

Background: GPI anchor is essential for virulence of C. albicans. Little is known about its GPI biosynthetic pathway. We explore roles of two GPI-N-acetylglucosaminyltransferase subunits catalyzing the first step.
Results: Subunits GPI2 and GPI19 are negatively co-regulated, affecting Ras1 activity and ERG11 levels, respectively.
Conclusion: GPI2/GPI19 levels affect morphogenesis and ergosterol biosynthesis.
Significance: C. albicans can be targeted by modulation of cross-talk among major pathways.
Keywords: Candida albicans, Fungi, Molecular Cell Biology, Ras, Sterol, Ergosterol Biosynthesis, Filamentation, GPI-N-Acetylglucosaminyltransferase Complex, Glycosylphosphatidyl Anchor Biosynthesis, Ras Signaling
Abstract
Candida albicans is a leading cause of fungal infections worldwide. It has several glycosylphosphatidylinositol (GPI)-anchored virulence factors. Inhibiting GPI biosynthesis attenuates its virulence. Building on our previous work, we explore the interaction of GPI biosynthesis in C. albicans with ergosterol biosynthesis and hyphal morphogenesis. This study is also the first report of transcriptional co-regulation existing between two subunits of the multisubunit enzyme complex, GPI-N-acetylglucosaminyltransferase (GPI-GnT), involved in the first step of GPI anchor biosynthesis in eukaryotes. Using mutational analysis, we show that the accessory subunits, GPI2 and GPI19, of GPI-GnT exhibit opposite effects on ergosterol biosynthesis and Ras signaling (which determines hyphal morphogenesis). This is because the two subunits negatively regulate one another; GPI19 mutants show up-regulation of GPI2, whereas GPI2 mutants show up-regulation of GPI19. Two different models were examined as follows. First, the two GPI-GnT subunits independently interact with ergosterol biosynthesis and Ras signaling. Second, the two subunits mutually regulate one another and thereby regulate sterol levels and Ras signaling. Analysis of double mutants of these subunits indicates that GPI19 controls ergosterol biosynthesis through ERG11 levels, whereas GPI2 determines the filamentation by cross-talk with Ras1 signaling. Taken together, this suggests that the first step of GPI biosynthesis talks to and regulates two very important pathways in C. albicans. This could have implications for designing new antifungal strategies.
Introduction
The ubiquitous GPI5 anchor in eukaryotes anchors a variety of proteins to the cell surface. These proteins in the human pathogenic fungus, Candida albicans, include virulence factors, enzymes involved in host immune response evasion, cell wall biogenesis, and maintenance proteins as well as hypha-specific proteins (1–3). The pathway appears to critically affect the functioning and viability of the organism (4, 5).
The elaborate pathway of roughly 10–12 steps that generates the precursor GPI anchor core in the endoplasmic reticulum involves mostly multisubunit membrane-bound enzymes. Based on studies in yeast and mammalian cells, the enzymatic subunit for each step has been identified, and its role in the catalytic step has been specified. However, the role of the accessory subunits in these steps is unclear. One hypothesis has been that the accessory proteins provide an opportunity for regulatory control on the pathway. However, very little experimental proof for such regulatory control was available until recently.
We recently demonstrated in a couple of reports, for the very first time, one such regulation occurring via the Gpi19 accessory subunit of the GPI-GnT complex that catalyzes the first step of GPI biosynthesis in C. albicans (6, 7). Down-regulation of GPI19 in conditional null mutants resulted not only in specific defects that could be attributed to GPI anchor deficiency, such as cell wall defects and increased cell clumping, but also to the following: (i) altered drug response phenotypes that appeared to be specifically linked to a down-regulation of the crucial rate-determining step of ergosterol biosynthesis via ERG11, and (ii) hyperfilamentation that was specifically associated with Ras hyperactivity.
In this study, we develop this theme further by examining the role of GPI2 (in both CAI4 as well as BWP17 strains of C. albicans), the gene encoding for another accessory subunit of the fungal GPI-GnT complex, and its effect on GPI biosynthesis, ergosterol biosynthesis, and filamentation. In addition, we seek to understand how Gpi2 affects Gpi19 and vice versa. We demonstrate that the mutants of GPI2 show phenotypes that are the exact opposite of the phenotypes of the GPI19 mutants. The GPI2 mutants are resistant to azole drugs due to a specific up-regulation of ERG11 and exhibit reduced filamentation specifically due to reduced Ras1 activity. We also demonstrate that GPI2 and GPI19 exhibit a mutual negative regulation and suggest that a fine balance between the two may be crucial to the proper functioning of the initiation step of the pathway as well as for both ergosterol biosynthesis and hyphal growth in C. albicans.
EXPERIMENTAL PROCEDURES
Materials
All chemicals used were of analytical grade and were purchased from Sigma, Qiagen, Merck, or SRL. Growth media were purchased from HiMedia. Enzymes and DNA markers were purchased from Fermentas or Bangalore Genei. The primers used in the study were synthesized by Sigma. UDP-[6-3H]GlcNAc was purchased from American Radiochemicals. Antibody against RNA pol II was purchased from Santa Cruz Biotechnology. Protein A-CL-agarose beads were purchased from Bangalore-Genei. Protease mixture inhibitor (P8340) was purchased from Sigma. cAMP-dependent PKA activity kit was purchased from Promega. Antibody for detection of phospho-p44/p42 MAPK (Thr202/Thr204) was purchased from Cell Signaling Technology.
Strains and Growth Conditions
The strains used in this study are listed in Table 1. The fungal strains were grown at 30 °C in YEPD or SD minimal medium supplemented with appropriate nutrients depending on the auxotrophic status of the strain. For the regulation of gene expression from MET3 promoter in the conditional mutants, the strains were grown in permissive (Met−/Cys−) or repressive (5–10 mm Met/Cys) minimal growth media. For hyphal induction, the strains were grown in either YEPD or Spider media at 37 °C. The bacterial strains were grown in LB medium at 37 °C.
TABLE 1.
List of C. albicans strains used in this study
| Strains | Reference in text | Source |
|---|---|---|
| CAI4 | CAI4 | 8 |
| CAI4-gpi2/GPI2 | GPI2 heterozygote | This study |
| CAI4-gpi2/PMET3-GPI2 | Conditional null GPI2 | This study |
| CAI4-gpi2/GPI2/PACT1-GPI2 | GPI2 revertant | This study |
| CAI4-PACT1-GFP | CAI4-URA3 | This study |
| CAI4-gpi2/GPI2/PACT1-GFP | GPI2 heterozygote carrying URA3 | This study |
| CAI4-PACT1-GPI2 | GPI2 overexpression | This study |
| CAI4-gpi2/GPI2::gpi3/GPI3 | GPI2/GPI3 double heterozygote | This study |
| BWP17 | BWP17 | 9 |
| BWP17-gpi2/GPI2 | GPI2 heterozygote in BWP17 strain background | This study |
| BWP17-gpi2/PMET3-GPI2 | Conditional null GPI2 mutant in BWP17 strain background | This study |
| BWP17-gpi2/GPI2/PACT1-GPI2 | GPI2 revertant in BWP17 strain background | This study |
| BWP17-gpi2/GPI2::gpi19/GPI19 | GPI2/GPI19 double heterozygote in BWP17 strain background | This study |
| BWP17-gpi2/GPI2::gpi15/GPI15 | GPI2/GPI15 double heterozygote in BWP17 strain background | This study |
| BWP17-gpi19/PMET3-GFP-GPI19 | Conditional null GPI19 mutant in BWP17 strain background | 6 |
| BWP17-gpi19/PMET3-GFP-GPI19::gpi2/GPI2 | Conditional null GPI19 mutant with a single allele disruption of GPI2 | This study |
| BWP17-erg11/ERG11 | ERG11 heterozygote in BWP17 strain background | 7 |
| BWP17-gpi2/GPI2/PACT1-RAS1 | GPI2 heterozygote with RAS1 overexpression | This study |
| BWP17-gpi19/PMET3-GFP-GPI19::ras1/RAS1 | Conditional null GPI19 mutant with a single allele disruption of RAS1 | This study |
| BWP17-ras1/ras1/PACT1-GFP | ras1 null carrying URA3 | This study |
| BWP17-ras1/ras1/PACT1-GPI2 | ras1 null with GPI2 overexpression | This study |
| BWP17-ras2/ras2/PACT1-GFP | ras2 null carrying URA3 | This study |
| BWP17-ras2/ras2/PACT1-GPI2 | ras2 null with GPI2 overexpression | This study |
Cloning and Generation of Mutants
The primer sequences used for generation and confirmation of different mutants are given in Table 2. The different genetic disruptions were carried out in C. albicans using URA3-blaster method (8) or PCR-mediated disruption strategy (9). The conditional null mutant was generated using PCR-mediated promoter replacement approach, thus replacing the wild type promoter with the MET3 promoter (10).
TABLE 2.
