

Background: CYCLIN A is degraded in early mitosis independent of spindle assembly checkpoint.
Results: APOLLON interacts with CYCLIN A and promotes its degradation in mitosis.
Conclusion: APOLLON is a novel regulator of CYCLIN A degradation in early mitosis.
Significance: This study expands our knowledge on the huge APOLLON protein known to regulate apoptosis and cytokinesis.
Keywords: Cell Cycle, Cyclins, Mitosis, Protein Degradation, Ubiquitin, APC/C, APPOLON, Cyclin A, Spindle Assembly Checkpoint
Abstract
In the mammalian cell cycle, both CYCLIN A and CYCLIN B are required for entry into mitosis, and their elimination is also essential to complete the process. During mitosis, CYCLIN A and CYCLIN B are ubiquitylated by the anaphase-promoting complex/cyclosome (APC/C) and then subjected to proteasomal degradation. However, CYCLIN A, but not CYCLIN B, begins to be degraded in the prometaphase when APC/C is inactivated by the spindle assembly checkpoint (SAC). Here, we show that APOLLON (also known as BRUCE or BIRC6) plays a role in SAC-independent degradation of CYCLIN A in early mitosis. APPOLON interacts with CYCLIN A that is not associated with cyclin-dependent kinases. APPOLON also interacts with APC/C, and it facilitates CYCLIN A ubiquitylation. In APPOLON-deficient cells, mitotic degradation of CYCLIN A is delayed, and the total, but not the cyclin-dependent kinase-bound, CYCLIN A level was increased. We propose APPOLON to be a novel regulator of mitotic CYCLIN A degradation independent of SAC.
Introduction
Cell cycle progression is regulated by the oscillatory activation of the cyclin-dependent kinases (CDKs),4 the activity of which is regulated by the cyclins (1–3). Among the cyclin family members, CYCLIN A predominantly interacts with CDK1 and -2 and CYCLIN B with CDK1 to activate the kinases, the activity of which is required for the entry into mitosis (4). During mitosis, these cyclins are rapidly degraded by the ubiquitin-proteasome system (5–11). Several lines of evidence have suggested that anaphase promoting complex/cyclosome (APC/C), along with CDC20 as a substrate recognition subunit, mediates the ubiquitylation of the mitotic cyclins in the M phase. These include the findings that APC/C promotes the ubiquitylation of CYCLIN A and CYCLIN B in vitro (12–14), the microinjection of antibodies against subunits of APC/C or CDC20 arrests the cells at metaphase and stabilizes CYCLIN A and CYCLIN B (12), and the genetic inactivation of fizzy, a homologue of CDC20 in Drosophila, arrests the cells with high levels of CYCLIN A and CYCLIN B (15). However, CYCLIN A and CYCLIN B are not degraded simultaneously. CYCLIN A begins to be degraded in the prometaphase immediately after nuclear envelope breakdown, whereas CYCLIN B begins to be degraded in the metaphase after all of the chromosomes are aligned on the metaphase plate (5, 8, 9, 12, 16).
The mechanism of CYCLIN B degradation has been extensively studied. CYCLIN B has a short sequence called the destruction box (D-box) that is recognized by CDC20 to induce ubiquitylation (17–19). CDC20 is inhibited by spindle assembly checkpoint (SAC) proteins, such as Mad2 and BubR1, until all of the chromosomes become bipolarly attached to the mitotic spindle in the metaphase (20–23). Therefore, CYCLIN B degradation is strictly held in check until the SAC is satisfied. The D-box of CYCLIN B promotes proteasomal degradation in the metaphase when it is grafted onto heterologous proteins (10, 24, 25), indicating the crucial role of the D-box in the SAC-dependent protein degradation.
Although CYCLIN A has a similar D-box-like motif (26, 27), it is degraded independently of the SAC (8, 9, 12, 16, 28, 29). The mechanism by which CYCLIN A is degraded in the presence of SAC has long been a mystery, and it has been speculated that CYCLIN A is recruited to the APC/C independently of the SAC. Recently, the amino terminus of CYCLIN A, including the D-box, was reported to directly bind to CDC20 and outcompete the SAC proteins (30). The CYCLIN A-CDK complex associated with CDC20 is recruited to APC/C by CKS1 and -2, thereby inducing SAC-independent degradation of CYCLIN A (31, 32). In line with this, deletions or mutations of the amino-terminal sequence of CYCLIN A stabilize the CYCLIN A protein in many species (27, 33). In Drosophila, however, mutation of the cyclin box, to which the CDKs bind, abolished the mitotic destruction of CYCLIN A, although the mutation did not affect CDK binding, suggesting a role for the cyclin box in the SAC-independent degradation of CYCLIN A besides CDK binding (34).
APPOLON is a huge protein that contains a baculoviral IAP repeat (BIR) and a ubiquitin-conjugating enzyme (UBC) domain (35–38). APPOLON ubiquitylates caspase-9, SMAC, and HtrA2 and inhibits apoptosis (35, 36, 39). Targeted disruption of Appolon in mice results in embryonic and neonatal lethality, but extensive apoptosis was not observed in these embryos, suggesting pleiotropic activities of APPOLON in the regulation of multiple cellular functions (36, 40). In this report, we show that APPOLON interacts with CYCLIN A that is not associated with CDKs. APPOLON also interacts with APC/C and facilitates the ubiquitylation of CYCLIN A without involving its UBC domain. An in situ proximity assay showed that APPOLON interacts with CYCLIN A in early mitotic cells. In addition, Appolon-deficient cells in early mitosis accumulate more CYCLIN A than do control cells, and the progression through mitosis is delayed in the Appolon-deficient cells. We propose APPOLON to be a novel regulator of CYCLIN A destruction in early mitotic cells independent of the SAC.
EXPERIMENTAL PROCEDURES
Cell Culture and Synchronization
Human cervical cancer HeLa and human embryonic kidney 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 100 μg/ml kanamycin at 37 °C in a humidified atmosphere of 5% CO2. Human fibrosarcoma HT-1080 cells were maintained in RPMI 1640 medium (Nissui Co., Ltd., Tokyo, Japan) containing 10% heat-inactivated FBS and 100 μg/ml kanamycin at 37 °C. MEFs were prepared from E13.5 embryos and cultured in DMEM as above. The genotypes of the MEFs were examined by PCR, as described (36). The senescent MEFs were maintained in the growth medium twice a week, and spontaneously immortalized MEFs were obtained after 2–3 months. To synchronize the cells in the G2 phase, cells were incubated in growth medium containing 9 μm RO-3306 for 20 h. The RO-3306-treated cells were released into fresh medium containing 5 μm MG132 or 100 ng/ml nocodazole for 2 h. For biochemical analysis, mitotic cells were collected by mild shake off. In some experiments, cells were treated with 1 μg/ml aphidicolin for 24 h or 100 ng/ml nocodazole for 16 h for synchronization in the S and M phases, respectively.
