO mice is less than that between the WT and KO mice. In Fig. 1C, the amount of Ninjurin1 expression is dependent on the dosage of the gene. These results imply that EAE pathogenesis is prohibited by the deficiency of Ninjurin1 in mice, which is closely correlated with its expression level.
FIGURE 2.

EAE susceptibility of Ninjurin1 Hetero and KO mice is ameliorated in a gene dosage-dependent manner. A, EAE was induced in WT (n = 12) and Ninjurin1 KO mice (n = 11) by immunization with 100 μg of MOG35–55 peptide per mouse. *, p < 0.05. B, Ninjurin1 KO mice had resistance to EAE with a difference in the peak clinical score (WT = 3.33 ± 0.08 and KO = 2.09 ± 0.12) but no difference in the onset time (WT = 11.9 ± 0.19 and KO = 11.6 ± 0.16 day after immunization). *, p < 0.05. C, EAE was induced in Ninjurin1 Hetero (n = 8) and KO mice (n = 8) by immunization with 100 μg of MOG35–55 peptide per mouse. D, statistical comparison of Ninjurin1 Hetero and KO mice with EAE.
Because some Ninjurin1 KO mice have developmental retardation and ataxia, we performed the rotarod test to examine the relevance between the basal motor functions and the EAE limb paralysis. The latency to fall off the rod in the Ninjurin1 KO mice was slightly reduced but showed no statistical significance to their WT counterparts (data not shown). This result indicates that the weak limb paralysis of EAE in Ninjurin1 KO mice mainly stems from the leukocyte-promoting demyelination regardless of the basal motor function with normal appearance.
Ninjurin1 KO Mice Had Reduced Recruitment of Leukocytes into the Lesion Sites of the Spinal Cord with EAE
Ninjurin1 is highly expressed in myeloid-lineage immune cells (15, 16, 18) and up-regulated in T lymphocytes (19) to promote their recruitment toward inflammatory sites. We, therefore, compared the number of leukocytes that infiltrated into the spinal cords of WT or Ninjurin1 KO mice with EAE. Consistent with previous results, a histological analysis of the spinal cords of mice revealed that Ninjurin1 KO mice had decreased accumulation of leukocytes with CD45, a pan-leukocyte marker, F4/80, a macrophage marker, and CD3, a lymphocyte marker (Fig. 3, A and B) and, consequently, results with a smaller lesion area and less demyelination (Fig. 3C). These results imply that Ninjurin1 KO mice protected against EAE due to the reduced amounts of leukocyte adhesion and infiltration into the injury lesions.
FIGURE 3.

Ninjurin1 KO mice have reduced leukocyte recruitment into the spinal cord injury with EAE. A, representative immunostaining images in the spinal cord tissues of WT or Ninjurin1 KO mice were taken with anti-CD45 (pan leukocytes), anti-F4/80 (macrophage), and anti-CD3 (T lymphocyte) antibodies. Scale bar = 50 μm. B, the number of infiltrated immune cells in the spinal cord was counted in three mice per group: **, p < 0.01. C, representative images of hematoxylin eosin (H&E) staining showing a lesion area and MOG staining (red) showing demyelination in the spinal cord of the WT or KO mice.
A recent study showed that Ninjurin1 depletion promotes cell apoptosis and senescence in a p53-dependent manner (21). Although the hemogram between WT and Ninjurin1 KO mice did not differ under normal conditions (Table 1), the inflammatory EAE induction may alter the homeostatic functions of leukocytes in Ninjurin1 KO mice. Therefore, the hemogram was compared when the clinical score after EAE induction was significantly different between each group (Table 2). As a result, we found no significant distinction in the number and proportion of leukocytes circulating inside blood vessels (Table 2). This result suggests that inflammatory EAE stimulation has no effect on leukocytes homeostasis between WT and Ninjurin1 KO mice.
TABLE 2.