List of primers used in this study
| Primer name | Primer sequence |
|---|---|
| GPI2 FP | 5′GCGGAGCTCATGGAAGAAATACATATAAGCTCT3′ |
| GPI2 RP | 5′GCGAAGCTTTCAGCTTTGTATACTTGACTTCAT3′ |
| GPI2 5′hz RP | 5′GCGAGATCTGTCCCATTGCTTCTCATATATT3′ |
| GPI23′hz FP | 5′GCGGTCGACACATTAGACGTATTTTTGTTTG3′ |
| GPI2-ARG4 FP | 5′ATGGAAGAAATACATATAAGCTCTTCCATTTTGCAAGATAGTGATTCAGCTCCCCCAAACCATTTTCAGTGTGGAATTGTGAGCGGATA3′ |
| GPI2-ARG4 RP | 5′TCAGCTTTGTATACTTGACTTCATTAACTTTGGTTTCGCCACATCCCATGGTCCTTGTATCTCGTTTTTTTTCCCAGTCACGACGTT3′ |
| GPI2 Up FP | 5′AAGTATTCTATATCTTGTGTTTTGTGGGTCGTGTTT3′ |
| GPI2 Down RP | 5′CACTGAAAAGAAAAGTGTATATTATCTTCTATAAGG3′ |
| GPI2-URA3-PMET3 FP | 5′AGTATAATTTTGAGTTTGTGTGTGTGTGTGTGGAAGTATTCTATATCTTGTGTTTTGTGGGTCGTGTCTAGAAGGACCACCTTTGATTG3′ |
| GPI2-URA3-PMET3 RP | 5′CATGCTGAAAATGGTTTGGGGGAGCTGAATCACTATCTTGCAAAATGGAAGAGCTTATATGTATTTCTTCCATTTTAATAAACGCGGATCC3′ |
| GPI19-HIS1 FP | 5′ATGATATTCCATTTTAACCAAAAAGAGAAAGCAGATTCAAGTAAATGTAAAACAATACATGTAAGAAAGACGGGGATCCTGGAGGATGAG3′ |
| GPI19-HIS1 RP | 5′TCATTCATATAGAACATCATTCACTAATGTAATAGGCAAATCCCAAACACCACTTGGTGCCTTATGAATACGGAATATTTATGAGAAACT3′ |
| GPI19 Up FP | 5′GAATTCGAAAACTAGATATTAATGTT3′ |
| GPI19 Down RP | 5′GTCGACTCTAGATCTCCTGCTTACTTTTTATTT3′ |
| GPI15-HIS1 FP | 5′ATGTCCTCATCGAAAAATTATAAATTGGAAATTTCCCCTAGTATCAACCAAACTAGTGCCACAGAATCGCGGGGATCCTGGAGGAATGAG3′ |
| GPI15-HIS1 RP | 5′CTAAATAACTTGTTTTAAACCTTGACCAGGCACTCTTCTCCAATAGCGTTTTGTGCTTCCAAATAGTAACGGAATATTTATGAGAAACT3′ |
| GPI15 Up FP | 5′TGGTAACCATTCATCAATCATATAG3′ |
| GPI15 Down RP | 5′GGTTTATACATAGATTTTATATACATTG3′ |
| PACT1-GFP-RAS1 FP | 5′ATTAGATATAATCAAAAAAATCAAAAAGAAACCCCGGGCAAACACAAATTCATATCCTCTAGAAGGACCACCTTTGATTG3′ |
| PACT1-GFP-RAS1 RP | 5′GGTTAAAGCGGATTTACCAACACCACCACCTCCAACAACAACTAATTTATATTCTCTCAATTTGTACAATTCATCCATAC3′ |
| RAS1-HIS1 FP | 5′ATGTTGAGAGAATATAAATTAGTTGTTGTTGGAGGTGGTGGTGTTGGTAAATCCGCTTTAACCATTCAACGGGGATCCTGGAGGATGAG3′ |
| RAS1-HIS1 RP | 5′TCAAACAATAACACAACATCCATTCTTTGATTTAGAGCTAGATTGTTTTTGACCAGAAAATTGATTTTCCGGAATATTTATGAGAAACT3′ |
| RAS1-ARG4 FP | 5′TTGATTCAATCCCATTTTGTTGATGAATATGACCCAACTATTGAGGATTCTTATCGTAAACAATGTACTTGTGGAATTGTGAGCGGAAG3′ |
| RAS1-ARG4 RP | 5′AGATTGTTGTTGTGGTTCTTGTTGTTGCTGTTGTTGTTGTCTTTGTGATTGTGATTGTGATTGATTTGGAATTTCCCAGTCACGACGTT3′ |
| RAS1 FP | 5′ATGTTGAGAGAATATAAATTAG3′ |
| RAS1 RP | 5′AACAATAACACAACATCCATTCT3′ |
| RAS2-HIS1 FP | 5′ATGTCATTACTACTAGATAGCTTACATGCATTAACAAAGAACTACGACATGTGTGTCATTGGCTCCAGTCGGGGATCCTGGAGGATGAG3′ |
| RAS2-HIS1 RP | 5′CTATGTTATTATGCAACATTTATTTTCAGCATGTATTTGACTGGATTTTGCCAGTGGGCCAGATGACTGCGGAATATTTATGAGAAACT3′ |
| RAS2-ARG4 FP | 5′AATGTCGGTAAATCAACCTTAGTCCTTCATTATGTGTATCATCATTTCGACGAAAGTTTGTATGACTTGTGTGGAATTGTGAGCGGAAG3′ |
| RAS2-ARG4 RP | 5′TTTTAATCGAGTTTTTCTTTGACGAGATGAAGTAGATGGTGTTTGTGCTATGGGTGTAGAACCATTCAAATTTTCCCAGTCACGACGTT3′ |
| RAS2 Up FP | 5′TCTAAATCCATAGTCTACATCAACTAATTT3′ |
| RAS2 Down RP | 5′AATAAGATTACATGTAACTCAAATTTTTGA3′ |
| GPI3 FP | 5′GCGGAGCTCATGGGATACAATATAGCAATGGTA3′ |
| GPI3 RP | 5′GAAGCTTCTAATTTACTTTGTTCGGAA3′ |
| GPI3 5′hz RP | 5′GGGCGAGATCTAGCAATTAGAAATTTCACTTTTGG3′ |
| GPI33′hz FP | 5′GGTCGACAGAAAATGTTTATAATTC3′ |
| GPI3 Up FP | 5′CACCACAAACGAATTACACAAGCA3′ |
| GPI3 Down RP | 5′ATAAATGGACTAAAAAGAAAGATCTG3′ |
| RPS10 RP | 5′TTTCTGGTGAATGGGTCAACGAC3′ |
| PACT1 FP | 5′GCGGGATCCGAGCTCCTCGATAATTCTAGTGGT3′ |
| GAPDH RT FP | 5′CAGCTATCAAGAAAGCTTCTG3′ |
| GAPDH RT RP | 5′GATGAGTAGCTTGAACCCAA3′ |
| ERG11 RT FP | 5′GGAACGAGATTGTAAGATAG3′ |
| ERG11 RT RP | 5′AGTACATTGGGTCATTTCAG3′ |
| ERG1 RT FP | 5′GGAACGAGATTGTAAGATAG3′ |
| ERG1 RT RP | 5′AGTACATTGGGTCATTTCAG3′ |
| GPI2 RT FP | 5′GGCCAATAGCATTTCTAAC3′ |
| GPI2 RT RP | 5′CCATCACAAAAACAGACAAA3′ |
| GPI19 RT FP | 5′CAAGAAGAAGAAGAAGGAGAA3′ |
| GPI19 RT RP | 5′AAACACCACTTGGTGCCTTA3′ |
| GPI15 RT FP | 5′CCACGATTATGGGCAGGTTA3′ |
| GPI15 RT RP | 5′CGCGGTAAGAATTCTGGAAA3′ |
| GPI3 RT FP | 5′TGCTGAACCGGAAGAAAACT3′ |
| GPI3 RT RP | 5′TACATCTTTGCAACCGCATC3′ |
| ERG11 PR FP | 5′ATGAAAGGGACCAATCTGTC3′ |
| ERG11 PR RP | 5′AAGTATTATCCTGTCTGATTCT3′ |
| ERG1 PR FP | 5′CATGTTTAAGAGACAAGTGTTT3′ |
| ERG1 PR RP | 5′TTCTCACCTCATCGTTCTCA3′ |
GPI2 was amplified from CAI4 genomic DNA using GPI2 FP and GPI2 RP. The amplified gene sequence was cloned into pET28a(+) between the SacI and HindIII restriction enzyme sites. Single knock-out of GPI2 gene for heterozygote generation in the CAI4 strain of C. albicans was done by using the URA3-blaster method (8). GPI2 disruption cassette was generated using the pMB7 vector. Briefly, 300 bp of GPI2 from the N and C termini were PCR-amplified using GPI2 FP/GPI2 5′hz RP and GPI2 3′hz FP/GPI2 RP primer combinations (Table 2) and cloned into pTZ57R/T vector. These fragments were then cloned into the multiple cloning site regions of the pMB7 vector (between SacI/BglII sites for the N-terminal fragment and SalI/HindIII sites for the C-terminal fragment), thus flanking the hisG-URA3-hisG sequence in pMB7 vector. The fragment for targeted disruption of GPI2 was released from the disruption cassette by restriction enzyme digestion using SacI and HindIII. The fragment for disruption was purified by agarose gel electrophoresis and used to transform CAI4 by the lithium acetate method. The colonies after transformation were selected on uridine-deficient SD minimal medium. The colonies obtained were streaked on YEPD plates for four generations, and then 5-fluoroorotic acid selection was done to select for the clones in which URA3 gene had been deleted by recombination between the hisG sequences. The colonies were confirmed by PCR using genomic DNA as template and the GPI2-flanking primers GPI2 Up FP/GPI2 Down RP.
Single knock-out mutant of GPI2 for heterozygote generation in C. albicans BWP17 strain was done using the PCR-mediated approach (10). The ARG4-selectable marker was amplified using primers with 60-bp homology of the N- and C-terminal sequence of GPI2 included in the GPI2-ARG4 FP and GPI2-ARG4 RP, in addition to 25-bp homology to the ARG4 marker (Table 2). The amplicon was used to transform the BWP17 strain, and the colonies after transformation were selected on arginine lacking growth medium. The genomic DNA from the colonies obtained after transformation was extracted and the successful disruption of GPI2 were confirmed by PCR using the GPI2-flanking primers GPI2 Up FP/GPI2 Down RP (Table 2).
Generation of conditional null mutants for GPI2 was achieved by placing the functional gene in the heterozygotes (in both CAI4 and BWP17 background strains), under the control of the MET3 promoter, using the PCR-mediated promoter replacement approach (9), using GPI2-URA3-PMET3FP and GPI2-URA3-PMET3RP. The targeted disruptions and introduction of the MET3 promoter were confirmed by PCR using GPI2 Up FP and GPI2 Down RP.
One allele of GPI2 was reintroduced into the GPI2 heterozygote mutants by cloning GPI2 under the PACT1 in pACT1-GFP vector, between the HindIII and NheI sites. The construct generated was linearized by the StuI enzyme and introduced into the GPI2 heterozygote. The transformants were selected on Ura− growth medium. The successful transformants were screened by PCR, using GPI2 FP and RPS10 RP for confirmation of integration of GPI2 at RPS10 locus. CAI4 was also transformed and confirmed similarly to generate a GPI2 overexpression strain. As controls for URA3, the strains were also transformed with StuI linearized empty vector pACT1-GFP and confirmed by PCR using PACT1 FP and RPS10 RP.
One allele of GPI19 and GPI15 was disrupted in GPI2 heterozygote strain created in the BWP17 strain background using PCR-mediated approach by amplifying the HIS1 selectable marker using GPI19-HIS1 FP/GPI19-HIS1 RP and GPI15-HIS1 FP/GPI15-HIS1 RP primer pairs, respectively (Table 2). The amplicons were purified and used for transformation. The successful transformants were confirmed by PCR using GPI19 Up FP/GPI19 Down RP and GPI15 Up FP/GPI15 Down RP for GPI19 and GPI15 disruptions, respectively.
Similarly, one allele of GPI2 was disrupted in the conditional null GPI19 mutant using GPI2-ARG4 FP/GPI2-ARG4 RP. The successful transformants were confirmed by PCR using GPI2 Up FP and GPI2 Down RP.
One allele of RAS1 was disrupted in the conditional null GPI19 mutant using RAS1-ARG4 FP/RAS1-ARG4 RP. The successful transformants were confirmed by PCR using RAS1 Up FP and RAS1 Down RP.
One allele of GPI3 was disrupted in the GPI2 heterozygote in CAI4 background using URA3-blaster method, similar to GPI2 disruption in CAI4. GPI3 was amplified from CAI4 genomic DNA using GPI3 FP and GPI3 RP. The amplified gene sequence was cloned into pET28a(+) vector between the NcoI and XhoI restriction enzyme sites. 708 bp from the N terminus and 262 bp from the C terminus of GPI3 were PCR-amplified using GPI3 FP/GPI3 5′hz RP and GPI3 3′hz FP/GPI3 RP primer combinations (Table 2) and cloned into pTZ57R/T vector. These fragments were then cloned into the multiple cloning site regions of the pMB7 vector (between SacI/BglII sites for the N-terminal fragment and SalI/HindIII sites for the C-terminal fragment), thus flanking the hisG-URA3-hisG sequence in the pMB7 vector. The fragment for targeted disruption of GPI3 was released from the disruption cassette using SacI and HindIII, purified by agarose gel electrophoresis and used for transforming the GPI2 heterozygote. The colonies after transformation were selected on uridine-deficient SD minimal medium. The colonies obtained were streaked on YEPD plates for four generations and then 5-fluoroorotic acid selection was done to select for the clones in which the URA3 gene had been deleted by recombination between the hisG sequences. The colonies were confirmed by PCR using genomic DNA as template and the GPI3-flanking primers GPI3 Up FP/GPI3 Down RP.
Null mutants for RAS1 and RAS2 were generated in BWP17 by disrupting both alleles using PCR-mediated disruption strategy. RAS1 alleles were disrupted using HIS1 and ARG4 selectable markers. Primers used for amplification of HIS1 and ARG4 were RAS1-HIS1 FP/RAS1-HIS1 RP and RAS1-ARG4 FP/RAS1-ARG4 RP combinations, respectively. The colonies were confirmed by PCR using genomic DNA as template and the RAS1-flanking primers RAS1 FP/RAS1 RP. Similarly, both alleles of RAS2 were disrupted using RAS2-HIS1 FP/RAS2-HIS1 RP and RAS2-ARG4 FP/RAS2-ARG4 RP primers and confirmed by PCR using RAS2-flanking primers, RAS2 Up FP/RAS2 Down RP.
Overexpression of GPI2 was done in ras1 and ras2 null mutants by transforming these strains with GPI2 overexpression vector, PACT1-GPI2. Transformants were confirmed by PCR using GPI2 FP and RPS10 RP for confirmation of integration of GPI2 at the RPS10 locus. As control, empty vector PACT1-GFP was also transformed in ras1 and ras2 null mutants and confirmed by PCR using PACT1 FP and the RPS10 RP.
Plate Assays
Plate assays were performed to observe the growth pattern and response of the mutants to the presence of various forms of stress inducers in the growth medium, as described previously (6).
Growth Rate Analysis
Growth curves were obtained to look for growth patterns in liquid growth media, as described previously (6). Briefly, cells were grown overnight in 10 ml of growth medium at 30 °C. A600 nm was observed for this saturated cell culture. Cells were added from this saturated culture to 50 ml of fresh growth medium to obtain an A600 nm of 0.1 and grown at 30 °C, 220 rpm. A600 nm was monitored every 2 h until saturation was attained. Growth curves were plotted using these A600 nm values, and the doubling times were calculated.