Plasmids and Transfection
The cDNAs encoding human CYCLIN A2, CYCLIN B1, CDK1, CDK2, CDK4, CDK6, and APC11 were amplified by PCR from U937 cDNA and cloned into pcDNA3myc, p3×FLAG-CMV10, pEGFP-N1, or pDsRed2-N1 vectors. APPOLON cDNA was cloned as described previously (36). All constructs generated from the PCR products were sequenced. Cells in 60-mm dishes were transfected with plasmid DNAs (6 μg) and siRNAs (240 pmol) using Lipofectamine 2000 and RNAiMAX reagents, respectively, according to the manufacturer's instruction. The siRNA oligonucleotides corresponding to the sequence of APPOLON (APPOLON siRNA-1, 5′-CAGACCAGUGCAAGAUCAG-3′; APPOLON siRNA-2, 5′-CUCAGGAGAGUACUGCUCA-3′), CDC20 (CDC20 siRNA-1, 5′-GUCCCCCCGGAAACCCACC-3′; CDC20 siRNA-2, 5′-CACAGCUGACCGCUGUAUCC-3′), and CYCLIN A (5′-AACUACAUUGAUAGGUUCCUG-3′) were synthesized with 3′-TT overhangs and duplexed before transfection. The control siRNA was from Qiagen (Allstar negative control).
Immunoprecipitation and Western Blotting
Cells were lysed with IP lysis buffer (10 mm Hepes, pH 7.4, 142.5 mm KCl, 5 mm MgCl2, 1 mm EGTA, and 1% Nonidet P-40), containing Complete Mini Protease Inhibitors (Roche Applied Science), rotated for 1 h at 4 °C, and centrifuged at 15,000 rpm for 10 min at 4 °C to obtain the supernatants. The lysates that had been precleared with naked protein G-Sepharose were immunoprecipitated with protein G-Sepharose conjugated with 2 μg of antibodies for 4 h at 4 °C. The precipitates were washed four times, and the proteins were separated by 4–20% gradient PAGE, transferred to PVDF membranes (Millipore), and Western-blotted using the appropriate antibodies. Protein bands were detected using the enhanced chemiluminescence detection method (ECL) or ECL Prime Western blotting detection reagents (GE Healthcare). In some experiments, cells were lysed in SDS lysis buffer (0.1 m Tris-HCl, pH 8.0, 10% glycerol, 1% SDS) for 10 min at 100 °C and cleared by centrifugation at 15,000 rpm for 10 min to prepare the whole cell lysate (41). The following antibodies were used: anti-APPOLON polyclonal antibody (pAb) prepared as described (36); anti-BRUCE monoclonal antibody (mAb) (BD Transduction Laboratories); anti-CYCLIN A mAb (NeoMarkers, MS-384); anti-CYCLIN A pAb (Santa Cruz Biotechnology, sc-751); anti-CYCLIN B mAb (Santa Cruz Biotechnology, sc-245); anti-CYCLIN B pAb (Pharmingen); anti-CDK1 mAb (Santa Cruz Biotechnology, sc-54); anti-CDK2 pAb (Upstate, 06-505); anti-CDK2 mAb (Upstate, 05-596); anti-CDC20 pAb (Santa Cruz Biotechnology, sc-8358); anti-SMAC pAb (Chemicon, AB3609); anti-APC3 (cdc27) mAb (Santa Cruz Biotechnology, sc-13154); anti-APC3 (cdc27) pAb (Santa Cruz Biotechnology, sc-6392); anti-γ-tubulin pAb (Sigma, T-5192); anti-α-tubulin mAb (Serotec, MCAP77); HRP-conjugated anti-actin mAb (Santa Cruz Biotechnology, sc-8432 HRP); HRP-conjugated anti-GAPDH pAb (Santa Cruz Biotechnology, sc-25778 HRP); anti-HSP90 mAb (BD Transduction Laboratories); HRP-conjugated anti-Myc mAb (Roche Applied Science); agarose-conjugated anti-Myc mAb (Santa Cruz Biotechnology, sc-40AC); HRP- and agarose-conjugated anti-FLAG mAb (Sigma, clone M2); HRP-conjugated anti-HA mAb (Roche Applied Science), and anti-VSV pAb (Sigma).
Ubiquitylation Assay
293T cells were transfected for 24 h with pcDNA3myc-Appolon, pcDNA3-HA-ubiquitin, and p3×FLAG-CMV10-cyclin A. The cells were then incubated with MG132 (10 μm) for 4 h before being harvested and lysed in 1% Nonidet P-40 buffer. The cell lysates were heated at 100 °C for 5 min in the presence of 1% SDS, diluted 10 times, and immunoprecipitated with anti-FLAG. The precipitates were Western-blotted using HRP-conjugated anti-HA mAb (Roche Applied Science).
Immunostaining and Proximity Ligation Assay
MEFs and HeLa cells were fixed in 4% paraformaldehyde in PBS for 5 min at RT or 100% methanol on ice for 10 min, washed twice with PBS, and blocked in PBS containing 3% BSA, 0.1% Triton X-100 (PBS-TB) for 1 h at RT (42). Cells were incubated for 2 h with anti-γ-tubulin, anti-CYCLIN A, or anti-APPOLON pAb as the first antibodies, for 1 h with Alexa Fluor 488-conjugated anti-rabbit IgG or Alexa Fluor 568-conjugated anti-mouse IgG (Molecular Probes) as the second antibodies, and for 3 min with Hoechst 33342 (Molecular Probes). For the proximity ligation assay (43), cells were fixed with methanol on ice and stained with anti-CYCLIN A mAb and anti-APPOLON pAb using the Duolink II fluorescence kit (Olink Biosciences) according to the manufacturer's user manual. The stained signal was measured by BZ-II analyzer (KEYENCE) and statistically analyzed by Student's t test or χ2 test.
Animals
Female C57Bl/6 mice were obtained from CLEA Japan, Inc., and crossed with Appolon heterozygote male mice to maintain the strain. Mouse colonies were maintained in a certified animal facility in accordance with national guidelines in Japan. The animal experiments were approved by the Institutional Review Board of the National Institute of Health Sciences, Japan.
RESULTS
Cell Cycle Perturbation in Appolon-deficient MEFs
MEFs were prepared from wild-type (WT) and Appolon-deficient embryos, and they were primarily cultured in vitro. The Appolon-deficient MEFs halted proliferation ∼10 days after cultivation, whereas the WT MEFs proliferated for ∼30 days (Fig. 1A). On day 11 the Appolon-deficient MEFs exhibited a flattened cell morphology and acid β-galactosidase activity, both of which are typical features of cellular senescence observed in the WT MEFs on day 34 (Fig. 1B). These observations indicate that Appolon-deficient MEFs undergo earlier replicative senescence in primary culture. This may account for the larger G1 population in Appolon-deficient MEFs than in WT MEFs reported previously (36). We also found that cells with an excess number of centrosomes, often coinciding with large or multiple nuclei (Fig. 1C), were more frequently observed in Appolon-deficient (28.6 ± 1.4%) than WT primary cultured MEFs (12.0 ± 1.5%).
FIGURE 1.

Appolon-deficient MEFs exhibit earlier replicative senescence, large nuclei with excess centrosome, and mitotic delay. A, cumulative growth curve of Appolon-deficient MEFs. WT (open circles) and Appolon-deficient (filled circles) MEFs were passaged twice a week. B, acid β-galactosidase staining of Appolon-deficient MEFs. C, excess number of centrosomes and a large nucleus observed in Appolon-deficient MEFs. MEFs were stained with γ-tubulin (green) and Hoechst 33342 (blue). D and E, Appolon-deficient MEFs required an extended time for mitosis. Images of the cultured MEFs were observed every 3 min. The time for mitosis (the time from rounding to respread) was estimated from the images of more than 100 cells. Bars, 20 μm.
