

Background: Hax-1 is a family of apoptotic regulators. The prototypical variant 1 has an antiapoptotic function.
Results: Hax-1 variant 2 promotes cell death and can abrogate the protective effect of variant 1.
Conclusion: The ratio of anti- and proapoptotic Hax-1 variants is likely to determine cell fate via sequestration or inactivation.
Significance: Different splice variants of Hax-1 have opposing roles in regulating apoptosis.
Keywords: Apoptosis, Bcl-2 Family Proteins, Cell Death, Oxidative Stress, Protein-Protein Interactions
Abstract
Studies on Hax-1 have mainly focused on variant (v) 1, demonstrating its antiapoptotic properties. However, HAX1 is heavily spliced, generating structurally distinct isoforms. We sought to characterize the Hax-1 isoforms expressed in rat heart before and after insult. We confirmed the presence of at least four Hax-1 transcripts in healthy rat cardiac muscle. These exhibited differential expression before and after induction of myocardial infarction, with v2 being up-regulated 12-fold at the transcript level and 1.5-fold at the protein level post-insult. Contrary to antiapoptotic rat and human v1, overexpression of rat v2 or human v4 (the human homologue of rat v2) in epithelial cells exacerbated cell death by 30% following H2O2 treatment compared with control vector. Coexpression of rat v1 and v2 or human v1 and v4 neutralized the protective effects of rat and human v1 and the proapoptotic effects of rat v2 and human v4 by modulating cytochrome c release. This is, at least partly, mediated by the ability of Hax-1 proteins to form homotypic and heterotypic dimers with binding affinities ranging from ∼3.8 nm for v1 dimers to ∼97 nm for v1/v2 dimers. The minimal binding region supporting these interactions lies between amino acids 97–278, which are shared by nearly all Hax-1 proteins, indicating that additional factors regulate the preferential formation of Hax-1 homo- or heterodimers. Our studies are the first to show that Hax-1 is a family of anti- and proapoptotic regulators that may modulate cell survival and death through homo- or heterodimerization.
Introduction
HS-1-associated protein X-1 (Hax-1) is a family of ubiquitously expressed proteins ranging in size from ∼26 to ∼35 kDa that result from alternative splicing of the single HAX-1 gene (1, 2). The prototypical Hax-1 variant (v)2 1 is an ∼35-kDa protein expressed in both humans and rodents. Early on, it was postulated that Hax-1 contains an NH2-terminal acidic box consisting of Asp and Glu residues, followed by two purported Bcl-2 homology domains, BH1 and BH2, a PEST motif, a predicted COOH-terminal transmembrane domain, and an integrin β6 binding domain (3, 4). Recently, though, the existence of the BH1, BH2, and transmembrane domains has been disputed on the basis of data obtained by sequence analysis and structure prediction (5).
The antiapoptotic role of Hax-1 v1 has been confirmed in different experimental and disease models. Consistent with this, ectopic expression of Hax-1 v1 in HeLa cells, HEK293 cells, and cardiomyocytes promotes cell survival following exposure to different apoptotic stimuli (6–9). More importantly, overexpression of Hax-1 v1 has been found in psoriasis, a severe inflammatory disease characterized by increased proliferation and diminished apoptosis of keratinocytes (10), as well as in melanoma and breast and lung cancers (11).
Hax-1 v1 has been reported to interact with an increasingly diverse array of proteins (3, 6, 7, 9, 12–15), indicating that it might exert its antiapoptotic activities through different pathways. Thus, it has been documented that Hax-1 directly binds initiator caspase 9, inhibiting its activation (8, 9), and the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) pump and its regulator phospholamban, modulating Ca2+ homeostasis (9, 14, 16, 17). Hax-1 is also involved in the processing and activation of the antiapoptotic factor HtrA2 by the mitochondrial protease PARL (18). Active HtrA2 prevents the accumulation of proapoptotic Bax in the outer mitochondrial membrane (18), which results in reduced cytochrome c release from the mitochondria and, thus, decreased apoptosis.
Although the importance of Hax-1 in regulating cell survival and death has been demonstrated, its exact mechanism of action still remains unclear. This is complicated by the presence of multiple, functionally diverse Hax-1 binding partners and the existence of many structurally distinct Hax-1 splice variants (4, 19). In this study, we examined the expression profile of Hax-1 variants in healthy and stressed hearts and studied their role in modulating cell fate following insult. We observed a significant increase in the transcript and protein levels of Hax-1 v2 in rat myocardium following induction of myocardial infarction. Overexpression of rat v2 or of its human homologue v4 confers a prodeath effect in epithelial cells after exposure to H2O2. Importantly, coexpression of rat v1 and v2 or human v1 and v4 abrogates the protective and prodeath effects of v1 and v2/v4, respectively, via regulation of cytochrome c release. This is modulated by the formation of homotypic and heterotypic dimers of Hax-1 proteins. Therefore, our findings document, for the first time, that Hax-1 comprises a family of antiapoptotic and proapoptotic proteins that may regulate cell fate under stress conditions via the formation of homo- or heterodimers.
EXPERIMENTAL PROCEDURES
Myocardial Infarction
Frozen lyophilized heart tissue from adult Sprague-Dawley rats was donated by Dr. William Stanley (University of Maryland, School of Medicine). Heart failure was induced by constriction of the left coronary artery via ligation to simulate myocardial infarction, as described previously (20, 21). The ligation clamp remained in place for 12 weeks. Control sham animals underwent the same surgery without artery ligation. Total heart tissue was collected from both groups at the end of the 12-week ligation period. For the myocardial infarction group, the heart tissue included infarcted and non-infarcted regions.
Quantitative RT-PCR
Poly-A mRNA was isolated from rat cardiac tissue subjected to myocardial infarction or sham surgery using the MicroPoly(A) Purist kit (Ambion, Life Technologies, Grand Island, NY). Quantitative RT-PCR reactions were set up in 50 μl of volume using Bio-Rad iScript and IQ SYBR Green Supermix (Bio-Rad). Hax-1 variant-specific primers were designed to span exon/intron junctions with an optimal Tm of 60 °C and a length of between 18 and 25 bases. Of the seven known rat Hax-1 variants, we were able to specifically amplify variants 1, 2, 4, and 6. The primers used were designed according to the published rat variant sequences (accession numbers NM_181627.2, AY919342.1, and AY291064.2) and were as follows: v1, 5′-GACCTCGGAGCCACAGAGATC-3′ (forward) and 5′-CCTGGAAGTTCAGGAGAGTGGG-3′ (reverse); v2, 5′-GACCTCGGAGCCACAGAGATC-3′ (forward) and 5′-CCTGGAAGTTCACTGTCTTCCTCC-3′ (reverse); v4, 5′-GCAAGTCATTGGCCACAGAGATC-3′ (forward) and 5′-CCTGGAAGTTCACTGTCTTCCTCC-3′ (reverse); and v6, 5′-CCTCCATTCTCAGCCACAGAGAT-3′ (forward) and 5′-CCTGGAAGTTCACTGTCTTCCTCC-3′ (reverse).