Propidium Iodide Staining
A primary culture of conditional null GPI2 mutant in CAI4 background (CAI4-gpi2/PMET3-GPI2) was set up overnight at 30 °C in permissive (Met−/Cys−) minimal medium for saturated growth. A secondary culture was set up in 10 ml of permissive (Met−/Cys−) or repressive (10 mm Met/Cys) minimal growth media, using primary culture inoculum corresponding to an A600 nm of 0.1. Samples were collected after 48 h of growth at 30 °C, 220 rpm. Cells at a density corresponding to A600 nm of 1.0 were taken in a microcentrifuge tube and washed twice with PBS. Propidium iodide was added to a final concentration of 5 μg/ml in a final volume of 200 μl. Cells were kept in dark at 25 °C for 30 min on a rocker. Excess stain was removed by washing with PBS. Cells were resuspended in 80% glycerol and observed under the microscope (Olympus IX71).
Preparation of Microsomes from C. albicans and GPI-GnT Activity Assay
10 ml of primary culture for each strain was grown overnight in minimal medium at 30 °C, 220 rpm. 200 ml of secondary culture was set up using 2% primary culture inoculum and grown overnight at 30 °C, 220 rpm. The cells were pelleted and washed using ice-cold TM buffer (50 mm Tris-Cl, pH 7.5; 2 mm MgCl2). The cells were resuspended in 4 ml of ice-cold TM buffer and lysed by vortexing using glass beads (12 rounds of vortexing: 1 min vortexing- 1 min on ice). The cell lysates were centrifuged at 1000 × g for 10 min, 4 °C. The supernatant collected was again centrifuged at 12,000 × g for 15 min, 4 °C. To the supernatant, 7.5 volumes of ice-cold 8.0 mm CaCl2 were added dropwise with continuous stirring on a magnetic stirrer, at 4 °C. Stirring was continued for 20 min at 4 °C. The supernatants were then centrifuged at 8000 × g for 10 min at 4 °C. The pellets obtained were resuspended in 300 μl of ice-cold TM buffer containing 10% glycerol. Aliquots were dispensed into microcentrifuge tubes and quickly frozen in liquid nitrogen before storage at −80 °C. The whole process was carried out under ice-cold conditions.
Protein concentration was quantified in the microsomes using a BCA kit (from Sigma). GPI-GnT assay was done using microsomes from each strain with 1000 μg of total protein, as described previously (11).
Chitin Estimation Using CFW Staining
Mid-log phase cells grown in SD minimal medium were pelleted down and washed with PBS. A600 nm was monitored for the different strains. 0.2 ml of 0.1 A600 nm cells were taken, stained with 100 μg/ml of CFW for 30 min at 30 °C, then washed with PBS, and observed under a microscope using 405 nm laser (Olympus IX71). Fluorescence intensity of the cells was quantified using Cell∧F software.
Monitoring Cell Wall Integrity Pathway Activation by PKC
PKC activation was monitored by measuring the levels of phosphorylated Mkc1 levels, as described previously (7).
MIC80 Determination
MIC80 values for azoles were determined using the broth dilution method, as done previously (12). Briefly, the different strains were grown for 24 h at 30 °C on YEPD plates. In a 96-well flat bottom microtiter plate, 2-fold serial dilutions of azoles were done in YEPD medium. Cells picked from the plates were suspended in 0.9% saline, diluted to an A600 nm equal to 0.1, and then further diluted 100-fold. 100 μl of cell suspensions were added to all the wells of the microtiter plate and incubated at 30 °C for 48 h. A600 nm was monitored using a microplate reader. MIC80 values were calculated by determining the concentration of azoles that lead to an inhibition of cell growth by 80% as compared with cell growth in the absence of azoles in the growth medium.
Rhodamine 6G (R6G) Influx/Efflux Assay
R6G influx/efflux was monitored for CAI4 and GPI2 heterozygote strains as described previously (12).
Sterol Analysis Using GC-MS
The total sterols were extracted and analyzed by GC-MS as described previously (7).
Transcript Level Analysis Using RT-PCR
10 ml of late log phase cells were taken, pelleted down, and washed twice with diethyl pyrocarbonate-treated water. Total RNA was extracted using TRIzol reagent (Sigma) and stored at −20 °C. cDNA was prepared using 3.0 μg of total RNA. The transcript levels were quantified using SYBR Green PCR Master Mix (Applied Biosystems) and RT primers are given in Table 2. GAPDH levels were taken as control in all experiments.
Chromatin Immunoprecipitation (ChIP) Assays
ChIP analysis was performed using a protocol design based on previously published protocols (13–15). Briefly, 2% inocula from 10 ml of overnight-grown saturated cultures at 30 °C were used to set up secondary cultures in 100 ml of growth media. To the cultures grown until mid-log phase, 1% v/v formaldehyde was added, and the cultures were incubated at 30 °C, 220 rpm for 30 min. 125 mm glycine was added, and the cultures were incubated again at 30 °C, 220 rpm for 5 min. The cells were harvested, washed twice with 1× PBS, and the pellet weights equalized. The cells were treated with 5 units of Lyticase (Sigma) at 30 °C for 2 h, pelleted down, and washed twice with 1× PBS before being resuspended in 700 μl of ice-cold ChIP lysis buffer (50 mm HEPES, pH 7.4; 140 mm NaCl; 1 mm EDTA; 1% Triton X-100; 1 mm PMSF). The cells were lysed by vortexing in the presence of glass beads (10 rounds: 1 min vortex and 1 min on ice) and centrifuged at 15,000 rpm for 15 min at 4 °C. The supernatant obtained was sonicated in a water bath sonicator (14 rounds: 30 s sonication and 30 s on ice) and centrifuged at 15,000 rpm for 15 min at 4 °C. The supernatant was collected and divided for use as input material and for immunoprecipitation. 50 μl of the supernatant was added to 200 μl of TE (10 mm Tris-Cl, pH 7.5; 1 mm EDTA), 1% SDS and used as input material. To another 300 μl of the supernatant, for immunoprecipitation, 10 μl of protein A-CL-agarose beads (pre-equilibrated in ChIP Lysis Buffer) were added and kept on a rotating wheel for 3 h at 4 °C. The supernatant was recovered by spinning the tubes at 1500 rpm for 5 min at 4 °C, and 1 μg of anti-RNA pol II antibody was added and kept on a rotating wheel for 12 h at 4 °C. The samples were then centrifuged at 15,000 rpm for 5 min at 4 °C, and the supernatants were transferred to fresh tubes. To the supernatants, 50 μl of protein A-CL-agarose beads (pre-equilibrated in ChIP Lysis Buffer) were added and kept on a rotating wheel for 4 h at 4 °C. The beads were then spun at 1500 rpm for 5 min and washed twice with ice-cold ChIP Lysis Buffer, twice with high salt buffer (50 mm HEPES, pH 7.4; 500 mm NaCl; 1.0 mm EDTA; 1% Triton X-100), twice with wash buffer (10 mm Tris-Cl, pH 8.0; 250 mm LiCl; 1 mm EDTA; 0.5% Nonidet P-40), and once with TE. 150 μl of elution buffer (50 mm Tris-Cl, pH 8.0; 10 mm EDTA; 1.0% SDS) was added to the beads and kept at 65 °C for 5 min, with occasional tapping, and the eluate was collected. One more eluate was collected similarly and pooled with the first eluate. The eluates and the previously stored input materials were kept overnight at 65 °C. The SDS concentration was reduced in the samples to 0.5% using TE, and 0.1 mg of RNase was added to each sample, followed by incubation at 37 °C for 2 h. 12.5 μg of proteinase K was next added to each sample and further incubated at 37 °C for 3 h. LiCl was then added to a final concentration of 0.4 m. DNA was separated using 1:1 phenol/chloroform mixture and purified using 1.0 ml of absolute ethanol and 20 μg of glycogen. The samples were precipitated overnight at −80 °C and recovered by centrifugation at 15,000 rpm for 30 min, 4 °C. The pellets were washed with 70% ethanol and resuspended in sterile water (60 μl for Input Material; 30 μl for immunoprecipitated DNA). The samples were analyzed by PCR using primers specific to the ERG11 promoter (ERG11 PR FP/ERG11 PR RP) or the ERG1 promoter regions (ERG1 PR FP/ERG1 PR RP) (Table 2).
Steady State Anisotropy Measurements
Change in membrane rigidity was monitored using DPH as a probe. Briefly, cells were grown to log phase, pelleted, and washed with PBS. 2 × 107 cells were taken in a glass tube, resuspended in 1.5 ml of PBS, and DPH added to a final concentration of 2 μm. The tubes were kept in dark at 30 °C, 220 rpm for 4 h. Fluorescence anisotropy was measured using a Cary-Varian spectrofluorimeter at excitation and emission wavelengths of 360 and 430 nm, respectively, with slit-widths of 10 and 5 nm respectively. The anisotropy values (r) were calculated using Equation 1,
where G is the instrument correction factor represented by Equation 2,
Here, IXY is the fluorescence intensity emitted in the Y plane when the sample is excited in the X plane. V and H denote the vertical and horizontal planes, respectively.
Morphological Studies
The cells were grown overnight in 10 ml of growth medium at 30 °C. 2% inocula from this saturated culture were transferred to 5 ml of fresh growth medium, and the cells were grown at 30 °C until they reached the mid-log phase. A600 nm was observed, and the culture was diluted to A600 nm of 0.1 using 0.9% saline. 10 μl of cell suspension was spotted on YEPD/Spider plates and incubated at 37 °C. The colony morphologies were photographed using AlphaImagerEC (AlphaInnotech). Hyphae formation at the edges of colonies was monitored at ×5 magnification under a Nikon Eclipse TiS microscope. For observing hyphae in liquid media, cells corresponding to A600 nm of 0.2 were grown at 37 °C in 5.0 ml of Spider medium. Aliquots were collected at 90 and 120 min, and cells were washed with PBS and observed under bright field microscope (Olympus IX71).
cAMP-dependent PKA Activity
cAMP-dependent protein kinase A (PKA) activity was monitored as described previously (7).
Heat Sensitivity Assays
The heat sensitivity of different strains was monitored as described previously (7). Briefly, plate assays were performed as mentioned above, before and after heat treatment at 48 °C for 8.0 min. The plates were incubated at 30 °C, and the growth was monitored at regular time intervals.
Statistical Significance of Data
Statistical significance of the data was calculated using Student's t test in SigmaPlot 8.0. The p values are indicated by asterisks in the figures with the following notations: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001. For all p values that are one or more orders of magnitude better than 0.0001, we continue to use ****.
RESULTS
GPI2 Is an Essential Gene Needed for Growth and Viability in C. albicans
GPI anchor biosynthesis has been shown to be essential for yeast and associated with growth and other cellular aberrations in C. albicans (5, 6, 16). GPI2 was cloned using primers (Table 2) based on the sequence information available at Prof. Eisenhaeber's site (17).
We generated the GPI2 heterozygous mutant (CAI4-gpi2/GPI2) first in C. albicans strain, CAI4 (Table 1), using the URA3-blaster strategy (8). The second allele disruption of GPI2 using the same strategy was not successful. Hence, we generated a conditional null mutant for GPI2 (CAI4-gpi2/PMET3-GPI2) using the regulatable MET3 promoter (10). The mutants were confirmed by PCR (data not shown).
The GPI2 mutants displayed GPI2 down-regulation vis-à-vis CAI4 (Fig. 1A). The growth of the strains was observed to see the effect of disruption of GPI2. GPI2 heterozygote showed growth similar to CAI4 in liquid as well as agar-YEPD media (data not shown) (Table 3). The growth of the conditional null GPI2 mutant depended on the concentration of Met/Cys used (Fig. 1, B and C; Table 3) and therefore on the extent of repression of the functional GPI2 allele from the MET3 promoter, thus indicating that GPI2 played an important role in the growth of C. albicans.
FIGURE 1.

GPI2 affects growth and viability in C. albicans. A, GPI2 transcript levels were decreased by 1.8 ± 0.03-fold (****, p value = 0.000018) in GPI2 heterozygote (CAI4-gpi2/GPI2) and 4.8 ± 0.1-fold (****, p value = 0.00016) in conditional null GPI2 mutant (CAI4-gpi2/PMET3-GPI2) as compared with CAI4. The average of two experiments done in duplicate along with standard deviations are plotted. B, conditional null GPI2 mutant showed growth defect in the presence of 5 mm Met/Cys, which was more severe in the presence of 10 mm Met/Cys. The average of three experiments done in duplicate along with standard deviations are plotted. C, CAI4, GPI2 heterozygote, and the conditional null GPI2 mutant strains were analyzed for growth patterns on SD-minimal medium plates as described previously (6). The plates shown here are after 48 h of incubation at 30 °C. CAI4 and GPI2 heterozygote showed similar growth. However, the conditional null GPI2 mutant showed a growth defect dependent on Met/Cys concentration, thus correlating the effect to GPI2 expression from MET3 promoter. The experiment was done twice, and each time similar results were obtained; a representative image is shown. False color has been assigned to the image (using GIMP software), to brighten the cell spots. D, conditional null GPI2 mutant was assessed for viability using propidium iodide staining. Cells grown in absence of Met/Cys exhibited higher viability as compared with cells grown in the presence of 10 mm Met/Cys. Although 24% of cells grown in permissive medium (absence of Met/Cys) were stained with propidium iodide, 84% cells grown in repressive medium (10 mm Met/Cys) were found to be propidium iodide-stained (*, p value = 0.032). The experiment was repeated twice.