From the primary cultured MEFs, we developed a pair of immortalized WT and Appolon-deficient MEFs. Again, centrosome overduplication was observed in the immortalized Appolon-deficient MEFs (∼10%) but not in the immortalized WT MEFs. The cell division of the immortalized MEFs was observed by time-lapse monitoring of the cells. Appolon-deficient MEFs require a longer time for the completion of mitosis than the WT MEFs (Fig. 1, D and E). Thus, Appolon deficiency results in an earlier replicative senescence, excess number of centrosomes, multiple nuclei, and cell cycle perturbation in MEFs, suggesting that APPOLON plays a key role in the regulation of cell cycle.
APPOLON Interacts with CYCLIN A
To study the mechanism by which APPOLON regulates the cell cycle, we screened for APPOLON-interacting proteins using an antibody array, and we found that CYCLIN A, but not CYCLIN B, strongly interacts with APPOLON (data not shown). Co-immunoprecipitation experiments confirmed that APPOLON strongly binds to CYCLIN A but only marginally to CYCLIN B (Fig. 2A). Endogenous APPOLON binds to endogenous CYCLIN A, especially when cells were treated with MG132, a proteasome inhibitor (Fig. 2B). Deletion analysis indicated that the carboxyl-terminal deletion down to an APPOLON(1–2690) fragment efficiently bound CYCLIN A, but further deletion seriously affected the binding adversely (Fig. 2, C and D). Mutations in the BIR or UBC domains did not affect the binding to CYCLIN A, although BIR mutation completely abolished the binding to SMAC (Fig. 2E), indicating that CYCLIN A and SMAC bind to APPOLON at different sites. We also examined the CYCLIN A domain responsible for binding to APPOLON, and we found that the cyclin-box1, to which CDK1 and CDK2 bind (44), interacts with APPOLON (Fig. 2, C and F). Consistent with this, expression of CDK1 and -2 strongly, and CDK4 and -6 weakly, competed with APPOLON for binding to CYCLIN A (Fig. 2G). Collectively, these results indicate that APPOLON binds to the CYCLIN A that is not associated with the CDKs.
FIGURE 2.

APPOLON binds CYCLIN A. A, APPOLON binds CYCLIN A but not CYCLIN B. HT-1080 cells were transfected with Myc-tagged APPOLON and FLAG-tagged cyclins for 36 h. Cell lysates were immunoprecipitated (IP) with anti-Myc antibody and Western-blotted using the indicated antibodies. B, endogenous APPOLON binds endogenous CYCLIN A. Lysates from HeLa cells were immunoprecipitated with anti-APPOLON or control antibodies and Western-blotted with the indicated antibodies. MG132 (10 μm) was added 4 h before harvesting the cells. C, domain structure of APPOLON and CYCLIN A. D and E, co-immunoprecipitation of CYCLIN A with APPOLON mutants. 293T cells were transfected with FLAG-tagged CYCLIN A and mutants of Myc-tagged APPOLON for 30 h and treated with MG132 (10 μm) for 4 h. Immunoprecipitates of anti-Myc antibody were Western-blotted with the indicated antibodies. F, co-immunoprecipitation of APPOLON with CYCLIN A mutants. 293T cells were transfected with FLAG-tagged APPOLON and deletion mutants of Myc-tagged CYCLIN A. Immunoprecipitates of anti-Myc antibody were Western-blotted with the indicated antibodies. G, expression of CDK1 and CDK2 inhibits the interaction of APPOLON and CYCLIN A. 293T cells were transfected with FLAG-tagged APPOLON, HA-tagged CYCLIN A, and Myc-tagged CDKs. Immunoprecipitates of anti-HA antibody were Western-blotted with the indicated antibodies.
APPOLON Ubiquitylates CYCLIN A in Collaboration with APC/C
Because APPOLON contains a UBC domain and ubiquitylates SMAC and caspase9 (35, 36), we examined the ubiquitylation of CYCLIN A by APPOLON. When cells were co-expressed with FLAG-CYCLIN A and HA-ubiquitin, APPOLON, but not survivin or UBC3, enhanced the ubiquitylation of CYCLIN A (Fig. 3A). Unexpectedly, however, the APPOLON C4638A mutant, in which a conserved cysteine residue in the UBC domain was substituted to alanine, enhanced the ubiquitylation of CYCLIN A as well as WT APPOLON (Fig. 3B, left panels). Because a homophilic interaction of APPOLON was observed (35), we carried out similar experiments with Appolon-deficient MEFs to rule out the possible involvement of endogenous wild-type APPOLON in the C4638A mutant-expressing cells. The C4638A mutant APPOLON again stimulated the ubiquitylation of CYCLIN A in the Appolon-deficient MEFs as well as the WT APPOLON (Fig. 3B, right panels). In accord with this, the APPOLON(1–2690) fragment lacking the UBC domain also stimulated CYCLIN A ubiquitylation (Fig. 3C). However, the APPOLON(1–2427) and APPOLON(1–1648) fragments only weakly stimulated CYCLIN A ubiquitylation (Fig. 3C), consistent with the reduced interaction with CYCLIN A (Fig. 2D). These results indicate that the ability to bind CYCLIN A is crucial, whereas the UBC domain of APPOLON is not, for the ubiquitylation of CYCLIN A in cells, implying a role for APPOLON as an E3 ubiquitin ligase rather than an E2 UBC for CYCLIN A ubiquitylation. The expression of CDK1 and -2 inhibited the ubiquitylation of CYCLIN A by APPOLON (Fig. 3D), which is consistent with the inhibition of APPOLON-CYCLIN A binding (Fig. 2G). Collectively, these results indicate that APPOLON ubiquitylates the CYCLIN A that is not associated with the CDKs, and the APPOLON UBC domain is not essential for CYCLIN A ubiquitylation.
FIGURE 3.

APPOLON ubiquitylates CYCLIN A. A, APPOLON ubiquitylates (Ubi) CYCLIN A. 293T cells were transfected with the indicated plasmids for 32 h and treated with MG132 (10 μm) for 4 h. Immunoprecipitates (IP) of the anti-FLAG antibody were Western-blotted with the indicated antibodies. B, APPOLON UBC mutant (C4638A) ubiquitylates CYCLIN A as wild-type APPOLON. 293T (left panels) or APPOLON-deficient MEFs (right panels) were transfected with Myc-tagged WT or C4638A mutant APPOLON together with FLAG-tagged CYCLIN A and HA-tagged ubiquitin and treated with MG132 before harvesting the cells. C, ubiquitylation of CYCLIN A by APPOLON deletion mutants. D, CDK1 and -2 inhibit the ubiquitylation of CYCLIN A mediated by APPOLON.
