

Background: Induced pluripotent stem cells (iPSCs) constitute an attractive source of cells for regenerative medicine.
Results: MicroRNA-21 mediates endothelial differentiation derived from iPSCs in presence of VEGF.
Conclusion: MicroRNA-21 and TGF-β2 signaling pathways regulate iPSC differentiation to endothelial lineage.
Significance: Elucidation of the molecular mechanisms underlying microRNA-21-regulated iPSC differentiation might provide the basic information for stem cell therapy of vascular diseases.
Keywords: Endothelial Cell, Induced Pluripotent Stem Cells, MicroRNA, Transforming Growth Factor Beta (TGFbeta), Vascular Endothelial Growth Factor (VEGF)
Abstract
Finding a suitable cell source for endothelial cells (ECs) for cardiovascular regeneration is a challenging issue for regenerative medicine. In this paper, we describe a novel mechanism regulating induced pluripotent stem cells (iPSC) differentiation into ECs, with a particular focus on miRNAs and their targets. We first established a protocol using collagen IV and VEGF to drive the functional differentiation of iPSCs into ECs and compared the miRNA signature of differentiated and undifferentiated cells. Among the miRNAs overrepresented in differentiated cells, we focused on microRNA-21 (miR-21) and studied its role in iPSC differentiation. Overexpression of miR-21 in predifferentiated iPSCs induced EC marker up-regulation and in vitro and in vivo capillary formation; accordingly, inhibition of miR-21 produced the opposite effects. Importantly, miR-21 overexpression increased TGF-β2 mRNA and secreted protein level, consistent with the strong up-regulation of TGF-β2 during iPSC differentiation. Indeed, treatment of iPSCs with TGFβ-2 induced EC marker expression and in vitro tube formation. Inhibition of SMAD3, a downstream effector of TGFβ-2, strongly decreased VE-cadherin expression. Furthermore, TGFβ-2 neutralization and knockdown inhibited miR-21-induced EC marker expression. Finally, we confirmed the PTEN/Akt pathway as a direct target of miR-21, and we showed that PTEN knockdown is required for miR-21-mediated endothelial differentiation. In conclusion, we elucidated a novel signaling pathway that promotes the differentiation of iPSC into functional ECs suitable for regenerative medicine applications.
Introduction
Endothelial cells (ECs)3 line the blood vessels of the entire circulatory system and represent the barrier between circulating blood and the rest of the vessel wall. The endothelium is a dynamic and heterogeneous organ with secretory, metabolic, synthetic, and immunological functions (1). There are two main mechanisms that lead to blood vessel formation, namely vasculogenesis and angiogenesis. Vasculogenesis is the formation of new vessels from progenitor cells and was historically believed to occur in embryo development, during the formation of the primitive vascular network from angioblasts (2). Angiogenesis is the formation of new blood vessels from pre-existing ones and occurs both during embryonic development and during physiological and pathological conditions in adult life. Angiogenesis in the infarcted tissue occurs through a complex link between extracellular matrix, ECs, and pericytes, in response to an imbalance of angiogenic factors, compared with angiostatic factors in the local environment (3, 4).
Atherosclerosis is a chronic inflammatory disease, started by endothelial dysfunction, that leads to the formation of an atherosclerotic plaque (5, 6). Balloon angioplasty and stenting are routinely used in clinical practice to treat these conditions. However, one of the major issues of stenting is restenosis, leading to the recurrence of symptoms through EC loss and subsequent smooth muscle proliferation and matrix deposition, thus originating luminal narrowing (7). It has been reported that after vascular injury, restenosis development can be prevented through accelerated re-endothelialization by mature ECs, which inhibits smooth muscle migration, proliferation, and neointima formation (8). In the past several years, accumulating evidence suggests that ES cells, a promising source of pluripotent stem cells with unlimited growth and self-renewal abilities, are able to differentiate into ECs in vitro and in vivo (9). The ethical and immunological problems associated with the use of ES cells have been recently bypassed using induced pluripotent stem cells (iPSCs), because they can be derived from somatic cell population isolated from the patient. In this study, we indeed aimed at understanding the molecular mechanisms of iPSC differentiation into ECs, to define new methods to produce large number of ECs with high purity from iPSCs: iPSC-derived ECs can be used to treat damaged vessels, to avoid restenosis, and to develop tissue engineered vascular grafts, now major challenges for the future of regenerative medicine (10).
miRNAs are single-stranded, noncoding molecules of RNA, 20–25 nucleotides long, able to regulate a wide range of cellular processes by binding to noncoding regions of mRNA (11). Recent studies focused on angiogenesis-associated miRNAs that are involved in ES cells differentiation toward ECs (12). It has been recently reported that miR-21 overexpression in human prostate cancer cells increased hypoxia-inducible factor-1α (HIF-1α) and VEGF expression, thereby inducing tumor angiogenesis (13). On the other hand, miR-21 has been shown to have antiangiogenic functions, targeting RhoB in ECs (14) and TGF-βRII in human adipose-derived stem cells (hADSCs), thus decreasing the tumor vascularity induced by the cells (15). In addition, in endothelial progenitor cells, miR-21 has been found to induce cell senescence and reduce angiogenesis in vitro and in vivo (16). In addition to the role of miR-21 in angiogenesis, miR-21 has been also shown to induce adipogenic differentiation of hADSCs via targeting TGF-βRII (17). However, so far there are no studies showing a link between miR-21 and endothelial differentiation from iPSCs.
TGF-β is a multifunctional cytokine that regulates proliferation, migration, differentiation, and survival of many different cell types. Deletion or mutation of different members of the TGF-β family have been shown to cause vascular remodeling defect and absence of mural cell formation, leading to embryonic lethality (18) or severe vascular disorders (19, 20). It has been previously demonstrated that stimulation of hADSCs with TGF-β2 increased the levels of VEGF and interleukin-6, thus promoting the proangiogenic action of the cells in a hind limb ischemia model (15). However, no previous report showed an involvement of TGF-β in endothelial differentiation of iPSCs. In particular, we focused on the study of one of the three known isoforms of TGF-β, TGF-β2. In the present study, we described a novel protocol for iPSC differentiation into ECs, and we elucidated the role of miR-21 in mediating this process, through targeting-specific pathways. We highlighted for the first time, to our knowledge, a connection between VEGF, miR-21/Akt, and TGF-β2 in the endothelial differentiation of iPSCs.
EXPERIMENTAL PROCEDURES
Materials
Cell culture media, serum, and cell culture supplements were purchased from ATCC, Millipore, Invitrogen, and PAA. Antibodies against VE-cadherin, Flk-1, eNOS, and GAPDH were purchased from Abcam. Antibodies against vWF, Akt 1/2, and Ser(P)-473 Akt were purchased from Santa Cruz. Antibodies against CD31 were purchased from Abbiotec and Santa Cruz. Antibody against PTEN was purchased from New England Biolabs. The secondary antibodies for immunostaining anti-goat Alexa 488, anti-goat Alexa 594, and anti-rabbit Alexa 488, and anti-rabbit Alexa 594 were purchased from Invitrogen. The secondary antibodies for Western blotting were purchased from Dakocytomation.
Cell Culture, Differentiation, and Treatment of iPSCs
iPSCs were generated in our laboratory starting from mouse embryonic fibroblasts isolated as stated in Ref. 21, using a similar method stated in Ref. 22. iPSCs were cultured on gelatin-coated flasks (PBS containing 0.04% of gelatin from bovine skin; Sigma) in DMEM (ATCC) supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin (Invitrogen); 10 ng/ml recombinant human leukemia inhibitory factor (Millipore); and 0.1 mm 2-mercaptoethanol (Invitrogen) in a humidified incubator supplemented with 5% CO2. The cells were passaged every 2 days at a ratio of 1:6. Differentiation of iPSCs was obtained by seeding the cells on type IV mouse collagen (5 μg/ml)-coated dishes in differentiation medium (DM) that contains α-MEM supplemented with 10% FBS (Invitrogen), 0.05 mm 2-mercaptoethanol, 100 units/ml penicillin, and 100 μg/ml streptomycin. The medium was supplemented with 50 ng/ml VEGF or 3 ng/ml TGFβ-2 (Peprotech), and the cells were maintained under these conditions for up to 7 days. 5 μm SMAD3 inhibitor SIS3 (Calbiochem), and 0.1 μg/ml VEGF neutralizing antibody (R&D System) were added to iPSCs cultured for 7 days with TGFβ-2; 1 μg/ml TGFβ-2 neutralizing antibody (R&D System) was added to the cells 24 h after transfection with pre-miR-21 (Pre-21) for further 24 h. The number of iPSCs seeded on collagen IV for the differentiation was 1.3 × 104/cm2 (d3), 6.6 × 103/cm2 (d5), and 3.3 × 103/cm2 (d7).