TABLE 3.
Doubling time (minutes) of the GPI2 mutants versus wild type in liquid synthetic dextrose medium without (M−C−) or with (M+C+) Met/Cys
Growth assays were done as described under “Experimental Procedures.” The values of doubling times reported are of two independent experiments done in duplicate. The p values for the mutant strains in the table are calculated with respect to the wild type strain grown under similar conditions. The GPI2 heterozygote (CAI4-gpi2/GPI2) shows no significant difference in doubling times versus CAI4. For repression of the MET3 promoter, 5 or 10 mm each of Met/Cys (M+C+) was used as shown. The doubling time of the conditional null GPI2 mutant (CAI4-gpi2/PMET3-GPI2) in repressive medium (M+C+) is significantly higher than that of the wild type or heterozygote. But in permissive medium (M−C−), where it should have behaved as a heterozygous strain, the doubling time of the conditional null GPI2 mutant is less than that for the GPI2 heterozygote (**, p value = 0.0012). It also shows a faster doubling time than the control CAI4. We have consistently noticed that the URA3 gene tends to improve growth rates in control strains possessing this gene. Differences in the strength of the MET3 promoter versus the endogenous promoter of GPI2 in the two strains could also contribute to this difference.
| YEPD | SD M−C− | SD M+C+ 5 mm | SD M+C+ 10 mm | |
|---|---|---|---|---|
| CAI4 | 189 ± 3 | 187 ± 6 | 206 ± 4 | 210 ± 5 |
| CAI4-gpi2/GPI2 | 193 ± 7 | 190 ± 4 | 201 ± 3 | 202 ± 3 |
| p value = 0.35 | p value = 0.43 | p value = 0.08 | *, p value = 0.035 | |
| CAI4-gpi2/PMET3-GPI2 | 163 ± 9 | 239 ± 7 | 318 ± 17 | |
| **, p value = 0.0035 | ****, p value = 0.00024 | ****, p value = 0.000020 |
The viability of the mutant cells upon the disruption of GPI2 was assessed by propidium iodide staining. Roughly 84% conditional null GPI2 cells grown in repressive medium (in medium containing 10 mm Met/Cys) were positively stained with propidium iodide as compared with 24% cells grown under permissive conditions (in absence of Met/Cys) (Fig. 1D). Considering the effect of GPI2 deletion on the growth and viability of the mutants, GPI2 can be said to be an essential gene in C. albicans.
GPI Anchor Biosynthesis Is Reduced in the GPI2 Mutants
Next, we monitored the GPI anchor levels in the GPI2 mutants. For this purpose, in the presence of tunicamycin, UDP-[6-3H]GlcNAc was provided to microsomes generated from these strains. We extracted the polar glycolipids at the end of the assay, ran them on an HPTLC plate, and scanned them using a TLC scanner. Microsomes from CAI4 generated a much higher amount of [6-3H]GlcNAc-PI as compared with GPI2 heterozygote or the conditional null GPI2 mutant grown under permissive conditions (absence of Met/Cys) (Fig. 2A). Microsomes from the conditional null GPI2 mutant grown under repressive conditions (10 mm Met/Cys) could generate very little [6-3H]GlcNAc-PI in comparison. Thus, disruption of GPI2 affects GPI anchor biosynthesis in C. albicans in a gene dosage-dependent manner.
FIGURE 2.

GPI2 affects GPI anchor biosynthesis and cell wall in C. albicans. A, microsomes generated from GPI2 mutants exhibited lower GPI-GnT activity as compared with CAI4. The experiment was repeated twice in duplicate for confirmation. The results shown are an average of data from a single experiment done in duplicate, along with the standard deviations. B, GPI2 heterozygote (CAI4-gpi2/GPI2) exhibits sensitivity to cell wall-perturbing agents CFW, congo red, and SDS as compared with the CAI4, suggesting an alteration in the cell wall of the mutant. The plates shown are after 48 h of incubation at 30 °C. The experiment was repeated twice. C, chitin levels were compared between CAI4 and GPI2 heterozygote using CFW staining. Panel i, image showing the lower staining in the GPI2 heterozygote as compared with CAI4 is shown. Panel ii, mean CFW fluorescence from 100 cells of each strain quantified using Cell∧F Software is shown. The average and standard deviations of data from two independent experiments are shown. GPI2 heterozygote showed lesser mean fluorescence (1997 ± 128) as compared with CAI4 (2479 ± 167); ****, p value = 0. D, cell wall integrity pathway activation by PKC was analyzed by comparing phosphorylated Mkc1 levels (7). The CFW-treated cells, used as positive control, showed higher PKC activity as compared with the control (*, p value = 0.026). No significant difference was observed between the CAI4 and GPI2 heterozygotes for the levels of phosphorylated Mkc1 (p value = 0.26). The experiment was done twice in duplicate; average values with standard deviations are shown.
GPI2 Mutation Leads to Altered Cell Wall in C. albicans
Almost 35% of cell surface proteins in C. albicans are GPI-anchored, and the GPI anchors cross-link with β-glucans on the cell surface (1). Cell wall alterations in C. albicans have been previously reported for other GPI biosynthesis mutants also (1, 5, 6, 18).
GPI2 heterozygote was sensitive to different cell wall perturbing agents (Fig. 2B) indicating an altered cell wall. The chitin level in GPI2 heterozygote was lower in comparison with CAI4 (Fig. 2C), as estimated by CFW staining. Notably, the reduced chitin content in GPI2 heterozygote was quite the opposite of what we had reported previously for the GPI19 mutants (6). No difference in CAI4 and GPI2 heterozygote strains (Fig. 2D) was observed with respect to activation of the cell wall integrity pathway mediated through PKC signaling. Given the inherent growth defect and inviability of the conditional null GPI2 mutant, we were unable to assess it for its response to cell wall perturbing agents.
GPI2 Mutant Shows an Up-regulation of Ergosterol Biosynthesis via Enhanced ERG11 Transcription
Previously, the conditional null GPI19 mutant has been shown to be resistant to amphotericin B (Amp B) and sensitive to azoles, the commonly used antifungals (6). These effects correlated with a reduction in total ergosterol levels due to down-regulation of ERG11 involved in the rate-determining step of ergosterol biosynthesis (7). Would down-regulation of GPI2 result in a similar response?
To understand this, we first examined the response of GPI2 heterozygote to azoles, which act by specifically targeting Erg11 in the ergosterol biosynthetic pathway (19). We found it to be azole-resistant (Fig. 3A) with a high MIC80 value for azole drugs (Table 4).
FIGURE 3.

GPI2 mutation alters ergosterol biosynthesis in C. albicans. A, GPI2 heterozygote (CAI4-gpi2/GPI2) exhibits azole resistance as compared with CAI4. The plates shown here are after 48 h of incubation at 30 °C. The experiment was done three times for confirmation. False color has been assigned to the image (using GIMP software) to brighten the cell spots. B, rhodamine 6G influx/efflux assays were performed in GPI2 mutant. Panel i, R6G influx assays were done to monitor the difference in R6G influx in CAI4 and GPI2 heterozygote. The drop in levels of extracellular R6G was an indication of the extent of influx of the dye into the cell. The two strains exhibited similar extents of R6G influx. The experiment was done twice in duplicate, and the average values with their standard deviations are reported. Panel ii, R6G efflux assay was done to see if the efflux of R6G varied in the two strains. R6G is a good substrate of the multidrug pumps that also efflux out azoles in C. albicans (21). The two strains exhibited similar levels of R6G efflux in the absence as well as presence of glucose (Glc). The experiment was done twice in duplicate, and the average values along with standard deviations are reported here. C, ERG11 transcript levels were enhanced in GPI2 heterozygote by 3.2-fold (±0.64) as compared with CAI4 (***, p value = 0.0004), although ERG1 transcript levels were decreased to 0.57-fold (±0.03) (****, p value = 0.000001). No difference was observed for transcription of ERG3 (p value = 0.32). The experiment was done twice in duplicate. The average values with standard deviation are plotted. D, GPI2 heterozygote exhibited sensitivity to the presence of terbinafine as well as Amp B when compared with CAI4. The plates shown here are after 48 h of incubation at 30 °C. The experiment was done twice. False color has been assigned to the image (using GIMP software), to brighten the cell spots. E, using ChIP study occupancy of ERG11 promoter by RNA, pol II was found to be higher in case of GPI2 heterozygote as compared with CAI4 (****, p = 0.00000015), although thr ERG1 promoter occupancy was found to be lower (****, p = 0.0000049 vis à vis CAI4). The experiment was done twice in duplicate, and the averages with standard deviations are shown. F, ChIP analysis was done to compare RNA pol II occupancy at ERG11 and ERG1 promoters in BWP17 and the conditional null GPI19 mutant (BWP17-gpi19/PMET3-GFP-GPI9). Occupancy of ERG11 promoter by RNA pol II was found to be lower in case of conditional null GPI19 mutant (***, p value = 0.00048). Conversely, ERG1 promoter occupancy by RNA pol II was found to be higher in conditional null GPI19 mutant (****, p value = 0.0000021). The experiment was done twice in duplicate. G, total ergosterol from CAI4 and GPI2 heterozygote was analyzed using GC-MS. The different sterol intermediates relative to squalene in CAI4 samples are shown. Squalene was observed to be insignificantly altered in GPI2 heterozygote as compared with CAI4 (p value = 0.11). However, GPI2 heterozygote showed lowered lanosterol levels (down to 0.24-fold) as compared with CAI4 (**, p value = 0.00276). Ergosta-5,24(28)-dien-3-ol was found to be unaltered in GPI2 heterozygote (p value = 0.10). However, the sterol intermediate ergosta-5,7,9(11),22-tetraen-3-ol, immediately upstream of ergosterol in the pathway, was detected only in the GPI2 heterozygote (p value for this could not be calculated due to its absence in CAI4). Ergosterol levels in GPI2 heterozygote were higher by 1.14-fold as compared with CAI4 (**, p value = 0.0069). The experiment was done twice and the average plotted with standard deviations.
TABLE 4.
MIC80 values (μg/ml) of azoles for the GPI2 heterozygous mutant
The values presented are average with standard deviations of two independent experiments done in duplicate.
| Miconazole | Ketoconazole | Fluconazole | |
|---|---|---|---|
| CAI4 | 0.13 ± 0.045 | 0.007 ± 0.0006 | 2.25 ± 0.30 |
| CAI4-gpi2/GPI2 | 1.12 ± 0.16 | 0.380 ± 0.075 | 80.0 ± 12.5 |
Up-regulation of drug efflux pumps is one of the causes of azole resistance in C. albicans. To test this possibility in the GPI2 heterozygote, we studied the efflux of the fluorescent dye, R6G. Although R6G and azoles do not share a common pathway for influx (20), they are effluxed out via the same mechanism (21). R6G influx was first monitored to confirm that variation in R6G influx does not lead to any observed difference in the efflux pattern of R6G. No significant difference was seen vis à vis CAI4 in the R6G influx of GPI2 heterozygote over a 3-h period (Fig. 3B, panel i). The pattern of R6G efflux from the GPI2 heterozygous mutant was also not significantly different from that of CAI4 (Fig. 3B, panel ii). Thus, we ruled out up-regulation of drug efflux mechanisms as a possible explanation for the observed azole resistance.
Inhibited PKC signaling can lead to azole susceptibility in C. albicans (22). As a corollary, one would expect enhanced PKC signaling to cause azole resistance. However, as seen in Fig. 2D, no difference in PKC mediated signaling was observed in GPI2 heterozygote vis à vis CAI4.