Western Blotting
Immunoblot analyses were performed as described previously (19). The only modification to this protocol was that 25 mg of heart tissue was homogenized in Nonidet P-40 buffer containing 10 mm NaPO4, 2 mm EDTA, 10 mm NaN3, 0.9% NaCl, and 2% Nonidet P-40 in the presence of a protease inhibitor mixture (Roche, catalog no. 11697498001) using a tissue grinder (VWR, Radnor PA, model no. 47747-366). Protein lysates from three independent experiments were prepared and analyzed. The following primary antibodies were used: Hax-1 (1:1000 dilution, BD Biosciences), His (1:1000 dilution, Santa Cruz Biotechnology, Dallas, TX), α tubulin (1:1000 dilution, Sigma-Aldrich, St. Louis, MO), cytochrome c (1:200 dilution, Abcam, Cambridge, MA), and GAPDH (1:1000 dilution, Santa Cruz Biotechnology). This was followed by goat anti-mouse or donkey anti-rabbit secondary antibody (1:2500 dilution, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Blots were developed using an alkaline phosphatase-based chemiluminescent system (Applied Biosystems, Foster City, CA). Relative levels of immunoreactive bands were quantified using ImageJ software and densitometric analysis (National Institutes of Health, Bethesda, MD).
Generation of Overexpression Plasmids
Overexpression constructs were generated by cloning the complete coding sequence of rat Hax-1 v1 and v2 and human Hax-1 v1 and v4 (accession numbers: NM_181627.2, AY919342.1, NM_006118.3, and EU190982.1, respectively) into the pIRES2-ZsGreen1 or pIRES2-ZsRed1 vector (Clontech, Mountain View, CA) using the Sac1 and Xma1 restriction sites. The primes used were as follows: rat v1 and v2, 5′-ACTGGAGCTCATGAGCGTCTTTGAT-3′ (forward) and 5′-ACTGCCCGGGCTATCGGGACCGAAACC-3′ (reverse). For human v1, the above rat forward primer was used with reverse primer 5′-ACTGCCCGGGTACCGGGACCGGAAAC-3′. The authenticity of the generated constructs was confirmed by sequencing (Genewiz Inc., South Plainfield, NJ).
Cell Culture and Transfections
HEK293 cells (ATCC) were cultured in growth medium (DMEM supplemented with 10% FBS and 1% penicillin/streptomycin) and maintained in a water-jacketed 5% CO2 incubator at 37 °C. Transfections were performed using Lipofectamine 2000 (Invitrogen) according to the protocol of the manufacturer, followed by medium change 5 h after transfection.
Measurement of Cell Viability using the XTT Assay
3 × 105 HEK293 cells were seeded in standard 6-well plates 24 h prior to transfection. 24 h after transfection, plasmid expression was confirmed by visualization of GFP or red fluorescent protein fluorescence. Cells were treated with either 2 μm thapsigargin for 24 h, 4 μm calcimycin for 24 h, or 0.33 mm H2O2 for 4 h. All treatments were performed in sodium pyruvate-free DMEM in the absence of serum or antibiotics. Cell viability was measured using XTT sodium salt (2,3-Bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide inner salt) (Sigma-Aldrich) according to the instructions of the manufacturer.
Evaluation of Apoptosis via FACS Analysis
Cells were plated, transfected, and treated with the indicated chemicals as for the XTT assay. Following treatment, cells were collected using 0.025% trypsin (Invitrogen) and prepared for FACS analysis using the annexin V phycoerythrin apoptosis detection kit (BD Biosciences) according to the protocol of the manufacturer. FACS analysis was performed on a BD FACS Scan Analyzer by the University of Maryland Flow Core staff. Only GFP-positive, red fluorescent protein-positive, or GFP/red fluorescent protein-positive cells were included in our analysis to ensure that only transfected cells were analyzed.
Production of Recombinant Hax-1 Proteins
His6- or GST-tagged recombinant proteins were generated for use in binding assays. Full-length rat Hax-1 v1 and v2 were cloned into the pet30a (Novagen, Darmstadt, Germany) and pGEX (GE Life Sciences, Pittsburgh, PA) vectors for expression of His6- and GST-tagged fusion proteins, respectively. Proteins were expressed in BL21 DE3 cells following standard techniques. For protein extraction, bacterial pellets from a 100-ml culture were resuspended in 20 ml STE buffer (10 mm Tris-HCl, 150 mm NaCl, and 1 mm EDTA (pH 7.5)). After one freeze-thaw cycle, lysozyme was added to the cell pellet to a final concentration of 100 μg/ml, followed by incubation on ice for 15 min. Subsequently, 5 mm DTT and 1% N-lauroylsarcosine were added to the cell slurry, followed by sonication for four 10-s bursts on medium power and centrifugation at 8900 × g at 4 °C for 30 min. The supernatant containing the recombinant protein of interest was removed, and 2% Triton X-100/20 mm CHAPS were added, followed by incubation with gentle rocking for 4 h at 4 °C. All samples were dialyzed in cold PBS in the presence of 10 mm NaN3 and 1 mm EDTA. His6-tagged proteins were purified using nickel-nitrilotriacetic acid beads (Qiagen, Hilden, Germany) and gravity flow columns, following the protocols of the manufacturer. GST-tagged proteins were incubated with glutathione-Sepharose beads (Novagen) according to the instructions of the manufacturer.
Protein Binding via GST Pulldown
GST-tagged rat Hax-1 v1 was bound to glutathione-Sepharose beads and incubated with 3 μg of His-tagged Hax-1 v1 or v2 overnight at 4 °C in pulldown buffer (50 mm Tris (pH 7.5), 120 mm NaCl, 10 mm NaN3, 2 mm DTT, and 0.5% Tween). Beads were washed five times with wash buffer (PBS with 10 mm NaN3 and 0.1% Tween), and bound proteins were eluted with 2× lithium dodecyl sulfate buffer (Invitrogen), followed by boiling at 95 °C for 10 min and separation on a 4–12% bis-Tris gel. Our standard Western blotting procedure was followed, using a His6 tag antibody (catalog no. SC-803, Santa Cruz Biotechnology).
Protein Binding via Surface Plasmon Resonance
Surface plasmon resonance was performed using a BiaCore 3000 instrument as described previously (22, 23). Analyte concentration ranged from 25–500 nm. Experiments were performed independently at least three times, yielding similar results.
Yeast Two-hybrid System
The Matchmaker Gold yeast two-hybrid system was used (Clontech). In brief, full-length rat Hax-1 v1 was cloned into the pGBKT7 vector and served as bait. The rat Hax-1 v1 transcript was split into eight different regions, on the basis of predicted domain structure, that were cloned into the pGADT7 vector and used as prey. The Gold-competent cells, provided by the manufacturer, were simultaneously transformed with the indicated bait and prey constructs and plated on the appropriate selective media. Positive interactants were then streaked on high-stringency plates for confirmation and evaluation of their relative strength, as described by the manufacturer.
Measurement of Intracellular Reactive Oxygen Species (ROS)
The OxiSelect intracellular ROS assay kit (Cell Biolabs, San Diego, CA) was used according to the protocol of the manufacturer. Cells overexpressing select Hax-1 constructs were treated with 0.33 mm H2O2, and intracellular ROS levels were measured after 1 and 4 h of treatment.
Measurement of Cytochrome c Release
The cytochrome c releasing apoptosis assay kit (Abcam, catalog no. ab65311) was used according to the instructions of the manufacturer. HEK293 cells were transfected with select Hax-1 constructs as described above and treated with 0.33 mm H2O2. A pilot time course was performed to determine the time point of maximum cytochrome c release after treatment, which was shown to be 2 h.
