

Background: Elevation of cerebral cholesterol turnover in mice due to increased CYP46A1 expression has palliative effects on hallmarks of Alzheimer disease.
Results: CYP46A1 could also be activated post-translationally by drugs in vitro and in vivo.
Conclusion: CYP46A1 is a viable therapeutic target.
Significance: Pharmacologic stimulation of CYP46A1 and cerebral cholesterol turnover may lead to new therapeutic strategies for the treatment of brain disorders.
Keywords: Alzheimer Disease, Brain Metabolism, Cholesterol, Cholesterol Metabolism, Cytochrome P450
Abstract
Cytochrome P450 46A1 (CYP46A1) is a brain-specific cholesterol 24-hydroxylase responsible for the majority of cholesterol elimination from the brain. Genetically increased CYP46A1 expression in mice leads to improved cognition and decreases manifestations of Alzheimer disease. We found that four pharmaceuticals (efavirenz (EFV), acetaminophen, mirtazapine, and galantamine) prescribed for indications unrelated to cholesterol maintenance increased CYP46A1 activity in vitro. We then evaluated the anti-HIV medication EFV for the mode of interaction with CYP46A1 and the effect on mice. We propose a model for CYP46A1 activation by EFV and show that EFV enhanced CYP46A1 activity and cerebral cholesterol turnover in animals with no effect on the levels of brain cholesterol. The doses of EFV administered to mice and required for the stimulation of their cerebral cholesterol turnover are a hundred times lower than those prescribed to HIV patients. At such small doses, EFV may be devoid of adverse effects elicited by high drug concentrations. CYP46A1 could be a novel therapeutic target and a tool to further investigate the physiological and medical significance of cerebral cholesterol turnover.
Introduction
The brain is perhaps the only organ in the human body that cannot acquire cholesterol from the systemic circulation because the blood-brain barrier is impermeable to cholesterol. Accordingly, the major source of cerebral cholesterol is local synthesis, and cholesterol removal relies mainly on enzymatic processing carried out by cytochrome P450 46A1 (CYP46A1) (1). CYP46A1 is normally localized in the endoplasmic reticulum (ER)2 of neurons, where it removes excess cholesterol by metabolism to a membrane-permeable (24S)-hydroxycholesterol (24-HC) (2, 3). Once 24-HC is formed, it rapidly diffuses to the systemic circulation and binds to LDL and HDL, which deliver this oxysterol to the liver for degradation to bile acids (4–6). Genetic ablation of Cyp46a1 expression in mice demonstrates that CYP46A1 controls not only cholesterol removal from the brain but also the rate at which cholesterol is turned over in this organ (7). The steady-state levels of cerebral cholesterol are unchanged in Cyp46a1−/− mice because there is a compensatory reduction by ∼40% in the rate of cerebral cholesterol biosynthesis (7). The rate of cholesterol turnover is thus decreased in the Cyp46a1−/− brain, affecting memory and learning of Cyp46a1−/− animals (8). Conversely, CYP46A1 transgenic mice have increased levels of a number of cholesterol precursors (1.5–1.7-fold), indicating increased cholesterol biosynthesis to balance an ∼2-fold increase in cholesterol elimination via cholesterol 24-hydroxylation (9, 10). Aged transgenic females also show an improvement in spatial memory compared with the matched wild-type mice (10). A positive effect, namely mitigation of cognitive deficits, was also observed in mouse models of Alzheimer disease when cerebral CYP46A1 expression in these animals was increased as a result of adenoviral CYP46A1 delivery or Acat1 ablation (11, 12). In addition, increased CYP46A1 expression reduces the formation of the amyloid-β peptide (11, 12). Collectively, animal data suggest that increasing Cyp46a1 levels or enzyme activity could be beneficial for humans of older age, leading to cognitive enhancements in healthy individuals and slowing the disease progression in patients with Alzheimer disease.
Predominant expression in the brain (13) makes CYP46A1 attractive as a new therapeutic target and a tool for studies of cerebral cholesterol homeostasis. This laboratory is investigating the potential of existing medications to stimulate CYP46A1 activity in the brain. We were encouraged by our previous structural studies demonstrating that the CYP46A1 active site is conformationally flexible (14, 15). We also found that in the reconstituted system in vitro, cholesterol 24-hydroxylation by purified recombinant CYP46A1 can be stimulated up to 145% by some of the therapeutic agents (14). Recently, we found additional pharmaceuticals that modulate CYP46A1 activity in vitro and are approved for human use in the United States by the Food and Drug Administration (16). Because the activation of purified CYP46A1 was only moderate in this screen, in the present work, we retested the identified P450 activators in isolated bovine brain microsomes, a physiologically more relevant system. We found that pharmacologic activation of CYP46A1 was strong (up to 11-fold) in this system. Therefore, we conducted additional studies in vitro and in vivo in mice. We demonstrate that the anti-HIV medication efavirenz (EFV) stimulates CYP46A1 in vitro and in vivo, leading to an increase in cholesterol turnover in mouse brain. We also propose a model for CYP46A1 activation by EFV. Our findings have direct medical relevance and could immediately be tested on elderly humans for enhancement of cognition and effect on progression of Alzheimer disease.
EXPERIMENTAL PROCEDURES
Chemicals
The S-isomer of EFV (brand name Sustiva) was purchased from Toronto Research Chemicals Inc. Phenacetin, acetaminophen, mirtazapine, galantamine, agomelatine, and huperzine were from Sigma-Aldrich. Drugs were dissolved in 100% methanol, whereas cholesterol was dissolved either in 4.5% aqueous 2-hydroxypropyl-β-cyclodextrin to prepare a 1 mm stock for spectral binding assay or in 45% aqueous 2-hydroxypropyl-β-cyclodextrin to prepare a 10 mm stock for optimized enzyme assay. No corrections were done for the affinity of cholesterol for 2-hydroxypropyl-β-cyclodextrin and other components in the assay buffer. Hence, the determined Km and Kd values represent the apparent values.
CYP46A1 Expression and Purification
Full-length and truncated (Δ(2–50)) recombinant human enzymes were heterologously expressed in Escherichia coli and purified as described (16, 17).
Enzyme Assay with Isolated Brain Microsomes
This assay was as described (16). Briefly, microsomes were obtained from the gray matter of bovine temporal lobe, one of the brain regions where CYP46A1 expression is abundant (13). Brain microsomes (1 mg of total protein) were used for reconstitution with 1 μm cytochrome P450 oxidoreductase (OR) and varying concentrations of a probe drug (10–60 μm) in a total volume of 1 ml of 50 mm potassium phosphate buffer (pH 7.2) containing 100 mm NaCl. CYP46A1-mediated 24-hydroxylation of endogenous cholesterol present in the microsomes (∼90 nmol/mg of protein) was initiated by the addition of a NADPH-regenerating system (5 mm NADPH, 50 mm glucose 6-phosphate, and 10 units of glucose-6-phosphate dehydrogenase) and proceeded for 30 min at 37 °C. The enzyme reaction was terminated by the addition of 6 ml of Folch reagent (2:1 (v/v) chloroform/methanol), followed by the determination of the 24-HC content in the organic extract by gas chromatography-mass spectrometry as described (18) using deuterated 24-HC as an internal standard.