Resistance to azoles could also suggest up-regulation of ERG11. A decrease in expression levels of ERG3 has also been reported as a mechanism for azole resistance in C. albicans (23). Hence, we examined the levels of both ERG11 as well as ERG3 in GPI2 heterozygote. The ERG11 transcripts were roughly 3-fold higher in GPI2 heterozygote as compared with CAI4 (Fig. 3C). However, no difference was observed in the levels of ERG3 transcripts (Fig. 3C). Thus, it appeared that the azole resistance was probably a result of up-regulation of ERG11.
To study whether the GPI2 heterozygote showed up-regulation at the earlier steps in the ergosterol biosynthesis pathway also, we monitored its response to terbinafine, a drug that specifically targets Erg1 in sterol biosynthesis. The GPI2 heterozygote mutant was sensitive to terbinafine (Fig. 3D), suggesting lowered Erg1 levels. This also corroborated well with the roughly 2-fold decrease in ERG1 transcripts in GPI2 heterozygote vis à vis CAI4 (Fig. 3C). The down-regulation of ERG1 suggests a feedback response to ERG11 up-regulation, a phenomenon that has also been reported previously (7, 24).
ChIP analysis confirmed higher RNA pol II occupancy of the ERG11 promoter in GPI2 heterozygote and lower occupancy of the ERG1 promoter (Fig. 3E). Exactly opposite results were seen for the conditional null GPI19 mutant (Fig. 3F), suggesting that the alteration in the ERG11 and ERG1 transcripts in GPI19 and GPI2 mutants was due to the difference in transcription from the respective promoters.
Levels of Ergosterol Are Higher in GPI2 Heterozygote
Does up-regulation of ERG11 also result in greater amounts of ergosterol production in the GPI2 heterozygote? To test this, we studied the response of the mutant to Amp B. It has been proposed that the more ergosterol content in the cell, the more target sites for Amp B in the membrane, and hence the cells will be more susceptible (25). The mutant was susceptible to Amp B as compared with CAI4 (Fig. 3D), suggesting that total ergosterol levels in our mutants were probably high. Steady state anisotropy (r) studies using the membrane probe DPH also suggested that the membrane of the GPI2 heterozygote was more rigid as compared with CAI4 (r = 0.198 ± 0.009 in GPI2 heterozygote as compared with 0.147 ± 0.017 in CAI4; ****, p value = 0.0000062), indicating higher ergosterol levels.
Sterol analysis of the GPI2 heterozygote was done by GC-MS. The mutant appeared to have 14% higher ergosterol (ergosta-5,7,22-trien-3-ol) levels as compared with CAI4 (Fig. 3G). The late sterol intermediate, ergosta-5,7,9(11),22-tetraen-2-diol, was also detected in the GPI2 heterozygote, which was normally not detectable in CAI4. Lanosterol was down by 76% in the mutant as compared with CAI4, correlating with enhanced ERG11 transcription. Despite a decrease in ERG1 transcription, a significant increase in squalene levels in the mutant was not observed. We infer that the downstream step involving Erg11 could be depleting its substrate rapidly and compelling the upstream steps toward more product formation, thereby keeping a more-or-less steady level of squalene in the cells.
Taken together, these experiments suggest higher ergosterol levels in the GPI2 heterozygote as compared with CAI4 via an up-regulation of ERG11 transcription. Interestingly, these alterations observed in the ergosterol biosynthesis pathway in GPI2 mutant are exactly opposite those observed in the GPI19 mutant (7).
GPI2 Disruption Leads to Defective Hyphal Morphogenesis Because of Altered Ras Signaling in C. albicans
GPI19 mutants of C. albicans showed hyperfilamentation (6). GPI2 mutants, however, were defective in filamentation as compared with CAI4 when grown at 37 °C on YEPD medium (Fig. 4A) or Spider medium in both solid (Fig. 4B) and liquid medium (Fig. 4C, panel I, and Table 5), suggesting a role for GPI2 in determining morphogenesis in C. albicans. Furthermore, treatment with sodium butyrate, a histone deacetylase inhibitor, improved filamentation in GPI2 heterozygotes, restoring it to wild type levels (Fig. 4C, panel ii). It has been previously shown that the Set3C complex is the sole histone deacetylase working downstream of cAMP-dependent PKA in the Ras signaling pathway and affecting filamentation; inhibiting Set3C results in hyperfilamentation (26). It would appear that the block introduced by GPI2 disruption in the heterozygous mutant can be overcome by inhibiting Set3C, indicating that the Ras signaling pathway is specifically affected in the GPI2 heterozygote.
FIGURE 4.

GPI2 mutation results in defective filamentation in C. albicans. A, CAI4, GPI2 heterozygote (CAI4-gpi2/GPI2) and conditional null GPI2 mutant (CAI4-gpi2/PMET3-GPI2) were compared for morphologies by spotting on a YEPD plate and incubating at 37 °C for 7 days. The mutants displayed defective filamentous morphology as compared with CAI4. The experiment was repeated twice for confirmation, and a representative image is shown. B, CAI4, GPI2 heterozygote, and conditional null GPI2 mutant were compared for filamentation pattern by spotting on a Spider plate and incubating at 37 °C for 7 days. The mutants showed defective filamentation as compared with CAI4. The experiment was repeated thrice for confirmation. C, CAI4, GPI2 heterozygote, and conditional null GPI2 mutant were grown in Spider medium at 37 °C, and hypha formation was monitored. Panel i, mutants were found to be defective in filamentation as compared with the wild type strain after 90 and 120 min. However, as shown in panel ii, when hypha formation was monitored in the presence of sodium butyrate (3 mm) after 90 min, the mutant displayed filamentation similar to the wild type strain, suggesting the impaired filamentation in the mutant to be an inhibitory effect of Set3/Hos2 histone deacetylase complex on Ras1/cAMP-PKA signaling (26). The experiment was done twice in duplicate, and a representative image is shown. D, CAI4 and GPI2 heterozygote were also compared for cAMP-dependent PKA activity (7). Lane 1 represents negative experimental control without cell lysate, and lane 2 represents positive experimental control with purified cAMP-dependent PKA, supplied by the kit manufacturer. GPI2 heterozygote was found to have lower PKA activity (lanes 5 and 6) as compared with CAI4 (lanes 3 and 4), correlating with lower Ras activity. E, GPI2 heterozygote was found to be resistant to heat shock when compared with CAI4. The plates shown here are after 48 h of incubation at 30 °C. The experiment was done twice for confirmation, and a representative image is shown. The conditional null GPI2 mutant could not be compared because of its inherent growth defect and reduced viability.
TABLE 5.
Percentage of different morphological forms observed after growing the strains in Spider medium at 37 °C for 120 min
GPI2 heterozygote (gpi2/GPI2) and conditional null GPI2 (gpi2/PMET3-GPI2) mutants in the different strain backgrounds as indicated showed lesser hyphae as compared with the wild type strains. The deletion of single alleles of GPI19 and GPI15 in the GPI2 heterozygous strain background does not affect hypha formation in the double heterozygote mutants GPI2/GPI19 (BWP17-gpi2/GPI2::gpi19/GPI19) and GPI2/GPI15 (BWP17-gpi2/GPI2::gpi15/GPI15). As published previously (6), the conditional null GPI19 mutant shows hyperfilamentation as compared with BWP17. However, disruption of a single GPI2 allele leads to reduced hyphae formation in the conditional null GPI19 mutant (BWP17-gpi19/PMET3-GFP-GPI19::gpi2/GPI2). Thus, reduced GPI2 levels seem to reduce filamentation in C. albicans. The experiment was done twice in duplicate.
| Yeast | Pseudo-hyphae | Hyphae | |
|---|---|---|---|
| % | % | % | |
| CAI4 | 12 ± 3 | 14 ± 4 | 74 ± 9 |
| CAI4-gpi2/GPI2 | 73 ± 6 | 18 ± 5 | 9 ± 4 |
| CAI4-gpi2/PMET3-GPI2 | 81 ± 9 | 17 ± 4 | 2 ± 2 |
| BWP17 | 10 ± 1 | 23 ± 4 | 67 ± 12 |
| BWP17-gpi2/GPI2 | 67 ± 9 | 19 ± 3 | 14 ± 2 |
| BWP17-gpi2/PMET3-GPI2 | 74 ± 8 | 16 ± 5 | 10 ± 3 |
| BWP17-gpi2/GPI2::gpi19/GPI19 | 69 ± 8 | 14 ± 2 | 17 ± 6 |
| BWP17-gpi2/GPI2::gpi15/GPI15 | 72 ± 7 | 16 ± 4 | 12 ± 4 |
| BWP17-gpi19/PMET3-GFP-GPI19 | 3 ± 1 | 3 ± 2 | 94 ± 8 |
| BWP17-gpi19/PMET3-GFP-GPI19::gpi2/GPI2 | 58 ± 12 | 24 ± 7 | 18 ± 4 |
Filamentation in C. albicans is predominantly dictated either by the Ras-mediated PKA pathway or by the cell wall integrity signaling pathway mediated by the PKC pathway (26). Because we did not see any significant difference in the PKC-dependent pathway between the mutant and wild type strains (Fig. 2D), we compared cAMP-dependent PKA activity between the GPI2 heterozygous mutant and CAI4, as an assessment of Ras signaling. We detected decreased PKA activity in the mutant as compared with CAI4 (Fig. 4D), further suggesting that the defective filamentation was due to reduced Ras activity in the mutant.
Strains possessing hyperactive Ras phenotypes tend to be heat shock-sensitive (27). Strains with lower Ras activity would be, by extension, more capable of withstanding heat shock. The GPI2 heterozygote was more resistant to heat shock as compared with CAI4 (Fig. 4E), thus confirming that it had lower Ras activity relative to CAI4.
Reintroduction of GPI2 into the GPI2 Heterozygous Strain Can Reverse the Phenotypes
The GPI2-revertant strain (CAI4-gpi2/GPI2/PACT1-GPI2) was generated to see if reintroduction of GPI2 in the GPI2 heterozygote could revert back to the phenotypes.
The GPI2 transcript analysis in the GPI2-revertant strain showed a 3.0 (±0.19)-fold increase in the GPI2 levels as compared with the GPI2 heterozygote, and a 1.7 (±0.19)-fold increase as compared with CAI4 (Fig. 5A), besides a restoration of GPI-GnT activity (Fig. 5B).
FIGURE 5.

Phenotype rescue in GPI2 revertant by reintroduction of GPI2 in GPI2 heterozygous strain background. A, GPI2 transcript levels in GPI2 revertant strain (CAI4-gpi2/GPI2/PACT1-GPI2) were found to be up-regulated (1.7-fold ±0.19; **, p value = 0.0019) as compared with CAI4 by RT-PCR analysis. The experiment was done twice in duplicate. B, GPI-GnT activity was quantified in microsomes generated from CAI4, GPI2 heterozygote (CAI4-gpi2/GPI2), and GPI2 revertant strain, as described under “Experimental Procedures.” The revertant strain exhibited GPI-GnT activity comparable with CAI4. The experiment was repeated twice for confirmation. The results shown are an average of data from a single experiment done in duplicate, along with the standard deviations. C, revertant strain displayed reversal of phenotypes in comparison with GPI2 heterozygote. The revertant strain was sensitive to azoles as compared with CAI4 and GPI2 heterozygote and resistant to the presence of SDS in the growth medium. The plates shown here are after 24 h of incubation at 30 °C. The experiment was done twice for confirmation. False color has been assigned to the image (using GIMP software) to brighten the cell spots. D, ERG11 transcript levels were found to be down to 0.47-fold (± 0.09) in the revertant strain (****, p value = 0.000022) as compared with CAI4 by RT-PCR analysis. The experiment was done twice in duplicate. E, revertant strain showed higher filamentation on a Spider plate after incubation at 37 °C for 7 days as compared with CAI4 and GPI2 heterozygote strains transformed with the empty vector (pACT1-GFP). Thus, reintroduction of GPI2 in GPI2 heterozygote led to restoration of filamentation in the mutant. GPI2-overexpressing strain (CAI4-PACT1-GPI2) was also seen to display hyperfilamentation suggesting higher GPI2 levels promote filamentation in C. albicans. The experiment was done twice for confirmation, and a representative image is shown. F, GPI2-overexpressing strain (CAI4-PACT1-GPI2) was assessed for growth in the presence of azoles in the growth medium as compared with the control CAI4 strain carrying the URA3 gene (CAI4-PACT1-GFP) and was found to be extremely sensitive to azole. The experiment was done twice, and a representative image is shown. False color has been assigned to the image (using GIMP software) to brighten the cell spots.