RESULTS
Expression Profile of Select Hax-1 Transcripts in the Rat Heart Prior to and Post-induction of Myocardial Infarction
Hax-1 v1 is expressed ubiquitously, albeit at different amounts among different tissues (10). Consistent with this, high transcript levels of Hax-1 v1 have been reported in the mammalian heart (4), whereas no such information is available for other Hax-1 variants. Using quantitative RT-PCR and variant-specific primers, we compared the expression levels of Hax-1 v1, v2, v4, and v6 transcripts in healthy and stressed rat hearts (Fig. 1A; no variant-specific primers can be designed for v3, v5, and v7). All four HAX-1 transcript variants were readily expressed in healthy rat myocardium (Fig. 1B, black bars). Interestingly, a significant increase of ∼12-fold was detected in the transcript levels of v2 12 weeks after transaortic ligation and induction of myocardial infarction (Fig. 1B, gray bars). No statistically significant changes in the transcript levels of v1, v4, and v6 were observed in the stressed rat heart (Fig. 1B). The up-regulation of v2 transcripts was corroborated by a ∼1.5-fold increase at the protein level in the treated hearts, compared with sham-operated control hearts, whereas the protein levels of Hax-1 v1 remained unchanged (Fig. 1, C and D).
FIGURE 1.

Expression profile of Hax-1 variants in the rat myocardium prior to and post-induction of myocardial infarction. A, schematic of Hax-1 variants showing predicted protein domains, corresponding exons (light gray boxes), and introns (dark gray boxes). MTS, mitochondrial targeting sequence; EE, acidic box; BH1 and BH2, Bcl-2 homology domains 1 and 2; PEST, proline (P), glutamic acid (E), serine (S), and threonine (T)-rich motif; TM, predicted transmembrane domain; β6, integrin β6 binding domain. The numbers on top indicate amino acid residues defining protein domains. The location of the primers that were used for amplification of Hax-1 variants in quantitative RT-PCR is denoted with arrows. Of note is that we adopted the Hax-1 structural organization presented by Suzuki and colleagues (3) for consistency purposes, although the presence of the BH1, BH2, and transmembrane domains has been disputed recently (5). B, quantitative RT-PCR analysis of the expression levels of Hax-1 v1, v2, v4, and v6 in rat myocardium prior to and 12 weeks post-induction of myocardial infarction (MI). Notably, the v2 transcript was up-regulated ∼12-fold following myocardial infarction (n = 3, Student's t test). *, p < 0.05. Error bars show mean ± S.E. C, immunoblot analysis of Hax-1 v1 and v2 in lysates prepared from rat myocardium prior to and 12 weeks post-induction of myocardial infarction using antibodies to Hax-1. Equal loading was ensured by probing for tubulin. D, quantification of the relative protein levels of Hax-1 v1 and v2 with densitometry and ImageJ software demonstrated that v2 levels are increased ∼1.5-fold 12 weeks post-induction of myocardial infarction (n = 3, Student's t test). *, p < 0.03. Error bars show mean ± S.E.
Hax-1 v1 and v2 Have Antagonistic Roles in Regulating H2O2-induced Apoptosis
Given the increased expression levels of Hax-1 v2 in the rat heart following myocardial infarction, we set forth to examine whether and how v2 regulates cell death. We used HEK293 epithelial cells, which have been used extensively and reliably for such studies (24). We transiently transfected HEK293 cells with rat Hax-1 v1, v2, v1, and v2 or control empty vector(s) (Fig. 2A) and subsequently subjected them to different apoptotic stimuli. Notably, we routinely observed >80% efficiency for single transfections and >70% efficiency for cotransfections. We used a variety of apoptotic stimuli, including thapsigargin, which raises cytosolic Ca2+ concentration by blocking the activity of SERCA (25); calcimycin, a divalent cation ionophore that allows Ca2+ to cross membranes (26)' serum starvation (27); and H2O2, which induces oxidative stress and causes cytochrome c release from the mitochondria, leading to caspase activation (28). We first measured metabolic activity using the XTT assay, which is indicative of cell viability (Fig. 2, B–F). Overexpression of Hax-1 v1 or v2 had no effect on cell viability, compared with control cells expressing the empty vector, following treatment with thapsigargin (Fig. 2B) or calcimycin (C) and after serum starvation (D). These findings are surprising given the direct association of Hax-1 v1 with major Ca2+ regulatory proteins (14, 16, 29). However, overexpression of Hax-1 v1 protected HEK293 cells from H2O2-induced death by ∼1.5-fold, whereas overexpression of Hax-1 v2 exacerbated cell death by ∼0.75-fold compared with control cells (Fig. 2, E and F). Importantly, cotransfection of Hax-1 v1 and v2 in HEK293 cells that yielded equivalent amounts of exogenous v1 and v2 proteins (Fig. 2A) abrogated their anti-apoptotic and prodeath effects, respectively, following exposure to H2O2 (F).
FIGURE 2.

Effects of Hax-1 variants on cell viability following exposure to different insults. A, immunoblot analyses of lysates collected from HEK293 cells 24 h post-transfection overexpressing rat Hax-1 v1 (RV1), v2 (RV2), RV1 and RV2, human v1 (HV1), v4 (HV4), or HV1 and HV4 and probed with a Hax-1 antibody (ab). Immunoreactive bands are denoted with colored dots for ease of identification. Red, endogenous human v1; green, exogenous rat v1; orange, exogenous rat v2; blue, endogenous and exogenous human v1, which comigrate; yellow, exogenous human v4. It is of interest to note that HEK293 cells to do not express detectable levels of endogenous Hax-1 v4 under normal conditions and that endogenous and exogenous HV1 migrate slightly faster than exogenous RV1 in our gel system. To ensure equal loading, a replica immunoblot analysis was probed with an antibody to tubulin. B–E, HEK293 cells overexpressing rat Hax-1 v1 or v2 were treated with 2 μm thapsigargin for 24 h (B) or 4 μm calcimycin for 24 h (C) or were serum-starved (-FBS, lacking FBS) for 24 h (D) or exposed to 0.33 mm H2O2 for 4 h (E). Cell viability was measured using the XTT assay. Rat Hax-1 v1 protected H2O2-treated cells from cell death, as evidenced by the ∼1.4-fold increase in cell viability compared with control cells (n = 3; Student's t test; *, p < 0.001; error bars show mean ± S.E.) but failed to protect thapsigargin- and calcimycin-treated or serum-starved cells. Conversely, rat Hax-1 v2 decreased cell viability of H2O2-treated cells by ∼0.8-fold compared with control cells (n = 3; Student's t test; *, p < 0.04; error bars show mean ± S.E.) but not of thapsigargin- and calcimycin-treated or serum-starved cells. F, HEK293 cells overexpressing RV1, RV2, or RV1 and RV2 and HV1, HV4, or HV1 and HV4 were treated with 0.33 mm H2O2 for 4 h as in D, followed by evaluation of cell viability using the XTT assay. Similarly to the rat variants, human v1 and v4 decreased and exacerbated cell death of H2O2-treated cells by 20 and 10%, respectively, compared with control cells. Importantly, coexpression of rat v1 and v2 or human v1 and v4 counteracted the antiapoptotic effect of v1 and the prodeath effect of v2/v4, resulting in cell viability levels similar to those observed in control cells expressing empty vectors (n = 3; Student's t test; **, p < 0.01; ***, p < 0.03; error bars show mean ± S.E.).