Optimized Enzyme Assay with Purified CYP46A1
Incubations were carried out as described (14), always in 1 ml of 50 mm potassium phosphate buffer (pH 7.2) containing 100 mm NaCl, 40 μg of dilauroylglycerol-3-phosphatidylcholine, and 0.02% CYMAL-6. Dilauroylglycerol-3-phosphatidylcholine-containing liposomes were added to the buffer first, followed by the addition of a drug, purified P450 (0.5 μm), OR (1.0 μm), 10 nm [3H]cholesterol, and unlabeled cholesterol (10, 20, or 40 μm when a fixed substrate concentration was used or varying concentrations when kinetic experiments were carried out). NADPH (final concentration of 1 mm) was added to initiate the enzyme reaction, which was carried out for 30 min at 37 °C. The reaction was terminated by the addition of CH2Cl2 (2 × 2 ml), which also served to extract reaction products and unused cholesterol. The organic extract was evaporated, dissolved in CH3CN, and analyzed by HPLC as described (19).
Spectral Binding Assay
The truncated form of CYP46A1 (CYP46A1Δ(2–50)) was used in this assay, as CYP46A1 activation was also observed with the truncated enzyme (Fig. 1). Spectral titrations were carried out as described (20), namely in 1 ml of 50 mm potassium phosphate buffer (pH 7.2) containing 100 mm NaCl and 0.5 μm CYP46A1Δ(2–50), the same P450 concentration that was used in the in vitro enzyme assays.
FIGURE 1.

EFV activation of CYP46A1Δ(2–50). Under identical assay conditions (see “Experimental Procedures” for optimized enzyme assay), full-length CYP46A1 and the truncated mutant (CYP46A1Δ(2–50)) showed similar activation of cholesterol 24-hydroxylation by EFV. The results of these and all other in vitro experiments represent the means ± S.D. of triplicate measurements. Statistical analysis is described under “Experimental Procedures”. ***, p < 0.001.
Computational Predictions of EFV Binding on the CYP46A1 Surface
An in silico search for potential allosteric binding sites on CYP46A1 was carried out as described (16) and included the protein surface in the gridded space. The IronTM simulation software (Pharmatrope Ltd.) was used, which employs the fragment-based approach (21).
Administration of EFV to Mice
All animal-handling procedures and experiments were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University. Mice (strain C57BL/6J, 3-month-old-males and females) were purchased from The Jackson Laboratory and housed in the Animal Resource Center at Case Western Reserve University. Drug administration was initiated after a 1-week adaptation period. When EFV was delivered by gavage, each mouse received daily 0.1 ml of the drug solution in 1% Tween 80 from one of the four stocks (10.1, 21.5, 43.0, or 86.0 μg/ml), corresponding to drug doses of 0.04, 0.08, 0.16, and 0.32 mg of EFV/kg of mouse body weight, respectively. These doses are 384–3075-fold lower than that given to HIV patients (600 mg/kg/day), if we use a 12.3 coefficient to account for a difference in the surface area/body weight ratio between mice and humans and assume that 60 kg is an average human body weight. The control group contained animals receiving the vehicle (0.1 ml of 1% Tween 80). Drug solutions were prepared freshly before each administration. When EFV was administered in drinking water, the drug was dissolved at 0.42 and 1.68 mg/liter concentrations and kept in light-protected bottles. This water was provided ad libitum. Each day, mice consumed 6.4 and 4.0 ml of 0.42 and 1.68 mg/liter of EFV-containing water, respectively. Thus, the EFV doses that were delivered were 0.09 and 0.22 mg/kg/day. No changes in body weight relative to the untreated animals or signs of toxicity were evident in EFV-treated mice for the duration of experiments, as assessed by lack of mortality, hunched posture, lethargy, fur ruffling, or respiratory distress. Prior to death, the animals were fasted overnight. The following morning, the animals were killed, and their brains were immediately isolated, with the left hemisphere used for sterol quantifications. Blood was collected as well via cardiac puncture and used for serum preparation as described (22).
Quantifications of Cerebral Sterols and Plasma Levels of 24-HC
This was carried out as described (18, 23) by isotope dilution gas chromatography-mass spectrometry using deuterated sterol analogs as internal standards.
Data Analysis
All data represent the mean ± S.D. All in vitro assays were carried out in triplicate. All quantifications in mice represent the average of the individual measurements in three different animals per one EFV dose and time point. Statistical significance of mean differences was determined either by Student's two-tailed unpaired t test (see Figs. 1, 3, 5, and 8) or by two-way repeated measures analysis of variance (see Fig. 9). If analysis of variance showed major interactions, post hoc contrasts between the same time points in different groups were performed using the t test. All statistical analyses were performed using GraphPad Prism software (GraphPad Software, San Diego, CA).
FIGURE 3.
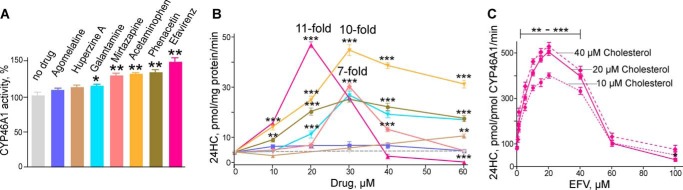
Effect of different drugs on CYP46A1 activity in the screening enzyme assay (A), assay with isolated brain microsomes (B), and optimized enzyme assay at three fixed cholesterol concentrations (C). Bars and lines of the same color pertain to the same drug. Conditions of the assays and statistical analysis are described under “Experimental Procedures.” *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 5.

The extent of CYP46A1 activation in the in vitro enzyme assay depends on the order in which the components of the reconstitution system are mixed. The solid line shows CYP46A1 activation when the enzyme is first mixed with EFV, followed by the addition of 20 μm cholesterol. The dashed line shows CYP46A1 activation when the P450 is first mixed with 20 μm cholesterol, followed by the addition of EFV. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 8.
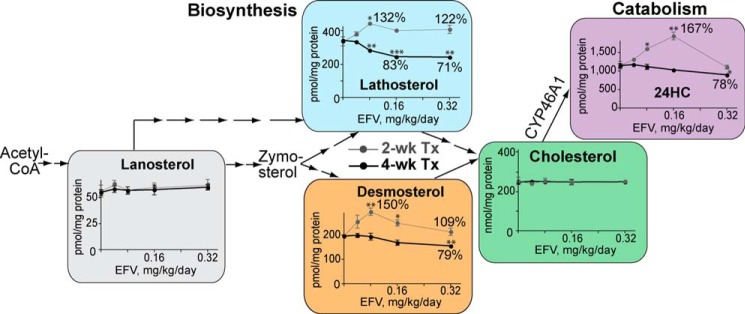
Effect of different doses of EFV on the cerebral sterol profile in mice (n = 3 per dose and time point) when the drug is delivered by gavage. Thin gray lines correspond to the 2-week treatment (2-wk Tx), whereas thick black lines reflect the 4-week treatment (4-wk Tx). The percentages of the sterol change relative to the untreated control are also shown, as is a scheme of the cholesterol biosynthesis pathway. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 9.