In the presence of azoles and SDS, the revertant strain displayed just the opposite phenotype of the GPI2 heterozygote (Fig. 5C), suggesting that the phenotypes seen in GPI2 heterozygotes were primarily due to depletion of the Gpi2 levels. Furthermore, ERG11 levels were reduced in the revertant mutant as compared with the wild type and the heterozygote strains (Fig. 5D). Filamentation was also restored in the revertant strain (Fig. 5E), when compared with CAI4 and GPI2 heterozygote transformed with the empty vector.
However, it must be pointed out that the revertant strain was more sensitive to azoles than the wild type strain itself probably because of higher levels of GPI2 in these strains (Fig. 5A). To test this, we introduced an additional copy of GPI2 in CAI4. This GPI2-overexpressing strain (CAI4-PACT1-GPI2) showed a 2.40 ± 0.48-fold increase in GPI2 transcription as compared with the control strain, CAI4-URA3 (CAI4-PACT1-GFP). The GPI2-overexpressing strain was also found to be azole-sensitive (Fig. 5F) and had lowered ERG11 levels (0.46 ± 0.034; ****, p value = 0.000024). The control strain showed no significant difference in ERG11 transcription in comparison with CAI4 (1.08 ± 0.04-fold). The GPI2-overexpressing strain also displayed more filamentation as compared with the control strain (Fig. 5E). Thus, the higher transcript levels of GPI2 correlated with azole sensitivity and hyperfilamentation in C. albicans.
Phenotypes of the GPI2 Mutants Are Independent of the Strain Background
We also probed whether the effects of the GPI2 mutation were similar in a different strain of C. albicans. For this, GPI2 mutants were generated in C. albicans BWP17 strain (details under “Experimental Procedures”). The conditional null mutant of GPI2 in the BWP17 background showed normal growth in permissive medium and reduced growth with increasing Met/Cys concentrations in the medium (Fig. 6A), similar to the conditional null mutant of GPI2 in CAI4 background. The GPI2 heterozygote in the BWP17 background was also found to be azole-resistant, which was reversed on reintroduction of a GPI2 allele (Fig. 6B). Furthermore, transcript levels of GPI2, ERG11, ERG1, and ERG3 (Fig. 6C), DPH anisotropy (Fig. 6D), and filamentation (Fig. 6, E and F) were found to be altered in this mutant similar to the GPI2 heterozygote created in the CAI4 strain background.
FIGURE 6.

GPI2 mutation affects growth and ergosterol biosynthesis in BWP17 strain of C. albicans. A, BWP17, GPI2 heterozygote (BWP17-gpi2/GPI2), and conditional null GPI2 mutant (BWP17-gpi2/PMET3-GPI2) in BWP17 strain background were analyzed for growth patterns on SD-minimal plates. BWP17 and the heterozygous mutant showed similar growth. However, conditional null GPI2 mutant showed a growth defect dependent on Met/Cys concentration. The experiment was done twice in duplicate, and a representative image is shown. B, BWP17 and GPI2 heterozygote were analyzed for growth pattern in the presence of ketoconazole. The heterozygous mutant exhibits resistance to the presence of ketoconazole as compared with BWP17. The experiment was done twice, and a representative image is shown. C, expression levels of GPI2, ERG11, ERG1, and ERG3 genes were quantified in BWP17 and GPI2 heterozygote. The strains were grown in YEPD, and total RNA was extracted as under “Experimental Procedures.” GPI2 levels were decreased in the mutant to about 0.59-fold (±0.011) (****, p value = 0.0000011). ERG11 transcript levels were enhanced in the heterozygote by almost 2.1-fold (±0.29) as compared with BWP17 (***, p value = 0.00014). However, the ERG1 transcript levels were decreased to 0.64-fold (±0.017) in the mutant as compared with BWP17 (****, p value = 0.0000044). No significant difference was observed for the transcription of ERG3 (p value = 0.14). The experiment was done twice in duplicate. D, steady state anisotropy was determined in BWP17 and GPI2 heterozygote strain using DPH. The higher anisotropy in the mutant (0.171 ± 0.019) as compared with BWP17 (0.121 ± 0.021) suggests a rigid membrane in the mutant (**, p value = 0.0041), similar to GPI2 heterozygote in CAI4 background. The experiment was done twice in duplicate. E, BWP17, GPI2 heterozygote, and conditional null GPI2 mutants were grown on YEPD plate and incubated at 37 °C for 7 days. The mutants displayed defective filamentous morphology as compared with the wild type strain. The experiment was done twice for confirmation and a representative image is shown. F, BWP17, GPI2 heterozygote and conditional null GPI2 mutant were also compared for filamentation pattern on Spider medium plate at 37 °C for 7 days. The mutants showed defective filamentation as compared with the wild type strain. The experiment was done twice, and a representative image is shown.
Thus, we infer that GPI2 is essential for growth and hyphal morphogenesis, and its mutation leads to an up-regulation of ergosterol biosynthesis via up-regulation of ERG11, in C. albicans, irrespective of the strain.
GPI2 Mutant Exhibits ERG11 Up-regulation Because of Up-regulation of GPI19
Because GPI2 mutants exhibited opposite morphogenetic phenotypes and opposite alterations in the ergosterol biosynthetic pathway, as compared with GPI19 mutant (7), we analyzed the GPI19 levels in the GPI2 mutants and vice versa to understand if there was a mutual regulation between the two genes. Mutants created in the BWP17 strain background were used in these studies.
GPI19 levels were found to be up-regulated in the GPI2 heterozygote by roughly 2-fold (Fig. 7A). Similarly, GPI2 levels were found to be up-regulated in the conditional null GPI19 mutant by roughly 2-fold and in the ERG11 heterozygote by nearly 1.5-fold (Fig. 7B). Because GPI2 and GPI19 levels were found to be up-regulated in GPI19 and GPI2 mutants, respectively, and also the mutants showed opposite phenotypes, we generated the double mutants for GPI19 and GPI2 in the BWP17 strain background (details under “Experimental Procedures”).
FIGURE 7.

GPI19 levels regulate ERG11 levels in C. albicans. A, transcript levels of GPI19 (2.34 ± 0.36; ***, p value = 0.00033) and GPI15 (1.58 ± 0.27; **, p value = 0.0016) were found to be up-regulated in GPI2 heterozygote (BWP17-gpi2/GPI2) as compared with BWP17. The experiment was done twice in duplicate. B, GPI2 transcript levels were up in the conditional null GPI19 mutant (BWP17-gpi19/PMET3-GFP-GPI19) (1.88 ± 0.27-fold; ***, p value = 0.00061) and ERG11 heterozygote (BWP17-erg11/ERG11) (1.46 ± 0.22; **, p value = 0.0059) as compared with BWP17. C, response of GPI2/GPI19 and GPI2/GPI15 double heterozygous mutants to azoles was monitored. GPI2/GPI15 double heterozygote (BWP17-gpi2/GPI2::gpi15/GPI15) displayed azole response similar to that of GPI2 heterozygote. However, GPI2/GPI19 double heterozygote (BWP17-gpi2/GPI2::gpi19/GPI19) was sensitive to azoles as compared with BWP17 and GPI2 heterozygote strains. The plates shown here are after 48 h of incubation at 30 °C. The experiment was done twice. False color has been assigned to the image (using GIMP software), to brighten the cell spots. D, GPI2/GPI15 double heterozygote continued to show an up-regulation of ERG11 transcript levels (1.96 ± 0.16; ****, p value = 0.000024) as compared with BWP17 and similar to GPI2 heterozygote. GPI2/GPI19 double heterozygote, however, had comparable ERG11 levels (0.83 ± 0.14; p value >0.05) as compared with BWP17. RT-PCR analysis also confirmed the allelic disruptions of GPI19 and GPI15. GPI2/GPI19 double heterozygote showed lower GPI19 levels (0.68-fold ± 0.18; **, p value = 0.0045), whereas GPI2/GPI15 double heterozygote showed lower GPI15 levels (0.56 ± 0.12; ****, p value = 0.000054). The experiments were done twice in duplicate. E, conditional null GPI19 mutant with a single allele disruption of GPI2 (BWP17-gpi19/PMET3-GFP-GPI19::gpi2/GPI2) showed lower ERG11 transcripts (0.63-fold ± 0.038; ****, p value = 0.0000037) than BWP17 and similar to conditional null GPI19 mutant (0.74 ± 0.027; ****, p value = 0.0000075 vis à vis BWP17). The experiment was done twice in duplicate. F, conditional null GPI19 mutant with a single allele disruption of GPI2 was more sensitive to ketoconazole than conditional null GPI19 mutant. The plates shown here are after 48 h of incubation at 30 °C. The experiment was done twice. False color has been assigned to the image (using GIMP software) to brighten the cell spots.
The GPI2/GPI19 double heterozygous mutant (BWP17-gpi2/GPI2::gpi19/GPI19) displayed a decrease in azole resistance as compared with the GPI2 heterozygous strain (Fig. 7C), suggesting that down-regulation of GPI19 (Fig. 7D) can decrease the observed azole resistance of the GPI2 heterozygote. The decrease in azole resistance correlated with the down-regulation of ERG11 in the GPI2/GPI19 double heterozygote as compared with GPI2 heterozygote (Fig. 7D). The transcript levels of ERG11 in this strain were comparable with that in BWP17.
The conditional null GPI19 mutant with a single allele disruption of GPI2 (BWP17-gpi19/PMET3-GFP-GPI19::gpi2/GPI2) was similarly generated and tested. GPI2 transcript levels were reduced by roughly 30% (Fig. 7E) in this mutant vis à vis BWP17. This strain was more sensitive to the azoles than the conditional null GPI19 mutant as well as the wild type (Fig. 7F). ERG11 transcript levels also remained low in this mutant as compared with BWP17 as well as the GPI2 heterozygote (Fig. 7E; p value for the data of this mutant in comparison with the GPI2 heterozygote is 0.009).
Previous studies from our group showed a mutual co-regulation of the expression of ERG11 and GPI19 (7). The above results suggest that disruption of a single allele of GPI19 in a GPI2-deficient mutant can lead to down-regulation of ERG11 levels in these mutants. Down-regulation of GPI2 in a conditional null GPI19 mutant is unable to push up the levels of ERG11. In other words, GPI19, rather than GPI2, appears to dictate the levels of ERG11. A corollary to this would be that the up-regulation of ERG11 levels in the GPI2 heterozygote mutants is possibly a consequence of up-regulation of GPI19 levels in these mutants.
As controls, we also looked at levels of two other GPI-GnT subunits, GPI15 and GPI3, in the GPI2 heterozygote. GPI15 transcript levels were found to be up-regulated in the GPI2 heterozygote (∼1.6-fold) (Fig. 7A). We also disrupted a single allele of GPI15 in the GPI2 heterozygote to generate the GPI2/GPI15 double heterozygous mutant (BWP17-gpi2/GPI2::gpi15/GPI15). This double heterozygous mutant displayed lowered GPI15 levels as expected (Fig. 7D). We have observed that GPI15 mutants display azole sensitivity in C. albicans.6 However, GPI15 disruption did not lead to any effect on the observed azole resistance in the GPI2/GPI15 double heterozygous mutant (Fig. 7C) or ERG11 levels (Fig. 7D). We also disrupted a single allele of the GPI-GnT catalytic subunit, GPI3, in the GPI2 heterozygote. GPI3 transcript levels were found to be unaltered in the GPI2 heterozygote (1.08 (±0.08)-fold in GPI2/GPI3 double heterozygote as compared with wild type strain; p value = 0.11). The GPI3 disruption did not lead to any effect on the observed azole resistance in GPI2 heterozygote (figure not shown), suggesting the up-regulation of ERG11 in the GPI2 heterozygote to be specifically associated with the up-regulation of GPI19.
GPI2 Is Needed for Filamentation
How does the mutual regulation between GPI2 and GPI19 affect hyphal morphogenesis? As mentioned above, GPI19 mutants displayed hyperfilamentation (6). Conditional null GPI19 mutant also had higher GPI2 transcript levels (Fig. 7B). We had also observed increased filamentation in the GPI2-overexpressing strain (Fig. 5E). Hence, we hypothesized that the up-regulation of GPI2 in the GPI19 mutants could be responsible for the hyperfilamentation. To test this hypothesis, the conditional null GPI19 mutant with single allele disruption of GPI2 was assessed for filamentation. It was seen to be more filamentous than the corresponding GPI2 heterozygote in YEPD medium at 37 °C but less filamentous as compared with conditional null GPI19 mutant in the same strain background (Fig. 8A). In liquid Spider medium also, the conditional null GPI19 mutant with a single allele disruption of GPI2 showed lesser filamentation as compared with the conditional null GPI19 mutant at 37 °C (Fig. 8B; Table 5). The GPI2/GPI19 and GPI2/GPI15 double heterozygotes did not show any difference in filamentation as compared with GPI2 heterozygote the (Fig. 8B; Table 5). Thus, down-regulating GPI2 in a GPI19 mutant can reduce filamentation, but lowering GPI19 levels in a GPI2 mutant cannot result in increased filamentation.