Hemogram of WT or Ninjurin1 KO mice after EAE induction
EAE was induced in 12-week-old mice (WT; n = 8, KO; n = 8). Peripheral blood was collected from the abdominal vein at 14 days after immunization when clinical scores were significantly different. Cell counts were performed using an automated cell counter with veterinary parameters and reagents. Significant differences of each value between the WT and Ninjurin1 KO mice were noted when p < 0.05. WBC, white blood cells.
| Variable | WT (EAE) |
KO (EAE) |
p Value | ||
|---|---|---|---|---|---|
| Mean | S.E. | Mean | S.E. | ||
| Clinical Score | 3.16 | 0.08 | 1.94 | 0.14 | 0.02* |
| WBC (103/μl) | 6.25 | 0.45 | 5.32 | 0.16 | 0.50 |
| Neutrophils (103/μl) | 3.46 | 0.35 | 2.95 | 0.07 | 0.62 |
| Lymphocytes (103/μl) | 2.28 | 0.14 | 2.10 | 0.09 | 0.71 |
| Monocytes (103/μl) | 0.43 | 0.03 | 0.29 | 0.02 | 0.20 |
| Eosinophils (103/μl) | 0.06 | 0.01 | 0.03 | 0.003 | 0.20 |
| Basophils (103/μl) | 0.02 | 0.003 | 0.01 | 0.001 | 0.09 |
| Erythrocytes (106/μl) | 8.18 | 0.06 | 8.64 | 0.09 | 0.14 |
| Platelets (103/μl) | 1013.25 | 26.41 | 898.50 | 24.95 | 0.28 |
Adherence of Leukocytes Is Decreased in the Ninjurin1 KO Mice with EIU
Attachment of leukocytes on the luminal side of the vessel is the initial event for TEM under inflammation (5). Retinal vasculature is a beneficial in vivo method to investigate leukocyte adhesion on the vessel network because of its two-dimensional property and easy visualization with a flat-mounted technique (22). Furthermore, inflammation in the retina can be easily generated by EIU, which is accompanied by acute and intense leukocyte infiltration into the vitreous in the retina within 24 h after an intraperitoneal injection of LPS at a sublethal dose (23). Using the retinal model coupled with EIU, we compared the number of leukocytes attached on the retinal vessel between Ninjurin1 KO mice and its WT counterpart. Ninjurin1 KO mice had less attachment of leukocytes on the vessel wall of the retina compared with the WT mice (Fig. 4, A and B), indicating that leukocyte-endothelium adhesions is directly regulated by Ninjurin1 in vivo.
FIGURE 4.
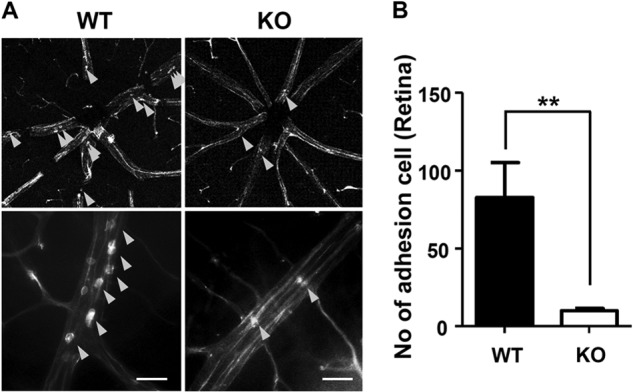
Adherence of leukocytes is decreased in the Ninjurin1 KO mice with EIU. A, EIU in WT (n = 7) and Ninjurin1 KO mice (n = 7). Mice were intraperitoneally injected with LPS at 9 mg/kg per mouse for 24 h, and the adherent leukocytes on the vessel wall of the retina were labeled with rhodamine-concanavalin (Con). A, representative images of adherent leukocytes (yellow arrowheads) are shown. Scale bar = 50 μm. B, the number of adherent cells per retina was counted. **, p < 0.01.
Selective Blockage of the Ninjurin1 Homophilic Binding Domain via Ab26–37 Antibody Reduces Leukocyte Trafficking and the EAE Clinical Score
Ninjurin1 can interact with itself through the homophilic binding domain, from Pro26 to Asn37, of the N-terminal ectodomain (15), which may mediate leukocyte-endothelium adhesion and subsequent TEM under inflammation. To examine the blockage effects of the Ninjurin1 homophilic binding domain, a custom-made antibody, Ab26–37, was generated using the peptide from Pro26 to Asn37 of the N-terminal ectodomain of Ninjurin1 as an antigen (Fig. 5A). The treatment with the Ab26–37 antibody decreased the adhesion (Fig. 5B) and TEM (Fig. 5C) of the Raw264.7 cells (murine peritoneal macrophage cells) on or across MBEC4 cell (mouse brain microvascular endothelial cells) monolayers in vitro (Fig. 5, B and C). Moreover, the intraperitoneal administration of the Ab26–37 antibody at 4, 6, 8, and 10 days after EAE induction significantly attenuated its clinical symptoms from 14 to 17 days after EAE immunization (Fig. 5D) with a significant difference in peak score and no difference in the onset time (Fig. 5E). Staining with anti-CD45 or anti-CD3 antibody revealed that the accumulation of leukocytes decreased in the Ab26–37-injected KO mice compared with an IgG control group (Fig. 5, F and G). These results mean that selective blockage corresponding to its homophilic binding domain inhibits the adhesion and TEM activity and, in turn, the accumulation of leukocytes, finally leading to protective effects against EAE progression.