Quantitative RT-PCR
Relative gene expression was determined by quantitative real time PCR, using 2 ng of cDNA (relative to RNA amount) for each sample with the SYBR Green Master Mix in a 20-μl reaction. Ct values were measured using ABI Prism 7000 sequence detector (Applied Biosystems). The 18 S ribosomal RNA served as the endogenous control to normalize the amounts of RNA in each sample. For each sample, PCR was performed in duplicate in a 96-well reaction plate (Eppendorf, twin.tec real time PCR plates). The gene was considered undetectable beyond 35 cycles. The primer sets used for this study are as follows: 18 S, forward, 5′-CCAGTAAGTGCGGGTCATAA-3′ and reverse, 5′-CCGAGGGCCTCACTAAACC-3′; VE-cadherin, forward, 5′-AAGAAACCGCTGATCGGCA-3′ and reverse, 5′-TCGGAAGAATTGGCCTCTGTC-3′; CD31, forward, 5′-CAAACAGAAACCCGTGGAGAT-3′ and reverse, 5′-ACCGTAATGGCTGTTGGCTTC-3′; Flk-1, forward, 5′-TGAAATTGAGCTATCTGCCGG-3′ and reverse, 5′-TTTGAAGGTGGAGAGTGCCAG-3′; vWF, forward, 5′-GGCTGTGCGGTGATTTTAACAT-3′ and reverse, 5′-CGTTTACACCGCTGTTCCTCA-3′; and TGF-β2, forward, 5′-CTTCGACGTGACAGACGCT-3′ and reverse, 5′-GCAGGGGCAGTGTAAACTTATT-3′.
miRNA Extraction, Reverse Transcription, Preamplification, and TaqMan Quantitative PCR Assay
Extraction of total RNA including miRNA was performed using the miRNeasy mini kit (Qiagen), according to the manufacturer's protocol. miRNA reverse transcription and amplification were performed using the Megaplex reverse transcription primers (rodent pools A and B, v3.0) and the Megaplex PreAmp primers (rodent primers A and B, v3.0) (Applied Biosystems) as previously described (23). TaqMan miRNA assays were used to assess the expression of individual miRNAs as described before (23).
miR-21 Transient Transfection
Manipulation of miR-21 levels in iPSCs differentiated in presence of VEGF for 3 days and cultured to 60–70% confluence was performed using Pre-21, or the nontargeting control (Pre-ctrl) (Ambion AB), whereas inhibition of miR-21 was performed using the locked nucleic acid (LNA) inhibitor of miR-21 (LNA-21) or a negative control (LNA-ctrl) (Exiqon). All transfections were performed using LipofectamineTM RNAiMAX (Invitrogen), in accordance with the manufacturer's protocol.
TaqMan miRNA Array
The expression profile of miRNAs in the samples was determined using the rodent TaqMan miRNA arrays A and B (Applied Biosystems), which is a set of two 384-well microfluidic cards (arrays A and B, v3.0). The arrays enable quantification of gene expression levels of 375 miRNAs per pool and are specific to human, mouse, or rat. Five endogenous controls and a negative control were included in each array for data normalization. PCRs were performed using 450 μl of the TaqMan Universal PCR Master Mix No AmpErase UNG (2×) and 9 μl of the diluted preamplification product to a final volume of 900 μl. 100 μl of the PCR mix were loaded to each port of the TaqMan miRNA array. The fluidic card was then centrifuged and mechanically sealed. Real time PCR was carried out on an Applied Biosystems 7900HT thermocycler using the manufacturer's recommended program.
Immunoblotting and Immunofluorescent Staining
For Western blot analysis, cell pellets were resuspended in radioimmune precipitation assay buffer and sonicated. The protein concentration was measured by the Bradford method. 20–50 μg of each sample was loaded on 6–8% polyacrylamide gels, and standard immunoblotting procedure was performed using the listed primary antibodies.
Immunostaining was performed as described previously (24). Adherent cells were fixed in 4% buffered paraformaldehyde and permeabilized (in the case of intracytoplasmatic antigens) with 0.1% Triton X prior and blocked in 1% BSA before incubation with primary antibody for VE-cadherin, CD31, vWF, and eNOS.
In Vitro and in Vivo Tube Formation Assay
iPSCs were differentiated in the presence of VEGF for 4 days and then transfected with Pre-21, Pre-ctrl, LNA-21, and LNA-ctrl. In vitro angiogenesis assays was performed after 48 h as described previously (25); in vitro angiogenesis assay was also performed after 7 days of differentiation in the presence of VEGF or TGF-β2 treatment. In vivo angiogenesis was performed 48 h after miR-21 transfection by mixing 5 × 105 cells with 200 μl of Matrigel and injecting it subcutaneously in mice (C57BL/6). The cryosections of the Matrigel plugs were stained with CD31 and VE-cadherin antibodies. Immunostaining was assessed by confocal imaging, and capillary density was calculated as the number of capillary number/mm2.
Mice
All procedures were performed according to protocols approved by the Institutional Committee for Use and Care of Laboratory Animals. All animals used in this study were inbred mice of C57BL/6 background.
Luciferase Reporter Assay
For the luciferase reporter assays, 3 × 104 iPSCs cells were seeded in each collagen-coated well of a12-well plate in DM containing VEGF. 72 h later, cells were transfected with the plasmid expressing luciferase under the control of the PTEN 3′-UTR (pGL3-PTEN-wt) and the miR-21 inhibitor and its control. As a control, the same experiment was performed with a plasmid expressing luciferase under the control of a mutated and inactive PTEN 3′-UTR (pGL3-PTEN-mut). Briefly, 0.33 μg/well of the reporter plasmids (AddGene, Joshua Mendell laboratory (26)) were co-transfected with the miR-21 inhibitor and the inhibitor negative control using jetPRIME® (2 μl/well) (Polyplus-transfection SA) according to the protocol provided. 48 h after transfection, luciferase activity was measured using the dual luciferase assay system (Promega). The firefly luciferase activity of each sample was normalized to Renilla luciferase activity.
shRNA Lentiviral Particle Transduction
Lentiviral particles were produced by transfecting HEK 293T with shPTEN pLKO.1 plasmid (Sigma Mission) or shTGF-β2 as described previously (27). Knockdown of PTEN and TGF-β2 was achieved by infecting iPSCs differentiated for 3 days in presence of VEGF with PTEN and TGF-β2 shRNA lentiviruses, respectively (shPTEN and shTGF-β2). Cells were infected with shPTEN, shTGF-β2, or the nontargeting control (shNT) (107 transfection units/ml) complete growth medium supplemented with 10 μg/ml of polybrene for 16–24 h. The viruses were then removed, and the cells were transfected with LNA-21 and LNA-Ctrl or with Pre-21 and Pre-Ctrl, as described before. Cells were harvested for further analysis after 48 h.
Enzyme-linked Immunosorbent Assay
The concentration of the VEGF and TGF-β2 released glycoprotein in the supernatant was detected by VEGF and TGF-β2 ELISA kits (Invitrogen and R&D, respectively) according to the manufacturers' protocol.
Statistical Analysis
Data expressed as the means ± S.E. were analyzed with a two-tailed Student's t test for two groups or pairwise comparisons. A value of p < 0.05 was considered significant.
RESULTS
VEGF Induces Functional Differentiation of iPSCs toward EC Lineage
We have adapted previous protocol (24) to the differentiation of mouse iPSCs by optimizing concentrations of VEGF and differentiation time points (data not shown). Stimulation of the cells with 50 ng/ml of VEGF rapidly induced a marked change in the morphology of iPSCs, which lost their three-dimensional organization and displayed a flat adherent phenotype (Fig. 1, A and B). Protein analysis showed a consistent up-regulation of endothelial markers such as CD31, Flk1, and VE-cadherin, starting after 3 and 5 days of differentiation and peaking at day 7 (Fig. 1C). Gene expression analysis confirmed this pattern of differentiation for VE-cadherin, Flk1, and vWF (Fig. 1, D–F). Furthermore, staining for VE-cadherin, eNOS, and vWF showed the localization of the proteins within the cells and their homogeneous expression in the population (Fig. 1, G–J). Finally, the functionality of ECs derived from iPSCs was tested in an in vitro angiogenesis assay, showing a significant increase in tubelike structure formation ability upon cell differentiation, as compared with the cells grown in absence of VEGF (Fig. 1, K–M). These findings indeed showed that the protocol established allows the generation of a homogeneous and functional population of endothelial cells from iPSCs.
FIGURE 1.