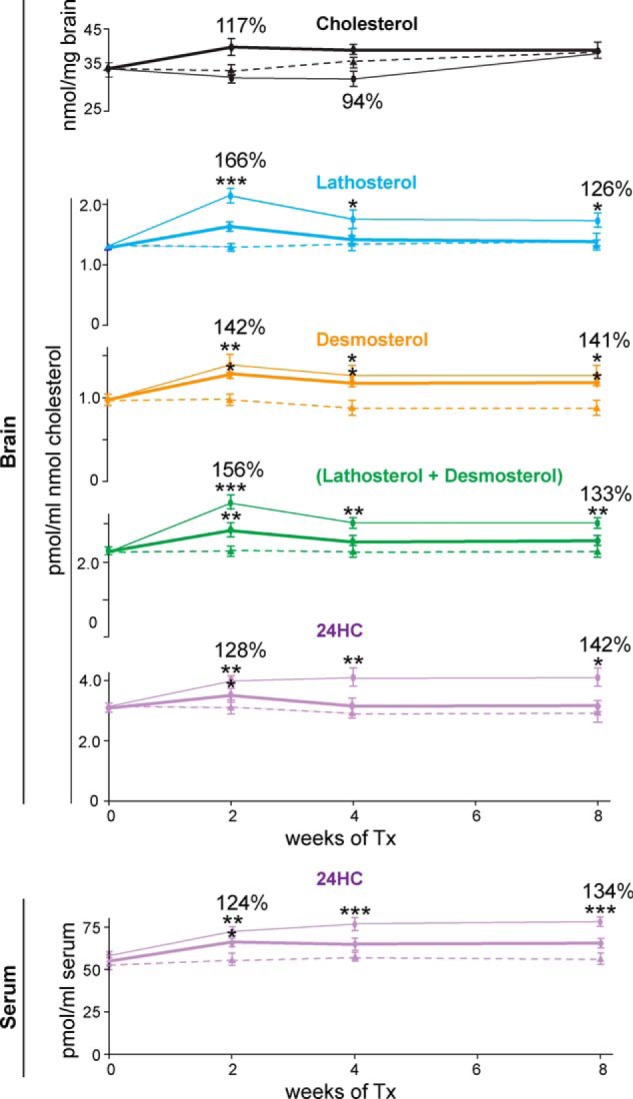
Effect of different doses of EFV on sterol content in mouse brain and serum (n = 3 per dose and time point) when the drug is delivered in drinking water. Dashed lines, untreated controls; thin lines, 0.09 mg/kg/day dose; thick lines, 0.22 mg/kg/day dose. Lines of the same color pertain to the same sterol. The percentages of the sterol increase relative to the untreated control are also shown. Tx, treatment. Please note that sterol normalizations are different compared with those in Fig. 8 and that one 1 mg of wet brain contains ∼115 μg of total protein. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
RESULTS
Pharmacologic Stimulation of Cholesterol 24-Hydroxylation in Isolated Brain Microsomes
CYP46A1 is the only known microsomal enzyme that can generate 24-HC (7, 13). Therefore, the production of this metabolite from ER cholesterol reflects solely the activity of CYP46A1. In this work, we re-evaluated seven drugs (Fig. 2) that were identified as potential P450 activators in our previous in vitro screening utilizing purified recombinant enzyme (14, 16). All of these compounds cross the blood-brain barrier, and the majority are used on a chronic treatment basis with the exception of phenacetin, which was withdrawn from the United States market. EFV is an anti-HIV medication; phenacetin and its metabolite acetaminophen are analgesics; mirtazapine and agomelatine are antidepressants; and galantamine and huperzine are prescribed for symptomatic treatments of Alzheimer disease. In the screening assay with purified CYP46A1, the seven selected pharmaceuticals activated the enzyme to different degrees: very weakly (by 110–115%, agomelatine, huperzine, and galantamine), weakly (by 130–132%, mirtazapine and acetaminophen), or moderately (by 145–150%, phenacetin and EFV) (Fig. 3A). In contrast, when the drugs were tested in isolated bovine brain microsomes, CYP46A1 activation was generally much stronger (Fig. 3B). Five of the seven drugs modulated CYP46A1 activity in a concentration-dependent manner, activating CYP46A1 at low concentrations (up to 30 μm) and inhibiting the enzyme at higher concentrations. The two strongest CYP46A1 activators were EFV and acetaminophen, increasing the formation of 24-HC by ∼10–11-fold. Mirtazapine, phenacetin, and galantamine stimulated CYP46A1 activity by 6–7-fold. The maximal CYP46A1 activation was observed at 30 μm drug concentrations, except for EFV, which activated CYP46A1 at a lower concentration (20 μm). Thus, we chose EFV for our subsequent studies.
FIGURE 2.

Chemical structures of the drugs tested in this study.
In Vitro Studies of the Mechanism of CYP46A1 Activation by EFV
Weak CYP46A1 activation in the screening assay relative to the strong P450 activation in isolated brain microsomes suggested that the enzyme activation probably depends on the ratio between CYP46A1, substrate (cholesterol), and activating drug. Indeed, each of the incubations with isolated brain microsomes contained ∼0.4 pmol of CYP46A1 (24), 90 nmol of endogenous cholesterol, and 10–60 nmol of the probe drug, i.e. cholesterol was at the saturating concentration for P450. In contrast, our screening assay contained 0.5 nmol of CYP46A1, 2.7 nmol of cholesterol, and 48 nmol of the probe drug. 2.7 μm cholesterol was the subsaturating concentration because the Km of CYP46A1 for cholesterol is 5.4 μm (14). Therefore, we investigated how varying amounts of EFV (5–100 nmol) affect CYP46A1 activity at three different saturating cholesterol concentrations (10, 20, and 40 nmol) while retaining the same 0.5 nmol of CYP46A1. In this assay, which we call the optimized in vitro assay, CYP46A1 activation by EFV was much stronger (up to 6-fold) (Fig. 3C) than in our screening assay (Fig. 3A). Moreover, the shape of the concentration dependence curves in the optimized in vitro assay was similar to that in the assay with isolated brain microsomes. The curve had a bell shape, with the maximal CYP46A1 activation observed at 20 μm EFV, with a subsequent decrease in the CYP46A1 stimulation and ultimately enzyme inhibition as the EFV concentrations were increased. The fact that the maximal activating concentration of EFV was the same at all three cholesterol concentrations suggested that there were no competition between cholesterol and EFV for binding to CYP46A1, possibly because the binding sites for cholesterol and EFV at activating EFV concentrations are different. These data also suggested that the 20 μm concentration is the saturating EFV concentration for binding to this site, different from that for cholesterol.
We also tested how EFV affects the kinetic parameters of CYP46A1-mediated cholesterol hydroxylation at activating (20 μm) and inhibiting (100 μm) drug concentrations (Fig. 4A and Table 1). At both concentrations, EFV did not significantly alter the CYP46A1 Km for cholesterol. The major change was in the kcat, which increased by 6.7-fold at the activating EFV concentration and decreased by 3.0-fold at the inhibiting drug concentration. Because this change could reflect the effect of EFV on the CYP46A1 interactions with the redox partner OR, we then measured the CYP46A1 Km and kcat for OR at the activating and inhibiting EFV concentrations (Fig. 4B and Table 1). As in kinetic experiments with cholesterol, the major effect was on the kcat, not Km.
FIGURE 4.