FIGURE 5.

Functional blockage of the Ninjurin1 homophilic binding domain via Ab26–37 reduces the adhesion activity and TEM of Raw264.7 cells and alleviates EAE susceptibility. A, schematic diagram of a custom-made antibody (Ab26–37). The Ab26–37 was made by immunizing rabbits with a synthetic peptide corresponding to the homophilic binding domain of Ninjurin1 as the antigen. B and C, impaired adhesion (B) or TEM activity (C) (Ab26–37, 40 μg/ml) of the CFSE (5 μm, 5 min)-labeled Raw264.7 cells on the MBEC4 monolayer with pretreatment of Ab26–37. The adhesion and transmigration index is shown relative to the IgG normalized to 100%. **, p < 0.01; ***, p < 0.001. D, 100 μg of Ab26–37 (n = 7) per mouse was administrated intraperitoneally on days 4, 6, 8, and 10 (black arrow) after MOG35–55 immunization. Control mice (n = 6) were injected with isotype IgG at an equivalent amount. *, p < 0.05. E, statistical comparison of Ninjurin1 Hetero and KO mice with EAE. *, p < 0.05. F, representative immunostaining images with CD45 (pan leukocytes) and CD3 (T lymphocyte) show the accumulation of leukocytes in the spinal cord of IgG or Ab26–37-injected mice. G, the number of infiltrated immune cells in the spinal cord was counted in three mice per group: *, p < 0.05; **, p < 0.01.
Ninjurin1 Is Up-regulated in the Myeloid Cells and Inflamed Endothelial Cells in the Spinal Cord of EAE Mice
Next, Western blotting with the Ab1–15 antibody showed Ninjurin1 up-regulation in the spinal cord with EAE according to clinical symptoms (Fig. 6, A and B).
FIGURE 6.

Ninjurin1 up-regulation in spinal cord of EAE mice. A, spinal cords of EAE mice with each clinical score were extracted. Western blot analysis was performed with Ab1–15 antibody. Actin was used as loading control (C) and arrowhead means nonspecific band. B, Ninjurin1 expression in the spinal cord of EAE mice was normalized to the actin level in each group. *, p < 0.05. C–F, spinal cord tissues were obtained from WT (C and E) and Ninjurin1 KO mice (D and F) with similar EAE clinical scores (3.5). The Ab1–15 (red) antibody was stained with each cell-type specific marker (green) in inflamed regions of WT (C) and Ninjurin1 KO tissues (D), anti-CD45 (pan-leukocytes), anti-F4/80 (macrophages/monocytes), and anti-CD3 (lymphoid cells) antibody. E and F, double immunostaining of the Ab1–15 antibody (red) with anti-CD31 (green, endothelial cells) antibody in the inflamed and non-inflamed regions of WT spinal cord tissues (E) and the inflamed regions of Ninjurin1 KO samples (F). Images were visualized by LSM700 microscopy, and representative images are shown. The arrowheads in (C and E) indicate the merged signal in the spinal cord of the WT mice. Scale bar = 50 μm.
To examine the Ninjurin1-expressing cell-types in the EAE spinal cord, we first tested our Ab1–15 antibody, which is specific to the mouse Ninjurin1 sequence. Strong and abundant Ninjuirn1-positive signals were detected in the lesion sites in which leukocytes were accumulated (Fig. 6C). These signals merged with the anti-CD45 (pan-leukocytes) antibody and the anti-F4/80 (macrophages/monocytes) antibody with a strong intensity level and high percentages and also merged with the anti-CD3 (lymphocytes) antibody with weak intensity and low proportions (Fig. 6C). Furthermore, the Ninjurin1-positive signals merged with the anti-CD31 (endothelial cells) antibody in the inflamed regions but not in non-inflamed regions (Fig. 6E). The signals of the Ab1–15 staining largely disappeared in the Ninjurin1 KO mice tissues, although some background signals remained (Fig. 6, D and F). Thus, these results show that Ninjurin1 is expressed in leukocytes and in inflamed endothelial cells (strong/abundant expression in myeloid-lineage cells and weak/partial expression in the lymphoid-lineage cells). These results are consistent with previous findings (16, 18, 19).
