

Background: Co-crystal structure of Fc-FcγRIII complex revealed that Fc binds to FcγRIII asymmetrically.
Results: We identified a panel of novel Fc heterodimers with enhanced ADCC activity.
Conclusion: Asymmetrical Fc engineering is an efficient approach for enhancing ADCC activity and stability of engineered antibodies.
Significance: The discovery could be applied in therapeutic antibodies for the treatment of cancers and infectious diseases.
Keywords: Antibody Engineering, Cancer Therapy, Cell Death, FCγ Receptors, NK Cells
Abstract
Antibody-dependent cellular cytotoxicity (ADCC) is mediated through the engagement of the Fc segment of antibodies with Fcγ receptors (FcγRs) on immune cells upon binding of tumor or viral antigen. The co-crystal structure of FcγRIII in complex with Fc revealed that Fc binds to FcγRIII asymmetrically with two Fc chains contacting separate regions of the FcγRIII by utilizing different residues. To fully explore this asymmetrical nature of the Fc-FcγR interaction, we screened more than 9,000 individual clones in Fc heterodimer format in which different mutations were introduced at the same position of two Fc chains using a high throughput competition AlphaLISA® assay. To this end, we have identified a panel of novel Fc variants with significant binding improvement to FcγRIIIA (both Phe-158 and Val-158 allotypes), increased ADCC activity in vitro, and strong tumor growth inhibition in mice xenograft human tumor models. Compared with previously identified Fc variants in conventional IgG format, Fc heterodimers with asymmetrical mutations can achieve similar or superior potency in ADCC-mediated tumor cell killing and demonstrate improved stability in the CH2 domain. Fc heterodimers also allow more selectivity toward activating FcγRIIA than inhibitory FcγRIIB. Afucosylation of Fc variants further increases the affinity of Fc to FcγRIIIA, leading to much higher ADCC activity. The discovery of these Fc variants will potentially open up new opportunities of building the next generation of therapeutic antibodies with enhanced ADCC effector function for the treatment of cancers and infectious diseases.
Introduction
Monoclonal antibodies have become an increasingly important class of therapeutic agents for the treatment of cancers, inflammatory diseases, and infectious diseases (1). In addition to binding to their specific targets, blocking or activating certain biochemical pathways, antibodies can recruit effector cells from the immune system through engagement of Fc receptors. There are two types of Fc receptors (FcγRs),3 for IgG activating FcγR and inhibitory FcγR (2, 3). Engagement of activating FcγRs can lead to a variety of antibody effector functions (2, 3) such as antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cell-mediated phagocytosis (ADCP).
Emerging clinical data demonstrate a correlation of better clinical response of therapeutic antibodies with high affinity FcγR alleles. Patients homozygous for the high affinity FcγRIIIA Val-158 allele showed higher response rates and improved progression-free survival by the treatment of anti-CD20 rituximab for non-Hodgkin lymphoma (4) and anti-Her2 trastuzumab for metastatic breast cancer (5). Another study (6) revealed that patients with the high affinity alleles FcγRIIIA 158 V/V and/or FcγRIIA 131 H/H had a significantly higher response rate and longer remission than patients without either genotype for rituximab-treated follicular lymphoma. Studies using knock-out mice showed that elimination of inhibitory receptor FcγRIIB resulted in significantly increased potency for anti-tumor antibodies (7–9). This leads to an hypothesis that the IgG subclass with a higher activating to inhibitory ratio, i.e. IgG with increased binding to FcγRIIIA or FcγRIIA but decreased/unchanged binding to FcγRIIB, could translate into significantly enhanced in vivo activity (9).
One of important strategies to improve the next generation of anti-cancer therapeutics is aiming to build antibodies with enhanced effector functions, mostly by increasing their binding capacities to FcγRIIIA. This has been accomplished by two general approaches. The fucose attached to the N-linked glycan at Asn-297 of Fc sterically hinders the interaction of Fc with FcγRIIIA, and removal of fucose by glyco-engineering can increase the binding to FcγRIIIA, which translates into >50-fold higher ADCC activity compared with wild type IgG1 controls (10, 11). Protein engineering, through amino acid mutations in the Fc portion of IgG1, has generated multiple variants that increase the affinity of Fc binding to FcγRIIIA (12–14). Notably, the triple alanine mutant S298A/E333A/K334A (12) displayed 2-fold increase binding to FcγRIIIA and ADCC function. S239D/I332E (2X) and S239D/I332E/A330L (3X) variants (13) demonstrated a significant increase in binding affinity to FcγRIIIA and augmentation of ADCC capacity in vitro and in vivo. Other Fc variants identified by yeast display also showed the improved binding to FcγRIIIA and enhanced tumor cell killing in mouse xenograft models (14). All these engineered Fc variants were built on homodimeric IgG molecules in which the same mutations were present at the same position of the two Fc chains. Analysis of the co-crystal structure of Fc-FcγRIII complex suggested that this may not be the most optimal approach.
Multiple structure studies revealed that the interaction of human FcγRIII with the Fc region of human IgG1 is asymmetric, i.e. FcγRIII comes into contact with different amino acid residues on the two Fc polypeptide chains (Fig. 1) (15). Thus, from a protein engineering point of view, the ideal way to maximally enhance the interaction of the Fc region of IgG1 with FcγRIIIA is to individually optimize the binding interface with FcγRIIIA at each side of the Fc chains by using different mutations. This asymmetrical engineering approach may allow us to address some issues associated with conventional homodimeric IgG. For example, both S239D/I332E (2X) and S239D/I332E/A330L (3X) variants led to decreased stability of the CH2 domain as indicated by the lowering of melting temperature (Tm) in differential scanning calorimetry (DSC) analysis, likely due to the presence of some destabilizing mutation(s) on both sides of Fc. In addition, the high affinity Fc variant S239D/I332E/A330L (3X) has increased binding to inhibitory FcγRIIB (13). An asymmetrical approach, using different mutations in the two Fc chains, may allow a better discrimination of FcγRIIB from FcγRIIA and/or FcγRIIIA and therefore increase the activating to inhibitory ratio of Fc variants, leading to better anti-tumor activities.
FIGURE 1.
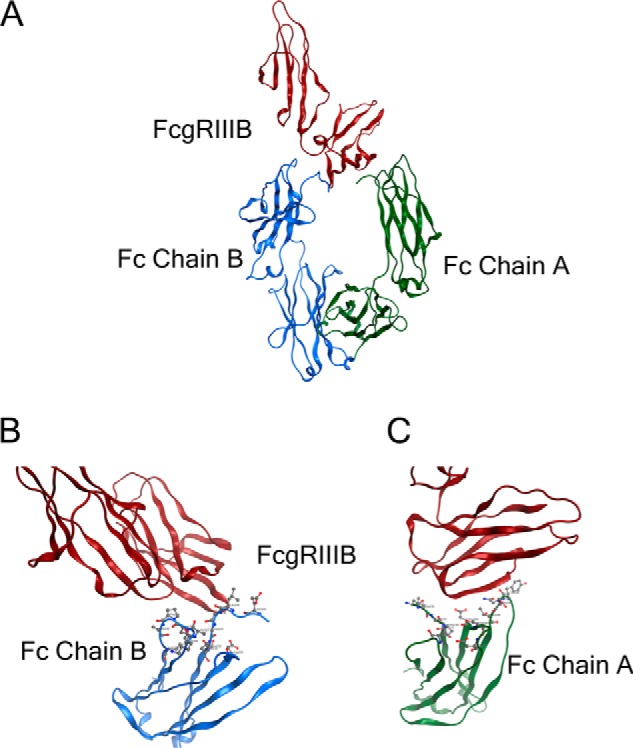
Fc of human IgG1 interacts asymmetrically with FcγRIII. A, representation of the x-ray co-crystal structure of the Fc-FcγRIIIB (Protein Data Bank code 1T83) complex. The FcγRIIIB structure is shown in red; the Fc chain B is shown in blue, and the Fc chain A is shown in green. B, zoomed-in interface between the FcγRIII and left Fc chain B. Contacting residues Leu-235, Ser-239, Asp-265, Leu-328, Pro-329, Ala-330, and Ile-332 are in proximity to the FcγRIII. C, zoomed-in interface between the FcγRIII and right Fc chain A. Contacting residues Leu-235, Gly-236, Pro-238, Ser-239, Asp-265, Ser-267, Asp-270, Tyr-296, Asn-297, Ser-298, Thr-299, and Ala-327 are in proximity to the FcγRIII.
To fully explore the asymmetrical nature of the Fc/FcγR interaction, we constructed heterodimeric Fc libraries in which the solvent-exposed residues in the lower hinge region and CH2 domain were targeted. We screened more than 9,000 individual clones using a high throughput competition AlphaLISA® assay. We identified 71 single mutants with improved binding to activating FcγRIIIA (Phe-158). We grouped these single mutants based on their positions and further combined beneficial mutations to seek synergy in the heterodimeric IgG1 format. To this end, we have identified a panel of novel Fc variants with significant binding improvement to FcγRIIIA (both Phe-158 and Val-158 allotypes), high CH2 stability, increased ADCC activity in tumor cell killing assays, and strong tumor growth inhibition in mice xenograft tumor models. Afucosylation of Fc variants further increases the affinity of Fc to FcγRIIIA, leading to a much higher ADCC activity. Some of the novel Fc variants exhibit higher binding to FcγRIIA but not to FcγRIIB. These Fc variants could be applied in building a new generation of heterodimeric monospecific and bispecific therapeutic antibodies with enhanced ADCC function for treatment of cancers and infectious diseases.