Effect of EFV on the enzymatic and spectral properties of CYP46A1. A and B, rates of cholesterol 24-hydroxylation versus increasing concentrations of cholesterol and OR, respectively, in the absence (black lines) and presence of 20 μm (gray lines) and 100 μm (red lines) EFV. The data were fit to the Michaelis-Menten equation. C and E, the amplitude (ΔA) of cholesterol- and EFV-induced changes, respectively, in the difference spectrum of CYP46A1. D, cholesterol-induced changes in the CYP46A1 ΔA in the presence of 20 μm EFV. F, EFV-induced changes in the CYP46A1 ΔA in the presence of 20 μm cholesterol. The spectral data were fit to a hyperbolic equation (when the apparent spectral Kd was higher than the enzyme concentration), to a quadratic equation (when the apparent spectral Kd was lower than the enzyme concentration assuming a 1:1 stoichiometry), or to the Hill equation (when cooperativity of binding was observed). Insets show ligand-induced P450 difference spectra indicating the positions of the spectral peaks and troughs. The schematic representation of ligand binding (green rectangles for cholesterol and magenta triangles for EFV) to CYP46A1 (open circles) is also shown and is discussed in detail in Fig. 7.
TABLE 1.
Effect of EFV on enzymatic and spectral properties of CYP46A1
The results represent the mean ± S.D. of triplicate measurements. AU, absorbance units.
| Assay/varying ligand/constant ligand(s) | Properties of CYP46A1 |
|
|---|---|---|
| Km | kcata | |
| μm | min−1 | |
| Enzyme activity/cholesterol/1 μm OR | 2.6 ± 0.4 | 0.09 ± 0.01 |
| Enzyme activity/cholesterol/20 μm EFV | 3.3 ± 0.2 | 0.6 ± 0.2 |
| Enzyme activity/cholesterol/100 μm EFV | 3.7 ± 0.5 | 0.03 ± 0.01 |
| Enzyme activity/OR/20 μm cholesterol | 0.5 ± 0.3 | 0.09 ± 0.02 |
| Enzyme activity/OR/20 μm cholesterol, 20 μm EFV | 0.6 ± 0.4 | 0.6 ± 0.2 |
| Enzyme activity/OR/20 μm cholesterol, 100 μm EFV | 0.5 ± 0.4 | 0.05 ± 0.01 |
| Kd | ΔAmax | λmax/λmin, difference spectrum | |
|---|---|---|---|
| μm | AU | ||
| Spectral assay/cholesterol | 0.08 ± 0.01 | 0.07 ± 0.01 | 390/422 |
| Spectral assay/cholesterol/20 μm EFV | 0.03 ± 0.01 | 0.003 ± 0.001 | 390/427 |
| Spectral assay/EFV | 5.0 ± 0.6 | 0.015 ± 0.001 | 434/400 |
| Spectral assay/EFV/20 μm cholesterol | K = 10.2 ± 0.3, H = 2.1 | 0.007 ± 0.001 | 430/394 |
a Normalized per nmol of P450.
An important finding from the enzyme activity measurements was that the extent of CYP46A1 activation by EFV depended on the order in which the components of the reconstituted system were mixed. Strong activation was observed only when EFV was added to CYP46A1 first, prior to the addition of cholesterol. If EFV was added to CYP46A1 after the addition of cholesterol, the maximal enzyme activation was ∼10-fold lower (Fig. 5). In contrast, CYP46A1 inhibition did not depend on the mixing order and always occurred at high EFV concentrations. The dependence of the CYP46A1 activation on the mixing order prompted an investigation of drug and cholesterol binding by spectral assay (25), which provides information about the coordination and microenvironment of the P450 heme iron.
Titrations were first carried out on resting CYP46A1. In this state, there is no substrate in the enzyme active site, and the P450 heme iron is hexacoordinated, with a water molecule being the distal (sixth) iron ligand (14). Both cholesterol and EFV elicited spectral shifts in the difference spectrum of resting CYP46A1 (Fig. 4, C and E), enabling determination of their apparent spectral Kd values: 0.08 μm for cholesterol and 5.0 μm for EFV (Table 1). However, the types of the CYP46A1 spectral responses upon the addition of these two ligands were different. Cholesterol induced the so-called type I spectral response (26, 27), occurring in P450s when a water molecule coordinating the heme iron is displaced, leaving the iron five-coordinated (28). Conversely, EFV induced a type II spectral response, reflecting the coordination of the heme iron by a nitrogen atom from the added compound (29, 30). Very often, but not always, type II ligands are inhibitors rather than substrates for P450s (15, 31). Next, we saturated CYP46A1 with 20 μm EFV and carried out P450 titrations with cholesterol (Fig. 4D). The cholesterol spectral Kd decreased to ∼0.03 μm, indicating tighter substrate binding at the activating drug concentrations. In a reverse experiment, CYP46A1 was first saturated with 20 μm cholesterol and then titrated with EFV (Fig. 4F). The drug binding became cooperative, with a Hill coefficient equal to 2.1, suggesting two binding sites (32). Thus, spectral titrations of CYP46A1 revealed the following. First, when added individually, both EFV and cholesterol bind to the CYP46A1 active site. However, the Kd values of the ligands for CYP46A1 differ significantly (5.0 and 0.08 μm, respectively), and EFV likely inhibits CYP46A1. Second, when added sequentially, the ligand that binds first affects binding of a second ligand. If EFV binds first, subsequent cholesterol binding becomes tighter (0.03 versus 0.08 μm Kd in the absence of EFV). When cholesterol is added first, EFV binding becomes cooperative and reveals the presence of a second EFV-binding site. To identify this site, the computational prediction of EFV binding to the CYP46A1 surface was carried out. This led to the identification of three putative allosteric sites, all in the P450 regions facing the cytosol (Fig. 6). On the basis of our data, we generated a model explaining CYP46A1 activation at low EFV concentrations and inhibition at high drug concentrations (Fig. 7).
FIGURE 6.

Computational analysis of EFV binding to the CYP46A1 surface. The CYP46A1 crystal structure is shown in light gray, cholesterol sulfate in the enzyme active site is shown in green, and EFV bound to each (A-C) of the three putative allosteric sites is shown in magenta. The heme group is shown in salmon, and amino acid residues are shown in dark gray. The black horizontal lines separate the cytosol (above) and the lipid bilayer (below). The proposed membrane orientation is taken from Ref. 49. The lowest energy EFV-binding site is near Lys-216 (A), whereas the sites near Phe-468 (B) and Pro-429 (C) have ∼3-fold higher relative free energies of EFV binding. The Lys-216-containing site is the most likely allosteric site for EFV.
FIGURE 7.