To further confirm these findings, we performed similar experiments using the human homologues of rat v1 and v2; i.e. v1 and v4, respectively (Fig. 2A). Notably, rat v1 and human v1 share ∼84% sequence identity, whereas rat v2 and human v4 share ∼75% identity (4). Similar to the rat variants, overexpression of human v1 in HEK293 cells increased viability by ∼1.7-fold following H2O2 treatment, whereas overexpression of human v4 promoted cell death by ∼0.7-fold (Fig. 2F). Moreover, coexpression of human v1 and v4 counteracted their individual effects on cell survival (Fig. 2F).
We then used FACS analysis to examine how ectopic expression of the rat Hax-1 variants 1 and 2 and human Hax-1 variants 1 and 4 affected apoptosis of HEK293 cells following exposure to H2O2. We performed measurements of annexin V PE and 7-amino-actinomycin D at 4 and 24 h post-treatment to capture early and late cell responses. Approximately 90% of HEK293 cells expressing rat v1, human v1, or a control empty vector were alive at 4 h following H2O2 treatment (Fig. 3A). Interestingly, only ∼80% of HEK293 cells expressing either rat v2 or human v4 were alive at the same time point (Fig. 3A). We then measured the levels of annexin V PE and 7ADD 24 h post-treatment with H2O2. Approximately 80% of HEK293 cells expressing rat or human v1 remained alive at 24 h compared with 50% of control cells, demonstrating the protective effects of rat and human v1 (Fig. 3B). In contrast, <40% of cells expressing rat v2 or human v4 were alive at 24 h, indicating the prodeath effects of rat v2 and human v4 (Fig. 3B). Similar measurements in HEK293 cells coexpressing equivalent amounts of rat v1 and v2 or human v1 and v4 (Fig. 2A) demonstrated that ∼90% of cells were alive at 4 h, whereas 50% of cells remained alive at 24 h, similar to control cells expressing empty vectors at the same time points (Fig. 3, A and B). Taken together, these results demonstrate that rat and human v1 promote cell survival under specific stress conditions (i.e. oxidative stress) by diminishing apoptosis, whereas rat v2 and human v4 exacerbate cell death under the same stress conditions by increasing apoptosis. Interestingly, coexpression of anti-apoptotic v1 and prodeath v2/v4 counteracts their individual effects, resulting in apoptotic levels similar to those of control cells.
FIGURE 3.
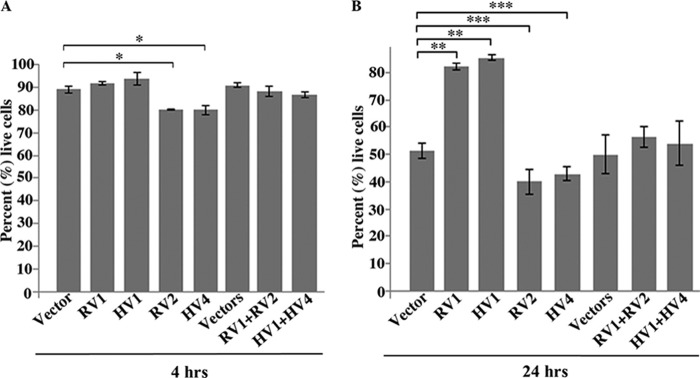
Effects of Hax-1 variants on early and late apoptosis following treatment with H2O2. A, HEK293 cells overexpressing rat Hax-1 v1 (RV1), human Hax-1 v1 (HV1), rat Hax-1 v2 (RV2), human Hax-1 v1 (HV4), RV1 and RV2, or HV1 and HV4 were treated with 0.33 mm H2O2 for 4 h. Apoptosis levels were evaluated by FACS analysis via staining with annexin V PE and 7AAD 4 h following treatment. Only fluorescing cells were included in the analysis to ensure that transfected cells were used. Cells expressing RV1, HV1, RV1 and RV2, or HV1 and HV4 showed similar levels of apoptosis to those of control cells transduced with empty vector(s). In contrast, cells expressing RV2 or HV4 exhibited a 10% increase in apoptosis levels relative to control cells (n = 3, Student's t test). *, p < 0.13. Error bars show mean ± S.E. B, HEK293 cells were transfected and treated as in A but were incubated for an additional 24 h following H2O2 treatment. Cells expressing RV1 or HV1 showed a ∼30% decrease in apoptosis levels compared with control cells expressing the empty vector (n = 3; Student's t test; **, p < 0.01; error bars show mean ± S.E.). In contrast, cells expressing RV2 or HV4 exhibited a ∼10% increase in apoptosis levels relative to control cells (n = 3; Student's t test; ***, p < 0.03; error bars show mean ± S.E.). Importantly, cells coexpressing RV1 and RV2 or HV1 and HV4 showed similar levels of apoptosis to control cells, indicating that coexpression of v1 and v2/v4 eliminated their protective and prodeath effects, respectively.
The Hax-1 Variants Can Form Homotypic and Heterotypic Dimers with High Affinity
To study the molecular mechanism through which Hax-1 v1 and v2/v4 counteract the effect of each other in regulating cell apoptosis, we examined their ability to form homotypic and heterotypic dimers, a property described previously for other members of the Bcl2 family (30, 31). We generated full-length recombinant rat Hax-1 v1 and v2 as GST- and His-tagged proteins (Fig. 4A) and performed a pulldown assay (B). Notably, both v1 and v2 were able to specifically and efficiently form homo- and heterodimers. To quantitate the binding affinity of these homo- and heterotypic interactions in real time, we performed surface plasmon resonance using a Biacore 3000 instrument (Fig. 4, C and D). v1 formed homodimers with a Kd of ∼4 nm and heterodimers with v2 with a Kd of ∼97 nm. Although the binding affinity of the v1 homodimer is ∼25-fold higher than the binding affinity of the v1/v2 heterodimer, both interactions are strong and, most likely, of physiological significance.
FIGURE 4.

Hax-1 variants can form homotypic and heterotypic dimers. A, Coomassie Blue-stained gel showing bacterially expressed rat Hax-1 v1 and v2 produced as GST- and His-tagged proteins. B, GST pulldown assays were performed using equivalent amounts of control GST protein, GST-Hax-1 v1, or GST-Hax-1 v2 bound to glutathione matrices and His-Hax-1 v1 or His-Hax-1 v2. Retention of His-Hax-1 v1 or His-Hax-1 v2 was determined by immunoblot analysis using antibodies (ab) to the His tag. GST-Hax-1 v1, but not control GST protein, specifically and efficiently precipitated both His-Hax-1 v1 and His-Hax-1 v2. Similarly, GST-Hax-1 v2 specifically and efficiently retained His-Hax-1 v2. C, evaluation of the binding affinity of the Hax-1 v1 homodimer in real time using surface plasmon resonance and a Biacore 3000 instrument. GST-Hax-1 v1 was used as a ligand, and His-Hax-1 v1 was used as analyte at five different concentrations ranging from 0–400 nm. A Kd of ∼4 nm was calculated with a χ2 = 0.2 for fitted and obtained sensograms. D, evaluation of the binding affinity of the Hax-1 v1/v2 heterodimer in real time using surface plasmon resonance and a Biacore 3000 instrument. GST-Hax-1 v1 was used as ligand, and His-Hax-1 v2 was used as analyte at five different concentrations, ranging from 0–200 nm. A Kd of ∼97 nm was calculated with a χ2 = 0.06 for fitted and obtained sensograms.