To confirm the specificity of the Ab1–15 antibody, liver tissues, which are known to show high Ninjurin1 expression levels, were tested (Fig. 1C) (20). Strong Ninjurin1 signals were observed in WT tissues, although these were mostly absent without any nonspecific background in the Ninjurin1 KO samples (data not shown), supporting the specificity and availability of our Ab1–15 antibody for tissue staining.
An IFNγ Treatment Increases Ninjurin1 Expression in Raw264.7 or MBEC4 Cells
Because some inflammatory cytokines regulate leukocyte recruitment toward legion sites in EAE pathogenesis (24), we examined the change in Ninjurin1 expression in macrophage and endothelial cells treated with TNFα or/and INFγ in vitro. In the Raw264.7 cells, INFγ treatment increased Ninjurin1 expression in a dose-dependent manner, whereas that of TNFα had no effect (Fig. 7, A and B). Ninjurin1 expression was also up-regulated by INFγ treatment in BMDMs (Fig. 7C), the purity (>90%) of which was evaluated by immunostaining with the anti-F4/80 antibody, a macrophage-specific marker (data not shown). In MBEC4 cells, INFγ treatment increased Ninjurin1 expression (Fig. 7D). These results indicate that inflammatory stimulation with INFγ up-regulates Ninjurin1 expression in both macrophage and endothelial cells and consequently might enhance the TEM. However, its regulation might vary according to the type of cytokines that require further investigation.
FIGURE 7.

Ninjurin1 is up-regulated in Raw264.7 or MBEC4 cells with IFNγ treatment in dose-dependent manner. A, Raw264.7 cells were cultured in the DMEM media with 10% FBS. After starvation for 6 h in serum-absent media, IFNγ was treated for 24 h with the indicated concentrations (ng/ml). Western blot analysis was performed with Ab1–15 antibody, anti-inducible nitric-oxide synthase antibody as the inflammatory indicator, and anti-actin antibody as the loading control. The arrowhead means a nonspecific band. B, Ninjurin1 expression in Raw264.7 cell was normalized to the actin level in each group. *, p < 0.05; **, p < 0.01. C, BMDMs were cultured in RPMI media with 10% FBS and 20 ng/ml macrophage-colony stimulating factor. After starvation for 6 h in both serum and macrophage-colony stimulating factor absent media, they were treated with IFNγ for 24 h with the indicated concentrations (ng/ml). Western blot analysis was performed with Ab1–15 antibody, anti-inducible nitric-oxide synthase antibody as an inflammatory indicator, and anti-actin antibody as a loading control. The arrowhead means a nonspecific band. D, MBEC4 cells were cultured in DMEM media with 10% FBS. After starvation for 6 h in serum-absent media, IFNγ was treated for 24 h with the indicated concentrations (ng/ml). Western blot analysis was performed with the Ab1–15 antibody and the anti-actin antibody as a loading control. The arrowhead means a nonspecific band.
Ninjurin1 Expression in the BMDMs and Raw264.7 Cells Is Directly Involved in Their TEM Activity across an MBEC4 Monolayer
Through previous and our current studies with blocking antibody or peptide, it has already been established that the Ninjurin1 homotypic adhesion property is crucial for the trafficking of leukocytes (14, 18, 19). However, direct evidence of whether the Ninjurin1 expression level might control the TEM activity has been lacking. Therefore, with an in vitro modified Boyden chamber assay, we compared the TEM activity levels penetrating a monolayer of MBEC4 cells with Ninjurin1 KO BMDMs and Raw264.7 cells suppressed with Ninjurin1 RNAi (siNinj1) or overexpressing the GFP-tagged mouse Ninjurin1 plasmid (GFP-mNinj1). The Ninjurin1 KO BMDMs (Fig. 8, A and B) and siNinj1 Raw264.7 cells (Fig. 8, C and D) showed less TEM activity than the WT BMDMs and the siCont Raw264.7 cells, respectively. In contrast, stable GFP-mNinj1 Raw264.7 cells showed increased TEM activity compared with the GFP Raw264.7 cells (Fig. 8, E and F). These results suggest that the Ninjurin1 expression level directly regulates the TEM of immune cells across the endothelial monolayer.
FIGURE 8.