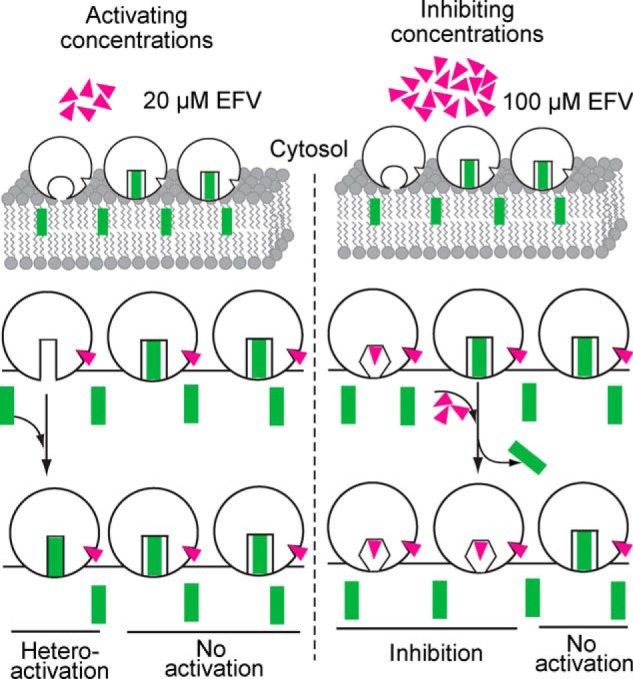
Model for CYP46A1 activation and inhibition by EFV. Molecules of the enzyme, drug, and substrate are shown as open circles (CYP46A1), magenta triangles (EFV), and green rectangles (cholesterol), respectively. A diagram for the lipid bilayer is also shown. We suggest that only one ligand, either cholesterol or EFV, can bind to the CYP46A1 active site, with ligand binding altering the shape of the active site cavity. Our previous crystallographic studies showed that when the substrate (cholesterol sulfate) is present, there is no space in the CYP46A1 active site for a second molecule the size of EFV (14). Also, only one drug molecule was found in the substrate-binding cavity in all seven CYP46A1-drug complexes crystallized so far (15, 16, 20, 50). Furthermore, in all ligand-bound CYP46A1 crystal structures, the P450 active site undergoes conformational changes to better fit the ligand. Our earlier studies indicated that the entrance to the CYP46A1 active site is embedded in the lipid bilayer, i.e. cholesterol enters CYP46A1 through the membrane (49). We envision that when EFV is at low concentrations (e.g. 20 μm) in vitro or in vivo, it binds to the CYP46A1 allosteric site, as this site is more accessible than the P450 active site embedded in the lipid bilayer. EFV binding to the allosteric site does not change the shape of the CYP46A1 active site when cholesterol is there, as this shape is rigidified by substrate-protein interactions. However, EFV alters the shape of the active site of cholesterol-free CYP46A1, making cholesterol binding tighter and enzyme catalysis more efficient. This would explain the enzyme's activation at low doses of EFV. The dependence of CYP46A1 activation on the addition of EFV prior to the addition of cholesterol in Fig. 5 supports this notion. However, when the concentration of EFV is high (e.g. 100 μm), not only does the drug bind to the allosteric site, but it also begins to bind to the active site and either prevents cholesterol from binding or displaces cholesterol from the active site. This causes CYP46A1 inhibition, which does not depend on whether EFV binds to substrate-free or substrate-bound CYP46A1. Our model is based on the assumption that despite its abundance in the ER, cholesterol may not always be present in the CYP46A1 active site when EFV binds to the allosteric site. This could be due to the low catalytic efficiency of CYP46A1 (its in vitro kcat for cholesterol is only ∼0.1 min−1 (14)), suggesting that the time between the CYP46A1 catalytic cycles may be sufficient for a drug (EFV) to occupy the CYP46A1 allosteric site. Alternatively, EFV could bind to the allosteric site when cholesterol still occupies the active site and elicit conformational changes in CYP46A1 after the substrate is metabolized to the product, and the product has left the active site.
EFV Activates CYP46A1 and Cerebral Cholesterol Turnover in Vivo in Mice
EFV was first administered by gavage each day for 2 or 4 weeks. Four drug doses (0.04, 0.08, 0.16, and 0.32 mg/kg/day) were tested for the effect on the sterol profile in the brain (Fig. 8). The measured sterols were cholesterol, three cholesterol precursors (lanosterol, lathosterol, and desmosterol), and the cholesterol elimination product 24-HC generated by CYP46A1. The cholesterol precursor zymosterol and the cholesterol metabolite 27-hydroxycholesterol were below the limits of detection (<0.5 pmol/mg of total tissue protein). Both males and females were evaluated. The pattern of changes was the same in both genders; hence, the data are shown for females only. All measured sterols, except lanosterol and cholesterol, were affected by EFV treatment, and the effect was dose- and time-dependent. After 2 weeks of treatment, the dose dependence curve for 24-HC had the same bell shape as in the assays with isolated brain microsomes and the optimized assay with purified CYP46A1 (Fig. 3, B and C). The sterol levels were up to 67% higher than those in the untreated controls at lower EFV doses (0.04–0.16 mg/kg) and the same as in the untreated controls at the highest drug dose (0.32 mg/kg). EFV also increased the levels of the cholesterol precursors lathosterol (a marker of cholesterol biosynthesis (33)) and desmosterol, and this increase was observed at all four drug doses. Thus, after 2 weeks of treatment, three doses of EFV (0.04, 0.08, and 0.16 mg/kg/day) increased both cholesterol biosynthesis and metabolism and did not affect cholesterol levels. Such a sterol profile suggested increased cholesterol turnover in mouse brain. However, after 4 weeks of drug treatment, neither of the measured sterols was at the higher levels relative to the untreated controls. In fact, sterol levels in the treatment groups decreased as the dose of EFV increased. This could be due to EFV accumulation in the brain within the additional 2 weeks of treatment, leading to the CYP46A1 inhibition and affecting cholesterol biosynthesis in turn. The alternative interpretation is that increased enzyme activity depleted the pool of cholesterol available for CYP46A1, and this depletion led to a decrease in the production of 24-HC and a compensatory decrease in cholesterol biosynthesis.
To account for possible CYP46A1 inhibition and depletion of the ER cholesterol, as well as to provide sufficient time for cerebral cholesterol homeostasis to reach steady state upon EFV treatment, we changed the EFV delivery method from gavage to drinking water and increased the treatment time to 8 weeks. Indeed, delivery via drinking water affords the same daily drug dose as gavage, but the drug is supplied in smaller portions. CYP46A1 activation may be smaller in this case, but cholesterol available to the enzyme may not be depleted. Furthermore, a smaller drug dose will be eliminated more efficiently than a larger dose, thus preventing accumulation of a lipophilic drug in the brain and inhibition of CYP46A1.
Two doses of EFV were administered in drinking water. They were planned to be equal to 0.08 and to 0.32 mg/kg/day. However, the consumption of EFV-containing water was not the same as that of regular water (see “Experimental Procedures”), and the animals ultimately received 0.09 and 0.22 mg of EFV/kg/day. The 0.09 mg/kg/day dose led to statistically significant increases in cholesterol precursor levels (lathosterol, desmosterol, and a sum of lathosterol and desmosterol) and cholesterol catabolite (24-HC) levels during all 8 weeks of EFV treatment (Fig. 9). However, the kinetics of this increase were different for cholesterol precursors and 24-HC. For this drug dose, the maximal increase in lathosterol and desmosterol content was observed after 2 weeks of EFV treatment. The sterol levels then decreased slightly and stayed at the same level during the subsequent 4 weeks of treatment. In contrast, the amounts of 24-HC were gradually increased during the first 4 weeks of treatment and leveled off within the next 4 weeks. During all 8 weeks of treatment with EFV at 0.09 mg/kg/day, changes in cholesterol biosynthesis were compensated for by changes in cholesterol catabolism, as cholesterol levels were not significantly altered. EFV at 0.22 mg/kg/day led to a parallel alteration in the amounts of cholesterol precursors and 24-HC. A small spike in the sterol concentrations after 2 weeks of treatment was followed by a decrease in the sterol amounts (except desmosterol) to essentially the same levels as in untreated animals. These decreased levels were observed from week 4 to week 8 of the treatment, indicating steady-state cerebral cholesterol homeostasis. The levels of 24-HC in serum changed in parallel to those in the brain, with the increase in the serum 24-HC content being more pronounced upon treatment with EFV at 0.09 mg/kg/day compared with 0.22 mg/kg/day (Fig. 9).