We were unable to efficiently capture Hax-1 v2 to CM5 or nitrilotriacetic acid chips as GST or His fusion proteins, respectively. Therefore, kinetic measurements for the v2 homodimer were not feasible.
The COOH Terminus of Hax-1 Supports Its Ability to Homo- and Heterodimerize
To determine the minimal region that supports the formation of Hax-1 homo- and heterodimers, we generated a series of deletion constructs and tested their ability to interact in the yeast two-hybrid system. We used full-length rat Hax-1 v1 as bait and three consecutive deletion constructs, referred to as A-C, as prey, containing amino acids 1–43, 35–102, and 97–278, respectively, and spanning the entire length of v1 (Fig. 5A). We also generated prey construct D, containing amino acid residues 79–106, which are unique to v7. Prey construct C, including amino acids 97–278, interacted specifically and efficiently with full-length v1 under high-stringency conditions in the yeast system (Fig. 5B). Further deletion analysis of fragment C (aa 97–278) and generation of constructs C1 (aa 97–193) and C2 (aa 189–278) demonstrated that both C1 and C2 were able to support binding to full-length v1. However, C1 did so more efficiently and to the same extent as construct C. Additional deletion analysis and generation of constructs C3 (aa 97–148), C4 (aa 127–202), C5 (aa 188–224), and C6 (aa 225–278) resulted in complete abolishment of binding to full-length v1 (Fig. 5B).
FIGURE 5.

Identification of the minimal binding site that supports the formation of Hax-1 homodimers and heterodimers. A, schematic of the Hax-1 bait and prey clones that were used in the yeast two-hybrid screen, denoting the amino acid residues that were included in each construct. MTS, mitochondrial targeting sequence; EE, acidic box; BH1 and BH2, Bcl-2 homology domains 1 and 2; PEST, proline (P), glutamic acid (E), serine (S), and threonine (T)-rich motif; TM, predicted transmembrane domain; β6, integrin β6 binding domain. B, prey clones C (aa 97–278), C1 (aa 97–193), and C2 (aa 189–278) were able to support binding to bait construct Hax-1 v1, although to different extents, with constructs C and C1 exhibiting a similar and higher affinity than construct C2. C, Coomassie Blue-stained gel showing bacterially expressed GST-Hax-1 v1 and His-Hax-1 clone C, His-Hax-1 clone C1, and His-Hax-1 clone C2 proteins. D, GST pulldown assays were performed using equivalent amounts of control GST-protein and GST-Hax-1 v1 bound to glutathione matrices and His-Hax-1 clone C, His-Hax-1 clone C1, or His-Hax-1 clone C2 proteins. Retention of His-tagged proteins was examined by immunoblot analysis using antibodies to the His tag. GST-Hax-1 v1, but not control GST protein, specifically precipitated all three His-Hax-1 proteins, although with different efficiencies.
We then tested the ability of constructs C (aa 97–278), C1 (aa 97–193), and C2 (aa 189–278) to directly bind to full-length Hax-1 v1 in a pulldown assay. Therefore, we generated recombinant GST-Hax-1 v1, which was immobilized to glutathione matrices and allowed to incubate with equivalent amounts of His-tagged C, C1, or C2 proteins (Fig. 5C). We observed that His-C, His-C1, and His-C2 proteins were able to specifically and directly bind to GST-Hax-1 v1 but not control GST protein (Fig. 5D). Similarly to the yeast two-hybrid data, we consistently observed that His-C and His-C1 were retained more efficiently by GST-Hax-1 v1 compared with His-C2. Thus, it appears that the C1 region containing residues 97–193 is the minimal site that mediates the ability of Hax-1 to form homotypic and heterotypic dimers, whereas the C2 region containing amino acids 189–278 may further contribute to the homo- or heterodimerization capability of Hax-1.
Notably, the COOH-terminal region of Hax-1 (aa 97–278), which also contains the homo- and heterodimerization site, as our experiments indicated, is shared by all known Hax-1 variants and supports binding to a number of other proteins, as reported previously, including phospholamban, SERCA2, HtrA2, PARL, caspase 9, and HS1 (4).
Overexpression of Antiapoptotic or Prodeath Hax-1 Variants Fails to Modulate the Levels of Intracellular ROS but Alters the Cytoplasmic Levels of Cytochrome c following H2O2 Treatment
We then investigated whether the antiapoptotic effects of rat and human v1 and the prodeath effects of rat v2 and human v4 are due to their ability to regulate intracellular ROS levels. HEK293 cells overexpressing rat Hax-1 v1, v2, or v1 and v2 and human Hax-1 v1, v4, or v1 and v4 were treated with H2O2, and the levels of intracellular ROS were measured 1 and 4 h post-treatment. As expected, all cell populations exposed to H2O2 exhibited significantly higher levels of intracellular ROS compared with untreated HEK293 cells expressing empty vector(s) (Fig. 6A). Interestingly though, we did not observe statistically significant differences in the levels of intracellular ROS at 1 h (Fig. 6A) or 4 h (not shown) following H2O2 treatment among cell groups overexpressing v1 or v2/v4 or cell groups coexpressing v1 and v2/v4 compared with control cells containing empty vector(s). Therefore, we then examined whether the opposing effects of v1 and v2/v4 are exerted via regulation of the levels of cytosolic cytochrome c, which we measured in the aforementioned HEK293 cell groups (Fig. 6B). We initially performed a time course (0.5–24 h, data not shown) to determine the time point of maximal cytochrome c release, which we found to be 2 h post-treatment. HEK293 cells overexpressing rat v2 or human v4 exhibited significantly higher levels of cytosolic cytochrome c (Fig. 6B, ∼1.4-fold increase) compared with cells overexpressing rat or human v1, rat v1 and v2, human v1 and v4, or empty vector(s), as evidenced by densitometry of the respective immunoreactive bands. Taken together, these findings indicate that the antagonistic roles of v1 and v2/v4 are not exerted via modulation of intracellular ROS levels but through regulation of cytochrome c release.
FIGURE 6.