EXPERIMENTAL PROCEDURES
Cell Lines and Reagents
The human tumor cell lines HEK-293 (kidney), NCI-N87 (gastric), OVCAR-8 (ovarian), CAPAN-2 (pancreas), JIMT-1 (breast), and SK-BR-3 (breast) were purchased from American Type Culture Collection (ATCC, Manassas, VA) and maintained in RPMI 1640 medium (for NCI-N87 and CAPAN-2 cells), DMEM for HEK-293 and JIMT-1 cells), or McCoy's 5A medium (for SK-BR-3 and OVCAR-8 cells) supplemented with 10% FBS, 4 mmol/liter glutamine, 10 mmol/liter HEPES, 50 μg/ml penicillin, and 100 μg/ml streptomycin. All cell lines were grown at 37 °C, 5% CO2. Human NK cell isolation kit (catalog no. 130-092-657) was purchased from Miltenyi Biotec (Auburn, CA). IGEPAL CA 630 (catalog no. I8896), 2,2′-azino-bis (3-ethylbenzthiazoline-6-sulfonic acid) (catalog no. A9941), calcein (catalog no. C1359), and goat anti-human IgG Fc-specific HRP-conjugated polyclonal antibody (catalog no. A0170) were purchased from Sigma. EZ-Link NHS-PEO4 biotinylation kit (catalog no. 21455), SuperSignal® West Pico chemiluminescent substrate (catalog no. 34080), CL-X PosureTM x-ray films (catalog no. 34091), and streptavidin-HRP (catalog no. 21130) were obtained from Thermo Fisher Scientific (Rockford, IL). CellTrace CFSE (catalog no. C34544) was purchased from Invitrogen. Anti-His antibodies (catalog no. 34698) were from Qiagen (Valencia, CA); polyethylenimine (PEI) 25-kDa linear (catalog no. 23966–2) was obtained from Polysciences Inc. (Warrington, PA). PfuTurbo® Hotstart DNA polymerase (catalog no. 600322) and QuikChange Lightning multisite-directed mutagenesis kit (catalog no. 210516) were purchased from Agilent Technologies (Santa Clara, CA). Restriction enzymes SalI, BamHI, NheI, BsiWI, NotI, and peptide:N-glycosidase F (catalog no. P0704S) were ordered from New England Biolabs Inc. (Ipswich, MA). Half-volume 96-well plates (catalog no. 6005569), streptavidin donor beads (catalog no. 6760002), and glutathione acceptor beads (catalog no. AL109M) were purchased from PerkinElmer Life Sciences. Female C.B-17/IcrHsd-Prkdcscid (CB-17/SCID) mice were purchased from Harlan Laboratories (Indianapolis, IN). All animal experiments were conducted in a Canadian Council on Animal Care or AAALAC International-accredited facilities, and all research protocols were approved by the Institutional Animal Care and Use Committees.
Computational Analyses
Sequence homology searches using BLAST (16) followed by sequence alignments using ClustalW (17) were performed to identify the most invariant regions in different FcγRs. X-ray crystal structure of the Fc-FcγRIIIB complex (Protein Data Bank code 1T83) was used to identify FcγRIIIB-contacting residues in the Fc chain A (R, right side) and Fc chain B (L, left side). The contacting residues are identified using 5.0-Å distance criterion; if any non-hydrogen atom of a residue from Fc is closer than 5.0 Å distance from any non-hydrogen atom of a residue from FcγRIIIB, then these two residues are considered to be interacting. We also compared the Fc-FcγRIIIB crystal structure with that of more recently published Fc-FcγRIIIA (Protein Data Bank code 3SGJ). The comparison showed the Fc interaction site in FcγRIIIA and FcγRIIIB is identical and well conserved. All of the six positions, which differ between the two receptors, are located away from the Fc interaction site in the structure. Therefore, our initial structural analysis of Fc-FcγRIIIB can be applied to study the residue positions influencing the affinity of Fc-FcγRIIIA interaction. A genetic algorithm for protein design (EGAD) (18) was used to search beneficial residue(s) at positions Ser-298, Ala-327, and Ala-330 in the Fc region, which could enhance the binding to FcγRIIIA and stabilize the Fc-FcγRIIIA complex using the available Fc-Fcγ receptor crystal structures. EGAD was also used to analyze some of combinations of mutations that had very favorable binding to Fcγ receptors. Examples of the EGAD-predicted mutations include S298C, S298I, S298V, S298T, A327Y, A327W, A327F, A327H, A330H, and A330F.
FcγRs Cloning and Expression
The amino acid sequences of the extracellular domain of human FcγRIIA (His-131) (NCBI accession number NP_001129691, FcγRIIB (NCBI accession number NP_001002274), and FcγRIIIA (Phe-158) (NCBI accession number NP_001121065) were used to design the DNA sequences that were codon-optimized for mammalian expression using GeneArt® program (Invitrogen). The DNAs encoding VkO1/O12 signal peptide at the 5′ end, FcγR in the middle, and FLAG-His6 or GST-His6 tag at the 3′ end with flanking sequences for restriction enzyme digestion at both ends were synthesized by Invitrogen. PCRs using PfuTurbo® Hotstart DNA Polymerase was carried out to amplify the inserts that were digested by SalI and NotI and then ligated with SalI- and NotI-treated pTT5 expression vector (19). Mutagenesis reactions were carried out to introduce the Arg-131 allele of FcγRIIA and the Val-158 allele of FcγRIIIA using QuikChange Lightning multisite-directed mutagenesis kit as directed by the manufacturer. All clones were verified by double strand DNA sequencing of plasmids. 2936E cells were transiently transfected with plasmid DNAs in the presence of PEI/DNA at a 3:1 (μg) ratio, and shaken for 6 days post-transfection. The conditioned medium was harvested and purified through immobilized metal affinity chromatography column to make the above FcγRs.
Construction of Libraries of Altered Fc Regions as Fc Heterodimers
Libraries of nucleic acids encoding either an scFv-Fc containing the charge pair substitutions E356K + D399K or an Fc polypeptide chain (“dummy Fc”) containing the charge pair substitutions K392D + K409D, with additional alterations at the designated site within the Fc-encoding regions, were created using PCR. For each residue selected for substitution, the nucleic acid was changed such that Fc regions with all 20 different amino acids at the selected site would be generated. One group of sites was entirely within the lower hinge region (residues 230, 231, 232, 233, 234, 235, 236, 237, and 238; Fig. 3A). The library containing nucleic acids with mutations at sites encoding these residues was referred to as the “Tier 1” library. Another group of sites were within the CH2 domain and were either close to or part of the area that contacts FcγRIIIB (239, 241, 255, 256, 258, 264, 265, 267, 268, 269, 270, 272, 276, 280, 285, 286, 290, 294, 295, 296, 298, 300, 307, 309, 315, 326, 327, 328, 330, 332, 333, 334, 337, and 339; Figs. 1 and 3A). The library containing nucleic acids with mutations at sites encoding these residues was referred to as the “Tier 2” library. A third group included sites within the CH2 domain that were solvent-exposed but were not close to or part of the area that contacts FcγRIIIB (243, 246, 248, 249, 251, 252, 253, 254, 260, 274, 275, 278, 279, 282, 283, 284, 287, 288, 289, 292, 293, 301, 302, 303, 305, 310, 311, 312, 313, 314, 317, 318, 320, 322, 324, 335, 336, 338, and 340; Fig. 3A). The library containing nucleic acids with mutations at the designated sites encoding these residues was referred to as the “Tier 3” library.
FIGURE 3.

Targeted residues in Fc region and screening process of Fc libraries. A, amino acid sequence of a human IgG1 Fc polypeptide to be targeted for the construction of Fc libraries. The amino acid sequence of a human IgG1 Fc region, starting from the hinge region and ending with the carboxyl terminus of the CH3 domain, is shown in single letter notation and is numbered according to the EU system of Edelman et al. (35). The amino acids underlined and in boldface type were randomized in constructing the libraries as described under “Experimental Procedures.” Below each of these amino acids is a 1, a 2, or a 3, which indicates that DNAs encoding variants at the corresponding site were included in a Tier 1, 2, or 3 library as described under “Experimental Procedures.” B, diagram to show the primary screening and initial combinatorial screening for substitutions that enhance binding to FcγRIIIA. The rectangle labeled SIG represents a polynucleotide encoding a signal sequence, which facilitates protein secretion from mammalian cells. A region encoding a hinge region is represented by a horizontal line labeled hinge. A rectangle labeled Fc polypeptide represents a polynucleotide encoding an Fc polypeptide chain. The five-pointed and four-pointed stars mean that the polynucleotides encoding the Fc polypeptide chains contain randomized codons at selected positions as explained under “Experimental Procedures.” The circles labeled with VH and VL represent the regions encoding a heavy chain variable region and a light chain variable region, respectively. The + + and − − in the rectangles labeled Fc polypeptide mean that these regions include mutations such that the encoded Fc polypeptide chain will have the substitutions E356K + D399K and K392D + K409D, respectively.