VEGF induces functional differentiation of iPSCs into ECs. A–J, iPSCs were cultured on collagen IV and in DM supplemented with 50 ng/ml VEGF, followed by morphological analysis (iPSCs undifferentiated on gelatin used as a control (A) and iPSC-derived ECs (B); scale bar, 25 μm), Western blotting (C), quantitative RT-PCR assay (D–F), (day 0 (d0) corresponds to the undifferentiated cells), and immunofluorescence staining (negative controls consisted of cells incubated with IgG followed by secondary antibody; scale bar, 50 μm) (G–J). The results show the expression levels of the EC markers CD31, Flk1, VE-cadherin (VE-cad), vWF, and eNOS. K–M, representative images of in vitro angiogenesis assay in iPSCs differentiated with or without VEGF for 7 days (DM and DM VEGF; scale bar, 25 μm) (K and L) and total tube length quantification (M). The data presented are representative of means ± S.E. (error bars) of three independent experiments. **, p < 0.01; ***, p < 0.001.
Characterization of miRNA Profile in iPSCs Differentiated to ECs
To investigate the miRNA profile during vascular differentiation of iPSCs, which might be involved in angiogenesis (28), we performed a miRNA array in undifferentiated and differentiated iPSCs for 3 days in the presence of 50 ng/ml VEGF; we obtained two pools of differentially expressed miRNAs (Table 1). Among the miRNAs that were most differentially expressed, we chose to focus on miRNA-21. miR-21 has been previously shown to be involved in angiogenesis (13–16) and adipogenic differentiation of stem cells (17), but its role in stem cell differentiation into endothelial cells remains to be elucidated. Real time PCR confirmed the overexpression of miR-21 starting at day 3 and peaking at days 5 and 7 (Fig. 2A). To study its role in differentiation, we transfected the cells after 3 days of culture in the presence of VEGF with miR-21 precursor and the precursor negative control (Pre-21 and Pre-Ctrl; Fig. 2B) or miR-21 inhibitor and the inhibitor control (LNA-21 and LNA-Ctrl; Fig. 2C) and analyzed the expression levels of endothelial markers. Overexpression of miR-21 significantly induced VE-cadherin and Flk1 gene expression (Fig. 2, D and E). Consistently, protein analysis confirmed the induction of the protein levels of VE-cadherin and CD31 (Fig. 2, F and G). Although miR-21 inhibition did not affect the gene expression of the EC markers (Fig. 2, H and I), it led to a significant decrease in the protein induction of these markers (Fig. 2, J and K). Even though we should expect a decrease in protein levels derived from RNA levels, the translational efficiency is differentially affected in the inhibitory system. Moreover, these differentiation markers are indirect targets of the miR-21; the effect on their expression is therefore mediated by other pathways.
TABLE 1.
MicroRNA array on iPSCs differentiated for 3 days with VEGF and undifferentiated cells
The microRNA arrays were performed on iPSCs differentiated for 3 days on collagen IV and in differentiation medium containing 50 ng/ml VEGF; undifferentiated cells were used as control. The results represent averages of the differential expression between two independent experiments. In the panel are shown 28 and 7 microRNAs respectively up- and down-regulated at least 2-fold relative to the undifferentiated iPSCs. miR-302a#, 34c#, and 92a# represent miR* passenger strands. The results were obtained after normalization with three different endogenous controls (mouse small nucleolar RNA snoRNA202 and 135 and mammalian MammU6). These microRNAs were initially characterized in mouse or rat (mmu, Mus musculus; rno, Rattus norvegicus).
| Target name | snoRNA202 | snoRNA135 | MammU6 |
|---|---|---|---|
| mmu-let-7c | 6.7 ± 3.4 | 4.9 ± 3.2 | 6.8 ± 1.9 |
| mmu-miR-129–3p | 33.9 ± 15 | 22.6 ± 6.2 | 39.1 ± 25.9 |
| mmu-miR-133a | 7.2 ± 4.4 | 5.3 ± 3.9 | 7.2 ± 2.8 |
| mmu-miR-139–5p | 3.6 ± 2.5 | 2.3 ± 1.3 | 4.2 ± 3.7 |
| mmu-miR-188–5p | 3.3 ± 0.5 | 2.3 ± 0.7 | 3.5 ± 0.4 |
| mmu-miR-20b | 3.1 ± 1.6 | 2.1 ± 0.7 | 3.6 ± 2.6 |
| mmu-miR-21 | 22.7 ± 27.3 | 17.5 ± 21.8 | 21.0 ± 23.4 |
| mmu-miR-218 | 2.9 ± 0.5 | 2.1 ± 0.7 | 3.1 ± 0.3 |
| mmu-miR-224 | 126.5 ± 170.7 | 98.5 ± 134.3 | 114.3 ± 151.0 |
| mmu-miR-26b | 23.0 ± 13.1 | 15.1 ± 6.3 | 26.9 ± 20.7 |
| mmu-miR-29b | 145.9 ± 189.9 | 113.2 ± 150.1 | 132.7 ± 166.7 |
| mmu-miR-302a | 88.1 ± 3.6 | 61.5 ± 13.4 | 95.7 ± 20.7 |
| mmu-miR-302b | 4,432 ± 4,226 | 2,820 ± 2,391 | 5,436 ± 5,862 |
| mmu-miR-302c | 27,395 ± 34,072 | 21,156 ± 27,080 | 25,158 ± 29,548 |
| mmu-miR-340–5p | 7.8 ± 2.0 | 5.6 ± 2.4 | 8.3 ± 0.0 |
| mmu-miR-34b-3p | 6.6 ± 1.1 | 4.7 ± 1.1 | 7.1 ± 0.6 |
| mmu-miR-367 | 65.9 ± 16.9 | 44.8 ± 3.6 | 74.4 ± 37.0 |
| mmu-miR-449a | 19.8 ± 12.9 | 14.5 ± 11.4 | 19.8 ± 8.6 |
| mmu-miR-685 | 2.9 ± 0.2 | 2.0 ± 0.2 | 3.2 ± 1.1 |
| rno-miR-224 | 9.3 ± 5.1 | 6.1 ± 2.4 | 10.8 ± 8.2 |
| hsa-miR-200b | 2.0 ± 0.6 | 2.0 ± 0.3 | 2.5 ± 0.6 |
| mmu-miR-212 | 4.4 ± 1.0 | 4.3 ± 0.5 | 5.6 ± 1.6 |
| mmu-miR-2138 | 6.5 ± 4.5 | 6.2 ± 3.8 | 7.1 ± 1.8 |
| mmu-miR-302a# | 108.1 ± 46.1 | 104.7 ± 34.8 | 129.4 ± 9.8 |
| mmu-miR-34c# | 7.0 ± 2.5 | 6.8 ± 1.7 | 8.5 ± 1.3 |
| mmu-miR-374–5p | 8.9 ± 7.1 | 8.4 ± 6.1 | 9.5 ± 3.6 |
| mmu-miR-449b | 370.8 ± 512.4 | 341.1 ± 469.7 | 326.7 ± 440.3 |
| mmu-miR-92a# | 4.1 ± 2.1 | 3.9 ± 1.7 | 4.7 ± 0.1 |
| mmu-miR-145 | 0.3 ± 0.1 | 0.2 ± 0.1 | 0.3 ± 0.1 |
| mmu-miR-467e | 0.5 ± 0.3 | 0.4 ± 0.3 | 0.5 ± 0.2 |
| mmu-miR-669a | 0.5 ± 0.2 | 0.3 ± 0.2 | 0.5 ± 0.1 |
| mmu-miR-409–3p | 0.3 ± 0.4 | 0.3 ± 0.3 | 0.3 ± 0.3 |
| mmu-miR-467a | 0.3 ± 0.1 | 0.3 ± 0.1 | 0.4 ± 0.0 |
| mmu-miR-690 | 0.4 ± 0.1 | 0.3 ± 0.1 | 0.4 ± 0.0 |
| mmu-miR-706 | 0.4 ± 0.2 | 0.4 ± 0.2 | 0.4 ± 0.0 |
FIGURE 2.

miR-21 is up-regulated in iPSCs differentiated with VEGF and is able to regulate this process. A–C, iPSCs were differentiated for up to 7 days in the presence of VEGF, followed by TaqMan quantitative PCR assay to analyze the expression of mature miR-21 (A). After 3 days of iPSC differentiation with VEGF, the levels of miR-21 were altered by transfecting the cells with precursor of miR-21 (Pre-21), negative control precursor (Pre-Ctrl) and inhibitor of miR-21 (LNA-21), and negative control inhibitor (LNA-Ctrl), followed by TaqMan quantitative PCR assay to assess the levels of mature miR-21 (B and C). D–G, quantitative RT-PCR assay (D and E) and Western blotting (F, representative image; G, quantitative analysis) were performed 48 h after miR-21 overexpression. H–K, quantitative RT-PCR assay (H and I) and Western blotting (J, representative image; K, quantitative analysis) were performed 48 h after miR-21 inhibition. The expression levels of the endothelial markers VE-cadherin (VE-cad), Flk1, and CD31 were assessed. The data presented are representative of means ± S.E. (error bars) of three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Importantly, miR-21 did not affect EC marker expression in the absence of VEGF stimulation (data not shown); this result indicates that VEGF stimulation is required to initiate the differentiation process toward an endothelial progenitor cell population in which miR-21 induction is able to further induce the EC marker up-regulation. Moreover, no effect of miR-21 overexpression was detected for smooth muscle markers and other mesoderm, endoderm, and ectoderm lineage markers (data not shown).