FIGURE 8.

GPI2 is needed for filamentation in C. albicans. A, BWP17, GPI2 heterozygote (BWP17-gpi2/GPI2), conditional null GPI19 mutant (BWP17-gpi19/PMET3-GFP-GPI19), and conditional null GPI19 mutant with a single allele disruption of GPI2 (BWP17-gpi19/PMET3-GFP-GPI19::gpi2/GPI2) were grown on a YEPD plate at 37 °C for 3 days. Conditional null GPI19 mutants with single allele disruption of GPI2 displayed more filamentation as compared with BWP17 and GPI2 heterozygote but defective filamentous morphology as compared with conditional null GPI19 mutant. The experiment was done twice for confirmation, and a representative image is shown. B, strains shown were analyzed for filamentation in liquid Spider medium at 37 °C for 90 and 120 min. GPI2 heterozygote displayed lesser filamentation as compared with BWP17. However, filamentation was restored in GPI2 heterozygote with overexpression of RAS1 (BWP17-gpi2/GPI2/PACT1-RAS1). The filamentation defect was more severe in conditional null GPI2 mutant (BWP17-gpi2/PMET3-GPI2). The double heterozygote mutants GPI2/GPI19 (BWP17-gpi2/GPI2::gpi19/GPI19) and GPI2/GPI15 (BWP17-gpi2/GPI2::gpi15/GPI15) showed filamentation similar to GPI2 heterozygote. Conditional null GPI19 mutant with single allele disruption of GPI2 showed reduced ability to filament as compared with conditional null GPI19 mutant. Also, filamentation was reduced in conditional null GPI19 mutant after deletion of a single allele of RAS1 (BWP17-gpi19/PMET3-GFP-GPI19::ras1/RAS1). Thus, the absence of Gpi2 affects filamentation in C. albicans. The experiment was done twice in duplicate. The statistics for this experiment are shown in Table 5. C, GPI2 was overexpressed in ras1 null and ras2 null mutants, and effect of GPI2 overexpression on filamentation pattern was monitored in these strains on Spider and YEPD medium at 37 °C. As a control, filamentation of these mutants transformed with empty vector pACT1-GFP was also assessed. Overexpression of GPI2 in ras1 null (BWP17-ras1/ras1/PACT1-GPI2) resulted in filamentation similar to control ras1 null carrying one copy of the URA3 gene (BWP17-ras1/ras1/PACT1-GFP). However, overexpression of GPI2 in ras2 null (BWP17-ras2/ras2/PACT1-GPI2) displayed higher filamentation as compared with ras2 null stain possessing one copy of the URA3 gene (BWP17-ras2/ras2/PACT1-GFP). Thus, overexpression of GPI2 in C. albicans can lead to hyperfilamentation but only in the presence of RAS1. The experiment was repeated twice for confirmation, and a representative image captured after 11 days is shown.
C. albicans has two Ras proteins that appear to work antagonistically to each other (28). Ras1, rather than Ras2, has been shown to be the major protein involved in filamentation (28). Overexpression of RAS1 in the GPI2 heterozygous mutant resulted in restoration of filamentous growth (Fig. 8B). Likewise, down-regulation of RAS1 in the conditional null GPI19 mutant could also reverse the hyperfilamentous phenotype of the strain (Fig. 8B), suggesting involvement of Ras1 in producing this phenotype. To further confirm whether the effect of GPI2 is via Ras1 or Ras2 proteins, we studied the effect of overexpressing GPI2 in the null mutants, ras1 (which has no Ras1 and expresses only Ras2) as well as ras2 (which expresses no Ras2, and has only Ras1) (Fig. 8C). Overexpression of GPI2 in the ras2 null mutant resulted in hyperfilamentation vis à vis the control strain carrying the empty vector, whereas its expression in ras1 null mutant did not result in any hyphal induction (Fig. 8C). This clearly indicates that the hyphal induction due to overexpression of GPI2 is dependent on Ras1. Thus, it appears that the GPI-GnT subunits cross-talk with Ras1 rather than Ras2 for triggering the morphogenetic switch and GPI2 is able to dictate the filamentation status of C. albicans.
DISCUSSION
Not much is known about the first step of GPI biosynthesis and its possible modes of interactions and regulations. Nevertheless, some studies have suggested a possible regulation of this step with other biochemical pathways. For example, in mammals the GPI-GnT complex has been shown to have regulatory contacts with Dpm2, a subunit that also regulates dolichol-phosphate mannose synthase (29). Likewise, in Saccharomyces cerevisiae, subunits of the GPI-GnT complex were shown to physically interact with and regulate Ras signaling (30, 31), as well as control intracellular sterol distribution via genetic interactions with ARV1 (32). A large scale split ubiquitin-based protein-protein interaction study also indicated that ScGpi2 in S. cerevisiae interacted with ScGpi19 as well as with ScErg11 and a large number of other proteins involved in many different biochemical pathways of the cell (33). Given that the GPI-GnT is involved in the initiating and committing step for a pathway that is essential for cell viability in lower eukaryotes and for embryogenesis and fetal development in higher eukaryotes (34, 35), the fact that this should be a step at which multiple regulations could happen should come as no surprise.
Our previous and the current studies have shown that the first step of GPI biosynthesis in C. albicans is indeed co-regulated with ergosterol biosynthesis via Erg11. As we demonstrate here, however, the cross-talk of different GPI-GnT complex subunits with the ergosterol pathway is not quite the same. Although our previous studies demonstrated that GPI19 down-regulation also down-regulates ERG11 levels and causes azole sensitivity (6, 7), this study shows that down-regulation of another subunit, GPI2, results in ERG11 up-regulation.
How does this regulation of sterol biosynthesis via the GPI-GnT subunits operate? One possible model would be that each of the subunits independently interacts with and regulates ERG11. Alternatively, the two subunits of the GPI-GnT complex mutually regulate one another and in doing so regulate the levels of ERG11. Indeed, our results suggest a mutual negative regulation between GPI19 and GPI2 (Fig. 9). Most interestingly, the two subunits do not appear to independently talk with the sterol biosynthetic pathway. Down-regulation of GPI19 in the GPI2 mutant results in ERG11 down-regulation, a feature typically seen in GPI19 mutants also. However, down-regulation of GPI2 in GPI19 mutant does not lead to ERG11 up-regulation as typically seen in the GPI2 mutants. In other words, GPI19, rather than GPI2, dictates the regulation of ERG11. The effects of GPI2 on ERG11 are merely a reflection of the cross-talk between the two subunits of the GPI-GnT complex. This is also corroborated by the fact that the ERG11 heterozygous mutant has an up-regulation of GPI2 but reduced GPI anchor biosynthesis (7), which is in keeping with the reduced GPI19 levels in this strain. When it comes to morphogenesis and cross-talk with the Ras signaling pathway, with very similar reasoning, our results suggest that GPI2 rather than GPI19 is the more important player. It would also follow that ergosterol levels are not critical to filamentation in C. albicans.
FIGURE 9.
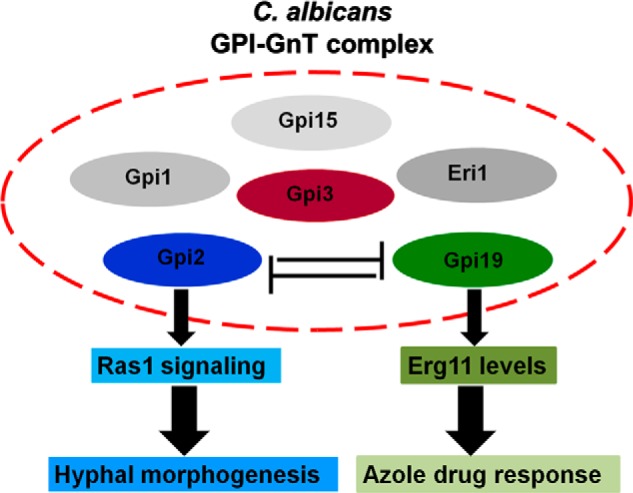
Proposed model. GPI-GnT complex includes different subunits. However, the interactions between the subunits are not known. Our model proposes that GPI2 and GPI19 subunits of GPI-GnT complex are negatively co-regulated. Thus, whereas GPI19 seems to directly affect ERG11 levels, and thereby alter azole response of C. albicans, GPI2 primarily affects Ras signaling and thus affects hyphal morphogenesis in C. albicans. The effect of GPI2 disruption on ERG11 appears to be via its interaction with GPI19, whereas the effect of GPI19 on Ras1 activity is dependent on its interaction with GPI2.
What could be the significance of these observations? C. albicans is the leading agent of fungal infections and mortality in immunocompromised patients (36). Several of its virulence factors are GPI-anchored, and impaired GPI biosynthesis leads to significant reduction in its infection and virulence (1, 5, 18, 37). What our previous and current studies show is that impairing this pathway can also affect ergosterol biosynthesis. Ergosterol is an important component of the fungal cell membrane and has been a useful drug target to combat fungal infections, not only because of its importance in fungal cells but also because of its specific fungal origin. Although most of the ergosterol in the plasma membrane can be replaced by other sterol intermediates, there are some functions within the cell, such as cell division, that necessarily require some amount of ergosterol to be present (23). Furthermore, membrane integrity and composition are not only essential for the viability of the fungal cells but also influence the localization and functioning of several membrane-localized proteins (38, 39). Unfortunately, with regular use, drug resistance against most antifungals has been observed in C. albicans (40, 41). Our work suggests that the first step of the GPI biosynthetic pathway in C. albicans could be a new target, which would allow for simultaneous down-regulation of GPI-dependent virulence factors as well as ergosterol levels and thereby provide an additional level of regulation and control of fungal infection. In addition, the first step of GPI anchor biosynthesis appears to concurrently regulate morphogenesis. Hyphal morphogenesis is closely linked to the ability of the organism to invade and infect the host (42, 43), although there are studies to indicate that once the organism gains entry into the bloodstream, hyphal formation may not be necessary for virulence (44). Several GPI-anchored proteins are expressed in a hypha-specific manner (45). That GPI anchor biosynthesis could dictate hyphal morphogenesis is therefore extremely exciting. It also suggests that targeting the first step of GPI anchor biosynthesis could allow for a parallel control of infection and virulence by controlling the morphogenetic switch of the organism as well.
Acknowledgments
We thank Lalremrauta Haunhar for RT-PCR of the CAI4 strain carrying the URA3 marker. We thank Prof. A. Mitchell for the C. albicans BWP17 strain, Dr. W. A. Fonzi for pMB7 vector, Dr. Cheryl Gale for URA3-MET3-GFP vector, and Prof. Alistair JP Brown for pACT1-GFP vector. The GC-MS data were recorded at the Advanced Instrument Research Facility, Jawaharlal Nehru University, with the help of Dr. Ajai Kumar. Microscopy, fluorescence spectroscopy, RT-PCR, and LiCOR Odyssey IR-Imaging facilities were used at the Central Instrumentation Facility, School of Life Sciences, Jawaharlal Nehru University.
This work was supported in part by the Department of Biotechnology (to S. S. K.), and DBT-BUILDER program, University Grants Commission-Resource Networking (UGC-RNW) funds from the School of Life Sciences, Jawaharlal Nehru University, and Department of Science and Technology-Promotion of University Research and Scientific Excellence (DST-PURSE) funds from Jawaharlal Nehru University.
L. Haunhar, K. Pawar, P. Kumar, B. Yadav, P. Jain, V. A. Pratyusha, S. Bhatnagar, R. Muthuswami, and S. S. Komath, unpublished data.
- GPI
- glycosylphosphatidylinositol
- GnT
- glucosaminyltransferase
- MIC
- minimum inhibitory concentration
- Amp B
- amphotericin B
- R6G
- rhodamine 6G
- CFW
- calcofluor white
- DPH
- 1,6-diphenyl-1,3,5-hexatriene.