In more detail, a DNA fragment encoding the scFv of a rat anti-mouse natural killer group 2D antibody fused to a human IgG1 Fc polypeptide with E356K + D399K charge pair mutations in CH3 domain was subcloned into the mammalian expression vector pTT5. A DNA fragment encoding a human IgG1 Fc polypeptide with K392D + K409D charge pair mutations in the CH3 domain was also subcloned into pTT5. The six small Fc libraries were made using splice overhang extension by PCR (20) as described below.
For each of 82 selected codons within the Fc-encoding region, an oligonucleotide randomized at the first two positions of the codon and having either a G or a C at the third position (“NN(G/C) codon”) was made (“NN(G/C) oligonucleotide”). This NN(G/C) codon was placed in the middle of the NN(G/C) oligonucleotide with about 21 bases extending both upstream and downstream. The NN(G/C) oligonucleotide was oriented such that its 5′ end was upstream of its 3′ end in the Fc-encoding region. Accordingly, “reverse oligonucleotides” that match the upstream 21 bases of the NN(G/C) oligonucleotides were synthesized individually. A universal downstream primer was combined with each of the NN(G/C) oligonucleotides and subjected to PCR amplifications to produce downstream fragments. Similarly, a universal upstream oligonucleotide and each of the reverse oligonucleotides were combined and subjected to PCR amplifications to make upstream DNA fragments. The upstream and downstream PCR fragments were purified from agarose gels, and the amounts of these PCR products were quantified by the Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). The same molar amount of individual upstream and downstream DNA fragments was combined with the universal upstream and downstream primers for a second round PCR to assemble the full-length PCR product. Full-length PCR fragments were purified from 1.8% agarose gel and quantified. Individual full-length PCR fragments at equal amounts were pooled, digested with restriction enzymes SalI and BamHI, and then inserted into an expression vector pTT5 that was treated with SalI and BamHI.
A total of six libraries were made. Three libraries, a Tier 1, a Tier 2, and a Tier 3 library, having mutations in a nucleic acid encoding a scFv-Fc were made. Similarly, a Tier 1, a Tier 2, and a Tier 3 library having mutations at the same positions in a nucleic acid encoding a dummy Fc was made. As illustrated diagrammatically in Fig. 3B, initial screening was performed as follows. The libraries were introduced into Escherichia coli, and enough individual colonies were picked such that at least three times as many colonies were picked as there were different variants in the library. For example, each Tier 1 library contained 20 different amino acids at each of nine sites, for a total of 180 different variants. In this case, 10 microtiter plates of colonies (96 wells per plate for a total of about 960) were picked and grown. Plasmid DNA was isolated. Each Tier 2 and Tier 3 library contained 20 different amino acids at each of 34 and 39 sites for a total of 680 and 780 different variants, respectively. Accordingly 45 plates of colonies (for a total of 4320) were picked for each Tier 2 and Tier 3 library, and plasmid DNA was isolated. These mutated plasmid DNAs were combined with unaltered plasmid DNAs (if the altered DNA was a scFv-Fc, then unaltered DNA was a dummy Fc, and vice versa, as shown in Fig. 3B) and used to transfect HEK-293 cells. The conditioned medium was tested 6 days post-transfection using a competition AlphaLISA® assay to screen Fc variants with higher binding affinity to GST-tagged FcγRIIIA (Phe-158).
Competition AlphaLISA® Assay to Screen Fc Libraries
Briefly, the AlphaLISA® assay was performed as follows. Glutathione-conjugated acceptor beads and GST-tagged human FcγRIIIA Phe-158 protein were incubated at RT for 2 h with shaking in 0.5-volume 96-well plates. Crude conditioned medium from transfected HEK-293 cells in 96-well plates was added. The plates were shaken at RT for 2 h. Biotinylated human IgG1 antibody and streptavidin-conjugated donor beads were added to the above wells. The plates were shaken at RT for over 4 h. Plates were spun at 2,500 rpm at RT for 10 min to collect any evaporated fluid. The plates were illuminated with light at 680 nm and emitted at about 615 nm in Envision 2012 Multilabel Reader (PerkinElmer Life Sciences). The wells with >20% signal lower than that of the wild type scFv-Fc/dummy Fc heterodimers were scored as primary hits. The plasmid DNAs of primary hits were repeatedly transfected in a 24-well plate; the Fc titer in conditioned medium was quantified by Fortebio Octet RED 96 System (Fortebio, Menlo Park, CA) and tested for two more rounds of competition AlphaLISA® assay after normalization. The hits that consistently showed more than 20% inhibition were considered positive. The plasmids of the positive hits were sequenced to decode the sequence variation in the Fc region.
Combinatorial Screening of Positive Hits
The individual positive hits identified from the dummy Fc libraries were combined with individual positive hits identified from scFv-Fc libraries. Plasmid DNAs for a total of 37 × 34 = 1,258 combinations were co-transfected in HEK-293 cells in 96-well plates, and the conditioned medium was tested by a high throughput competition AlphaLISA® assay to identify combinations that could bind to FcγRIIIA more effectively. The 21 combinations (described under “Results”), which showed significantly higher binding affinity to FcγIIIA, were re-tested for confirmation as anti-Her2 heterodimeric IgG1 form. Specifically, the five beneficial mutations from dummy Fc libraries (R255S, E294L, T307P, Q311M, and K334V) were separately incorporated into a humanized anti-Her2 IgG1 heavy chain containing K392D + K409D in the CH3 domain by PCR splice overhang extension technology. Similarly, the 11 beneficial mutations from scFv-Fc libraries (L234Y, K290G, K290S, K290Y, E294L, Y296L, Y296W, S298A, S298C, T307G, and L309E) were separately integrated into a humanized anti-Her2 IgG1 heavy chain containing E356K + D399K in the CH3 domain. Then the plasmid DNAs of two different heavy chains and one common light chain all in mammalian expression vector pTT5 were co-transfected in HEK-293 cells to express the 21 heterodimeric anti-Her2 IgG1 antibodies. Mutations located at the lower hinge regions, around Pro-329, close to Asp-265, and around Asn-297, were preferentially selected to make a new panel of variants for further binding improvement with the aid of EGAD.
Mammalian Expression of Anti-TuAg and Anti-Her2 IgG1 Variants
For 500-ml medium scale expression of antibodies, a total of 250 μg of plasmid DNAs using a 2:1:1 ratio (125 μg of light chain and 62.5 μg of heavy chain 1 and 62.5 μg of heavy chain 2) were mixed in a 50-ml tube; 25 ml of 293 serum-free medium containing 250 μl of 3 mg/ml PEI, pH 7.0, was added in the above 50-ml tube, and the mixture was incubated at RT for 20 min. The mixture of DNA/PEI was loaded into 475 ml of 2936E cells at 1–2 × 106/ml in a 1-liter shaking flask. The next day, 12.5 ml of 20% Yeastolate was added to each flask to a 0.5% final concentration. Cells were shaken for 6 more days. The supernatant was harvested by centrifuging cells at 4,000 rpm for 15 min and then filtered through a 0.22-μm membrane. The supernatant was purified in a protein A column followed by size exclusion column to get rid of high molecular weight species and half-antibodies. The purified antibodies were subject to further characterizations such as DSC analysis, competition AlphaLISA® assay, mass spectrometry analysis, Biacore analysis, cell-based ADCC assays, and mouse xenograft studies.
Mass Spectrometry Analysis
Treatment for deglycosylation and reduction of all antibodies was carried out as follows. Deglycosylation reaction was carried out by incubating 20 μg of antibodies with 1 μl of peptide:N-glycosidase F in 20 μl of 50 mm Tris buffer, pH 7.2, at 37 °C for 18 h. Deglycosylated or nondeglycosylated antibody was completely reduced with 9 mm DTT in 20 μl of 4 m guanidine HCl, 50 mm Tris buffer, pH 8.0, at 55 °C for 15 min. Intact mass analysis of deglycosylated and nondeglycosylated whole antibodies was done in an HPLC-ESI-TOF system (Agilent 6210 TOF mass spectrometer in combination with an Agilent 1200 liquid chromatography system (Agilent/Varian Inc, Santa Clara, CA)). A 2.1 × 150-mm Pursuit Diphenyl column with 5 μm particle size (Agilent/Varian Inc, Santa Clara, CA) was connected to the liquid chromatography system and operated at 400 ml/min. The column temperature was 75 °C; solvent A was 0.1% TFA in water, and solvent B was 0.1% TFA in acetonitrile. The gradient started at 25% solvent B and increased linearly to 80% B over 30 min. The TOF mass spectrometer was tuned and calibrated in the range of 100 to 4,500 m/z. The capillary voltage was set at 4,500 V, drying gas at 12 liter/min, drying gas temperature at 300 °C, nebulizer gas flow at 40 liter/min, and fragmentor voltage at 375 V for intact antibodies and 300 V for reduced antibodies.