Next, miR-21 transfected cells were tested in in vitro and in vivo angiogenesis assays. Overexpression of miR-21 induced the formation of tubelike structures in vitro (Fig. 3, A–C). Inclusion of miR-21-overexpressing cells in in vivo Matrigel plugs led to the formation of a significantly higher number of CD31+ and VE-cadherin+ capillaries, as compared with the plugs seeded with precursor control transfected cells (Fig. 3, D–I). Accordingly, miR-21 inhibition reduced the number of capillary structures both in vitro (Fig. 4, A–C) and in vivo (Fig. 4, D–I). These results suggest that miR-21 is able to mediate a lineage-specific effect on iPSCs treated with VEGF to boost their differentiation into functional ECs.
FIGURE 3.
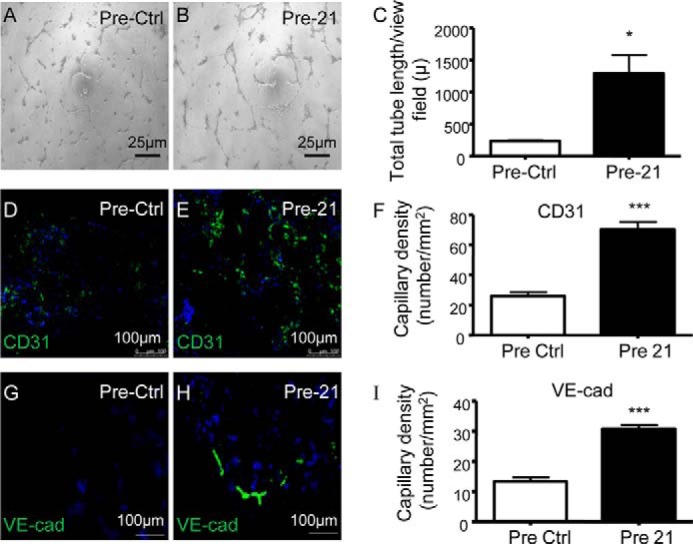
miR-21 overexpression induces functional iPSC differentiation into ECs in vitro and in vivo. A–I, iPSCs differentiated for 4 days in the presence of VEGF were transfected with miR-21 precursor (Pre-21) and the negative control (Pre-Ctrl). A–C, representative images of in vitro angiogenesis assay (scale bar, 25 μm) (A and B) and tube length quantification (C). D–I, representative images (scale bar, 100 μm) of in vivo angiogenesis assay and quantification of capillaries positive for the endothelial markers VE-cadherin (VE-cad) (D–F) and CD31 (G–I). The data presented are representative of means ± S.E. (error bars) of three independent experiments. *, p < 0.05; ***, p < 0.001.
FIGURE 4.
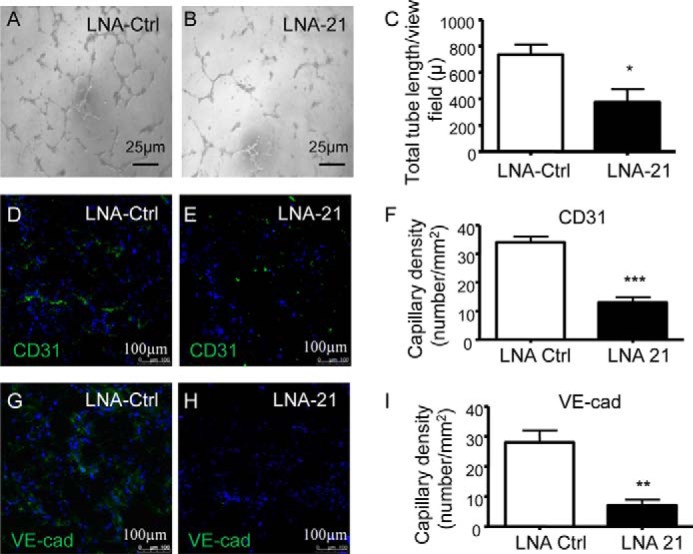
miR-21 inhibition decreases functional differentiation of iPSCs into ECs in vitro and in vivo. A–I, iPSCs differentiated for 4 days in presence of VEGF were transfected with miR-21 inhibitor (LNA-21) and the negative control (LNA-Ctrl). A–C, representative images of in vitro angiogenesis assay (scale bar, 25 μm) (A and B) and tube length quantification (C). D–I, representative images (scale bar, 100 μm) of in vivo angiogenesis assay and quantification of capillaries positive for the endothelial markers VE-cadherin (VE-cad) (D–F) and CD31 (G–I). The data presented are representative of means ± S.E. (error bars) of three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Identification of the Molecular Targets of miRNA-21: TGF-β2 Pathway Is a Downstream Target of miRNA-21 and Drives iPSC Differentiation into ECs
Next, we aimed at elucidating the molecular mechanisms involved in miR-21-mediated differentiation of iPSCs. In silico predicted targets of miR-21 included some proteins involved in the angiogenic process. Transfected cells were analyzed for the expression of genes known to regulate EC functions and vessel growth, such as transforming growth factor β receptor II (TGF-βRII), regulator of ERK activation Sprouty 1 (SPRY1), RhoGTPase RhoB and Sox7, and genes encoding regulators of cell migration such as vinculin. It has been previously shown that in human adipose tissue-derived mesenchymal stem cells miR-21 directly targets the 3′-UTR of TGF-βRII, thus inducing adipogenic differentiation (17) or inhibiting tumor angiogenesis induced by the cells (15). In particular, increased expression of miR-21 in hADSCs inhibits VEGF up-regulation induced by TGF-β and therefore angiogenesis (15). On the contrary, TGF-βRII gene expression was not decreased by miR-21 overexpression or increased by its inhibition (data not shown). This result indicates that TGF-βRII is not directly targeted by miR-21 in ECs derived from iPSCs. Furthermore, we tested expression of genes of the Wnt and the HIF-1α/VEGF signaling pathways, which are known regulators of EC differentiation during embryogenesis and tumor growth (29–31) (data not shown). Our results demonstrated that VEGF expression was not inhibited by miR-21 overexpression or increased by its inhibition (data not shown); therefore, VEGF is not a direct target of miR-21 during the endothelial differentiation of iPSCs.
Additionally, we investigated the expression pattern of other genes belonging to the TGF-β family and found that TGF-β2 was up-regulated by miR-21 overexpression (Fig. 5A), although not significantly down-regulated by miR-21 inhibition (Fig. 5B). Furthermore, ELISA result showed increased levels of secreted TGF-β2 in cells transfected with Pre-21 and reduced in cells treated with miR-21 inhibitor (Fig. 5, C and D).
FIGURE 5.
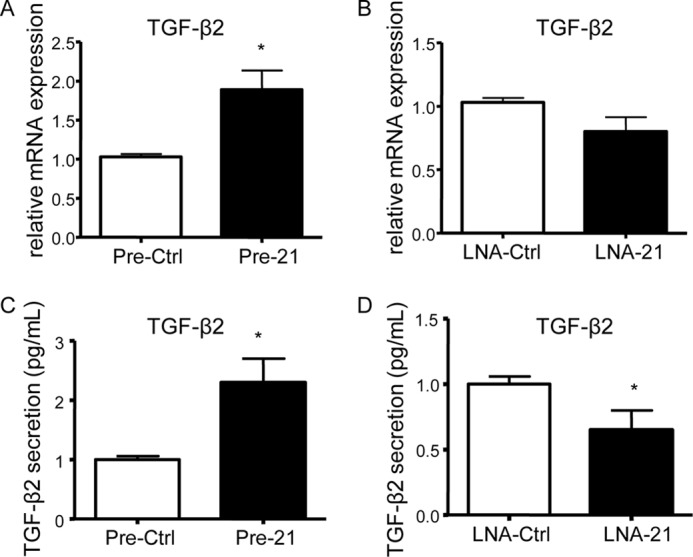
TGF-β2 is a downstream target of miR-21. A–D, iPSCs differentiated for 3 days with VEGF were transfected with miR-21 precursor (Pre-21), inhibitor (LNA-21), and the negative controls of the miR-21 precursor (Pre-Ctrl) and inhibitor (LNA-Ctrl), followed by quantitative RT-PCR assay (A and B) and ELISAs (C and D) 48 h after transfection to assess TGF-β2 expression and secretion levels, respectively. The data presented are representative of means ± S.E. (error bars) of three independent experiments. *, p < 0.05.