We next examined how APPOLON ubiquitylates CYCLIN A without involving its UBC domain. Because CYCLIN A ubiquitylation was mostly mediated by APC/C (5–9), we examined the interaction of APPOLON with APC3, a core component of APC/C. A co-immunoprecipitation experiment showed that endogenous APPOLON interacts with endogenous APC3 (Fig. 4A). APPOLON also interacts with APC11, a RING-containing subunit in APC/C recruiting E2-UBC (Fig. 4B). In line with this, co-precipitation of CYCLIN A with APC11 was enhanced by the full-length and the 1–2690-residue fragment of APPOLON to which CYCLIN A binds, but not by the APPOLON fragments (residues 1–1648 and 1–677) to which CYCLIN A does not bind (Fig. 4C). These results strongly suggest that APPOLON recruits CYCLIN A to APC/C for ubiquitylation.
FIGURE 4.

APPOLON interacts with subunits of APC/C. A, endogenous APPOLON physically interacts with endogenous APC3. Lysates from HeLa cells were immunoprecipitated with anti-APPOLON antibody, and the precipitates were Western-blotted with the indicated antibodies. Anti-myc antibody was used as a control antibody. B, APPOLON interacts with APC11. 293T cells were transfected with the indicated plasmids, and cell lysates were prepared. Immunoprecipitates of anti-Myc antibody were Western-blotted with the indicated antibodies. C, APPOLON increased the interaction of CYCLIN A and APC/C. 293T cells were transfected with Myc-tagged APPOLON, FLAG-tagged APC11, and HA-tagged CYCLIN A. Cell lysates were immunoprecipitated (IP) with an anti-FLAG antibody, and the precipitates were Western blotted with the indicated antibodies.
APPOLON Interacts with CYCLIN A in Early Mitotic Cells
Immunostaining of WT MEFs with rabbit anti-APPOLON pAb showed that APPOLON predominantly localizes in cytoplasm in the interphase cells. When WT MEFs were arrested at the G2/M boundary with a CDK inhibitor RO-3306 (45) and released in fresh medium containing MG132, a substantial number of the cells entered into early mitosis, although CYCLIN A degradation was inhibited. Double staining of APPOLON and CYCLIN A showed the presence of both proteins in the mitotic cells (Fig. 5A, upper panels). When MEFs were released in the medium containing nocodazole after the RO-3306 treatment, CYCLIN A was reduced in the mitotic cells (Fig. 5A, lower panels), showing the degradation of CYCLIN A independent of the SAC.
FIGURE 5.
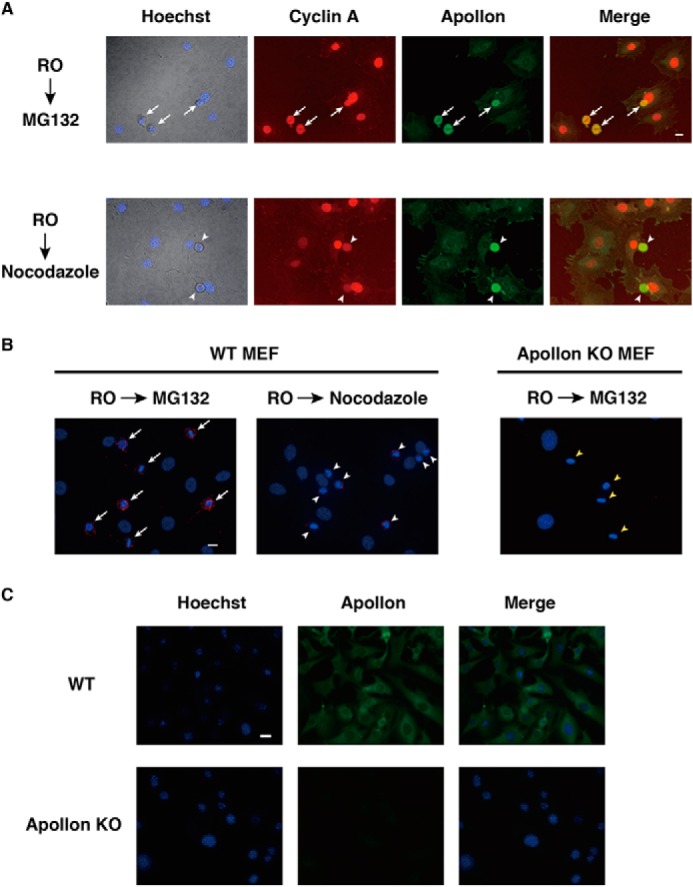
APPOLON interacts with CYCLIN A in early mitotic cells. A, immunostaining of APPOLON and CYCLIN A in early mitotic cells. WT MEFs were arrested at the G2 phase by RO-3306, released into fresh medium containing MG132 or nocodazole for 2 h, and then immunostained with the indicated antibodies. The white arrows in merged upper panel indicate mitotic cells expressing a large amount of CYCLIN A, which appear yellow. The white arrowheads in merged lower panel indicate mitotic cells expressing a reduced amount of CYCLIN A that appear greenish. B, close proximity of APPOLON and CYCLIN A in early mitotic cells. WT and Appolon-deficient MEFs were treated as above, and close proximity ligation assay was performed with antibodies against APPOLON and CYCLIN A. Nuclei were stained with Hoechst 33342. The white arrows in the left panel indicate a robust signal of close proximity (244,635 ± 69,311, n = 17), and the white arrowheads in the middle panel indicate a reduced interaction signal (154,047 ± 36,545, n = 19; p < 0.001, t test) that is consistent with reduced CYCLIN A in the early mitotic cells. The yellow arrowheads in the right panel indicate that there are no signals in the early mitotic Appolon-deficient MEFs. Representative data of two independent experiments are shown. C, specificity of the anti-APPOLON antibodies. Wild-type and Appolon-deficient MEFs were immunostained with rabbit pAb against APPOLON, and nuclei were stained with Hoechst 33342. Bars, 20 μm.
To investigate the possible interaction between APPOLON and CYCLIN A in the mitotic cells, we employed an in situ proximity ligation assay, which is highly specific and sensitive in detecting the close proximity of cellular molecules (43). Robust interaction signals were observed in the mitotic but not the interphase WT MEFs released in the presence of MG132 (Fig. 5B, left panel). The interaction signal was greatly reduced in the mitotic cells released with nocodazole (Fig. 5B, middle panel), which is consistent with the reduction of CYCLIN A (Fig. 5A, lower panels). No interaction signals were detected in mitotic Appolon-deficient MEFs released with MG132 (Fig. 5B, right panel). We also carried out the in situ proximity ligation assay in HeLa cells. The interaction signals were similarly detected in the mitotic HeLa cells released with MG132, but no signals were observed when either one of the two primary antibodies was omitted (data not shown). Fig. 5C shows that WT but not Appolon-deficient MEFs were stained with anti-APPOLON antibody, indicating the specificity of this antibody. Taken together, these results indicate that APPOLON interacts with CYCLIN A in early mitotic cells.
Appolon-deficient Cells Accumulate More CYCLIN A and Exhibit a Delayed Progression of Mitosis
We next examined the role of APPOLON in the stability of CYCLIN A. Because CYCLIN A is degraded independent of SAC, mitotic cells containing large amounts of CYCLIN A were hardly observed in normal culture conditions. However, when HeLa cells were treated with siRNA against APPOLON, mitotic cells with strong staining of CYCLIN A were observed (Fig. 6A). To further study the role of APPOLON in CYCLIN A stability in mitosis, WT and Appolon-deficient MEFs were treated with nocodazole. Nocodazole activates SAC and arrests the cells in early mitosis, but CYCLIN A is degraded in the nocodazole-treated cells independent of SAC (12). Immunocytochemical analysis shows that cells with strong CYCLIN A staining were found in the mitotic APPOLON-deficient MEFs, but not in WT MEFs (Fig. 6B). These results indicate a role of APPOLON in the regulation of CYCLIN A stability in mitosis.