Biacore Analysis to Measure the Affinity of Fc Variants to FcγRs
Binding of the Fc variants to recombinant human and murine FcγRs was tested on a Biacore T100 instrument (GE Healthcare). Human FcγRIIA with a histidine at position 131, FcγRIIA with an arginine at position 131, human FcγRIIB and human FcγRIIIA with a valine at position 158, FcγRIIIA with a phenylalanine at position 158, and mouse FcγRIV were tested. Briefly, mouse anti-His antibody was immobilized on all four flow cells of a CM5 chip using standard amine coupling to a density around 7,000 response units. His6-tagged FcγRs were captured on flow cell 2, 3, or 4 and reached 40–80 response units. Flow cell 1 was used as a background control. Anti-TuAg or anti-Her2 IgG1 variants (ranged from 0.78 to 800 nm in PBS plus 0.1 mg/ml BSA, 0.005% P20) were injected over the FcγR surfaces at 50 μl/min for a 3-min association and a 3-min dissociation. The binding kinetics, kon (1/ms), koff (1/s), and KD (nm) was calculated from global fittings using 1:1 kinetics binding model on BIAevaluation software (GE Healthcare). In the case of very fast on-rate and very fast off-rate, the KD value was calculated from steady state affinity using 1:1 binding model on the BIAevaluation software.
Biacore Analysis to Detect the Binding of Engineered Antibody Variants to Neonatal Fc Receptor
Binding of anti-Her2 IgG1 variants to human neonatal Fc receptor (FcRn) was tested on a Biacore T100 instrument. Briefly, CHO-produced human Fc was immobilized on flow cell 2 of a CM5 chip using standard amine coupling to a density around 6,000 response units. Flow cell 1 was used as a background control. 10 nm of human FcRn was mixed with serial dilutions of the antibodies (ranged from 0.1 to 2,000 nm in 10 mm sodium acetate, pH 5.5, 150 mm NaCl, 0.005% P20, 0.1 mg/ml BSA) and incubated for 1 h at RT. Binding of the free FcRn to immobilized huFc was measured by injecting the mixture over the CM5 chip surfaces. 100% FcRn binding signal was determined in the absence of antibodies in solution. A decreased FcRn-binding response with increasing concentrations of antibodies indicated that FcRn bound to the antibodies in solution, which sequestered FcRn from binding to the immobilized Fc surfaces. By plotting the FcRn binding signal versus antibody concentrations, FcRn binding activity can be compared between anti-Her2 IgG1 WT and engineered anti-Her2 IgG1 variants.
Thermal Stability Analysis by DSC
The DSC measurements were obtained using a VP-Capillary DSC system (Microcal Inc., Northampton, MA) equipped with tantalum 61 cells, each with an active volume of 125 μl. Protein samples were diluted to 0.5 mg/ml, and the corresponding buffer was used as a reference. The samples were scanned from 20 to 110 °C at a rate of 20 °C/h with an initial 15 min of equilibration at 20 °C. A filtering period of 16 s was used, and data were analyzed using Origin 7.0 software (OriginLab Corp., Northampton, MA). Thermograms were corrected by subtraction of buffer-only blank scans. The corrected thermograms were normalized for protein concentration. The melting temperatures reported represent peaks in the experimental thermograms.
In Vitro Cell-based ADCC Assays
For ADCC assay with OVCAR-8 and CAPAN-2 cells, peripheral blood mononuclear cells were recovered from human blood by layering over Ficoll-Paque (GE Healthcare) and centrifuging at 400 × g for 40 min. Peripheral blood mononuclear cells were washed twice in PBS, and NK cells were purified by negative selection using the Miltenyi AutoMacs Pro negative selection system as per the manufacturer's instructions. NK cells were held overnight at 4 °C on a rocker, then washed, counted, and resuspended at 4 × 106 cells/ml in RPMI 1640 medium for use in the ADCC assay. OVCAR-8 and CAPAN-2 cells expressing a specific tumor antigen were harvested with trypsin/EDTA, washed, and counted. 6 × 106 targets were resuspended in RPMI 1640 medium and labeled with calcein at a final concentration of 10 μm for 40 min at 37 °C, 5% CO2. Cells were washed twice in PBS, resuspended in RPMI 1640 medium, and incubated at 37 °C, 5% CO2 for 2 h. After labeling, target cells were washed, recounted, and resuspended at 0.4 × 10e6 cells/ml in RPMI 1640 medium. The ADCC assay was performed in a sterile 96-well round bottom tissue culture plate (Corning Glass). Antibodies were titrated from 2 μg/ml to 0.00002 μg/ml at 1:10 series dilutions in RPMI 1640 medium containing 10% FBS. 50 μl of 20,000 calcein-labeled target cells and 50 μl of various concentrations of antibody were incubated for 40 min at 4 °C, and 50 μl containing 200,000 NK cells were added (effector/target (E/T) = 10:1). Cultures were incubated for 4 h at 37 °C, and then supernatants were harvested and assayed for calcein release by measuring fluorescence at 485–535 nm in Wallac Victor II 1420 Multilabel HTS counter. 100% lysis values were determined by lysing six wells of calcein-labeled targets with IGEPAL CA 630 detergent (3 μl per well). Percent (%) specific lysis was defined as follows: (sample fluorescence) − (spontaneous lysis fluorescence)/(100% lysis − spontaneous lysis fluorescence)·100. Percent specific lysis values were transformed, and sigmoidal dose-response curve fits were done in GraphPad Prism 5.0.
Her2-expressing SK-BR-3 and JIMT-1 cells were labeled with CellTrace CFSE at a final concentration 0.25 μm at 37 °C for 15 min, incubated with NK cells (FcγR IIIA 158 F/F genotype) at E/T = 5:1 at 37 °C for 4 h, washed with 1× PBS, pH 7.4, 2% FBS, resuspended in 1× PBS, pH 7.4, 2% FBS, 10 μg/ml 7-aminoactinomycin D at 4 °C for 30 min, washed again with 1× PBS, pH 7.4, 2% FBS, and then subjected to FACS analysis. At least 2,000 CFSE+ tumor cells were gated for each sample; the % specific lysis was defined as positive for 7-aminoactinomycin D+ CFSE+ cells/CFSE+ cells·100.
NCI-N87 and JIMT-1 Xenograft Mouse Models
Female CB-17/SCID mice (7–8 weeks old) were implanted subcutaneously with 2 × 106 cells of the human gastric cancer cell line NCI-N87 mixed 1:1 with reduced growth factor Matrigel (BD Biosciences) in a total volume of 200 μl. Three days post-tumor implantation, mice were randomized to control and treatment groups of 10 mice each, so that the mean tumor size was similar across groups at the start of treatment. Mice were treated with 10 mg/kg anti-Her2 IgG1 (WT), anti-Her2 IgG1 (W165), anti-Her2 IgG1 (afuco-W117), anti-TuAg (clone H158) IgG1 (S239D/I332E/A330L), anti-TuAg (clone H158) IgG1 (W165), or isotype control anti-SAv IgG1 antibodies. The antibodies were administered intraperitoneally in a volume of 100 μl two times per week for 3 weeks (days 3, 7, 10, 14, 17, and 21).
Female CB-17/SCID mice (7–8 weeks old) were injected subcutaneously with 5 × 106 JIMT-1 cells mixed 1:1 with Matrigel. Seven days post-tumor implantation, tumor volume was measured, and mice were randomized into control and treatment groups of 10 mice each. The mice were treated with 250 μg of anti-Her2 IgG1 (WT), anti-Her2 IgG1 (W165), anti-Her2 IgG1 (afuco-W117), or isotype control anti-SAv IgG1 antibodies. The antibodies were administered intraperitoneally in a volume of 200 μl once per week for 4 weeks. For all studies, tumor-bearing mice (implanted with electronic bar-coded chips) were monitored for weight and for tumor volume two times per week by obtaining perpendicular measurements of the tumor with an electronic caliper. Tumor size was calculated using Equation 1,
Percent tumor growth inhibition (%TGI) was calculated based on Equation 2,
where ΔC or ΔT indicates the difference between the average tumor volume on the last day and day of initial measurement for the control (ΔC) or treatment groups (ΔT). Animals receiving isotype control IgG1 served as the control group for these calculations. Anti-tumor activity was defined as percent tumor growth inhibition ≥50%.
Statistical Analysis
Tumor growth was expressed as the means ± S.E. and plotted as a function of time. Statistical comparison of groups was done using the analysis of variance test followed by Dunnett's test adjusted for multiple comparisons. Statistical calculations were made through the use of JMP software version 7.0 interfaced with SAS version 9.2 (SAS Institute, Inc., Cary, NC) or GraphPad Prism 5.0 in the case of NCI-N87 xenograft studies.
RESULTS
Heterodimeric IgG1 Antibodies Containing Asymmetrical Fc Mutants Enhance ADCC Effector Function
The crystal structure studies revealed that the interaction of human FcγRIII with IgG1 Fc region is asymmetric, i.e. FcγRIII comes into contact with different amino acid residues on the two Fc polypeptide chains that make up the Fc region (15). On one Fc chain, the lower hinge region and residues around Pro-329 contribute to the binding with FcγRIII. On another Fc chain, the lower hinge region, residues close to Asp-265, residues around Asn-297, and the glycan attached on Asn-297 are involved in the binding to FcγRIII. From a protein engineering point of view, asymmetric alterations, which mimic the natural Fc/FcγRIII interaction, may be the ideal way to maximally increase the binding of the Fc region to FcγRIII, thus enhancing ADCC effector function. We tested this hypothesis by making asymmetric Fc variants in the context of an anti-TuAg (clone H158) human IgG1 in heterodimeric form.