Analysis of TGF-β2 expression in ECs derived from in iPSCs showed a strong and significant induction of TGF-β2 gene expression, after 7 days of differentiation (Fig. 6A). This increase was matched by the increase of its receptors TGF-βRII and III (Fig. 6, B and C). Treatment of iPSCs with TGF-β2 induced a statistically significant up-regulation of VE-cadherin and Flk1 expression (Fig. 6, D and E). Western blotting confirmed these results for CD31 and VE-cadherin at the protein level (Fig. 6, F and G). Moreover, iPSCs treated with TGF-β2 for 7 days showed a significantly increased tube formation capacity, as compared with untreated cells (Fig. 6, H–J). Importantly TGF-β2 treatment did not affect the smooth muscle marker gene expression (data not shown).
FIGURE 6.

TGF-β2 is increased in ECs derived from iPSCs and promotes this process. A–C, iPSCs were differentiated with VEGF for up to 7 days, followed by quantitative RT-PCR assay to assess the expression of TGF-β2 (A), TGF-βRII (B), and TGF-βRIII (C). D–L, iPSCs were treated for 7 days with 3 ng/ml TGF-β2 (DM + TGF-β2) or not (DM), followed by quantitative RT-PCR assay for the endothelial markers VE-cadherin and Flk1 (VE-cad, D and E), Western blotting for the endothelial markers VE-cadherin (VE-cad) and CD31 (F, representative image; G, quantitative analysis), in vitro angiogenesis assay (representative images; scale bar, 25 μm) (H and I), and quantification of tube formation (J). The data presented are representative of means ± S.E. (error bars) of three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To confirm the contribution of TGF-β2 in mediating miR-21 effect on EC differentiation, Pre-21 transfected iPSCs were treated with TGFβ-2 neutralizing antibody. TGF-β2 neutralization reduced the miR-21-induced VE-cadherin and CD31 expression at both gene and protein expression levels (Fig. 7, A and B). To further confirm this result, we established a stable knockdown of TGF-β2 in iPSCs differentiated for 3 days in presence of VEGF, using specific shRNAs. Following the knockdown, cells were transfected with Pre-21, and EC marker expression was analyzed. TGF-β2 gene expression showed a significant repression after infection with shTGF-β2, confirming the efficiency of the shRNA knockdown of TGF-β2 (Fig. 7C). Importantly, in Pre-21 transfected cells, the gene expression levels of VE-cadherin and Flk1 showed a significant reduction after shTGF-β2 infection (Fig. 7, D and E). Remarkably, to our knowledge, these findings are the first to suggest a role for TGF-β2 in the miR-21-mediated endothelial differentiation of iPSCs.
FIGURE 7.
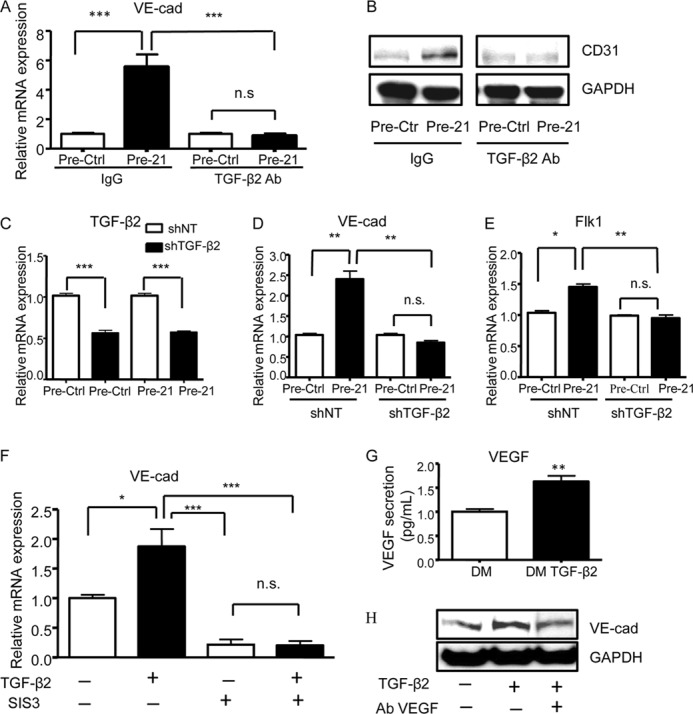
The effect of miR-21/TGF-β2 on EC differentiation is mediated by SMAD3 and VEGF secretion. A and B, iPSCs transfected with miR-21 precursor (Pre-21) and precursor control (Pre-Ctrl) after 3 days of differentiation in presence of VEGF were treated with 1 μg/ml TGF-β2 neutralizing antibody (TGF-β2 Ab) or IgG as a control; quantitative RT-PCR assay (A) and Western blotting (B) were performed 5 h after transfection to assess the expression levels of the EC markers VE-cadherin (VE-cad) and CD31, respectively. C–E, iPSCs transfected with miR-21 precursor (Pre-21) and precursor control (Pre-Ctrl) after 3 days of differentiation in presence of VEGF were infected with shRNA specific for TGF-β2 (shTGF-β2) or nontargeting shRNAs (shNT) as a control; quantitative RT-PCR assay was performed 48 h after infection to assess the expression of TGF-β2 (C), VE-cadherin (VE-cad) (D), and Flk1 (E). F, iPSCs differentiated with (+) or without (−) TGF-β2 for 7 days were treated with SMAD3 inhibitor (SIS3) or DMSO as control, followed by quantitative RT-PCR assay to assess the expression of VE-cadherin (VE-cad). G, ELISAs showing VEGF secretion in iPSCs treated for 7 days with TGF-β2. H, iPSCs differentiated with (+) or without (−) TGF-β2 for 7 days were simultaneously treated with VEGF neutralizing antibody (Ab VEGF +) or IgG as a control (Ab VEGF −), followed by Western blotting to assess VE-cadherin (VE-cad) expression. The data presented are representative of means ± S.E. (error bars) of three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To clarify the molecular mechanisms through which TGF-β2 is able to drive iPSC differentiation into ECs, we focused on the role of the TGF-β2/SMAD3 pathway, which has been previously shown to mediate TGF-β-dependent ES cell differentiation (32). Treatment of iPSCs with the SMAD3 inhibitor SIS3 significantly reduced VE-cadherin expression, in the presence or absence of TGF-β2 (Fig. 7F). Furthermore, we analyzed the effect of TGF-β2 treatment on VEGF levels, based on previous studies showing that VEGF secretion is induced by TGF-β2 (33, 34). ELISAs performed on iPSCs treated with TGF-β2 for 7 days showed a significant induction in the VEGF secretion, as compared with untreated cells (Fig. 7G). Additionally, in iPSCs differentiated for 7 days with TGF-β2 treatment, VEGF-blocking antibody reduced the TGF-β2-dependent EC marker expression (Fig. 7H). These data suggest that TGF-β2 induces EC differentiation in iPSCs through SMAD-dependent pathway and by inducing VEGF secretion. However, TGF-β2 treatment of iPSCs was not able to induce miR-21 up-regulation (data not shown). The result indicates that TGF-β2 does not act upstream of miR-21. These findings could indicate that additional pathways may also act together with TGF-β2 to contribute to VEGF secretion, leading to maintenance of endothelial differentiation state.
Identification of the Molecular Targets of miRNA-21: miR-21 Targets the PTEN/Akt Pathway, Which Regulates iPSC Differentiation into ECs
One of the in silico predicted targets for miR-21 is phosphatase and tensin homolog (PTEN). Recent studies indicated that miR-21 inhibited the tumor suppressor PTEN by binding to its 3′-UTR (35). Inhibition of PTEN by miR-21 has been reported to induce tumor angiogenesis through Akt and ERK activation and HIF-1α expression (13). Moreover, miR-21 has been shown to target PTEN/Akt and partly mediate TGF-β-induced endothelial to mesenchymal transition (36). However, so far the role of PTEN in miR-21-mediated endothelial differentiation of iPSCs has not been elucidated. We first confirmed PTEN as a direct target of miR-21 at a protein expression level. In iPSCs differentiated with VEGF for 3 days, transfection with Pre-21 caused a significant reduction of PTEN protein expression, whereas transfection with LNA-21 led to a significant induction of PTEN (Fig. 8A). Because PTEN is known to be the antagonist of PI3K, which removes the 39 phosphate of phosphatidylinositol 1,4,5-trisphosphate, resulting in inhibition of the Akt signaling pathway (37), we aimed to assess whether PTEN inhibition is required for the activation of Akt during the miR-21-mediated iPSC differentiation process. Concomitantly, previous data from our group showed that the PI3K/Akt signaling pathway drives the shear-induced ES cell differentiation into ECs (24). Our results show that miR-21 overexpression increased the phosphorylation of Akt at the serine 473 site, whereas inhibition of miR-21 reduced Akt phosphorylation, as compared with the relative negative controls. The total level of Akt was not significantly altered by miR-21 overexpression or inhibition (Fig. 8B). In parallel, to further confirm PTEN as a direct target of miR-21, we performed a luciferase assay to assess the binding of miRNA to mRNA. Co-transfection of the WT PTEN 3′-UTR plasmid and LNA-21 in iPSCs differentiating with VEGF resulted in a significant increase in luciferase activity (Fig. 8C). Importantly, mutations in the sequence targeted by miR-21 in PTEN 3′-UTR abolished the observed up-regulation of luciferase activity by miR-21 (Fig. 8D). These results confirmed PTEN as a direct target of miR-21 and validated its specific binding to the predicted binding site.