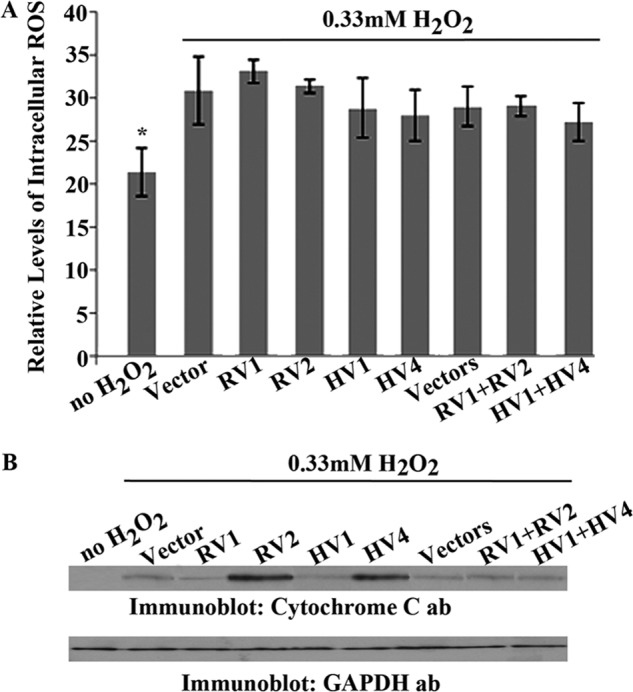
Measurements of intracellular ROS and cytoplasmic cytochrome c levels in HEK293 cells expressing distinct Hax-1 variants or combinations thereof following H2O2 treatment. A, HEK293 cells overexpressing rat Hax-1 v1 (RV1), v2 (RV2), and RV1 and RV2 or human v1 (HV1), v4 (HV4), and HV1 and HV4 were treated with 0.33 mm H2O2 for 1 and 4 h (the 4-h data are not shown because they are statistically equivalent to the 1-h data), and intracellular ROS levels were measured using the Oxiselect ROS assay. Intracellular ROS levels were significantly higher in cells treated with H2O2 compared with untreated control cells (n = 3; Student's t test; *, p < 0.03; error bars show mean ± S.E.). However, there was no statistically significant difference in intracellular ROS levels between treated cells expressing empty vector(s) and treated cells expressing the different Hax-1 variants or combinations thereof (n = 3; Student's t test; *, p > 0.3; error bars show mean ± S.E.). These results indicate that Hax-1 v1 and v2/v4 do not regulate H2O2-induced apoptosis by modulation of intracellular ROS levels. B, HEK293 cells overexpressing RV1, RV2, and RV1 and RV2 or HV1, HV4, and HV1 and HV4 or empty vector(s) were treated with 0.33 mm H2O2 for 2 h. Cell groups were then subjected to subcellular fractionation to collect the cytoplasmic fractions, using the Abcam cytochrome c release kit, which were analyzed by immunoblotting using antibodies (ab) to cytochrome c. As expected, untreated cells contain undetectable levels of cytoplasmic cytochrome c, whereas treated cells expressing empty vector(s), RV1, HV1, RV1 and RV2 or HV1 and HV4 have similar, yet low, amounts of cytoplasmic cytochrome c. In contrast, treated cells expressing RV2 or HV4 show notably higher levels of cytosolic cytochrome c (∼1.4-fold increase compared with control cells), indicating its release from the mitochondria and the initiation of apoptosis. GAPDH served as a loading control.
DISCUSSION
Hax-1 v1 is a ubiquitously expressed protein with established antiapoptotic activity that has been studied extensively in humans and rodents (4, 19). Contrary to the mouse gene, the human and rat HAX-1 genes are heavily spliced, giving rise to at least seven distinct variants that primarily differ in the NH2 terminus (1). In this study, we examined the expression profile of Hax-1 variants in normal and stressed rat myocardia. We observed a significant increase in the transcript and protein levels of Hax-1 v2 12 weeks post-induction of myocardial infarction. Consistent with this, rat v2 and its human homologue v4 exacerbated cell death under select stress conditions that led to oxidative stress and abrogated the antiapoptotic effect of rat and human v1, respectively. Rat v2 and human v4 are structurally similar to the rat and human v1. They only differ in the retention of exon 2, which is present in all known v1 proteins but is partly spliced out in the rat v2 and human v4 isoforms (1). The spliced-out portion of exon 2 encodes 61 amino acids in rats and 48 amino acids in humans, comprising the predicted BH1 and BH2 domains and flanking sequence. Therefore, it appears that the BH1 and BH2 domains present in Hax-1 v1 confer their antiapoptotic activity, as reported previously for antiapoptotic members of the BCL2 family (32–34). Notably, although the existence of the BH1 and BH2 domains in Hax-1 is controversial, our findings highlight the functional importance of this region of Hax-1 encoded by exon 2.
Previous studies have demonstrated that Hax-1 v1 can interact specifically and directly with several proteins that are intimately involved in the regulation of apoptosis via distinct mechanisms. These include phospholamban (9) and SERCA (14), which are major modulators of Ca2+ cycling; mitochondrial proteases presenilins-associated rhomboid-like protein (PARL); high temperature-regulated A2 (HtrA2) (18); and cytoplasmic procaspase 9 (5, 18). Consistent with these findings, it has been postulated that direct binding of Hax-1 v1 to phospholamban and SERCA modulates Ca2+ homeostasis (14, 17, 35). Alternatively, direct binding of Hax-1 v1 to HtrA2 and PARL facilitates the PARL-mediated activation of HtrA2 (18). Activated HtrA2 prevents the accumulation of proapoptotic BAX (BCL2-associated X protein) in the mitochondrial outer membrane, thus inhibiting mitochondrial pore opening, loss of membrane potential, and cytochrome c release (18). Moreover, direct binding of Hax-1 v1 to initiator procaspase 9 precludes its activation, leading to failure of activation of death procaspase 3 (8, 9).
Our studies demonstrate that Hax-1 proteins can also form homotypic and heterotypic dimers with affinities in the low nanomolar range, suggesting that these are strong physiological interactions that take place within the cell. Whether Hax-1 v1 interacts with the battery of identified binding partners as a monomer, homodimer, or heterodimer is currently unknown. Interestingly, though, nearly all known binding interactions of Hax-1 v1, including the formation of homo- and heterodimers, are mediated by amino acid residues present in the COOH-terminal region of the molecule (amino acids 97–278). This finding has two important implications. First, there is likely competition among the partners of Hax-1 v1 for binding, which may be regulated by their relative expression levels, activation status, or subcellular distribution, given that v1 may localize to and possibly oscillate between the sarco/endoplasmic reticulum and the mitochondria (19, 36). Secondly, the COOH-terminal region that contains all currently identified binding sites is shared by all known Hax-1 isoforms, indicating that other Hax-1 variants with potentially distinct, if not opposing, roles from v1 (e.g. v2/v4) may interact directly with the same proteins and, thus, antagonize the anti-apoptotic activity of v1. Therefore, it is possible that Hax-1 v2/v4 may directly bind to HtrA2 and PARL, leading to their inactivation.
Conversely, Hax-1 homodimers or heterodimers may act independently of other Hax-1 binding partners. Thus, homodimerization may be required for v1 to exert its antiapoptotic effect, and for v2/v4 to potentiate cell death following insult. Alternatively, heterodimerization may neutralize the individual effects of v1 or v2/v4 via sequestration. Thus, overexpression of rat v2 or human v4 may promote cytochrome c release following H2O2 treatment by heterodimerizing and sequestering endogenous v1, thus preventing it from facilitating PARL-mediated activation of HtrA2 and BAX removal from the outer mitochondrial membrane (18). Therefore, it becomes apparent that there is a high degree of previously unforeseen complexity that characterizes the homotypic, heterotypic, and allotypic binding interactions of the Hax-1 proteins, which may depend on their ratio and subcellular localization as well as the presence and type of stress exerted to the cell.
The antagonistic roles of Hax-1 v1 and v2/v4 are reminiscent of the opposing roles of the BCL-X splice variants, BCL-Xl and BCL-Xs (37, 38). BCL-Xl binds to and sequesters BAX and BAK (BCL2 homologous antagonist/killer), inhibiting them from forming pores in the outer mitochondrial membrane and, thus, preventing cytochrome c release (33, 37). BCL-Xs, similarly to Hax-1 v2/v4, lacks the BH1 and BH2 domains present in BCL-Xl and Hax-1 v1 and promotes cell death in the presence of a stress stimulus (39). Although the exact mechanism of action of BCL-Xs is still unknown, it has been shown that it can bind to and inhibit or sequester antiapoptotic proteins BCL-Xl and BCL2 (32, 38, 40–42). Notably, the proapoptotic effect of BCL-Xs is counteracted by overexpression of BCL2 or BCL-Xl (30), similarly to Hax-1 v2/v4 and v1.