Human IgG1 variant with S239D/A330L/I332E (3X) mutations has been shown to have significantly increased binding affinity to FcγRIIIA, but it requires three mutations on both Fc chains. However, examination of Fc/FcγRIII co-crystal structure suggested that not all of Ser-239, Ala-330, and Ile-332 on both Fc chains are simultaneously engaged in interacting with the corresponding residue(s) of FcγRIII (Fig. 2A) (13, 15).
FIGURE 2.

Asymmetrically engineered Fc variants in the context of anti-TuAg (clone H158) IgG1 antibodies have enhanced ADCC effector function. A, interface between the Fc chains (left chain (B) and right chain (A)) and FcγRIII based on x-ray crystal structure of the Fc-FcγRIIIB complex (Protein Data Bank code 1T83). On chain B, Ala-330 is within 4 Å of receptor residue Ile-88 of FcγRIII; Ile-332 is close to Lys-161 of FcγRIII, and Ser-239 and Ser-298 are not engaged. On chain A, Ser-239 interacts with Lys-120 of FcγRIII; Ser-298 is in proximity to residue Tyr-132 of FcγRIII; Ala-330 and Ile-332 is far away from any residues of FcγRIII. B, DSC analysis to measure the domain melting temperature of anti-TuAg (clone H158) IgG1 antibodies in heterodimer format. The sequence alternations in each antibody variant are indicated in Table 1. C, ADCC activity of anti-TuAg (clone H158) IgG1 antibody variants with OVCAR-8 cells in the presence of purified human NK cells (FcγRIIIA F/F genotype). D, ADCC activity of anti-TuAg (clone H158) IgG1 antibody variants with CAPAN-2 cells in the presence of purified human NK cells (FcγRIIIA F/F genotype). Irrelevant human IgG1 was used as isotype control, and anti-TuAg (clone H158) WT IgG1 was used as a base line. Effects of concentrations at 50% of maximal killing (EC50, pm) are shown beside the designated Fc variants. Ab, antibody.
To test whether the asymmetrical engineering strategy can allow us to achieve a greater Fc/FcγRIII interaction, we generated four heterodimeric IgG1 variants (Table 1). In variant V1, on chain A, the S239D (near the hinge region) and S298A (near the residue Asn-297 and glycan) were introduced in CH2 domain; on chain B, the A330L and I332E (both near residue Pro-329) were introduced in the CH2 domain; charge pair residues for heterodimerization K392D + K409D and E356K + D399K were incorporated in the CH3 domain, respectively. It was predicted that each mutation in CH2 domain would contribute to the binding improvement by interacting with its corresponding residue in the FcγRIIIA. To test whether the charge pair residues for heterodimerization have an impact on the binding of Fc to FcγRIIIA, we switched the charge pairs in CH3 domain to make variant V2. Similarly, we made variants V3 and V4 by introducing S239D in both Fc chains, because S239D in one Fc chain could interact with Lys-120 of FcγRIIIA, whereas S239D in the other Fc chain could interact with Lys-161 of FcγRIIIA.
TABLE 1.
Biacore measurement of anti-TuAg (clone H158) IgG1 variants binding to monomeric human FcγRIIA (Arg-131), FcγRIIA (His-131), FcγRIIB and FcγRIIIA (Val-158), and FcγRIIIA (Phe-158)
| Antibody | Type | Sequence variations |
FcγR (KD) (μm) |
|||||
|---|---|---|---|---|---|---|---|---|
| Fc chain A | Fc chain B | IIA 131R | IIA 131H | IIB | IIIA 158V | IIIA 158F | ||
| Anti-TuAg IgG1 (WT) | Homodimer | No change | No change | 9.2 | 6.11 | 13.9 | 1.64 | 7.56 |
| Anti-TuAg IgG1 (3×) | Homodimer | S239D + A330L + I332E | S239D + A330L + I332E | 9.8 | 6.98 | 6.9 | 0.046 | 0.059 |
| Anti-TuAg IgG1 (V1) | Heterodimer | S239D + S298A + E356K + D399K | A330L + I332E + K392D + K409D | 6.71 | 5.53 | 12.4 | 0.028 | 0.04 |
| Anti-TuAg IgG1 (V2) | Heterodimer | S239D + S298A + K392D + K409D | A330L + I332E + E356K + D399K | 7.5 | 5.82 | 14.7 | 0.03 | 0.047 |
| Anti-TuAg IgG1 (V3) | Heterodimer | S239D + S298A + E356K + D399K | S239D + A330L + I332E + K392D + K409D | 6.9 | 5.23 | 8.6 | 0.019 | 0.022 |
| Anti-TuAg IgG1 (V4) | Heterodimer | S239D + S298A + K392D + K409D | S239D + A330L + I332E + E356K + D399K | 3.88 | 2.96 | 6.3 | 0.025 | 0.031 |
The homodimeric wild type human IgG1 or variant IgG1 containing S239D/I332E/A330L (3X) or heterodimeric variants V1, V2, V3, or V4 was tested by Biacore to measure their binding affinity to FcγRIIA (His-131), FcγRIIA (Arg-131), FcγRIIB, FcγRIIIA (Phe-158), and FcγRIIIA (Val-158) (Table 1). Wild type human IgG1 binds to low affinity allotype FcγRIIIA (Phe-158) with a KD of 7.56 μm and to high affinity allotype FcγRIIIA (Val-158) with a KD of 1.64 μm, around a 5-fold difference that is in line with the data from the literature (13). The homodimeric IgG1 S239D/I332E/A330L variant significantly improves the binding to FcγRIIIA (Val-158) with a KD of 0.046 μm and FcγRIIIA (Phe-158) with a KD of 0.059 μm. The four heterodimer variants showed further improvement of binding to both FcγRIIIA allotypes, with the variant V3 being the top binder of FcγRIIIA (Val-158) at KD of 0.019 μm and FcγRIIIA (Phe-158) at KD of 0.022 μm. The variants V1 and V2 have comparable binding affinity to FcγRIIIA and so do the variants V3 and V4, suggesting that the charge pair residues in the CH3 domain of the Fc region for heterodimerization have little impact on the Fc/FcγRIIIA interaction. The homodimeric IgG1 S239D/I332E/A330L variant showed 2-fold higher binding affinity to FcγRIIB than the wild type IgG1; however, the heterodimeric variants V1 and V2 had comparable binding to FcγRIIB as wild type IgG1. Heterodimeric variants V3 and V4 had comparable binding to FcγRIIB as the homodimeric IgG1 S239D/I332E/A330L variant, in line with the fact that S239D substitutions in both Fc chains improve the binding to FcγRIIB (13). Interestingly, all four heterodimer variants showed improved binding to both FcγRIIA His-131 and Arg-131 allotypes to some extent, indicating that the heterodimeric variants might be able to distinguish the highly homologous FcγRs, i.e. FcγRIIA and FcγRIIB.
The above homodimeric and heterodimeric IgG1 variants along with wild type IgG1 were tested by DSC to assess their stability (Fig. 2B). The wild type IgG1 showed a typical Tm of Fab/CH3 at ∼85 °C and Tm of CH2 domain at ∼72 °C. The homodimeric IgG1 S239D/I332E/A330L variant keeps the Tm for the Fab region at ∼85 °C, slightly decreases the Tm of CH3 domain to ∼78 °C, and significantly decreases the Tmof CH2 domain more than 20 °C, to 50 °C. All four heterodimer IgG1 variants exhibit a Tm of Fab at ∼85 °C, while slightly decreasing the Tm of CH2/CH3 to ∼65 °C due to the introduction of two charge pair residues in the CH3 domain for heterodimerization. The data suggested that heterodimeric IgG1 variants have an improved FcγRIIIA binding capacity and have less impact on the CH2 stability than the homodimeric IgG1 S239D/I332E/A330L variant, the strongest Fc variant containing S239D/A330L/I332E mutations in both Fc chains.
To test whether the above heterodimeric IgG1 variants retain potent ADCC capacity, OVCAR-8 cells (Fig. 2C) expressing a moderate level of specific tumor antigen and CAPAN-2 cells (Fig. 2D) expressing a lower level of specific tumor antigen were used for an in vitro cell-based killing assay. In the presence of human NK cells (FcγRIIIA 158 F/F genotype) at E/T = 10:1 ratio, neither the control IgG1 nor the wild type anti-TuAg IgG1 showed dose-dependent killing of OVCAR-8 cells. The S239D/I332E/A330L variant showed a high maximal lysis level and an increased potency exhibiting an EC50 of 27 pm. The four heterodimeric IgG1 variants all exhibited enhanced potency relative to wild type IgG1 with EC50 values of 20, 16, 11, and 44 pm for V1, V2, V3, and V4, respectively, but not enhanced maximal lysis when compared with the S239D/I332E/A330L variant. A similar trend of CAPAN-2 cell killing by ADCC was observed with these heterodimers. These results indicated that heterodimeric IgG1 variants could effectively induce potent ADCC activity against tumor cells expressing different levels of specific tumor antigen.
Taken together, the results showed that asymmetrical Fc engineering is a viable and efficient approach to enhance the ADCC effector function of the targeted antibodies. To further explore other sequence space involved in Fc/FcγRIIIA interaction using the asymmetrical engineering strategy, we built and screened Fc libraries in a heterodimeric format.