DISCUSSION
The major finding of this work is that pharmacologic stimulation of CYP46A1 and cerebral cholesterol turnover is possible in principle, at least in mice. This was a challenge because only very few drugs on the market act as enzyme activators; the majority of the therapeutic agents are enzyme inhibitors (34–36). Nevertheless, we found a number of CYP46A1 activators among currently used drugs. We have demonstrated that administration of one of them (EFV) in drinking water (0.09 mg/kg/day) to mice results in a simultaneous increase in cholesterol biosynthesis and cholesterol catabolism, with no effect on cholesterol levels. We thus obtained a tool, additional to genetic manipulations and gene therapy, to investigate the benefits of enhanced cerebral cholesterol turnover on cognition in aged healthy mice and disease manifestations in mouse models of Alzheimer disease. Perhaps EFV could even be tested on patients with Alzheimer disease because the maximal suggested drug dose for HIV patients (600 mg/day) is >300 times higher than that (0.09 mg/kg/day) necessary for the activation of cerebral cholesterol turnover in mice. It is likely that at the very low doses that are required for stimulation of cerebral cholesterol turnover, EFV will not elicit the side effects (dizziness, confusion, anxiety, or depression) occurring upon administration of the high HIV dose of 600 kg/day (37). In mice, the maximal EFV-dependent increase in steady-state levels of cerebral 24-HC was 42%. In humans, more cholesterol is eliminated from the brain via the CYP46A1-dependent mechanism than in mice (∼75% versus ∼40%) (6, 38). Hence, it is possible that in humans, administration of EFV will lead to higher increases in the cerebral levels of 24-HC and greater stimulation of cerebral cholesterol turnover. However, to begin to use EFV off-label, a clinical trial is required to establish the best way to monitor the increased production of 24-HC in human brain, the range of EFV concentrations causing stimulation of CYP46A1 in humans, and whether these drug doses elicit any neurotoxicity.
Previously, cholesterol homeostasis in mouse brain was targeted pharmacologically with T0901317, the liver X receptor agonist (39, 40). When this agonist was administered to a mouse model of Niemann-Pick disease type C, cholesterol excretion from the brain increased, whereas synthesis of brain cholesterol and expression of Cyp46a1 remained unaltered (39). In another study, T0901317 was administered to aged wild-type mice and mice with the APPSLxPS1 mutations modeling Alzheimer disease (40). In both genotypes, T0901317 increased the levels of several cerebral cholesterol precursors and the expression of a number of liver X receptor target genes; 24-HC levels were not increased with this treatment (30). Thus, EFV seems to be the first identified drug that simultaneously enhances the production of both cholesterol precursors and 24-HC; our in vitro data suggest that EFV may not be the only such drug. Furthermore, CYP46A1 is expressed predominantly in the brain (13). Accordingly, unlike liver X receptor agonists that affect the entire body's cholesterol homeostasis, as well as other processes (41), the effects of small doses of EFV should be limited mainly to the brain.
Our enzyme assays with purified CYP46A1 and isolated brain microsomes strongly suggest that the primary reason for the in vivo stimulation of cerebral cholesterol turnover by EFV is activation of CYP46A1 at low drug doses. This activation likely proceeds via an allosteric mechanism and involves EFV binding outside the CYP46A1 active site. The mechanism of CYP46A1 inhibition is different and is probably based on a competition between the substrate (cholesterol) and EFV for the P450 active site. However, alternative explanations are possible for the increased production of 24-HC in mouse brain upon EFV treatment. The first is that in mice, EFV stimulates primarily the pathway of cholesterol input (cholesterol biosynthesis and/or transport between the cells), and CYP46A1 activation is secondary to this stimulation. We do not believe this is the case because CYP46A1 expression is not regulated by cholesterol levels (42). Also, enzyme activity is unlikely to be limited by cholesterol availability, as cholesterol is at the saturating level for the enzyme in the ER (∼250,000:1 cholesterol/CYP46A1 molar ratio) (43). These arguments are consistent with the results of the study with the liver X receptor agonist T0901317 described above, when stimulation of cerebral cholesterol biosynthesis did not lead to an increase in 24-HC levels (44).
The second explanation is that EFV has a dual effect on mouse brain. The drug not only activates CYP46A1 and hence cerebral cholesterol biosynthesis but also acts as a transcriptional activator affecting cholesterol efflux from brain cells and transport within this organ via lipoproteins. In this scenario, an increase in cholesterol precursor levels represents a summary response to the enhanced cholesterol elimination via CYP46A1-dependent and CYP46A1-independent mechanisms. Indeed, EFV has been reported to be an agonist for the pregnane X receptor, which appears to regulate the expression of apolipoprotein A1 in mice, the major component of plasma HDL (45, 46). It has also been suggested that the pregnane X receptor is activated in EFV-treated mice to explain the transient elevation in plasma HDL cholesterol (47). An increase in plasma HDL cholesterol along with plasma total and LDL cholesterol is also observed in EFV-treated HIV patients (48). Although much higher doses of EFV are prescribed to HIV patients (up to 600 mg/day) compared with that used in the present work (up to 0.32 mg/kg/day), we cannot exclude that the increase in the levels of cholesterol precursors upon EFV delivery by gavage could be due in part to transcriptional activation of the genes encoding lipoproteins. The presence of additional mechanism(s) that stimulate cholesterol biosynthesis in EFV-gavaged mice, independent of CYP46A1 activation, is also supported by the dissimilar shapes of the dose dependence curves for lathosterol and desmosterol compared with that for 24-HC (Fig. 8). After 2 weeks of gavage with the 0.32 mg/kg/day dose, cholesterol biosynthesis was still stimulated in the brain, as indicated by the lathosterol levels, yet cholesterol catabolism began to be inhibited compared with the lower 0.16 mg/kg/day dose. The mechanism(s) underlying this uncoupling of cholesterol biosynthesis and catabolism are unclear, but they do not appear to be operative when the drug is delivered in drinking water. In this treatment paradigm, EFV at 0.09 mg/kg/day resulted in an increase in lathosterol, desmosterol, and 24-HC levels during all treatment times, unequivocally demonstrating an increase in cerebral cholesterol turnover. Thus, our experiments strongly suggest that CYP46A1 has good potential to be a novel therapeutic target for the development of either cognitive enhancers or anti-Alzheimer disease medications. Moreover, novel applications of existing medications such as EFV could yield faster, cheaper, and more effective therapies for Alzheimer disease than conventional drug development processes.