Several antiapoptotic BCL2 family members, such as BCL2 and BCL-Xl, have been shown to prevent the accumulation of ROS within cells, thus protecting against ROS-induced apoptosis (43, 44). Interestingly, cells overexpressing Hax-1 v1 failed to reduce the levels of intracellular ROS following H2O2 treatment, compared with control cells, but exhibited reduced levels of cytoplasmic cytochrome c. Conversely, cells overexpressing Hax-1 v2/v4 failed to increase the levels of intracellular ROS but displayed increased levels of cytoplasmic cytochrome c. These findings indicate that the antiapoptotic and prodeath effects of Hax-1 proteins are not mediated via regulation of intracellular ROS levels but through modulation of cytochrome c release, possibly via the HtrA2/PARL pathway (18, 45).
Taken together, our findings demonstrate that Hax-1 comprises a family of apoptotic regulators with antagonistic roles in response to oxidative stress. The ability of Hax-1 proteins to form homo- and heterodimers and to interact with diverse proteins involved in the regulation of cell fate indicates that modulation of cell survival or death via the Hax-1 family is highly complex and manifold. Further work is required to delineate the precise molecular mechanisms via which Hax-1 proteins may promote cell survival or apoptosis and to establish how their ratio; subcellular distribution; and preferential homotypic, heterotypic, or allotypic binding interactions contribute to the determination of cell fate under stress conditions.
Acknowledgments
We thank the late Dr. William Stanley and Dr. Peter Hecker (University of Maryland, School of Medicine) for samples of normal and stressed rat myocardium and Dr. Solomon Yap (University of Maryland, School of Medicine) for initial observations on the molecular complexity of the Hax-1 subfamily.
This work was supported, in whole or in part, by NHLBI, National Institutes of Health Grant 1R21HL106197 to (A. K. K.). This work was also supported by a Science, Mathematics, and Research for Transformation (SMART) scholarship (to J. K.).
- v
- variant
- SERCA
- sarco/endoplasmic reticulum Ca2+ ATPase
- PARL
- presenilins-associated rhomboid-like protein
- ROS
- reactive oxygen species
- aa
- amino acids.
REFERENCES
- 1. Grzybowska E. A., Sarnowska E., Konopiński R., Wilczyńska A., Sarnowski T. J., Siedlecki J. A. (2006) Identification and expression analysis of alternative splice variants of the rat Hax-1 gene. Gene 371, 84–92 [DOI] [PubMed] [Google Scholar]
- 2. Lees D. M., Hart I. R., Marshall J. F. (2008) Existence of multiple isoforms of HS1-associated protein X-1 in murine and human tissues. J. Mol. Biol. 379, 645–655 [DOI] [PubMed] [Google Scholar]
- 3. Suzuki Y., Demoliere C., Kitamura D., Takeshita H., Deuschle U., Watanabe T. (1997) HAX-1, a novel intracellular protein, localized on mitochondria, directly associates with HS1, a substrate of Src family tyrosine kinases. J. Immunol. 158, 2736–2744 [PubMed] [Google Scholar]
- 4. Fadeel B., Grzybowska E. (2009) HAX-1. A multifunctional protein with emerging roles in human disease. Biochim. Biophys. Acta 1790, 1139–1148 [DOI] [PubMed] [Google Scholar]
- 5. Jeyaraju D. V., Cisbani G., De Brito O. M., Koonin E. V., Pellegrini L. (2009) Hax1 lacks BH modules and is peripherally associated to heavy membranes. Implications for Omi/HtrA2 and PARL activity in the regulation of mitochondrial stress and apoptosis. Cell Death. Differ. 16, 1622–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sharp T. V., Wang H. W., Koumi A., Hollyman D., Endo Y., Ye H., Du M. Q., Boshoff C. (2002) K15 protein of Kaposi's sarcoma-associated herpesvirus is latently expressed and binds to HAX-1, a protein with antiapoptotic function. J. Virol. 76, 802–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yedavalli V. S., Shih H. M., Chiang Y. P., Lu C. Y., Chang L. Y., Chen M. Y., Chuang C. Y., Dayton A. I., Jeang K. T., Huang L. M. (2005) Human immunodeficiency virus type 1 Vpr interacts with antiapoptotic mitochondrial protein HAX-1. J. Virol. 79, 13735–13746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Han Y., Chen Y. S., Liu Z., Bodyak N., Rigor D., Bisping E., Pu W. T., Kang P. M. (2006) Overexpression of HAX-1 protects cardiac myocytes from apoptosis through caspase-9 inhibition. Circ. Res. 99, 415–423 [DOI] [PubMed] [Google Scholar]
- 9. Vafiadaki E., Sanoudou D., Arvanitis D. A., Catino D. H., Kranias E. G., Kontrogianni-Konstantopoulos A. (2007) Phospholamban interacts with HAX-1, a mitochondrial protein with anti-apoptotic function. J. Mol. Biol. 367, 65–79 [DOI] [PubMed] [Google Scholar]
- 10. Mirmohammadsadegh A., Tartler U., Michel G., Baer A., Walz M., Wolf R., Ruzicka T., Hengge U. R. (2003) HAX-1, identified by differential display reverse transcription polymerase chain reaction, is overexpressed in lesional psoriasis. J. Invest Dermatol. 120, 1045–1051 [DOI] [PubMed] [Google Scholar]
- 11. Trebinska A., Rembiszewska A., Ciosek K., Ptaszynski K., Rowinski S., Kupryjanczyk J., Siedlecki J. A., Grzybowska E. A. (2010) HAX-1 overexpression, splicing and cellular localization in tumors. BMC Cancer 10, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gallagher A. R., Cedzich A., Gretz N., Somlo S., Witzgall R. (2000) The polycystic kidney disease protein PKD2 interacts with Hax-1, a protein associated with the actin cytoskeleton. Proc. Natl. Acad. Sci. U.S.A. 97, 4017–4022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ortiz D. F., Moseley J., Calderon G., Swift A. L., Li S., Arias I. M. (2004) Identification of HAX-1 as a protein that binds bile salt export protein and regulates its abundance in the apical membrane of Madin-Darby canine kidney cells. J. Biol. Chem. 279, 32761–32770 [DOI] [PubMed] [Google Scholar]
- 14. Vafiadaki E., Arvanitis D. A., Pagakis S. N., Papalouka V., Sanoudou D., Kontrogianni-Konstantopoulos A., Kranias E. G. (2009) The anti-apoptotic protein HAX-1 interacts with SERCA2 and regulates its protein levels to promote cell survival. Mol. Biol. Cell 20, 306–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lam A. K., Galione A., Lai F. A., Zissimopoulos S. (2013) Hax-1 identified as a two-pore channel (TPC)-binding protein. FEBS Lett. 587, 3782–3786 [DOI] [PubMed] [Google Scholar]
- 16. Vafiadaki E., Papalouka V., Arvanitis D. A., Kranias E. G., Sanoudou D. (2009) The role of SERCA2a/PLN complex, Ca2+ homeostasis, and anti-apoptotic proteins in determining cell fate. Pflugers Arch. 457, 687–700 [DOI] [PubMed] [Google Scholar]