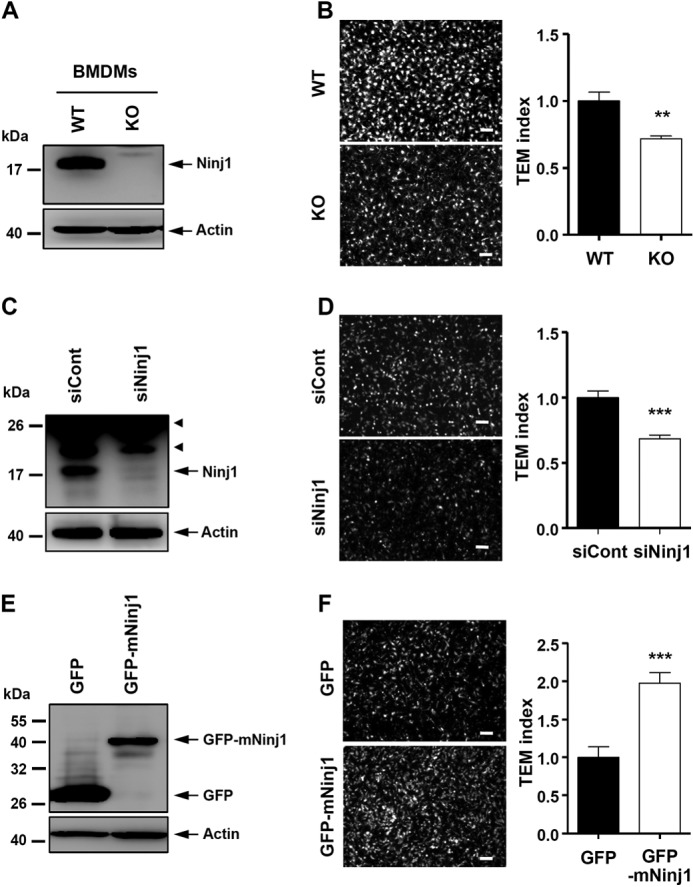
Change of Ninjurin1 expression in BMDMs and Raw264.7 cell directly regulates the TEM activity across the MBEC4 monolayer. A, Western blot of BMDMs isolated from WT and Ninjurin1 KO mice. The arrowhead means a nonspecific band. B, TEM activity of Ninjurin1-deficient BMDMs across the MBEC4 cell monolayer. C, Western blot of Raw264.7 cells knock-downed with Ninjurin1 RNAi interference (siNinj1). D, TEM activity of siNinj1 Raw264.7 cells across the MBEC4 cell monolayer. E, Western blot of Raw264.7 cells stably overexpressing GFP or GFP-tagged mouse Ninjurin1 plasmid (GFP-mNinj1). F, TEM activity of siNinj1 Raw264.7 cells across the MBEC4 cell monolayer. A, C, and E, Western blot is performed with Ab1–15 antibody (A and C) and anti-GFP antibody for Ninjurin1 detection (E), and actin is used as a loading control. B, D, and F, the TEM activity was analyzed by a modified Boyden chamber assay. Each cell, labeled with 5 μm CFSE for 5 min, was added onto the MBEC4 monolayer containing 10 ng/ml TNFα and INFγ in lower chamber as chemoattractants. After 12 h, the trans-well fixed at 4% paraformaldehyde, and its upper side was cleaned using cotton, after which it was mounted. The migrated cells were visualized using microscopy. Representative images (left) are shown and a transmigration index (right, n = 3) is shown relative to the WT BMDMs, siCont, or GFP Raw264.7 cells normalized to 100% in each case: **, p < 0.01; ***, p < 0.001. Scale bar = 50 μm.
DISCUSSION
Herein we clarified the contributions of Ninjurin1 using gene-deficient mice (Fig. 1) and antibody-neutralized mice coupled with the EAE and EIU animal model in vivo. Ablation of Ninjurin1 in mice alleviated EAE susceptibility (Fig. 2) with reduced leukocyte infiltration (Fig. 3), and decreased adherence of leukocytes on retinal vessels was shown in EIU mice (Fig. 4). In addition, a custom-made antibody, Ab26–37, specific to the homophilic binding domain of the Ninjurin1 N-terminal ectodomain, effectively attenuated EAE susceptibility (Fig. 5). Furthermore, we discovered that direct control via Ninjurin1 expression is crucial for the TEM of immune cells (Fig. 8). With primary cultured BMDMs, RNAi, and overexpressing Raw264.7 cells (Fig. 8), we showed the relationship between the Ninjurin1 expression level and the TEM (Fig. 8).