Clustering of Positions for Mutagenesis
X-ray crystal structure of the Fc-FcγRIIIB (Protein Data Bank code 1T83) complex was used to identify FcγRIII contacting residues in the Fc chain A (R, right side) and Fc chain B (L, left side). Residues in the lower hinge region of both Fc chains are critical to the FcγRIII binding. Another group of residues in the CH2 domain are either close to or part of the area that contacts FcγRIII. Residues in the third group within the CH2 are solvent-exposed but are not close to or part of the area that contacts FcγRIIIA. Fig. 3A shows the positions of these sites within a human IgG1 Fc segment. Oligonucleotides with NN(G/C) codon at the center to randomize the 82 selected positions in the above three groups were synthesized to make Fc libraries (see under “Experimental Procedures”).
Construction of Fc Libraries and Expression of Heterodimer Fc Molecules for Screening
To express Fc variants as heterodimer, mammalian HEK-293 cells were co-transfected with plasmid DNAs encoding an scFv of rat anti-mouse natural killer group 2D antibody that was fused to a human IgG1 Fc polypeptide containing E356K + D399K charge pair mutations in the CH3 domain and plasmid DNAs encoding a human IgG1 Fc polypeptide containing K392D + K409D charge pair mutations in the CH3 domain. Two polypeptides were translated and secreted mainly as heterodimeric molecules that can be separated by size in the nonreduced SDS-polyacrylamide gel (21). Six small Fc libraries representing three tiers of mutants from both Fc chains were generated using PCR splice overhang extension technology for a quick and more focused screening. Depending on the designed complexity of each library, three times more colonies were picked to ensure the full coverage of libraries. 95 randomly picked colonies from each library were sequenced to verify that libraries had good diversity by having different mutations at the designated positions (data not shown). Analysis of the conditioned media from HEK-293 cells suggested co-transfection of two plasmids encoding scFv-Fc and Fc chain yielded mostly scFv-Fc/Fc heterodimer (data not shown). The successful construction of Fc heterodimer libraries allowed us to initiate screening with a high throughput competition AlphaLISA® assay.
Identification of Fc Mutants with Enhanced Binding to FcγRIIIA
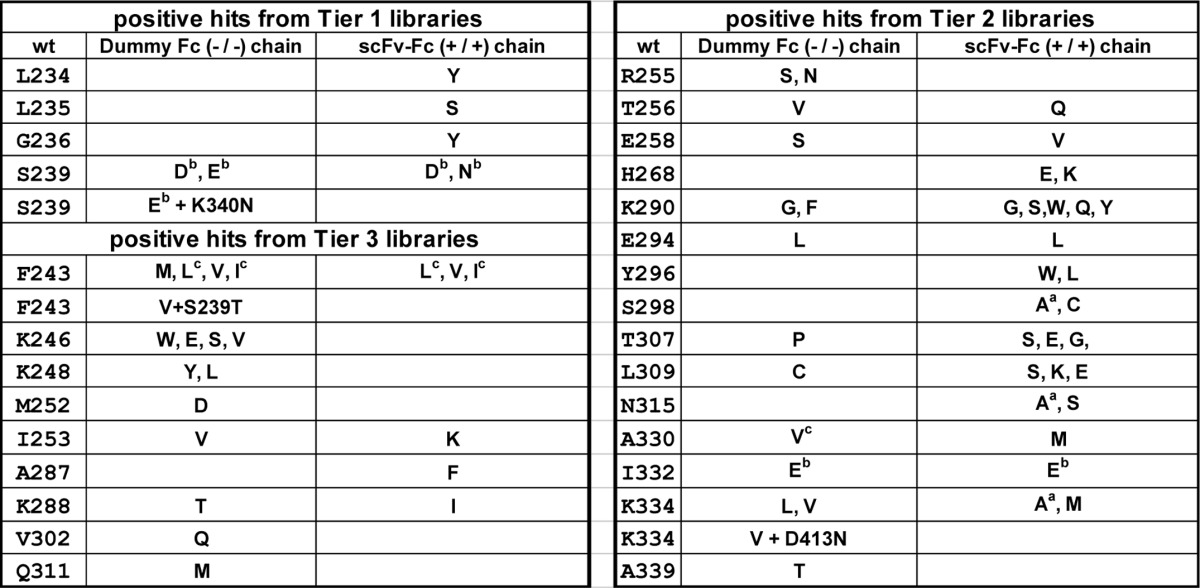
Conditioned medium was harvested 6 days post-transfection and screened to identify Fc variants with higher binding affinity to GST-His6-tagged FcγRIIIA Phe-158 (Fig. 3B). Hits with >20% inhibition to the binding of regular human IgG1 to FcγRIIIA Phe-158 were scored as positive in the primary screening and then went through two additional rounds of screening for confirmation. By calculation, a total of 1,640 different mutants were included in the scFv-Fc-encoding Tier 1, 2, and 3 libraries combined. The same number of variants was included in the dummy Fc-encoding Tier 1, 2, and 3 libraries combined. Overall 9,600 mutants in 100 plates (96-well) were tested to cover the complexity of Fc libraries for ∼3 times (see “Experimental Procedures”). 37 mutants from scFv-Fc libraries and 34 mutants from dummy Fc libraries repeatedly showed strong competition to the binding of regular human IgG1 to FcγRIIIA Phe-158. Many of these variants were discovered multiple times. Thus, in total, about 2% of the 1,640 different variants included in the libraries yielded positive signals. Of these 71 hits (Table 2), some were identified previously by others, but most of the hits were novel. Interestingly, only a small number of beneficial mutants in both Fc chains were found from six Fc libraries. Three mutants had double substitutions at different positions, likely due to PCR errors during the amplifications.
TABLE 2.
Summary of primary positive hits identified from Fc libraries
The Fc libraries were screened by a high throughput competition AlphaLISA® assay to find Fc mutants with improved binding to FcγRIIIA (Phe-158), after the first round of primary screen; Fc mutants with >20% inhibition to the interaction of regular IgG1/FcγRIIIA (Phe-158) were scored as positive hits. Two more rounds of competition AlphaLISA® assay were conducted to confirm the activity. Left panel, positive hits from Tier 1 and Tier 3 Fc libraries. Right panel, positive hits from Tier 2 Fc libraries. Amino acid residues with superscript “a” indicate they were identified previously by Shields et al. (12). Amino acid residues with superscript “b” indicate they were identified previously by Lazar et al. (13). Amino acid residues with superscript “c” indicate they were identified previously by Stavenhagen et al. (14).
Construction and Characterization of Combinational Mutants in IgG1 Format
All the primary hits were mapped out on the Fc/FcγRIIIA co-crystal structure using molecular operating environment, a molecular modeling program from Chemical Computing Group Inc. (Montreal, Canada). To identify the combinations that could improve the binding affinity to FcγRIIIA, a total of 1,258 (37 × 34) combinations of individual positive hits were made and tested by the competition AlphaLISA® assay. We found 21 combinations that contain our novel Fc mutations did significantly compete the interaction of biotinylated regular human IgG1 with FcγRIIIA as follows: E294L/E294L; E294L/Y296L; E294L/K290G; E294L/K290S; E294L/S298A; E294L/T307G; T307P/T307G; T307P/K290G; T307P/Y296L; T307P/K290S; R255S/S298C; T307P/S298C; E294L/S298C; K334V/K290Y; T307P/L309E; E294L/L309E; T307P/L234Y; E294L/L234Y; Q311M/Y296W; Q311M/L234Y; and K334V/Y296W in the Fc chain containing K392D + K409D/E356K + D399K respectively (data not shown). All of these 21 combinations were re-tested and confirmed as anti-Her2 heterodimeric IgG1 antibodies (see “Experimental Procedures”). IgG1 variants with mutations located at the hinge regions, around Pro-329, close to Asp-265, and around Asn-297, for example, K334V in one Fc chain and Y296W in another Fc chain, were selected for further improvement by adding more beneficial mutations. Two leading variants M61 and M63 were picked based on their lower EC50 value in competition AlphaLISA® assay and good manufacturability profile. M61 has K334V and L234Y + Y296W; M63 has K334V and L234Y + E294L in Fc chain containing K392D + K409D and E356K + D399K charge pair residues in the CH3 domain, respectively. Tests of more combinations identified K290Y as an enhancing mutation when added in Fc chain containing L234Y + Y296W.
Substitutions within the N-glycosylation site (Asn-297–Ser-298–Thr-299) and/or near the Pro-329 site in either Fc chain were explored using molecular modeling. Substitutions within both of these areas (i.e. S298C, S298A, A330M, and A330V) had been found in the primary screen. To arrive at candidate combinations of substitutions and to eliminate substitutions that might create manufacturability issues (e.g. replacing a cysteine with another amino acid), structural analyses were performed using the Fc-FcγRIII crystal structures (Protein Data Bank codes 1T83, 1T89, and 1E4K), and binding energy was calculated using EGAD (18). EGAD is a computational protein design algorithm that predicts changes in protein stability upon substitution of one or more amino acid residues in a protein.