REFERENCES
- 1. Richard M. L., Plaine A. (2007) Comprehensive analysis of glycosylphosphatidylinositol anchored proteins in Candida albicans. Eukaryot. Cell 6, 119–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Plaine A., Walker L., Da Costa G., Mora-Montes H. M., McKinnon A., Gow N. A., Gaillardin C., Munro C. A., Richard M. L. (2008) Functional analysis of Candida albicans GPI-anchored proteins: roles in cell wall integrity and caspofungin sensitivity. Fungal. Genet. Biol. 45, 1404–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chaffin W. L. (2008) Candida albicans cell wall proteins. Microbiol. Mol. Biol. Rev. 72, 495–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Richard M., Ibata-Ombetta S., Dromer F., Bordon-Pallier F., Jouault T., Gaillardin C. (2002) Complete glycosylphosphatidylinositol anchors are required in Candida albicans for full morphogenesis, virulence, and resistance to macrophages. Mol. Microbiol. 44, 841–853 [DOI] [PubMed] [Google Scholar]
- 5. Grimme S. J., Colussi P. A., Taron C. H., Orlean P. (2004) Deficiencies in the essential Smp3 mannosyltransferase block glycosylphosphatidylinositol assembly and lead to defects in growth and cell wall biogenesis in Candida albicans. Microbiology 150, 3115–3128 [DOI] [PubMed] [Google Scholar]
- 6. Victoria G. S., Kumar P., Komath S. S. (2010) The Candida albicans homologue of PIG-P, CaGpi19p: gene dosage and role in growth and filamentation. Microbiology 156, 3041–3051 [DOI] [PubMed] [Google Scholar]
- 7. Victoria G. S., Yadav B., Hauhnar L., Jain P., Bhatnagar S., Komath S. S. (2012) Mutual coregulation between GPI-N-acetylglucosaminyltransferase and ergosterol biosynthesis in Candida albicans. Biochem. J. 443, 619–625 [DOI] [PubMed] [Google Scholar]
- 8. Fonzi W. A., Irwin M. Y. (1993) Isogenic strain construction and gene mapping in Candida albicans. Genetics 134, 717–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilson R. B., Davis D., Mitchell A. P. (1999) Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J. Bacteriol. 181, 1868–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gerami-Nejad M., Hausauer D., McClellan M., Berman J., Gale C. (2004) Cassettes for the PCR-mediated construction of regulatable alleles in Candida albicans. Yeast 21, 429–436 [DOI] [PubMed] [Google Scholar]
- 11. Ashraf M., Yadav B., Perinthottathil S., Kumar K. S., Vats D., Muthuswami R., Komath S. S. (2011) N-Acetyl-d-glucosaminylphosphatidylinositol de-N-acetylase from Entamoeba histolytica: metal alters catalytic rates but not substrate affinity. J. Biol. Chem. 286, 2543–2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Singh P., Kaur J., Yadav B., Komath S. S. (2009) Design, synthesis and evaluations of acridone derivatives using Candida albicans–search for MDR modulators led to the identification of an anti-candidiasis agent. Bioorg. Med. Chem. 17, 3973–3979 [DOI] [PubMed] [Google Scholar]
- 13. Sanyal K., Baum M., Carbon J. (2004) Centromeric DNA sequences in the pathogenic yeast Candida albicans are all different and unique. Proc. Natl. Acad. Sci. U.S.A. 101, 11374–11379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nobile C. J., Nett J. E., Hernday A. D., Homann O. R., Deneault J.-S., Nantel A., Andes D. R., Johnson A. D., Mitchell A. P. (2009) Biofilm matrix regulation by Candida albicans Zap1. PLoS Biol. 7, e1000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shukla S., Yadav V., Mukhopadhyay G., Prasad R. (2011) Ncb2 is involved in activated transcription of CDR1 in azole-resistant clinical isolates of Candida albicans. Eukaryot. Cell 10, 1357–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fujita M., Yoko-o T., Okamoto M., Jigami Y. (2004) GPI7 involved in glycosylphosphatidylinositol biosynthesis is essential for yeast cell separation. J. Biol. Chem. 279, 51869–51879 [DOI] [PubMed] [Google Scholar]
- 17. Eisenhaber B., Maurer-Stroh S., Novatchkova M., Schneider G., Eisenhaber F. (2003) Enzymes and auxiliary factors for GPI lipid anchor biosynthesis and post-translational transfer to proteins. BioEssays 25, 367–385 [DOI] [PubMed] [Google Scholar]
- 18. Martinez-Lopez R., Monteoliva L., Diez-Orejas R., Nombela C., Gil C. (2004) The GPI-anchored protein CaEcm33p is required for cell wall integrity, morphogenesis and virulence in Candida albicans. Microbiology 150, 3341–3354 [DOI] [PubMed] [Google Scholar]
- 19. Hitchcock C. A., Dickinson K., Brown S. B., Evans E. G., Adams D. J. (1990) Interaction of azole antifungal antibiotics with cytochrome P-450-dependent 14 α-sterol demethylase purified from Candida albicans. Biochem. J. 266, 475–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mansfield B. E., Oltean H. N., Oliver B. G., Hoot S. J., Leyde S. E., Hedstrom L., White T. C. (2010) Azole drugs are imported by facilitated diffusion in Candida albicans and other pathogenic fungi. PLoS Pathog. 6, e1001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maesaki S., Marichal P., Vanden Bossche H., Sanglard D., Kohno S. (1999) Rhodamine 6G efflux for the detection of CDR1-overexpressing azole-resistant Candida albicans strains. J. Antimicrob. Chemother. 44, 27–31 [DOI] [PubMed] [Google Scholar]
- 22. LaFayette S. L., Collins C., Zaas A. K., Schell W. A., Betancourt-Quiroz M., Gunatilaka A. A., Perfect J. R., Cowen L. E. (2010) PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog. 6, e1001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kelly S. L., Lamb D. C., Corran A. J., Baldwin B. C., Kelly D. E. (1995) Mode of action and resistance to azole antifungals associated with the formation of 14 α-methylergosta-8,24(28)-dien-3β,6α-diol. Biochem. Biophys. Res. Commun. 207, 910–915 [DOI] [PubMed] [Google Scholar]
- 24. Sanglard D., Ischer F., Parkinson T., Falconer D., Bille J. (2003) Candida albicans mutations in the ergosterol biosynthetic pathway and resistance to several antifungal agents. Antimicrob. Agents Chemother. 47, 2404–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sanglard D., Odds F. C. (2002) Resistance of Candida species to antifungal agents: molecular mechanisms and clinical consequences. Lancet Infect. Dis. 2, 73–85 [DOI] [PubMed] [Google Scholar]
- 26. Hnisz D., Majer O., Frohner I. E., Komnenovic V., Kuchler K. (2010) The Set3/Hos2 histone deacetylase complex attenuates cAMP/PKA signaling to regulate morphogenesis and virulence of Candida albicans. PLoS Pathog. 6, e1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shapiro R. S., Uppuluri P., Zaas A. K., Collins C., Senn H., Perfect J. R., Heitman J., Cowen L. E. (2009) Hsp90 orchestrates temperature-dependent Candida albicans morphogenesis via Ras1-PKA signaling. Curr. Biol. 19, 621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu Y., Fang H.-M., Wang Y.-M., Zeng G.-S., Zheng X.-D., Wang Y. (2009) Ras1 and Ras2 play antagonistic roles in regulating cellular cAMP level, stationary-phase entry and stress response in Candida albicans. Mol. Microbiol. 74, 862–875 [DOI] [PubMed] [Google Scholar]
- 29. Maeda Y., Tomita S., Watanabe R., Ohishi K., Kinoshita T. (1998) DPM2 regulates biosynthesis of dolichol phosphate-mannose in mammalian cells: correct subcellular localization and stabilization of DPM1, and binding of dolichol phosphate. EMBO J. 17, 4920–4929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sobering A. K., Watanabe R., Romeo M. J., Yan B. C., Specht C. A., Orlean P., Riezman H., Levin D. E. (2004) Yeast Ras regulates the complex that catalyzes the first step in GPI-anchor biosynthesis at the ER. Cell 117, 637–648 [DOI] [PubMed] [Google Scholar]
- 31. Sobering A. K., Romeo M. J., Vay H. A., Levin D. E. (2003) A novel Ras inhibitor, Eri1, engages yeast Ras at the endoplasmic reticulum. Mol. Cell. Biol. 23, 4983–4990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kajiwara K., Watanabe R., Pichler H., Ihara K., Murakami S., Riezman H., Funato K. (2008) Yeast ARV1 is required for efficient delivery of an early GPI intermediate to the first mannosyltransferase during GPI assembly and controls lipid flow from the endoplasmic reticulum. Mol. Biol. Cell 19, 2069–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miller J. P., Lo R. S., Ben-Hur A., Desmarais C., Stagljar I., Noble W. S., Fields S. (2005) Large-scale identification of yeast integral membrane protein interactions. Proc. Natl. Acad. Sci. U.S.A. 102, 12123–12128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leidich S. D., Kostova Z., Latek R. R., Costello L. C., Drapp D. A., Gray W., Fassler J. S., Orlean P. (1995) Temperature-sensitive yeast GPI anchoring mutants gpi2 and gpi3 are defective in the synthesis of N-acetylglucosaminyl phosphatidylinositol. Cloning of the GPI2 gene. J. Biol. Chem. 270, 13029–13035 [DOI] [PubMed] [Google Scholar]
- 35. Kawagoe K., Kitamura D., Okabe M., Taniuchi I., Ikawa M., Watanabe T., Kinoshita T., Takeda J. (1996) Glycosylphosphatidylinositol-anchor-deficient mice: implications for clonal dominance of mutant cells in paroxysmal nocturnal hemoglobinuria. Blood 87, 3600–3606 [PubMed] [Google Scholar]
- 36. Sardi J. C., Scorzoni L., Bernardi T., Fusco-Almeida A. M., Mendes Giannini M. J. (2013) Candida species: current epidemiology, pathogenicity, biofilm formation, natural antifungal products and new therapeutic options. J. Med. Microbiol. 62, 10–24 [DOI] [PubMed] [Google Scholar]
- 37. Hayek P., Dib L., Yazbeck P., Beyrouthy B., Khalaf R. A. (2010) Characterization of Hwp2, a Candida albicans putative GPI-anchored cell wall protein necessary for invasive growth. Microbiol. Res. 165, 250–258 [DOI] [PubMed] [Google Scholar]
- 38. Morioka S., Shigemori T., Hara K., Morisaka H., Kuroda K., Ueda M. (2013) Effect of sterol composition on the activity of the yeast G-protein-coupled receptor Ste2. Appl. Microbiol. Biotechnol. 97, 4013–4020 [DOI] [PubMed] [Google Scholar]
- 39. Pasrija R., Panwar S. L., Prasad R. (2008) Multidrug transporters CaCdr1p and CaMdr1p of Candida albicans display different lipid specificities: both ergosterol and sphingolipids are essential for targeting of CaCdr1p to membrane rafts. Antimicrob. Agents Chemother. 52, 694–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kathiravan M. K., Salake A. B., Chothe A. S., Dudhe P. B., Watode R. P., Mukta M. S., Gadhwe S. (2012) The biology and chemistry of antifungal agents: a review. Bioorg. Med. Chem. 20, 5678–5698 [DOI] [PubMed] [Google Scholar]
- 41. Ostrosky-Zeichner L., Casadevall A., Galgiani J. N., Odds F. C., Rex J. H. (2010) An insight into the antifungal pipeline: selected new molecules and beyond. Nat. Rev. Drug Discov. 9, 719–727 [DOI] [PubMed] [Google Scholar]
- 42. Lo H. J., Köhler J. R., DiDomenico B., Loebenberg D., Cacciapuoti A., Fink G. R. (1997) Nonfilamentous C. albicans mutants are avirulent. Cell 90, 939–949 [DOI] [PubMed] [Google Scholar]
- 43. Jacobsen I. D., Wilson D., Wächtler B., Brunke S., Naglik J. R., Hube B. (2012) Candida albicans dimorphism as a therapeutic target. Expert Rev. Anti Infect Ther. 10, 85–93 [DOI] [PubMed] [Google Scholar]
- 44. Noble S. M., French S., Kohn L. A., Chen V., Johnson A. D. (2010) Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat. Genet. 42, 590–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nantel A., Dignard D., Bachewich C., Harcus D., Marcil A., Bouin A.-P., Sensen C. W., Hogues H., van het Hoog M., Gordon P., Rigby T., Benoit F., Tessier D. C., Thomas D. Y., Whiteway M. (2002) Transcription profiling of Candida albicans cells undergoing the yeast-to hyphal transition. Mol. Biol. Cell 13, 3452–3465 [DOI] [PMC free article] [PubMed] [Google Scholar]