For a gene depletion study, we used Ninjurin1 KO mice with a normal phenotype that exhibits similar hemograms (Table 1 and Table 2) as well as BMDMs F4/80+ proportional to the WT counterpart (data not shown). However, a recent paper indicated that Ninjurin1 KO mice died prematurely (21). Consistent with this result, we also observed developmental defects in some of the Ninjurin1 KO mice. Therefore, this novel mechanism of Ninjurin1 on p53 activity and cell survival could explain these features of Ninjurin1 KO mice with severe developmental defects.
The weaker inflammatory response from the Ninjurin1 KO mice with EAE is thought to stem from the impairment of leukocyte-endothelium adhesions (Figs. 2 and 3). However, Ninjurin1 depletion in our KO mice is not restricted to myeloid cells or endothelial cells; moreover, MOG35–55-induced EAE mice show chronic inflammation that lasts for a long time (25). These limitations left us to debate whether the anti-inflammatory effect seen in the Ninjurin1 KO mice was from specific events related to the leukocyte-endothelium interactions. We resolved this argument somewhat using the EIU animal model (Fig. 4), which shows acute inflammation properties that induce rapid and intense leukocyte adhesion on the vessel wall of the retina within 24 h after LPS administration (23). This short time response in EIU mice might exclude the contributions of other effectors, except for leukocyte-endothelium interactions. Therefore, our study with the EIU model supports the role of Ninjurin1 on leukocyte-endothelium adhesions under inflammation (Fig. 4).
With the neutralizing study using the Ab26–37 antibody (Fig. 5), we confirmed that the homotypic binding property of Ninjurin1 is crucial for leukocyte trafficking and its targeting is beneficial for treating EAE. Indeed, Ifergan et al. (18) previously tried to block the functions of Ninjurin1 in EAE with a commercial antibody produced from immunization with the N-terminal regions of the mouse Ninjurin1. On the other hand, our custom-made Ab26–37 antibody was generated by immunization with a peptide corresponding to the homophilic binding domain of Ninjurin1 (Fig. 5). Thus, our antibody is thought to be an improvement for Ninjurin1 targeting strategies against its homophilic domain with a more specific interaction in EAE compared with the previous trial.
Because Ninjurin1 is known as an adhesion molecule (15), its homotypic binding properties can aptly explain how Ninjurin1 mediates leukocyte trafficking. However, it is likely that Ninjurin1 fulfils additional functional and regulatory complexities in leukocyte trafficking beyond its well established adhesion properties. Indeed, both leukocytes and endothelial cells undergo diverse cellular events during diapedesis (4, 5). For example, dynamic morphological alternations like “podosomes” (26) or “filopodia” (27) in leukocytes and like “migratory cup” in endothelial cells (28) are found at the docking portions facing the penetrating leukocytes on the endothelial cell monolayer. Furthermore, the activity of numerous effectors including chemokines and their cognate receptors (24), cytoskeletal and vesicular regulations (29, 30), extracellular matrix components (31), and intercellular signaling molecules (32) should be well orchestrated for the completion of TEM in a stage-restrictive or non-restrictive manner. It is, therefore, notable that Ninjurin1 might possibly play a role in the processes of each of the steps during diapedesis described above. In the future, whether Ninjurin1 might have other stage-specific roles and, if it does, how it interacts with the homotypic binding activity in the leukocyte-endothelium interaction would be interesting subjects to further investigate.
Finally, because Ninjurin1 was also detected in various other cell types, including neurons, oligodendrocytes, and astrocytes in the CNS (14, 16), an investigation of the coordinated interactions between Ninjurin1-expressing cells under inflammation will be helpful when seeking to understand CNS diseases. The growing evidence suggests that many diseases share inflammation as a convergent downstream signal after injury despite the divergent initial triggers (33). Therefore, our results are applicable for the treatments of inflammatory CNS diseases, including atherosclerosis (34), stroke (35), and trauma (36) as well as other diseases such as rheumatoid arthritis and various wounds.
This work was supported by a grant from a National Research Foundation of Korea funded by the Ministry of Education, Science, and Technology through the Global Research Laboratory Program (2011-0021874), Brain Korea 21 Program (2013-036038), and the Global Core Research Center Program (2012-0001187). This work was also supported by National Institutes of Health Grants R37-NS37074, R01-76694, and P01-NS55104.
- EAE
- experimental autoimmune encephalomyelitis
- TEM
- transendothelial migration
- EIU
- endotoxin-induced uveitis
- BMDM
- bone marrow-derived macrophage
- MOG
- myelin oligodendrocyte glycoprotein
- Hetero
- heterozygous
- CFSE
- carboxyfluorescein succinimidyl ester.