In summary, we have identified a marketed drug (efavirenz) that activates the enzyme (CYP46A1) controlling the major pathway of cholesterol elimination from the brain. We have shown that EFV increases CYP46A1 activity in vitro in enzyme assays as well as in vivo in mice. We also proposed a model for CYP46A1 activation by the drug. Our data reveal the strong potential of CYP46A1 as a drug target and may lead to new therapeutic strategies for the treatment of neurodegenerative disorders or age-dependent deteriorations of memory and cognition.
This work was supported in part by United States Public Health Service Grant GM62882 (to I. A. P). Computational predictions were made using Iron software sold commercially by Pharmatrope Ltd., which was founded by Matthew Clark and Jeffrey Wiseman.
- ER
- endoplasmic reticulum
- 24-HC
- (24S)-hydroxycholesterol
- EFV
- efavirenz
- OR
- cytochrome P450 oxidoreductase.
REFERENCES
- 1. Russell D. W., Halford R. W., Ramirez D. M., Shah R., Kotti T. (2009) Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annu. Rev. Biochem. 78, 1017–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ramirez D. M., Andersson S., Russell D. W. (2008) Neuronal expression and subcellular localization of cholesterol 24-hydroxylase in the mouse brain. J. Comp. Neurol. 507, 1676–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Björkhem I. (2006) Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J. Intern. Med. 260, 493–508 [DOI] [PubMed] [Google Scholar]
- 4. Meaney S., Bodin K., Diczfalusy U., Björkhem I. (2002) On the rate of translocation in vitro and kinetics in vivo of the major oxysterols in human circulation: critical importance of the position of the oxygen function. J. Lipid Res. 43, 2130–2135 [DOI] [PubMed] [Google Scholar]
- 5. Babiker A., Diczfalusy U. (1998) Transport of side-chain oxidized oxysterols in the human circulation. Biochim. Biophys. Acta 1392, 333–339 [DOI] [PubMed] [Google Scholar]
- 6. Björkhem I., Lütjohann D., Diczfalusy U., Ståhle L., Ahlborg G., Wahren J. (1998) Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 39, 1594–1600 [PubMed] [Google Scholar]
- 7. Lund E. G., Xie C., Kotti T., Turley S. D., Dietschy J. M., Russell D. W. (2003) Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J. Biol. Chem. 278, 22980–22988 [DOI] [PubMed] [Google Scholar]
- 8. Kotti T. J., Ramirez D. M., Pfeiffer B. E., Huber K. M., Russell D. W. (2006) Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc. Natl. Acad. Sci. U.S.A. 103, 3869–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shafaati M., Olin M., Båvner A., Pettersson H., Rozell B., Meaney S., Parini P., Björkhem I. (2011) Enhanced production of 24S-hydroxycholesterol is not sufficient to drive liver X receptor target genes in vivo. J. Intern. Med. 270, 377–387 [DOI] [PubMed] [Google Scholar]
- 10. Maioli S., Båvner A., Ali Z., Heverin M., Ismail M. A., Puerta E., Olin M., Saeed A., Shafaati M., Parini P., Cedazo-Minguez A., Björkhem I. (2013) Is it possible to improve memory function by upregulation of the cholesterol 24S-hydroxylase (CYP46A1) in the brain? PLoS ONE 8, e68534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hudry E., Van Dam D., Kulik W., De Deyn P. P., Stet F. S., Ahouansou O., Benraiss A., Delacourte A., Bougnères P., Aubourg P., Cartier N. (2010) Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer's disease. Mol. Ther. 18, 44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bryleva E. Y., Rogers M. A., Chang C. C., Buen F., Harris B. T., Rousselet E., Seidah N. G., Oddo S., LaFerla F. M., Spencer T. A., Hickey W. F., Chang T. Y. (2010) ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc. Natl. Acad. Sci. U.S.A. 107, 3081–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lund E. G., Guileyardo J. M., Russell D. W. (1999) cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc. Natl. Acad. Sci. U.S.A. 96, 7238–7243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mast N., White M. A., Björkhem I., Johnson E. F., Stout C. D., Pikuleva I. A. (2008) Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc. Natl. Acad. Sci. U.S.A. 105, 9546–9551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mast N., Charvet C., Pikuleva I. A., Stout C. D. (2010) Structural basis of drug binding to CYP46A1, an enzyme that controls cholesterol turnover in the brain. J. Biol. Chem. 285, 31783–31795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mast N., Linger M., Clark M., Wiseman J., Stout C. D., Pikuleva I. A. (2012) In silico and intuitive predictions of CYP46A1 inhibition by marketed drugs with subsequent enzyme crystallization in complex with fluvoxamine. Mol. Pharmacol. 82, 824–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. White M. A., Mast N., Björkhem I., Johnson E. F., Stout C. D., Pikuleva I. A. (2008) Use of complementary cation and anion heavy-atom salt derivatives to solve the structure of cytochrome P450 46A1. Acta Crystallogr. D. Biol. Crystallogr. 64, 487–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mast N., Reem R., Bederman I., Huang S., DiPatre P. L., Björkhem I., Pikuleva I. A. (2011) Cholestenoic acid is an important elimination product of cholesterol in the retina: comparison of retinal cholesterol metabolism with that in the brain. Invest. Ophthalmol. Vis. Sci. 52, 594–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mast N., Andersson U., Nakayama K., Björkhem I., Pikuleva I. A. (2004) Expression of human cytochrome P450 46A1 in Escherichia coli: effects of N- and C-terminal modifications. Arch. Biochem. Biophys. 428, 99–108 [DOI] [PubMed] [Google Scholar]
- 20. Mast N., Zheng W., Stout C. D., Pikuleva I. A. (2013) Binding of a cyano- and fluoro-containing drug bicalutamide to cytochrome P450 46A1: unusual features and spectral response. J. Biol. Chem. 288, 4613–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clark M., Meshkat S., Talbot G. T., Carnevali P., Wiseman J. S. (2009) Fragment-based computation of binding free energies by systematic sampling. J. Chem. Inf. Model. 49, 1901–1913 [DOI] [PubMed] [Google Scholar]
- 22. Mast N., Shafaati M., Zaman W., Zheng W., Prusak D., Wood T., Ansari G. A., Lövgren-Sandblom A., Olin M., Björkhem I., Pikuleva I. (2010) Marked variability in hepatic expression of cytochromes CYP7A1 and CYP27A1 as compared to cerebral CYP46A1. Lessons from a dietary study with omega 3 fatty acids in hamsters. Biochim. Biophys. Acta 1801, 674–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dzeletovic S., Breuer O., Lund E., Diczfalusy U. (1995) Determination of cholesterol oxidation products in human plasma by isotope dilution-mass spectrometry. Anal. Biochem. 225, 73–80 [DOI] [PubMed] [Google Scholar]