FIGURE 8.
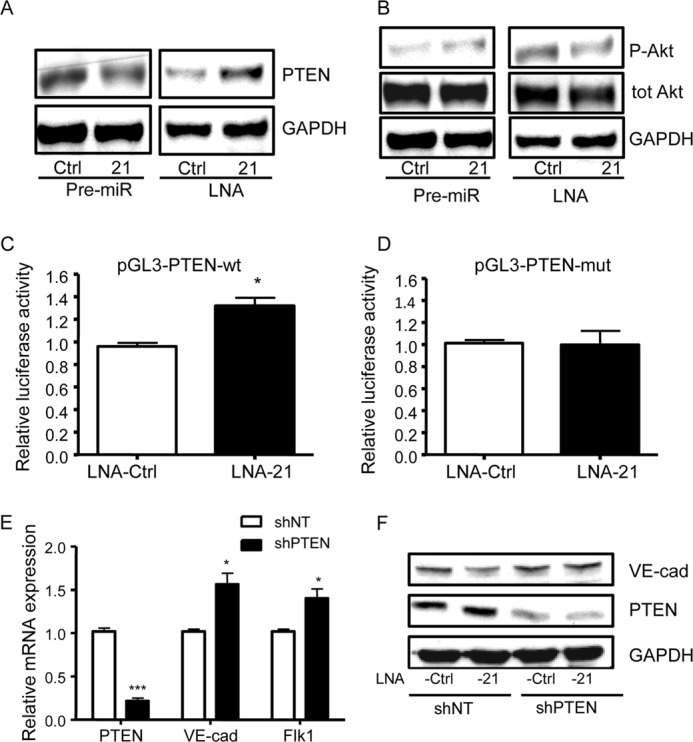
miR-21 targets the PTEN/Akt pathway, which regulates iPSC differentiation into ECs. A and B, iPSCs differentiated for 3 days with VEGF were transfected with miR-21 precursor (Pre-21), inhibitor (LNA-21), and the negative controls of the miR-21 precursor (Pre-Ctrl) and inhibitor (LNA-Ctrl), followed by Western blotting 48 h after transfection to assess PTEN expression (A) and Ser(P)-473 Akt (P-Akt) and total Akt (tot Akt) expression (B). C and D, the wild type or mutated reporter plasmid pGL3 for PTEN 3′-UTR (pGL3-PTEN-wt, C or pGL3-PTEN-mut, D) was co-transfected with miR-21 inhibitor (LNA-21) and its control (LNA-Ctrl) in iPSCs differentiated for 3 days with VEGF, followed by luciferase assay 48 h after transfection. E, iPSCs differentiated for 3 days with VEGF, were infected with shRNA specific for PTEN (shPTEN) or nontargeting shRNAs (shNT) as a control, followed by quantitative RT-PCR assay to assess the expression of PTEN, VE-cadherin (VE-cad), and Flk1 48 h after infection. F, iPSCs transfected with miR-21 inhibitor (LNA-21) and inhibitor control (LNA-Ctrl) after 3 days of differentiation in the presence of VEGF were infected with shRNA specific for PTEN (shPTEN) or nontargeting shRNAs (shNT) as a control; Western blotting was performed 48 h after infection to assess the expression of PTEN and VE-cadherin (F). The data presented are representative of means ± S.E. (error bars) of three independent experiments. *, p < 0.05; ***, p < 0.001.
We therefore decided to assess whether the PTEN/Akt pathway was essential in driving miR-21-mediated iPSC differentiation into EC. We indeed aimed to establish a stable knockdown of PTEN in iPSCs differentiated with VEGF using specific shRNAs, to then analyze the EC marker expression. PTEN gene expression showed a significant repression in iPSCs infected with shPTEN, confirming the efficiency of the shRNA knockdown of PTEN. Importantly, VE-cadherin and Flk1 showed a statistically significant increase in the gene expression after shPTEN infection (Fig. 8E). We then analyzed the expression of the EC markers in iPSCs treated with VEGF and transfected with LNA-21, using shPTEN. PTEN protein expression showed a significant down-regulation in iPSCs infected with shPTEN, confirming the efficiency of the shRNA knockdown of PTEN (Fig. 8F). Our results also showed that in LNA-21 transfected cells, the protein levels of VE-cadherin were significantly increased after shPTEN infection (Fig. 8F).
These data suggest that during EC differentiation of iPSCs, miR-21 inhibits PTEN expression, which in turn increases Akt phosphorylation. The results presented in this study also highlight that PTEN inhibition is required for miR-21 mediated up-regulation of EC markers in differentiated iPSCs.
DISCUSSION
Regeneration of damaged vessels and promotion of new vessel formation in infarcted tissues require the identification of a suitable source of ECs that can be derived from the patient in adequate number for clinical use. Somatic cells from patients have the potential to be reprogrammed to iPSCs, which can be expanded in an undifferentiated state, or to be subjected to lineage specific differentiation in response to a stimulus. In the present study, we established a differentiation protocol for iPSCs, based on the use of collagen IV and 50 ng/ml VEGF. We demonstrated that miR-21 is overexpressed in ECs derived from iPSCs, as compared with the undifferentiated cells in gelatin, and mediates endothelial differentiation through targeting-specific pathways. Thus, our findings provide the basic information for production of endothelial cells from iPSCs that has a great potential in regenerative medicine.
Previously, Zeng et al. (24) stimulated Sca-1+ vascular progenitor cells with medium containing 10 ng/ml VEGF, and Narazaki et al. (38) cultivated Flk1+ progenitor cells with 100 ng/ml VEGF. In the present study, we tested several concentrations ranging between these two extremes by using undifferentiated iPSCs without predifferentiating the cells or selecting for a specific progenitor population. We established that the treatment of iPSCs with increasing concentrations of VEGF had an effect on endothelial differentiation up to 50 ng/ml, which led to a faster differentiation. Importantly, compared with previously described differentiation protocols, our protocol shows a higher scale of EC marker induction, up to a 100-fold increase in Flk1 expression, thus indicating better differentiation efficiency. For instance, Narazaki et al. (38) showed that, in Flk1+ progenitor cells, the effect of VEGF only led to a 20-fold increase in EC marker expression. In conclusion, our protocol allows the direct and efficient EC differentiation from undifferentiated iPSCs in a relative short time and at the highest level, as compared with the previously published protocols.
One of the main aims in this study was to establish the molecular mechanisms driving the differentiation of iPSCs to ECs. Previous work on ES cells has shown that pluripotent cells and cells differentiated to vascular ECs express different pools of miRNAs (28). To our knowledge, there are no studies investigating the miRNA profile during vascular differentiation of iPSCs though recent publications (39). We decided to perform miRNA arrays on iPSCs differentiated for 3 days with VEGF and undifferentiated cells. We obtained a pool of differentially expressed miRNAs and validated five of the most interesting candidates. Among those, we decided to focus on miR-21, a molecule previously reported in tumor angiogenesis and adipogenic differentiation of stem cells (13–17). It was found that miR-21 could induce EC marker expression in iPSCs predifferentiated with VEGF, thus boosting the efficiency of differentiation. These results were confirmed at a protein level and through in vivo and in vitro functional angiogenesis assays. In the study of Liu et al. (13), Matrigel plugs seeded with Pre-21- and LNA-21-transfected human prostate cancer cells were applied onto the chicken chorioallantoic membrane of 9-day-old embryos to assess tumor angiogenesis. Supporting our findings, the number of branches of microvessels were increased by miR-21 overexpression and vice versa decreased by miR-21 inhibition, indicating a pro-angiogenic function of miR-21. In our study, the in vivo angiogenesis process appears to be promoted by miR-21 transfected cells. These results suggest that miR-21 is a key modulator in the endothelial differentiation of iPSCs. The screening for miR-21 potential targets is a challenging process, because prediction algorithms generate a high percentage of false positives.