FIGURE 6.

Accumulation of CYCLIN A in Appolon-deficient cells. A, immunostaining of CYCLIN A and APPOLON in HeLa cells treated with APPOLON siRNA. Arrows indicate mitotic cells. The signal intensities of CYCLIN A were 67,328 ± 48,264 (n = 35) and 163,219 ± 139,467 (n = 92) (p < 0.001, t test) in control and APPOLON siRNA-treated cells, respectively. The signal intensities of APPOLON were 172,178 ± 39,865 (n = 35) and 77,933 ± 25,181 (n = 92) (p < 0.001, t test) in control and APPOLON siRNA-treated cells, respectively. B, immunostaining of CYCLIN A in MEFs. MEFs were treated with 100 ng/ml nocodazole for 16 h and then stained with anti-CYCLIN A and Hoechst 33342. The numbers of mitotic cells strongly stained with CYCLIN A (larger than average signal + 2Δ in WT-MEFs) were 0/31 and 4/28 in WT and Appolon-deficient MEFs, respectively (p < 0.05, χ2 test). Bars, 20 μm. C, CYCLIN A levels before and after cells enter into mitosis. HeLa cells were treated with the indicated siRNA, arrested at G2 by RO-3306 (RO) and released into the medium containing nocodazole for 2 h. Mitotic cells were collected by mild shake off, and the cell lysates were analyzed by Western blot. D, depletion of APPOLON increases the total but not the CDK-bound CYCLIN A in early mitotic cells. HeLa cells were treated as above, and the cell lysates were prepared. The total CYCLIN A in the whole cell lysates and the CDK-bound CYCLIN A co-precipitated with anti-CDK2 antibody were evaluated. Representative data of three independent experiments are shown.
Next, we compared the level of CYCLIN A before and after the entry into mitosis. The siRNA-treated HeLa cells were synchronized with RO-3306 and released with nocodazole for 2 h, and then mitotic cells were harvested by a mild shake off. APPOLON depletion increased the level of mitotic CYCLIN A as well as CDC20 depletion (Fig. 6C, lanes 4–6), confirming the role of APPOLON on mitotic degradation of CYCLIN A.
Because APPOLON binds the CYCLIN A that is not associated with the CDKs, we also examined the effect of APPOLON depletion on the amount of CDK-bound CYCLIN A in mitotic cells. APPOLON depletion increased the total amount of CYCLIN A as well as CDC20 depletion (Fig. 6D, lanes 5–7). However, APPOLON depletion did not increase the CYCLIN A co-precipitated with CDK, although CDC20 depletion elevated the level of CDK-bound CYCLIN A (Fig. 6D, lanes 2–4). These observations strongly suggest that the depletion of APPOLON increases the CYCLIN A that is not associated with CDKs in early mitotic cells.
To monitor the CYCLIN A degradation in living mitotic cells, 293T cells were co-transfected with CYCLIN A-GFP, CYCLIN B-DsRed, and an siRNA against APPOLON. After synchronization with aphidicolin, cells were released and monitored for the degradation of CYCLIN A-GFP. In the G2 phase, CYCLIN A-GFP localized in the nucleus and CYCLIN B-DsRed in the cytoplasm (Fig. 7A, −90 min). The CYCLIN B-DsRed then translocated to the nucleus, giving a yellow/orange signal (Fig. 7A, 0 min), after which the CYCLIN A-GFP was degraded within 30 min in the control siRNA-treated cells. In the APPOLON siRNA-treated cells, however, CYCLIN B-DsRed translocated to the nucleus at time 0, but the degradation of CYCLIN A-GFP was delayed to 210 min (Fig. 7A). Thus, APPOLON depletion delays the degradation of CYCLIN A in early mitotic cells.
FIGURE 7.
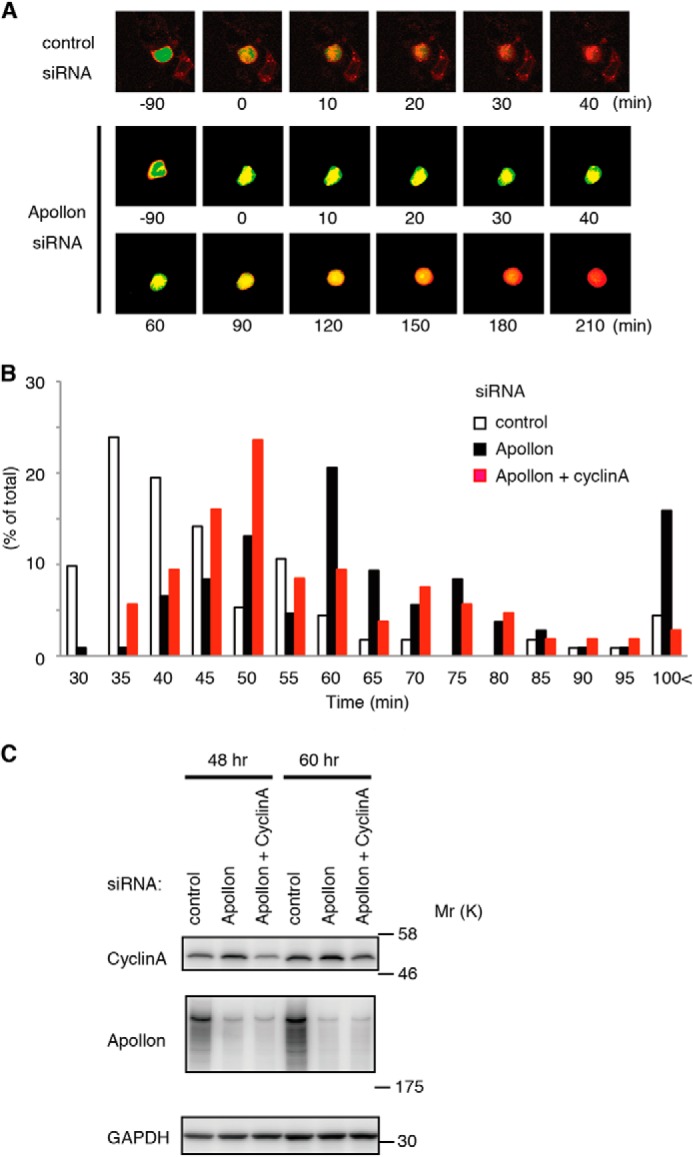
CYCLIN A degradation and progression through mitosis were delayed in APPOLON-depleted cells. A, delayed degradation of CYCLIN A in APPOLON-depleted mitotic cells. Cells transfected with CYCLIN A-GFP and CYCLIN B-DsRed2 were synchronized by aphidicolin and released into fresh medium. Live cell imaging was obtained every 10 min under fluorescent microscopy equipped with a time-lapse recording system. Time 0 was assigned when CYCLIN B-DsRed2 translocated into the nuclei. B, mitotic delay due to depletion of APPOLON was partially corrected by the down-regulation of CYCLIN A. HeLa cells were transfected with siRNA against control (white), APPOLON (black), or APPOLON plus CYCLIN A (red) for 48 h, and then the cell division was monitored. The time from nuclear envelope breakdown to telophase was estimated for more than 100 cells in each group. C, down-regulation of APPOLON and CYCLIN A by siRNA. Cells were treated as above, and the cell lysates were analyzed by Western blot. The down-regulation of APPOLON increased CYCLIN A, which was corrected by adding one-eighth the amount of CYCLIN A siRNA.