- 24. Wang M., Heo G.-Y., Omarova S., Pikuleva I. A., Turko I. V. (2012) Sample prefractionation for mass spectrometry quantification of low-abundance membrane proteins. Anal. Chem. 84, 5186–5191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schenkman J. B., Jansson I. (2006) Spectral analyses of cytochromes P450. Methods Mol. Biol. 320, 11–18 [DOI] [PubMed] [Google Scholar]
- 26. Remmer H., Schenkman J., Estabrook R. W., Sasame H., Gillette J., Narasimhulu S., Cooper D. Y., Rosenthal O. (1966) Drug interaction with hepatic microsomal cytochrome. Mol. Pharmacol. 2, 187–190 [PubMed] [Google Scholar]
- 27. Schenkman J. B., Remmer H., Estabrook R. W. (1967) Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol. Pharmacol. 3, 113–123 [PubMed] [Google Scholar]
- 28. Poulos T. L., Finzel B. C., Howard A. J. (1987) High-resolution crystal structure of cytochrome P450cam. J. Mol. Biol. 195, 687–700 [DOI] [PubMed] [Google Scholar]
- 29. Dawson J. H., Andersson L. A., Sono M. (1982) Spectroscopic investigations of ferric cytochrome P-450-CAM ligand complexes. Identification of the ligand trans to cysteinate in the native enzyme. J. Biol. Chem. 257, 3606–3617 [PubMed] [Google Scholar]
- 30. Poulos T. L., Howard A. J. (1987) Crystal structures of metyrapone- and phenylimidazole-inhibited complexes of cytochrome P-450cam. Biochemistry 26, 8165–8174 [DOI] [PubMed] [Google Scholar]
- 31. Gigon P. L., Gram T. E., Gillette J. R. (1968) Effect of drug substrates on the reduction of hepatic microsomal cytochrome P-450 by NADPH. Biochem. Biophys. Res. Commun. 31, 558–562 [DOI] [PubMed] [Google Scholar]
- 32. Weiss J. N. (1997) The Hill equation revisited: uses and misuses. FASEB J. 11, 835–841 [PubMed] [Google Scholar]
- 33. Lund E., Sisfontes L., Reihner E., Björkhem I. (1989) Determination of serum levels of unesterified lathosterol by isotope dilution-mass spectrometry. Scand. J. Clin. Lab. Invest. 49, 165–171 [DOI] [PubMed] [Google Scholar]
- 34. Häberle J. (2011) Role of carglumic acid in the treatment of acute hyperammonemia due to N-acetylglutamate synthase deficiency. Ther. Clin. Risk Manag. 7, 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matschinsky F. M., Zelent B., Doliba N. M., Kaestner K. H., Vanderkooi J. M., Grimsby J., Berthel S. J., Sarabu R. (2011) Research and development of glucokinase activators for diabetes therapy: theoretical and practical aspects. Handb. Exp. Pharmacol. 203, 357–401 [DOI] [PubMed] [Google Scholar]
- 36. Blum C. A., Ellis J. L., Loh C., Ng P. Y., Perni R. B., Stein R. L. (2011) SIRT1 modulation as a novel approach to the treatment of diseases of aging. J. Med. Chem. 54, 417–432 [DOI] [PubMed] [Google Scholar]
- 37. Treisman G. J., Kaplin A. I. (2002) Neurologic and psychiatric complications of antiretroviral agents. AIDS 16, 1201–1215 [DOI] [PubMed] [Google Scholar]
- 38. Dietschy J. M., Turley S. D. (2004) Thematic review series: brain lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 45, 1375–1397 [DOI] [PubMed] [Google Scholar]
- 39. Repa J. J., Li H., Frank-Cannon T. C., Valasek M. A., Turley S. D., Tansey M. G., Dietschy J. M. (2007) Liver X receptor activation enhances cholesterol loss from the brain, decreases neuroinflammation, and increases survival of the NPC1 mouse. J. Neurosci. 27, 14470–14480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vanmierlo T., Rutten K., Dederen J., Bloks V. W., van Vark-van der Zee L. C., Kuipers F., Kiliaan A., Blokland A., Sijbrands E. J., Steinbusch H., Prickaerts J., Lütjohann D., Mulder M. (2011) Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol. Aging 32, 1262–1272 [DOI] [PubMed] [Google Scholar]
- 41. Cao G., Liang Y., Jiang X. C., Eacho P. I. (2004) Liver X receptors as potential therapeutic targets for multiple diseases. Drug News Perspect. 17, 35–41 [DOI] [PubMed] [Google Scholar]
- 42. Ohyama Y., Meaney S., Heverin M., Ekström L., Brafman A., Shafir M., Andersson U., Olin M., Eggertsen G., Diczfalusy U., Feinstein E., Björkhem I. (2006) Studies on the transcriptional regulation of cholesterol 24-hydroxylase (CYP46A1). Marked insensitivity toward different regulatory axes. J. Biol. Chem. 281, 3810–3820 [DOI] [PubMed] [Google Scholar]
- 43. Liao W.-L., Heo G.-Y., Dodder N. G., Reem R. E., Mast N., Huang S., DiPatre P. L., Turko I. V., Pikuleva I. A. (2011) Quantification of cholesterol-metabolizing P450s CYP27A1 and CYP46A1 in neural tissues reveals a lack of enzyme-product correlations in human retina but not human brain. J. Proteome Res. 10, 241–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vanmierlo T., Bloks V. W., van Vark-van der Zee L. C., Rutten K., Kerksiek A., Friedrichs S., Sijbrands E., Steinbusch H. W., Kuipers F., Lütjohann D., Mulder M. (2010) Alterations in brain cholesterol metabolism in the APPSLxPS1mut mouse, a model for Alzheimer's disease. J. Alzheimers Dis. 19, 117–127 [DOI] [PubMed] [Google Scholar]
- 45. Bachmann K., Patel H., Batayneh Z., Slama J., White D., Posey J., Ekins S., Gold D., Sambucetti L. (2004) PXR and the regulation of apoA1 and HDL-cholesterol in rodents. Pharmacol. Res. 50, 237–246 [DOI] [PubMed] [Google Scholar]
- 46. Faucette S. R., Zhang T. C., Moore R., Sueyoshi T., Omiecinski C. J., LeCluyse E. L., Negishi M., Wang H. (2007) Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J. Pharmacol. Exp. Ther. 320, 72–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tohyama J., Billheimer J. T., Fuki I. V., Rothblat G. H., Rader D. J., Millar J. S. (2009) Effects of nevirapine and efavirenz on HDL cholesterol levels and reverse cholesterol transport in mice. Atherosclerosis 204, 418–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gotti D., Cesana B. M., Albini L., Calabresi A., Izzo I., Focà E., Motta D., Bellagamba R., Fezza R., Narciso P., Sighinolfi L., Maggi P., Brianese N., Quiros-Roldan E., Guaraldi G., Torti C. (2012) Increase in standard cholesterol and large HDL particle subclasses in antiretroviral-naive patients prescribed efavirenz compared to atazanavir/ritonavir. HIV Clin. Trials 13, 245–255 [DOI] [PubMed] [Google Scholar]
- 49. Mast N., Liao W.-L., Pikuleva I. A., Turko I. V. (2009) Combined use of mass spectrometry and heterologous expression for identification of membrane-interacting peptides in cytochrome P450 46A1 and NADPH-cytochrome P450 oxidoreductase. Arch. Biochem. Biophys. 483, 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mast N., Zheng W., Stout C. D., Pikuleva I. A. (2013) Antifungal azoles: structural insights into undesired tight binding to cholesterol-metabolizing CYP46A1. Mol. Pharmacol. 84, 86–94 [DOI] [PMC free article] [PubMed] [Google Scholar]