Each of the three positions at Ser-298, Ala-327, and Ala-330 was changed to all other 19 amino acids in silico, and the change in the stability of the Fc/FcγRIII interaction was predicted. Substitutions that EGAD predicted to enhance binding to FcγRIIIA include S298C, S298I, S298V, S298T, A327Y, A327W, A327F, A327H, A330H, A330F, and A330M. AlphaLISA® assay confirmed that addition of some mutations at positions Ser-298 and Ala-330 did improve binding to the FcγRIIIA. For example, the combination designated “W23,” which has L234Y, K290Y, and Y296W mutations in one Fc chain and S298T and K334V mutations in the other Fc chain, resulted from this approach. When A330M was added in the Fc chain containing S298T and K334V of variant W23, the new variant that results in W165 binds more effectively to FcγRIIIA than wild type huIgG1 and slightly better than variant W23. In such a “step-by-step” approach, a new panel of Fc variants containing a total of 5 or 6 beneficial substitutions (Table 3) were identified as showing stronger binding to FcγRIIIA, high expression, and good manufacturability (data not shown).
TABLE 3.
Binding of anti-Her2 IgG1 containing the combinatorial beneficial Fc variants to human and murine FcγRs by competition AlphaLISA® assay and Biacore analysis
The amino acid substitutions in Fc chain A and Fc chain B, which contain the heterodimerization mutations K392D + K409D and E356K + D399K, respectively, are shown under the Sequence Variations column. The effect of concentrations at 50% inhibition (EC50, nm) of anti-Her2 IgG1 containing the designated Fc variants to the interaction of wild type IgG1 and human FcγRIIIA (Val-158) or FcγRIIIA (Phe-158) are indicated under the AlphaLISA® Assay column. Measurements of kon (1/ms), koff (1/s), and KD (nm) of anti-Her2 IgG1 containing the designated Fc variants are summarized under the Biacore Analysis column. a, homodimeric IgG1; b, ND, not determined. M01 is an anti-Her2 wild type IgG1; M02 (or 2X) is an anti-Her2 IgG1 containing S239D + I332E in both Fc chains; M03 (or 3X) is an anti-Her2 IgG1 containing S239D + I332E + A330L in both Fc chains; M04 is an anti-Her2 IgG1 containing only charge pair residues for Fc heterodimerization.
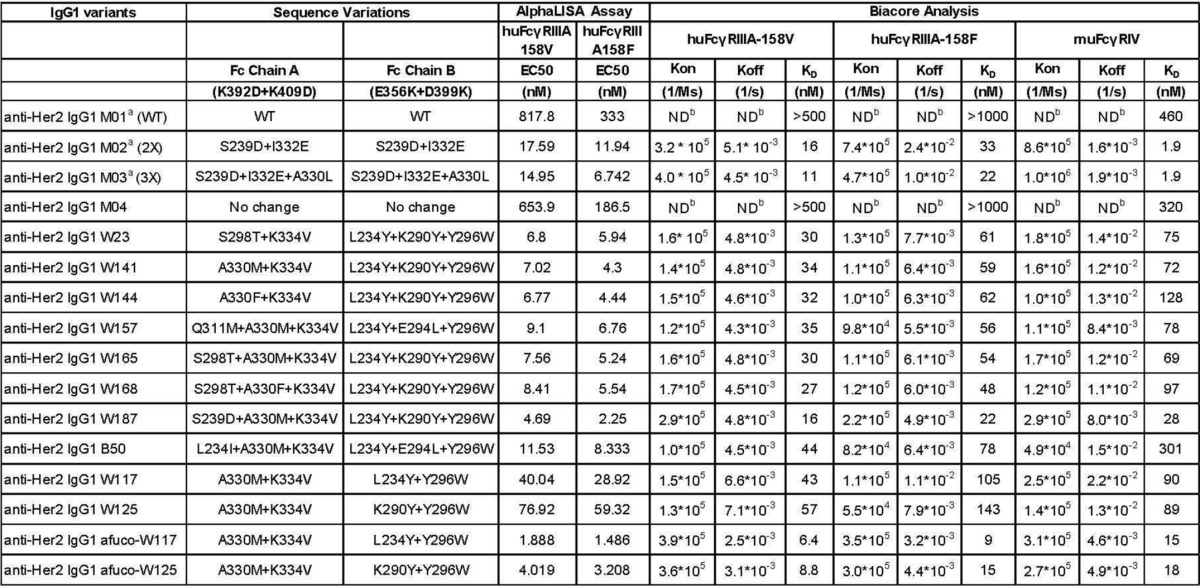
As shown in Fig. 4A, M01, a humanized anti-Her2 wild type IgG1, inhibits the IgG1 binding to FcγRIIIA (Phe-158) by only about 25% at the highest concentration tested (360 nm). M04, a humanized anti-Her2 IgG1 antibody that contains heterodimerization charge pairs K392D + K409D in one Fc chain and E356K + D399K in the other Fc chain, was slightly more effective than M01. Compared with M01 and M04, other heterodimeric variants such as W23, W141, W144, W157, W165, W168, W187, and B50 competed much more strongly to the interaction between regular human IgG1 and FcγIIIA, as evidenced by a left-shift of the competition curves and a lower EC50 value in the AlphaLISA® assay (Table 3). A similar pattern was observed when the anti-Her2 IgG1 variants were tested as competitors between regular human IgG1 and FcγIIIA Val-158 (Fig. 4B). Of the variants tested, W187 was the strongest binder to both allotypes of FcγRIIIA.
FIGURE 4.

Percent inhibition of AlphaLISA® signal by full-length IgG1 antibodies containing variant Fc regions. A, percent inhibition of IgG1 containing the designated Fc variants to the interaction of IgG1 containing regular Fc and FcγRIIIA (Phe-158). B, percent inhibition of IgG1 containing the designated Fc variants to the interaction of IgG1 containing regular Fc and FcγRIIIA (Val-158). C, percent inhibition of IgG1 containing the designated regular and afucosylated Fc variants to the interaction of IgG1 containing regular Fc and FcγRIIIA (Phe-158). D, percent inhibition of IgG1 containing the designated regular or afucosylated Fc variants to the interaction of IgG1 containing regular Fc and FcγRIIIA (Val-158). The graphs show the percent inhibition of an AlphaLISA® signal as a function of concentration of competitor. The various competitors, which are human IgG1 antibodies, are indicated by alias in the graph, and the substitutions contained in each competitor and effect concentrations at 50% maximal inhibition (EC50) are indicated in Table 3.
Asymmetrically Engineered IgG1 Variants Have Strong Binding to FcγRIIIA
All anti-Her2 IgG1 variants in Table 3 were produced by mammalian 2936E cells and confirmed to have the correct mass by mass spectrometry analysis. No significant change in Asn-297 glycan was observed (data not shown). To test their binding to FcγRIIIA, Biacore analysis was carried out to measure the kon, koff, and KD values of anti-Her2 IgG1 antibodies that were embedded with the ADCC enhancement Fc mutations (Table 3 and Fig. 5). The results showed that introduction of the charge pair residues in the CH3 domain for heterodimerization made no big difference in KD for binding to any of the FcγRs tested as illustrated by the comparison of M04 and M01. Furthermore, the various asymmetric Fc mutants improved the on-rate and drastically have a slower off-rate when compared with anti-Her2 wild type IgG1 M01 or IgG1 variant M04 (Fig. 5, A and B, and Table 3), leading to a reduced (more than 10-fold in all cases) KD value (ranging from 6.4 to 143 nm) for binding to both allotypes of FcγRIIIA. In addition, the various asymmetric Fc mutants also significantly improved the binding to mouse FcγRIV compared with wild type IgG1 (Fig. 5C and Table 3). These data, combined with the results of ADCC assay discussed below, show that increased cell killing correlates with increased on rates and decreased off-rates, i.e. a decreased KD or higher affinity.
FIGURE 5.

Biacore analysis of humanized anti-Her2 human IgG1 antibody variants binding to human and mouse FcγRs. A, sensorgram of humanized anti-Her2 IgG1 antibody variants binding to monomeric human FcγRIIIA (Phe-158 allotype) aligned before association. Note that all variants have improved on-rate with the afuco-W117 variant being the top one when compared with wild type IgG1. B, sensorgram of humanized anti-Her2 IgG1 antibody variants binding to monomeric human FcγRIIIA (Val-158 allotype) aligned before association. Note that all variants have improved on-rate with the afuco-W117 variant being the top one when compared with wild type IgG1. C, sensorgram of humanized anti-Her2 IgG1 antibody variants binding to monomeric mouse FcγRIV aligned before association. Note that all variants have improved on-rate with the S239D/I332E variant being the top one when compared with wild type IgG1. D, sensorgram of humanized anti-Her2 IgG1 antibody variants binding to monomeric human FcγRIIB aligned before association. Note that homodimeric S239D/I332E variant has significant increase of on-rate when compared with wild type IgG1; W187 variant has some increase of on-rate; other heterodimeric variants have comparable binding to human FcγRIIB as wild type IgG1. E, sensorgram of humanized anti-Her2 IgG1 antibody variants binding to monomeric human FcγRIIA (H131 allotype) aligned before association. Note that all heterodimeric variants have improved on-rate with the W23 variant being the top one when compared with wild type IgG1. F, sensorgram of humanized anti-Her2 IgG1 antibody variants binding to monomeric human FcγRIIA (R131 allotype) aligned before association. Note that homodimeric S239D/I332E variant is the only one having improved on-rate when compared with wild type IgG1. G, percent of free human FcRn when the binding of 10 nm of human FcRn to regular human IgG1 was competed by different concentrations of humanized anti-Her2 IgG1 containing the Fc variants. Note all variants have comparable competition to FcRn as wild type IgG1. The various human IgG1 antibodies used in the Biacore analysis are indicated by alias in parentheses, and their substitutions contained in each variant are shown in Table 3. RU, response units.