We then examined the role of the APPOLON-CYCLIN A axis in the regulation of mitotic progression. Cells were treated with an siRNA against APPOLON for 48 h, and the cell division was monitored using time-lapse microscopy with 5-min intervals. Depletion of APPOLON delayed mitotic progression and greatly increased the number of cells that require more than 100 min (Fig. 7B and supplemental Movies 1–3). This coincided with an increased accumulation of CYCLIN A (Fig. 7C). When the increased CYCLIN A level was reduced to the normal level in the APPOLON-depleted cells by co-transfecting one-eighth the amount of siRNA against CYCLIN A, the population that requires more than 100 min for mitosis was reverted to normal levels, and the delayed progression of mitosis was partially corrected. These results indicate that CYCLIN A regulation by APPOLON plays a significant role in mitotic progression.
DISCUSSION
Mitotic regulators such as CYCLIN A, Nek2A, CYCLIN B, and securin are degraded by the ubiquitin-proteasome system during the course of mitosis. The ubiquitin ligase APC/C plays a major role in the ubiquitylation of the mitotic regulator proteins (5–9, 11, 13, 46, 47). For the ubiquitylation of CYCLIN B and securin, a WD40 family protein CDC20 recruits the substrate proteins to APC/C to activate ubiquitylation (48, 49). Later in mitosis, another WD40 member, CDH1, replaces CDC20 in the ubiquitylation of many proteins (50). Thus, the activity of APC/C is regulated by CDC20 in early mitosis, and by CDH1 in late mitosis and G1 phase. In early mitosis, SAC proteins inactivate CDC20 until all of the chromosomes are bipolarly attached on the metaphase plate (50–56). Therefore, the SAC prevents the degradation of CYCLIN B and securin until metaphase, which ensures the precise distribution of each chromosome into two daughter cells.
In contrast to CYCLIN B and securin, CYCLIN A and Nek2A are degraded independently of the SAC (12, 16, 57). SAC-independent Nek2A degradation requires the carboxyl-terminal di-peptide of Nek2A (Met-Arg), and the di-peptide mediates the recruitment of Nek2A to APC/C to activate ubiquitylation (58, 59). Thus, Nek2A is degraded independently of the SAC. Recently, it was reported that CYCLIN A is recruited to APC/C by CDC20 and CKS proteins independently of the SAC (30–32). In this process, significant amounts of CDC20 bind to CYCLIN A even in the presence of SAC proteins, although CKS proteins interact with CDKs, and thus the CYCLIN A-CDK complex was efficiently recruited to APC/C and degraded independently of the SAC.
In this study, we showed that APPOLON binds and ubiquitylates CYCLIN A that is not associated with CDKs (Figs. 2 and 3). The UBC domain of APPOLON is not required for CYCLIN A ubiquitylation, but APPOLON appeared to recruit CYCLIN A to APC/C (Figs. 3 and 4). In addition, depletion of APPOLON increases CYCLIN A in mitotic cells. Although not formally proven, the available data are consistent with a model in which APC/C in conjunction with APPOLON, even in the absence of its UBC domain, promotes CYCLIN A ubiquitylation for mitotic degradation.
Depletion of APPOLON increases the total amount of CYCLIN A in early mitotic cells without increasing CDK-bound CYCLIN A, whereas CDC20 depletion increases both the total and the CDK-bound CYCLIN A (Fig. 6). This suggests that the CDC20-CKS pathway targets CDK-bound CYCLIN A, although APPOLON targets free CYCLIN A, for proteasomal degradation. These two pathways probably constitute complementary mechanisms for SAC-independent CYCLIN A degradation. The mechanism by which APPOLON binds CYCLIN A in the presence of CDKs is currently unclear. A possible explanation is that APPOLON may outcompete the CDKs in early mitosis. However, we speculate that a subset of CYCLIN A dissociates from the CDKs during mitosis, and then APPOLON recognizes the free CYCLIN A. The increase in the total amount of CYCLIN A, but not the CDK-bound form, in APPOLON-depleted cells (Fig. 6D) suggests the presence of free CYCLIN A in early mitosis. The dissociation of CYCLIN A could be a mechanism to inactivate CDKs prior to the degradation of CYCLIN A.
Although APPOLON has a functionally active UBC domain (35, 36), it is not essential for CYCLIN A ubiquitylation (Fig. 3). Instead, APPOLON facilitates the ubiquitylation of CYCLIN A, probably by recruiting it to APC/C (Fig. 4), suggesting a role as a substrate recognition subunit in a complex of ubiquitin ligase. The role of APPOLON as an E3 ligase was suggested by the way its overall structure resembles the HECT-type E3 ligase bearing a long amino-terminal extension from the HECT domain at the carboxyl terminus (38). In addition, in vitro ubiquitylation experiments suggest a role for APPOLON as an E3 ligase as well as an E2 UBC in the ubiquitylation of SMAC (35, 36). Thus, APPOLON appears to play a double role as an E2 UBC and an E3 ligase for ubiquitylation.
The UBC domain of APPOLON is highly conserved in humans, mice, and Drosophila. Gene-trap mouse strains expressing truncated APPOLON proteins lacking the UBC domain are homozygously lethal and display impaired placental and embryonic development as Appolon null mice (60, 61). In Drosophila, however, homozygous mutant strains of dBRUCE lacking the UBC domain are viable but the male are sterile, exhibiting nuclear hyper-condensation and degeneration of the spermatids (62, 63). The genetic evidence suggests that the E2 function operated by the UBC domain of APPOLON plays an indispensable role in embryonic development in mammals and spermatogenesis in Drosophila. It is not known at this point whether the E3 ligase function of APPOLON is conserved in Drosophila.
Appolon-deficient MEFs are vulnerable to the cell death induced by various stimuli (36), and they also exhibit earlier replicative senescence, large and multiple nuclei, centrosome overduplication, and delay in mitosis (Fig. 1). Among these abnormalities, CYCLIN A regulation by APPOLON could be, at least in part, instrumental in the mitotic delay, because the delay due to the depletion of APPOLON was partially corrected by the reduction of CYCLIN A (Fig. 7B). The vulnerability to cell death is likely to involve the regulation of caspase9, SMAC, and HtrA2, for the binding of which the BIR domain of APPOLON is required, as reported previously (35, 36, 39). With respect to the large and multiple nuclei with excess centrosomes, APPOLON localizes in the midbody of the cells, where ubiquitylated proteins and de-ubiquitylating enzymes accumulate during the cytokinesis in late mitosis (64). Down-regulation of APPOLON in U2OS cells results in cytokinesis failure and an increase in polynucleic cells. These observations indicate a critical role of APPOLON in the regulation of cytokinesis, although the substrate protein ubiquitylated by APPOLON remains to be identified. Because of its huge molecular size, APPOLON interacts with many proteins. Further studies will be needed to uncover the pleiotropic functions of APPOLON, which could link the E2 and E3 activity to the abnormality found in the Appolon-deficient cells.