- 17. Zhao W., Waggoner J. R., Zhang Z. G., Lam C. K., Han P., Qian J., Schroder P. M., Mitton B., Kontrogianni-Konstantopoulos A., Robia S. L., Kranias E. G. (2009) The anti-apoptotic protein HAX-1 is a regulator of cardiac function. Proc. Natl. Acad. Sci. U.S.A. 106, 20776–20781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chao J. R., Parganas E., Boyd K., Hong C. Y., Opferman J. T., Ihle J. N. (2008) Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature 452, 98–102 [DOI] [PubMed] [Google Scholar]
- 19. Yap S. V., Koontz J. M., Kontrogianni-Konstantopoulos A. (2011) HAX-1. A family of apoptotic regulators in health and disease. J. Cell. Physiol. 226, 2752–2761 [DOI] [PubMed] [Google Scholar]
- 20. O'Shea K. M., Khairallah R. J., Sparagna G. C., Xu W., Hecker P. A., Robillard-Frayne I., Des Rosiers C., Kristian T., Murphy R. C., Fiskum G., Stanley W. C. (2009) Dietary omega-3 fatty acids alter cardiac mitochondrial phospholipid composition and delay Ca2+-induced permeability transition. J. Mol. Cell Cardiol. 47, 819–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morgan E. E., Chandler M. P., Young M. E., McElfresh T. A., Kung T. A., Rennison J. H., Tserng K. Y., Hoit B. D., Stanley W. C. (2006) Dissociation between gene and protein expression of metabolic enzymes in a rodent model of heart failure. Eur. J. Heart Fail. 8, 687–693 [DOI] [PubMed] [Google Scholar]
- 22. Kontrogianni-Konstantopoulos A., Jones E. M., Van Rossum D. B., Bloch R. J. (2003) Obscurin is a ligand for small ankyrin 1 in skeletal muscle. Mol. Biol. Cell 14, 1138–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bowman A. L., Catino D. H., Strong J. C., Randall W. R., Kontrogianni-Konstantopoulos A., Bloch R. J. (2008) The Rho-guanine nucleotide exchange factor domain of obscurin regulates assembly of titin at the Z-disk through interactions with Ran binding protein 9. Mol. Biol. Cell 19, 3782–3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thomas P., Smart T. G. (2005) HEK293 cell line. A vehicle for the expression of recombinant proteins. J. Pharmacol. Toxicol. Methods 51, 187–200 [DOI] [PubMed] [Google Scholar]
- 25. Lytton J., Westlin M., Hanley M. R. (1991) Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 266, 17067–17071 [PubMed] [Google Scholar]
- 26. Reed P. W., Lardy H. A. (1972) A23187. A divalent cation ionophore. J. Biol. Chem. 247, 6970–6977 [PubMed] [Google Scholar]
- 27. Kulkarni G. V., McCulloch C. A. (1994) Serum deprivation induces apoptotic cell death in a subset of Balb/c 3T3 fibroblasts. J. Cell Sci. 107, 1169–1179 [DOI] [PubMed] [Google Scholar]
- 28. Stridh H., Kimland M., Jones D. P., Orrenius S., Hampton M. B. (1998) Cytochrome c release and caspase activation in hydrogen peroxide- and tributyltin-induced apoptosis. FEBS Lett. 429, 351–355 [DOI] [PubMed] [Google Scholar]
- 29. Simmen T. (2011) Hax-1. A regulator of calcium signaling and apoptosis progression with multiple roles in human disease. Expert. Opin. Ther. Targets 15, 741–751 [DOI] [PubMed] [Google Scholar]
- 30. Youle R. J., Strasser A. (2008) The BCL-2 protein family. Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 [DOI] [PubMed] [Google Scholar]
- 31. O'Neill J. W., Manion M. K., Maguire B., Hockenbery D. M. (2006) BCL-XL dimerization by three-dimensional domain swapping. J. Mol. Biol. 356, 367–381 [DOI] [PubMed] [Google Scholar]
- 32. Gross A., McDonnell J. M., Korsmeyer S. J. (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 13, 1899–1911 [DOI] [PubMed] [Google Scholar]
- 33. Cheng E. H., Wei M. C., Weiler S., Flavell R. A., Mak T. W., Lindsten T., Korsmeyer S. J. (2001) BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 8, 705–711 [DOI] [PubMed] [Google Scholar]
- 34. Aouacheria A., Rech de Laval V., Combet C., Hardwick J. M. (2013) Evolution of Bcl-2 homology motifs. Homology versus homoplasy. Trends Cell Biol. 23, 103–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lam C. K., Zhao W., Cai W., Vafiadaki E., Florea S. M., Ren X., Liu Y., Robbins N., Zhang Z., Zhou X., Jiang M., Rubinstein J., Jones W. K., Kranias E. G. (2013) Novel role of HAX-1 in ischemic injury protection involvement of heat shock protein 90. Circ. Res. 112, 79–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yap S. V., Vafiadaki E., Strong J., Kontrogianni-Konstantopoulos A. (2010) HAX-1. A multifaceted antiapoptotic protein localizing in the mitochondria and the sarcoplasmic reticulum of striated muscle cells. J. Mol. Cell Cardiol. 48, 1266–1279 [DOI] [PubMed] [Google Scholar]
- 37. Kim R. (2005) Unknotting the roles of Bcl-2 and Bcl-xL in cell death. Biochem. Biophys. Res. Commun. 333, 336–343 [DOI] [PubMed] [Google Scholar]
- 38. Tao W., Kurschner C., Morgan J. I. (1998) Bcl-xS and Bad potentiate the death suppressing activities of Bcl-xL, Bcl-2, and A1 in yeast. J. Biol. Chem. 273, 23704–23708 [DOI] [PubMed] [Google Scholar]
- 39. Willimott S., Merriam T., Wagner S. D. (2011) Apoptosis induces Bcl-XS and cleaved Bcl-XL in chronic lymphocytic leukaemia. Biochem. Biophys. Res. Commun. 405, 480–485 [DOI] [PubMed] [Google Scholar]
- 40. Farrow S. N., Brown R. (1996) New members of the Bcl-2 family and their protein partners. Curr. Opin. Genet. Dev. 6, 45–49 [DOI] [PubMed] [Google Scholar]
- 41. Michels J., Kepp O., Senovilla L., Lissa D., Castedo M., Kroemer G., Galluzzi L. (2013) Functions of BCL-X L at the interface between cell death and metabolism. Int. J. Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lindenboim L., Yuan J., Stein R. (2000) Bcl-xS and Bax induce different apoptotic pathways in PC12 cells. Oncogene 19, 1783–1793 [DOI] [PubMed] [Google Scholar]
- 43. Esposti M. D., Hatzinisiriou I., McLennan H., Ralph S. (1999) Bcl-2 and mitochondrial oxygen radicals. New approaches with reactive oxygen species-sensitive probes. J. Biol. Chem. 274, 29831–29837 [DOI] [PubMed] [Google Scholar]
- 44. Ouyang Y. B., Carriedo S. G., Giffard R. G. (2002) Effect of Bcl-x(L) overexpression on reactive oxygen species, intracellular calcium, and mitochondrial membrane potential following injury in astrocytes. Free Radic. Biol. Med. 33, 544–551 [DOI] [PubMed] [Google Scholar]
- 45. Yoshioka H., Katsu M., Sakata H., Okami N., Wakai T., Kinouchi H., Chan P. H. (2013) The role of PARL and HtrA2 in striatal neuronal injury after transient global cerebral ischemia. J. Cereb. Blood Flow Metab. 33, 1658–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]