REFERENCES
- 1. Ousman S. S., Kubes P. (2012) Immune surveillance in the central nervous system. Nat. Neurosci. 15, 1096–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goverman J. (2009) Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 9, 393–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lopez-Diego R. S., Weiner H. L. (2008) Novel therapeutic strategies for multiple sclerosis. A multifaceted adversary. Nat. Rev. Drug Discov. 7, 909–925 [DOI] [PubMed] [Google Scholar]
- 4. Ley K., Laudanna C., Cybulsky M. I., Nourshargh S. (2007) Getting to the site of inflammation. The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689 [DOI] [PubMed] [Google Scholar]
- 5. Nourshargh S., Hordijk P. L., Sixt M. (2010) Breaching multiple barriers. Leukocyte motility through venular walls and the interstitium. Nat. Rev. Mol. Cell Biol. 11, 366–378 [DOI] [PubMed] [Google Scholar]
- 6. Imhof B. A., Aurrand-Lions M. (2004) Adhesion mechanisms regulating the migration of monocytes. Nat. Rev. Immunol. 4, 432–444 [DOI] [PubMed] [Google Scholar]
- 7. Finger E. B., Puri K. D., Alon R., Lawrence M. B., von Andrian U. H., Springer T. A. (1996) Adhesion through L-selectin requires a threshold hydrodynamic shear. Nature 379, 266–269 [DOI] [PubMed] [Google Scholar]
- 8. Smith C. W., Marlin S. D., Rothlein R., Toman C., Anderson D. C. (1989) Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J. Clin. Invest. 83, 2008–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barreiro O., Yanez-Mo M., Serrador J. M., Montoya M. C., Vicente-Manzanares M., Tejedor R., Furthmayr H., Sanchez-Madrid F. (2002) Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J. Cell Biol. 157, 1233–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O'Brien C. D., Lim P., Sun J., Albelda S. M. (2003) PECAM-1-dependent neutrophil transmigration is independent of monolayer PECAM-1 signaling or localization. Blood 101, 2816–2825 [DOI] [PubMed] [Google Scholar]
- 11. Schenkel A. R., Mamdouh Z., Chen X., Liebman R. M., Muller W. A. (2002) CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat. Immunol. 3, 143–150 [DOI] [PubMed] [Google Scholar]
- 12. Constantin G., Majeed M., Giagulli C., Piccio L., Kim J. Y., Butcher E. C., Laudanna C. (2000) Chemokines trigger immediate β2 integrin affinity and mobility changes. Differential regulation and roles in lymphocyte arrest under flow. Immunity 13, 759–769 [DOI] [PubMed] [Google Scholar]
- 13. Ostermann G., Weber K. S., Zernecke A., Schröder A., Weber C. (2002) JAM-1 is a ligand of the β2 integrin LFA-1 involved in transendothelial migration of leukocytes. Nat. Immunol. 3, 151–158 [DOI] [PubMed] [Google Scholar]
- 14. Araki T., Milbrandt J. (1996) Ninjurin, a novel adhesion molecule, is induced by nerve injury and promotes axonal growth. Neuron 17, 353–361 [DOI] [PubMed] [Google Scholar]
- 15. Araki T., Zimonjic D. B., Popescu N. C., Milbrandt J. (1997) Mechanism of homophilic binding mediated by ninjurin, a novel widely expressed adhesion molecule. J. Biol. Chem. 272, 21373–21380 [DOI] [PubMed] [Google Scholar]
- 16. Ahn B. J., Lee H. J., Shin M. W., Choi J. H., Jeong J. W., Kim K. W. (2009) Ninjurin1 is expressed in myeloid cells and mediates endothelium adhesion in the brains of EAE rats. Biochem. Biophys. Res. Commun. 387, 321–325 [DOI] [PubMed] [Google Scholar]
- 17. Lee H. J., Ahn B. J., Shin M. W., Jeong J. W., Kim J. H., Kim K. W. (2009) Ninjurin1 mediates macrophage-induced programmed cell death during early ocular development. Cell Death Differ. 16, 1395–1407 [DOI] [PubMed] [Google Scholar]
- 18. Ifergan I., Kebir H., Terouz S., Alvarez J. I., Lécuyer M. A., Gendron S., Bourbonnière L., Dunay I. R., Bouthillier A., Moumdjian R., Fontana A., Haqqani A., Klopstein A., Prinz M., López-Vales R., Birchler T., Prat A. (2011) Role of Ninjurin-1 in the migration of myeloid cells to central nervous system inflammatory lesions. Ann. Neurol. 70, 751–763 [DOI] [PubMed] [Google Scholar]