We showed that miR-21 induced TGF-β2 expression and secretion during EC differentiation. Importantly, TGF-β2 neutralization and knockdown inhibited the miR-21-mediated EC marker induction, thus indicating the requirement of TGF-β2 during this process. Accordingly, expression of TGF-β2 and its receptors appeared to be highly increased in iPSCs differentiated with VEGF. TGF-β2 secretion from iPSCs transfected with Pre-21 appeared to be only in the scale of pg/ml. However, because iPSCs continuously secrete TGF-β2, which probably exerts a paracrine action on neighboring cells, that concentration was sufficient to be effective in iPSC differentiation.
TGF-β has been so far shown to induce smooth muscle differentiation via Notch or SMAD2 and SMAD3 signaling in ES cells (32) or in a neural crest stem cell line (40). Here, we report that TGF-β2 treatment is able to induce iPSC differentiation specifically toward EC lineage, without affecting smooth muscle gene expression. TGF-β has been shown to bind to TGF-βRI and to induce phosphorylation of SMAD2/3, thereby inhibiting proliferation, tube formation, and migration of ECs (41). However, so far there are no studies reporting the role of SMAD3 in stem cell differentiation into ECs. The results reported in the present study suggest the involvement of the TGF-β2/SMAD3 pathway in the iPSC differentiation process; SMAD3 inhibition not only abolished TGF-β2 induction of EC markers, but also reduced the baseline expression of VE-cadherin, in the absence of TGF-β2 stimulation. This result may be explained by considering that SMAD3 is at the center of many other molecular pathways, and it is not only a TGF-β2 downstream effector. For instance, it has been reported that SMAD2/3 is a downstream effector of Activin/Nodal signaling that shares some biological activities with TGF-β (42); moreover, SMAD2/3 signaling has been shown to keep vascular integrity through regulating N-cadherin, VE-cadherin, and S1PR1 expression (43).
miR-21 has been previously shown to target PTEN through Akt and ERK activation to induce tumor angiogenesis (13) and through Akt to partly mediate TGF-β-induced endothelial to mesenchymal transition (36). Our findings showed that miR-21 directly and specifically targets the PTEN/Akt pathway in EC differentiation derived from iPSCs. Interestingly, it has been shown that the PI3K/Akt signaling pathway plays a crucial role in many intracellular cascade events including tumor angiogenesis and tumor growth (44), and in particular, the PI3K/Akt pathway has been reported to drive the shear-induced stem cell differentiation into ECs (24). These results highlight that PTEN knockdown is required to induce EC marker expression in iPSCs differentiated to ECs and transfected with miR-21 inhibitor.
In a recently published study, it has been shown that Akt-mediated Twist1 phosphorylation promotes epithelial-mesenchymal transition and breast cancer metastasis by modulating its transcriptional target TGF-β2; this leads to enhanced TGF-β receptor signaling, which in turn maintains hyperactive PI3K/Akt signaling (45). It is therefore possible that miR-21 activation of the PTEN/Akt pathway may induce Twist1 phosphorylation. Activation of Twist1 may target and induce TGF-β2 to mediate the endothelial differentiation of iPSCs. Phosphorylated Twist1 may indeed have a key role in mediating cross-talk between the miR-21/Akt and TGF-β/Smad signaling axes that supports endothelial differentiation of iPSCs. Other additional pathways might be also involved in this process and will be investigated in future.
In summary, the data presented in this study suggest that in iPSCs predifferentiated with VEGF, miR-21 targets PTEN/Akt and induces TGF-β2, therefore mediating endothelial differentiation (Fig. 9). Indeed, we established a new link between VEGF, miR-21, and TGF-β2 during the endothelial differentiation of iPSCs. Elucidation of the molecular pathways involved in the miR-21-mediated iPSC differentiation into ECs might provide the basic information for stem cell therapy of vascular diseases, e.g., tissue engineering and endothelial repair in damaged vessels.
FIGURE 9.
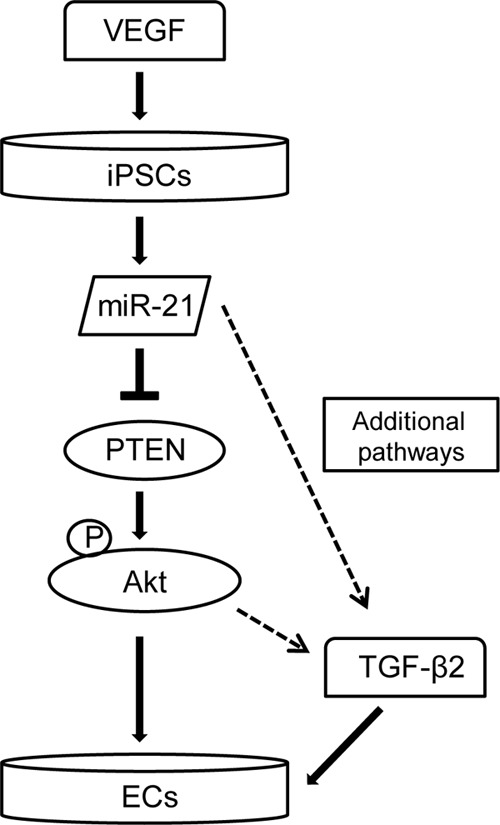
Schematic representation of the mechanisms of iPSC differentiation into ECs. In ECs, differentiation derived from iPSCs VEGF induces miR-21, which targets PTEN/Akt and results in TGF-β2 activation and robust induction of EC differentiation.
This work was supported by a Marie Curie initial training network fellowship from the European Union and a grant from British Heart Foundation.
- EC
- endothelial cell
- iPSC
- induced pluripotent stem cell
- miRNA/miR
- microRNA
- LNA
- locked nucleic acid
- shPTEN
- PTEN shRNA lentivirus
- shNT
- nontarget shRNA lentivirus
- HIF
- hypoxia-inducible factor
- hADSC
- human adipose-derived stem cell
- DM
- differentiation medium
- TGF-βRII
- transforming growth factor β receptor II.
REFERENCES
- 1. Fishman A. P. (1982) Endothelium. A distributed organ of diverse capabilities. Ann. N.Y. Acad. Sci. 401, 1–8 [DOI] [PubMed] [Google Scholar]
- 2. Gilbert S. F. (2000) Developmental Biology, Sinauer Associates, Sunderland, MA [Google Scholar]
- 3. Frangogiannis N. G., Smith C. W., Entman M. L. (2002) The inflammatory response in myocardial infarction. Cardiovasc. Res. 53, 31–47 [DOI] [PubMed] [Google Scholar]
- 4. Ferrara N., Alitalo K. (1999) Clinical applications of angiogenic growth factors and their inhibitors. Nat. Med. 5, 1359–1364 [DOI] [PubMed] [Google Scholar]
- 5. Sima A. V., Stancu C. S., Simionescu M. (2009) Vascular endothelium in atherosclerosis. Cell Tissue Res. 335, 191–203 [DOI] [PubMed] [Google Scholar]
- 6. Sandoo A., van Zanten J. J., Metsios G. S., Carroll D., Kitas G. D. (2010) The endothelium and its role in regulating vascular tone. Open Cardiovasc Med. J. 4, 302–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xiao Q., Zeng L., Zhang Z., Margariti A., Ali Z. A., Channon K. M., Xu Q., Hu Y. (2006) Sca-1+ progenitors derived from embryonic stem cells differentiate into endothelial cells capable of vascular repair after arterial injury. Arterioscler. Thromb. Vasc. Biol. 26, 2244–2251 [DOI] [PubMed] [Google Scholar]
- 8. Nowak G., Karrar A., Holmén C., Nava S., Uzunel M., Hultenby K., Sumitran-Holgersson S. (2004) Expression of vascular endothelial growth factor receptor-2 or Tie-2 on peripheral blood cells defines functionally competent cell populations capable of reendothelialization. Circulation 110, 3699–3707 [DOI] [PubMed] [Google Scholar]
- 9. Levenberg S., Golub J. S., Amit M., Itskovitz-Eldor J., Langer R. (2002) Endothelial cells derived from human embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 99, 4391–4396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hibino N., Duncan D. R., Nalbandian A., Yi T., Qyang Y., Shinoka T., Breuer C. K. (2012) Evaluation of the use of an induced puripotent stem cell sheet for the construction of tissue-engineered vascular grafts. J. Thorac. Cardiovasc. Surg. 143, 696–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Daubman S. (2010) MicroRNAs in angiogenesis and vascular smooth muscle cell function. Circ. Res. 106, 423–425 [DOI] [PubMed] [Google Scholar]