Acknowledgments
We thank Drs. T. Tsuruo, A. Tomida, N. Fujita, K. Okuhira, N. Shibata, and T. Hattori for helpful discussions. Pacific Edit reviewed the manuscript prior to submission.
This work was supported by the Ministry of Education, Science, Sports and Culture, Japan (grants-in-aid for cancer research and scientific research), the NOVARTIS Foundation (Japan) for the Promotion of Science, and the Cosmetology Research Foundation.

This article contains supplemental Movies 1–3.
- CDK
- cyclin-dependent kinase
- APC/C
- anaphase promoting complex/cyclosome
- BIR
- baculoviral IAP repeat
- MEF
- mouse embryonic fibroblast
- SAC
- spindle assembly checkpoint
- UBC
- ubiquitin-conjugating enzyme
- pAb
- polyclonal antibody.
REFERENCES
- 1. Nigg E. A. (2001) Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2, 21–32 [DOI] [PubMed] [Google Scholar]
- 2. Malumbres M., Barbacid M. (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 [DOI] [PubMed] [Google Scholar]
- 3. Murray A. W. (2004) Recycling the cell cycle: cyclins revisited. Cell 116, 221–234 [DOI] [PubMed] [Google Scholar]
- 4. Merrick K. A., Larochelle S., Zhang C., Allen J. J., Shokat K. M., Fisher R. P. (2008) Distinct activation pathways confer cyclin-binding specificity on Cdk1 and Cdk2 in human cells. Mol. Cell 32, 662–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peters J. M. (2002) The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol. Cell 9, 931–943 [DOI] [PubMed] [Google Scholar]
- 6. van Leuken R., Clijsters L., Wolthuis R. (2008) To cell cycle, swing the APC/C. Biochim. Biophys. Acta 1786, 49–59 [DOI] [PubMed] [Google Scholar]
- 7. Morgan D. O. (1999) Regulation of the APC and the exit from mitosis. Nat. Cell Biol. 1, E47–53 [DOI] [PubMed] [Google Scholar]
- 8. Peters J. M. (2006) The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol. 7, 644–656 [DOI] [PubMed] [Google Scholar]
- 9. Pines J. (2006) Mitosis: a matter of getting rid of the right protein at the right time. Trends Cell Biol. 16, 55–63 [DOI] [PubMed] [Google Scholar]
- 10. Glotzer M., Murray A. W., Kirschner M. W. (1991) Cyclin is degraded by the ubiquitin pathway. Nature 349, 132–138 [DOI] [PubMed] [Google Scholar]
- 11. Pines J. (2011) Cubism and the cell cycle: the many faces of the APC/C. Nat. Rev. Mol. Cell Biol. 12, 427–438 [DOI] [PubMed] [Google Scholar]
- 12. Geley S., Kramer E., Gieffers C., Gannon J., Peters J. M., Hunt T. (2001) Anaphase-promoting complex/cyclosome-dependent proteolysis of human Cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J. Cell Biol. 153, 137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sudakin V., Ganoth D., Dahan A., Heller H., Hershko J., Luca F. C., Ruderman J. V., Hershko A. (1995) The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell 6, 185–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rape M., Kirschner M. W. (2004) Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature 432, 588–595 [DOI] [PubMed] [Google Scholar]
- 15. Sigrist S., Jacobs H., Stratmann R., Lehner C. F. (1995) Exit from mitosis is regulated by Drosophila fizzy and the sequential destruction of cyclins A, B and B3. EMBO J. 14, 4827–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. den Elzen N., Pines J. (2001) Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J. Cell Biol. 153, 121–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kraft C., Vodermaier H. C., Maurer-Stroh S., Eisenhaber F., Peters J. M. (2005) The WD40 propeller domain of Cdh1 functions as a destruction box receptor for APC/C substrates. Mol. Cell 18, 543–553 [DOI] [PubMed] [Google Scholar]
- 18. Raff J. W., Jeffers K., Huang J. Y. (2002) The roles of Fzy/Cdc20 and Fzr/Cdh1 in regulating the destruction of CYCLIN B in space and time. J. Cell Biol. 157, 1139–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zur A., Brandeis M. (2002) Timing of APC/C substrate degradation is determined by fzy/fzr specificity of destruction boxes. EMBO J. 21, 4500–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kops G. J. (2008) The kinetochore and spindle checkpoint in mammals. Front. Biosci. 13, 3606–3620 [DOI] [PubMed] [Google Scholar]
- 21. Kulukian A., Han J. S., Cleveland D. W. (2009) Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev. Cell 16, 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sczaniecka M. M., Hardwick K. G. (2008) The spindle checkpoint: how do cells delay anaphase onset? SEB Exp. Biol. Ser. 59, 243–256 [PubMed] [Google Scholar]
- 23. Nilsson J., Yekezare M., Minshull J., Pines J. (2008) The APC/C maintains the spindle assembly checkpoint by targeting Cdc20 for destruction. Nat. Cell Biol. 10, 1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. King R. W., Glotzer M., Kirschner M. W. (1996) Mutagenic analysis of the destruction signal of mitotic cyclins and structural characterization of ubiquitinated intermediates. Mol. Biol. Cell 7, 1343–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Klotzbücher A., Stewart E., Harrison D., Hunt T. (1996) The 'destruction box' of Cyclin A allows B-type cyclins to be ubiquitinated, but not efficiently destroyed. EMBO J. 15, 3053–3064 [PMC free article] [PubMed] [Google Scholar]
- 26. Jacobs H. W., Keidel E., Lehner C. F. (2001) A complex degradation signal in Cyclin A required for G1 arrest, and a C-terminal region for mitosis. EMBO J. 20, 2376–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaspar M., Dienemann A., Schulze C., Sprenger F. (2001) Mitotic degradation of Cyclin A is mediated by multiple and novel destruction signals. Curr. Biol. 11, 685–690 [DOI] [PubMed] [Google Scholar]
- 28. Fry A. M., Yamano H. (2006) APC/C-mediated degradation in early mitosis: how to avoid spindle assembly checkpoint inhibition. Cell Cycle 5, 1487–1491 [DOI] [PubMed] [Google Scholar]
- 29. Fung T. K., Poon R. Y. (2005) A roller coaster ride with the mitotic cyclins. Semin. Cell Dev. Biol. 16, 335–342 [DOI] [PubMed] [Google Scholar]
- 30. Di Fiore B., Pines J. (2010) How cyclin A destruction escapes the spindle assembly checkpoint. J. Cell Biol. 190, 501–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van Zon W., Ogink J., ter Riet B., Medema R. H., te Riele H., Wolthuis R. M. (2010) The APC/C recruits CYCLIN B1-Cdk1-Cks in prometaphase before D box recognition to control mitotic exit. J. Cell Biol. 190, 587–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wolthuis R., Clay-Farrace L., van Zon W., Yekezare M., Koop L., Ogink J., Medema R., Pines J. (2008) Cdc20 and Cks direct the spindle checkpoint-independent destruction of cyclin A. Mol. Cell 30, 290–302 [DOI] [PubMed] [Google Scholar]