- 19. Odoardi F., Sie C., Streyl K., Ulaganathan V. K., Schläger C., Lodygin D., Heckelsmiller K., Nietfeld W., Ellwart J., Klinkert W. E., Lottaz C., Nosov M., Brinkmann V., Spang R., Lehrach H., Vingron M., Wekerle H., Flügel-Koch C., Flügel A. (2012) T cells become licensed in the lung to enter the central nervous system. Nature 488, 675–679 [DOI] [PubMed] [Google Scholar]
- 20. Ahn B. J., Le H., Shin M. W., Bae S. J., Lee E. J., Wee H. J., Cha J. H., Park J. H., Lee H. S., Lee H. J., Jung H., Park Z. Y., Park S. H., Han B. W., Seo J. H., Lo E. H., Kim K. W. (2012) The N-terminal ectodomain of Ninjurin1 liberated by MMP9 has chemotactic activity. Biochem. Biophys. Res. Commun. 428, 438–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cho S. J., Rossi A., Jung Y. S., Yan W., Liu G., Zhang J., Zhang M., Chen X. (2013) Ninjurin1, a target of p53, regulates p53 expression and p53-dependent cell survival, senescence, and radiation-induced mortality. Proc. Natl. Acad. Sci. U.S.A 110, 9362–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ishida S., Yamashiro K., Usui T., Kaji Y., Ogura Y., Hida T., Honda Y., Oguchi Y., Adamis A. P. (2003) Leukocytes mediate retinal vascular remodeling during development and vaso-obliteration in disease. Nat. Med. 9, 781–788 [DOI] [PubMed] [Google Scholar]
- 23. Rosenbaum J. T., McDevitt H. O., Guss R. B., Egbert P. R. (1980) Endotoxin-induced uveitis in rats as a model for human disease. Nature 286, 611–613 [DOI] [PubMed] [Google Scholar]
- 24. Sallusto F., Baggiolini M. (2008) Chemokines and leukocyte traffic. Nat. Immunol. 9, 949–952 [DOI] [PubMed] [Google Scholar]
- 25. Zamvil S., Nelson P., Trotter J., Mitchell D., Knobler R., Fritz R., Steinman L. (1985) T-cell clones specific for myelin basic protein induce chronic relapsing paralysis and demyelination. Nature 317, 355–358 [DOI] [PubMed] [Google Scholar]
- 26. Carman C. V., Sage P. T., Sciuto T. E., de la Fuente M. A., Geha R. S., Ochs H. D., Dvorak H. F., Dvorak A. M., Springer T. A. (2007) Transcellular diapedesis is initiated by invasive podosomes. Immunity 26, 784–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shulman Z., Shinder V., Klein E., Grabovsky V., Yeger O., Geron E., Montresor A., Bolomini-Vittori M., Feigelson S. W., Kirchhausen T., Laudanna C., Shakhar G., Alon R. (2009) Lymphocyte crawling and transendothelial migration require chemokine triggering of high-affinity LFA-1 integrin. Immunity 30, 384–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carman C. V., Springer T. A. (2004) A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J. Cell Biol. 167, 377–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vicente-Manzanares M., Sánchez-Madrid F. (2004) Role of the cytoskeleton during leukocyte responses. Nat. Rev. Immunol. 4, 110–122 [DOI] [PubMed] [Google Scholar]
- 30. Mamdouh Z., Kreitzer G. E., Muller W. A. (2008) Leukocyte transmigration requires kinesin-mediated microtubule-dependent membrane trafficking from the lateral border recycling compartment. J. Exp. Med. 205, 951–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sorokin L. (2010) The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 10, 712–723 [DOI] [PubMed] [Google Scholar]
- 32. Laudanna C., Campbell J. J., Butcher E. C. (1996) Role of Rho in chemoattractant-activated leukocyte adhesion through integrins. Science 271, 981–983 [DOI] [PubMed] [Google Scholar]
- 33. Lo E. H. (2010) Degeneration and repair in central nervous system disease. Nat. Med. 16, 1205–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moore K. J., Tabas I. (2011) Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lo E. H. (2009) T time in the brain. Nat. Med. 15, 844–846 [DOI] [PubMed] [Google Scholar]
- 36. David S., Kroner A. (2011) Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 12, 388–399 [DOI] [PubMed] [Google Scholar]