- 12. Wu F., Yang Z., Li G. (2009) Role of specific microRNAs for endothelial function and angiogenesis. Biochem. Biophys. Res. Commun. 386, 549–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu L.-Z., Li C., Chen Q., Jing Y., Carpenter R., Jiang Y., Kung H.-F., Lai L., Jiang B.-H. (2011) MiR-21 induced angiogenesis through AKT and ERK activation and HIF-1α expression. PLoS One 6, e19139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sabatel C., Malvaux L., Bovy N., Deroanne C., Lambert V., Gonzalez M.-L. A., Colige A., Rakic J.-M., Noël A., Martial J. A., Struman I. (2011) MicroRNA-21 exhibits antiangiogenic function by targeting RhoB expression in endothelial cells. PLoS One 6, e16979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shin K. K., Lee A. L., Kim J. Y., Lee S. Y., Bae Y. C., Jung J. S. (2012) miR-21 modulates tumor outgrowth induced by human adipose tissue-derived mesenchymal stem cells in vivo. Biochem. Biophys. Res. Commun. 422, 633–638 [DOI] [PubMed] [Google Scholar]
- 16. Zhu S., Deng S., Ma Q., Zhang T., Jia C., Zhuo D., Yang F., Wei J., Wang L., Dykxhoorn D. M., Hare J. M., Goldschmidt-Clermont P. J., Dong C. (2013) MicroRNA-10A* and MicroRNA-21 modulate endothelial progenitor cell senescence via suppressing high-mobility group A2. Circ. Res. 112, 152–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim Y. J., Hwang S. J., Bae Y. C., Jung J. S. (2009) MiR-21 regulates adipogenic differentiation through the modulation of TGF-β signaling in mesenchymal stem cells derived from human adipose tissue. Stem Cells 27, 3093–3102 [DOI] [PubMed] [Google Scholar]
- 18. Pardali E., Ten Dijke P. (2009) Transforming growth factor-β signaling and tumor angiogenesis. Front. Biosci. 14, 4848–4861 [DOI] [PubMed] [Google Scholar]
- 19. Bertolino P., Deckers M., Lebrin F., ten Dijke P. (2005) Transforming growth factor-β signal transduction in angiogenesis and vascular disorders. Chest 128, 585S–590S [DOI] [PubMed] [Google Scholar]
- 20. Lebrin F., Deckers M., Bertolino P., Ten Dijke P. (2005) TGF-β receptor function in the endothelium. Cardiovasc. Res. 65, 599–608 [DOI] [PubMed] [Google Scholar]
- 21. Takahashi K., Okita K., Nakagawa M., Yamanaka S. (2007) Induction of pluripotent stem cells from fibroblast cultures. Nat. Protoc. 2, 3081–3089 [DOI] [PubMed] [Google Scholar]
- 22. Kaji K., Norrby K., Paca A., Mileikovsky M., Mohseni P., Woltjen K. (2009) Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 458, 771–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zampetaki A., Kiechl S., Drozdov I., Willeit P., Mayr U., Prokopi M., Mayr A., Weger S., Oberhollenzer F., Bonora E., Shah A., Willeit J., Mayr M. (2010) Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ. Res. 107, 810–817 [DOI] [PubMed] [Google Scholar]
- 24. Zeng L., Xiao Q., Margariti A., Zhang Z., Zampetaki A., Patel S., Capogrossi M. C., Hu Y., Xu Q. (2006) HDAC3 is crucial in shear- and VEGF-induced stem cell differentiation toward endothelial cells. J. Cell Biol. 174, 1059–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Campagnolo P., Cesselli D., Al Haj Zen A., Beltrami A. P., Kränkel N., Katare R., Angelini G., Emanueli C., Madeddu P. (2010) Human adult vena saphena contains perivascular progenitor cells endowed with clonogenic and proangiogenic potential. Circulation 121, 1735–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Donnell K. A., Wentzel E. A., Zeller K. I., Dang C. V., Mendell J. T. (2005) c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435, 839–843 [DOI] [PubMed] [Google Scholar]
- 27. Margariti A., Zampetaki A., Xiao Q., Zhou B., Karamariti E., Martin D., Yin X., Mayr M., Li H., Zhang Z., De Falco E., Hu Y., Cockerill G., Xu Q., Zeng L. (2010) Histone deacetylase 7 controls endothelial cell growth through modulation of β-catenin. Circ. Res. 106, 1202–1211 [DOI] [PubMed] [Google Scholar]
- 28. Kane N. M., Meloni M., Spencer H. L., Craig M. A., Strehl R., Milligan G., Houslay M. D., Mountford J. C., Emanueli C., Baker A. H. (2010) Derivation of endothelial cells from human embryonic stem cells by directed differentiation. Analysis of microRNA and angiogenesis in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 30, 1389–1397 [DOI] [PubMed] [Google Scholar]
- 29. Logan C. Y., Nusse R. (2004) The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 [DOI] [PubMed] [Google Scholar]
- 30. Dejana E. (2010) The role of Wnt signaling in physiological and pathological angiogenesis. Circ. Res. 107, 943–952 [DOI] [PubMed] [Google Scholar]
- 31. Kong J., Kong J., Pan B., Ke S., Dong S., Li X., Zhou A., Zheng L., Sun W.-B. (2012) Insufficient radiofrequency ablation promotes angiogenesis of residual hepatocellular carcinoma via HIF-1α/VEGFA. PLoS One 7, e37266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kurpinski K., Lam H., Chu J., Wang A., Kim A., Tsay E., Agrawal S., Schaffer D. V., Li S. (2010) Transforming growth factor-β and Notch signaling mediate stem cell differentiation into smooth muscle cells. Stem Cells 28, 734–742 [DOI] [PubMed] [Google Scholar]
- 33. Bian Z.-M., Elner S. G., Elner V. M. (2007) Regulation of VEGF mRNA expression and protein secretion by TGF-β2 in human retinal pigment epithelial cells. Exp. Eye Res. 84, 812–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma J., Zhang Q., Moe M. C., Zhu T. (2012) Regulation of cell-mediated collagen gel contraction in human retinal pigment epithelium cells by vascular endothelial growth factor compared with transforming growth factor-β2. Clin. Experiment. Ophthalmol. 40, e76–e86 [DOI] [PubMed] [Google Scholar]
- 35. Meng F., Henson R., Wehbe-Janek H., Ghoshal K., Jacob S. T., Patel T. (2007) MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133, 647–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kumarswamy R., Volkmann I., Jazbutyte V., Dangwal S., Park D.-H., Thum T. (2012) Transforming growth factor-β-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler. Thromb. Vasc. Biol. 32, 361–369 [DOI] [PubMed] [Google Scholar]
- 37. Jiang B.-H., Liu L.-Z. (2008) PI3K/PTEN signaling in tumorigenesis and angiogenesis. Biochim. Biophys. Acta 1784, 150–158 [DOI] [PubMed] [Google Scholar]
- 38. Narazaki G., Uosaki H., Teranishi M., Okita K., Kim B., Matsuoka S., Yamanaka S., Yamashita J. K. (2008) Directed and systematic differentiation of cardiovascular cells from mouse induced pluripotent stem cells. Circulation 118, 498–506 [DOI] [PubMed] [Google Scholar]
- 39. Okamoto H., Matsumi Y., Hoshikawa Y., Takubo K., Ryoke K., Shiota G. (2012) Involvement of microRNAs in regulation of osteoblastic differentiation in mouse induced pluripotent stem cells. PLoS One 7, e43800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen S., Lechleider R. J. (2004) Transforming growth factor-β-induced differentiation of smooth muscle from a neural crest stem cell line. Circ. Res. 94, 1195–1202 [DOI] [PubMed] [Google Scholar]
- 41. Goumans M.-J., Valdimarsdottir G., Itoh S., Lebrin F., Larsson J., Mummery C., Karlsson S., ten Dijke P. (2003) Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol. Cell 12, 817–828 [DOI] [PubMed] [Google Scholar]
- 42. Massagué J., Chen Y.-G. (2000) Controlling TGF-β signaling. Genes Dev. 14, 627–644 [PubMed] [Google Scholar]
- 43. Itoh F., Itoh S., Adachi T., Ichikawa K., Matsumura Y., Takagi T., Festing M., Watanabe T., Weinstein M., Karlsson S., Kato M. (2012) Smad2/Smad3 in endothelium is indispensable for vascular stability via S1PR1 and N-cadherin expressions. Blood 119, 5320–5328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xia C., Meng Q., Cao Z., Shi X., Jiang B.-H. (2006) Regulation of angiogenesis and tumor growth by p110α and AKT1 via VEGF expression. J. Cell. Physiol. 209, 56–66 [DOI] [PubMed] [Google Scholar]
- 45. Xue G., Restuccia D. F., Lan Q., Hynx D., Dirnhofer S., Hess D., Rüegg C., Hemmings B. A. (2012) Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-β signaling axes. Cancer Discov. 2, 248–259 [DOI] [PubMed] [Google Scholar]