- 33. Fung T. K., Yam C. H., Poon R. Y. (2005) The N-terminal regulatory domain of cyclin A contains redundant ubiquitination targeting sequences and acceptor sites. Cell Cycle 4, 1411–1420 [DOI] [PubMed] [Google Scholar]
- 34. Ramachandran V., Matzkies M., Dienemann A., Sprenger F. (2007) Cyclin A degradation employs preferentially used lysines and a cyclin box function other than Cdk1 binding. Cell Cycle 6, 171–181 [DOI] [PubMed] [Google Scholar]
- 35. Bartke T., Pohl C., Pyrowolakis G., Jentsch S. (2004) Dual role of BRUCE as an antiapoptotic IAP and a chimeric E2/E3 ubiquitin ligase. Mol. Cell 14, 801–811 [DOI] [PubMed] [Google Scholar]
- 36. Hao Y., Sekine K., Kawabata A., Nakamura H., Ishioka T., Ohata H., Katayama R., Hashimoto C., Zhang X., Noda T., Tsuruo T., Naito M. (2004) Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat. Cell Biol. 6, 849–860 [DOI] [PubMed] [Google Scholar]
- 37. Chen Z., Naito M., Hori S., Mashima T., Yamori T., Tsuruo T. (1999) A human IAP-family gene, apollon, expressed in human brain cancer cells. Biochem. Biophys. Res. Commun. 264, 847–854 [DOI] [PubMed] [Google Scholar]
- 38. Hauser H. P., Bardroff M., Pyrowolakis G., Jentsch S. (1998) A giant ubiquitin-conjugating enzyme related to IAP apoptosis inhibitors. J. Cell Biol. 141, 1415–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sekine K., Hao Y., Suzuki Y., Takahashi R., Tsuruo T., Naito M. (2005) HtrA2 cleaves Apollon and induces cell death by IAP-binding motif in Apollon-deficient cells. Biochem. Biophys. Res. Commun. 330, 279–285 [DOI] [PubMed] [Google Scholar]
- 40. Lotz K., Pyrowolakis G., Jentsch S. (2004) BRUCE, a giant E2/E3 ubiquitin ligase and inhibitor of apoptosis protein of the trans-Golgi network, is required for normal placenta development and mouse survival. Mol. Cell. Biol. 24, 9339–9350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Naito M., Katayama R., Ishioka T., Suga A., Takubo K., Nanjo M., Hashimoto C., Taira M., Takada S., Takada R., Kitagawa M., Matsuzawa S., Reed J. C., Tsuruo T. (2004) Cellular FLIP inhibits β-catenin ubiquitylation and enhances Wnt signaling. Mol. Cell. Biol. 24, 8418–8427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ishioka T., Katayama R., Kikuchi R., Nishimoto M., Takada S., Takada R., Matsuzawa S., Reed J. C., Tsuruo T., Naito M. (2007) Impairment of the ubiquitin-proteasome system by cellular FLIP. Genes Cells 12, 735–744 [DOI] [PubMed] [Google Scholar]
- 43. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 44. Jeffrey P. D., Russo A. A., Polyak K., Gibbs E., Hurwitz J., Massagué J., Pavletich N. P. (1995) Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376, 313–320 [DOI] [PubMed] [Google Scholar]
- 45. Vassilev L. T., Tovar C., Chen S., Knezevic D., Zhao X., Sun H., Heimbrook D. C., Chen L. (2006) Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. U.S.A. 103, 10660–10665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. King R. W., Peters J. M., Tugendreich S., Rolfe M., Hieter P., Kirschner M. W. (1995) A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 81, 279–288 [DOI] [PubMed] [Google Scholar]
- 47. Thornton B. R., Toczyski D. P. (2006) Precise destruction: an emerging picture of the APC. Genes Dev. 20, 3069–3078 [DOI] [PubMed] [Google Scholar]
- 48. Fang G., Yu H., Kirschner M. W. (1998) Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol. Cell 2, 163–171 [DOI] [PubMed] [Google Scholar]
- 49. Yu H. (2007) Cdc20: a WD40 activator for a cell cycle degradation machine. Mol. Cell 27, 3–16 [DOI] [PubMed] [Google Scholar]
- 50. Zachariae W., Schwab M., Nasmyth K., Seufert W. (1998) Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science 282, 1721–1724 [DOI] [PubMed] [Google Scholar]
- 51. Bharadwaj R., Yu H. (2004) The spindle checkpoint, aneuploidy, and cancer. Oncogene 23, 2016–2027 [DOI] [PubMed] [Google Scholar]
- 52. Fang G., Yu H., Kirschner M. W. (1998) The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 12, 1871–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sudakin V., Chan G. K., Yen T. J. (2001) Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J. Cell Biol. 154, 925–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tang Z., Bharadwaj R., Li B., Yu H. (2001) Mad2-Independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev. Cell 1, 227–237 [DOI] [PubMed] [Google Scholar]
- 55. Tang Z., Shu H., Oncel D., Chen S., Yu H. (2004) Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol. Cell 16, 387–397 [DOI] [PubMed] [Google Scholar]
- 56. Vanoosthuyse V., Hardwick K. G. (2005) Bub1 and the multilayered inhibition of Cdc20-APC/C in mitosis. Trends Cell Biol. 15, 231–233 [DOI] [PubMed] [Google Scholar]
- 57. Hames R. S., Wattam S. L., Yamano H., Bacchieri R., Fry A. M. (2001) APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box. EMBO J. 20, 7117–7127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hayes M. J., Kimata Y., Wattam S. L., Lindon C., Mao G., Yamano H., Fry A. M. (2006) Early mitotic degradation of Nek2A depends on Cdc20-independent interaction with the APC/C. Nat. Cell Biol. 8, 607–614 [DOI] [PubMed] [Google Scholar]
- 59. Sedgwick G. G., Hayward D. G., Di Fiore B., Pardo M., Yu L., Pines J., Nilsson J. (2013) Mechanisms controlling the temporal degradation of Nek2A and Kif18A by the APC/C-Cdc20 complex. EMBO J. 32, 303–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ren J., Shi M., Liu R., Yang Q. H., Johnson T., Skarnes W. C., Du C. (2005) The Birc6 (Bruce) gene regulates p53 and the mitochondrial pathway of apoptosis and is essential for mouse embryonic development. Proc. Natl. Acad. Sci. U.S.A. 102, 565–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hitz C., Vogt-Weisenhorn D., Ruiz P., Wurst W., Floss T. (2005) Progressive loss of the spongiotrophoblast layer of Birc6/Bruce mutants results in embryonic lethality. Genesis 42, 91–103 [DOI] [PubMed] [Google Scholar]
- 62. Arama E., Agapite J., Steller H. (2003) Caspase activity and a specific cytochrome c are required for sperm differentiation in Drosophila. Dev. Cell 4, 687–697 [DOI] [PubMed] [Google Scholar]
- 63. Vernooy S. Y., Chow V., Su J., Verbrugghe K., Yang J., Cole S., Olson M. R., Hay B. A. (2002) Drosophila Bruce can potently suppress Rpr- and Grim-dependent but not Hid-dependent cell death. Curr. Biol. 12, 1164–1168 [DOI] [PubMed] [Google Scholar]
- 64. Pohl C., Jentsch S. (2008) Final stages of cytokinesis and midbody ring formation are controlled by BRUCE. Cell 132, 832–845 [DOI] [PubMed] [Google Scholar]