Afucosylation of our Novel Fc Variants Further Improves the Binding to FcγRIIIA
To minimize the probability of immunogenicity of engineered antibodies while keeping their potent ADCC activity, we made afucosylated preparations of W117 and W125 (afuco-W117 and afuco-W125) in which fewer (only 2) beneficial mutations are present in either Fc chain (Table 3). As shown in Fig. 4C, M01 and M04 exhibited weak competition to the binding of regular human IgG1 to FcγRIIIA Phe-158. Afuco-W117 and afuco-W125 were the most effective competitors, followed by W165, M03 (S239D/I332E/A330L), W157, W117, afuco-IgG1, and W125. Similar results for binding to FcγRIIIA Val-158 are shown in Fig. 4D. These results along with the data of Biacore analysis (Table 3 and Fig. 5) showed that an IgG1 antibody comprising an afucosylated wild type Fc did compete more effectively than an antibody comprising a wild type Fc; an IgG1 antibody comprising the afucosylated Fc variants did compete more effectively than an antibody comprising an afucosylated wild type Fc, suggesting that glyco-engineering and protein engineering work together to further improve the binding of engineered Fc to FcγRIIIA.
Asymmetrically Engineered IgG1 Variants Do Not Change the Binding to Inhibitory FcγRIIB
It was established that anti-Her2 and anti-E-cadherin mAb had amplified therapeutic activities in mice deficient for inhibitory FcγRIIB compared with the effect observed in wild type mice (7, 8). From an efficacy point of view, it is desirable to engineer mAbs that can enhance the interaction with activating FcγRs, but not with inhibitory FcγRIIB, leading to a high activating to inhibitory ratio. By Biacore analysis (Fig. 5D) and competition AlphaLISA® assay (data not shown), all asymmetrically engineered IgG1 variants except W187 have comparable binding capacity to FcγRIIB as the wild type IgG1 (M01) and IgG1 containing only charge pair residues in CH3 domain of Fc chains (M04), suggesting that the asymmetrically engineered IgG1 antibodies may have a high activating to inhibitory ratio, leading to a better in vivo anti-tumor efficacy. However, the homodimeric anti-Her2 IgG1 S239D/I332E variant containing S239D/I332E in both Fc chains and heterodimeric anti-Her2 IgG1 variant W187 containing S239D in both Fc chains increased the binding to FcγRIIB, in line with the previously reported data (13).
Asymmetrically Engineered IgG1 Variants Could Distinguish Highly Homologous FcγRs Such as Activating FcγRIIA and Inhibitory FcγRIIB
Because the asymmetrically engineered Fc variants introduce different alterations in Fc chains and could fine-tune the binding between Fc and FcγRs, it is possible that they can distinguish the highly homologous FcγRIIA and FcγRIIB. By Biacore analysis (Fig. 5, E and F) and competition AlphaLISA® assay (data not shown), all anti-Her2 heterodimeric IgG1 variants showed enhanced binding to FcγRIIA (His-131) with W23 variant being the top one when compared with wild type IgG1, whereas no increase of binding to FcγRIIA (R131) or FcγRIIB (Fig. 5D) was observed. The homodimeric IgG1 S239D/I332E variant (2X) increased the binding to FcγRIIA (both His-131 and Arg-131 allotypes) and significantly increased the binding to inhibitory FcγRIIB. The results suggested that the asymmetrically engineered IgG1 variants can distinguish FcγRIIA (His-131) from highly homologous FcγRIIB and may potentiate ADCP activity on top of ADCC enhancement.
Asymmetrically Engineered IgG1 Variants Do Not Alter the Binding to FcRn
Fc neonatal receptor (FcRn) mediates the recycling of antibodies, conferring the long half-life of antibodies. It is preferred for engineered antibodies to retain the half-life as the wild type antibody. This was assessed by Biacore analysis. After human Fc was immobilized on CM5 chips, 10 nm human FcRn was flowed through the surface of chips, and the interaction between Fc and FcRn generated response units. When 0.1–2,000 nm of anti-Her2 IgG1 variants were present, they competed the interaction of Fc/FcRn, knocking down the response units (Fig. 5G). All heterodimeric anti-Her2 IgG1 variants (W23, W141, W144, W157, W165, W168, W187, B50, W117, W125, afuco-W117, and afuco-W125) showed comparable levels of competition as the wild type IgG1 (M01), suggesting that the ADCC enhancement substitutions in the lower hinge region and CH2 domain do not change the binding of engineered Fc to FcRn. More in vivo studies in cynomolgus monkeys and humans to analyze the pharmacokinetic of engineered antibodies will elucidate whether the Fc variants described in this work could have an impact on the half-life of antibodies in the body.
Homodimeric IgG1 Variants Do Not Improve the Binding to FcγRIIIA as Effectively as the Asymmetrical Engineered Heterodimeric IgG1 Variants
To answer the question whether the same set of mutations placed symmetrically (conventional format) into IgG1 could also enhance the ADCC activity as well as asymmetrically engineered IgG1 variants, W144, which has strong binding to both FcγRIIIA allotypes and has a total of five beneficial mutations (Table 3), was selected for comparison as homodimer or heterodimer by competition AlphaLISA® assay (Fig. 6 and Table 4). Anti-Her2 IgG1 (W144) is a heterodimeric antibody that was embedded with A330F and K334V in the CH2 domain and charge pair mutations K392D and K409D in the CH3 domain of one Fc chain, L234Y and K290Y and Y296W in the CH2 domain, and charge pair mutations E356K and D399K in the CH3 domain of the other Fc chain. For comparisons, homodimeric anti-Her2 IgG1 variants were made; W199 contains A330F and K334V in both Fc chains; W211 contains L234Y and K290Y and Y296W in both Fc chains. Heterodimeric variant W144 had EC50 at 6.77 and 4.44 nm when binding to FcγRIIIA Val-158 and FcγRIIIA Phe-158, respectively. However, homodimeric variants W199 and W211 possessed 10–20-fold lower binding affinity for both FcγRIIIA allotypes. W202, which contains all five mutations (A330F, K334V, L234Y, K290Y, and Y296W) in both Fc chains as a homodimeric IgG1, did not improve the binding affinity to FcγRIIIA as effectively as the heterodimeric IgG1 variant W144.
FIGURE 6.

Percent inhibition of AlphaLISA® signal by full-length anti-Her2 IgG1 antibodies containing variant Fc regions as homodimer or heterodimer. A, percent inhibition of AlphaLISA® signal by anti-Her2 IgG1 variants M01, M02, M03, M04, W144, W190, W211, and W202 to the interaction between wild type human IgG1 and monomeric human FcγRIIIA (Phe-158). B, percent inhibition of AlphaLISA® signal by anti-Her2 IgG1 variants M01, M02, M03, M04, W188, W203, W211, and W204 to the interaction between wild type human IgG1 and monomeric human FcγRIIIA (Phe-158). The graphs show the percent inhibition of AlphaLISA® signal as a function of competitor concentration. The various competitors, which are humanized anti-Her2 IgG1 antibody containing the Fc variants, are indicated by alias in the graph; the substitutions contained in each competitor and 50% of maximal effective concentration (EC50) are indicated in Table 3. Note that variants M04, W144, and W188 are heterodimeric IgG1s, and others are homodimeric IgG1s.
TABLE 4.
Homodimeric IgG1 variants do not improve the binding to FcγRIIIA as effectively as the asymmetrically engineered heterodimeric IgG1 variant
Homodimeric and heterodimeric anti-Her2 IgG1 variants were made and compared with a competition AlphaLISA® assay. The variant alias, protein type, sequence variations in each Fc chain, and EC50 values for binding to FcγRIIIA V158 and FcγRIIIA F158 are summarized.
| Variants | Type | Sequence variation |
huFcγRIIIa Val-158, EC50 (nm) | huFcγRIIIa Phe-158, EC50 (nm) | |
|---|---|---|---|---|---|
| Chain A | Chain B | ||||
| Anti-Her2 IgG1 (M01) | Homodimer | WT | WT | 817.8 | 333 |
| Anti-Her2 IgG1 (M02) | Homodimer | S239D + I332E | S239D + I332E | 17.59 | 11.94 |
| Anti-Her2 IgG1 (M03) | Homodimer | S239D + I332E + A330L | S239D + I332E + A330L | 14.95 | 6.742 |
| Anti-Her2 IgG1 (M04) | Heterodimer | K392D + K409D | E356K + D399K | 653.9 | 186.5 |
| Anti-Her2 IgG1 (W144) | Heterodimer | K392D + K409D + A330F + K334V | E356K + D399K + L234Y + K290Y + Y296W | 6.77 | 4.44 |
| Anti-Her2 IgG1 (W199) | Homodimer | A330F + K334V | A330F + K334V | 115.3 | 48.54 |
| Anti-Her2 IgG1 (W211) | Homodimer | L234Y + K290Y + Y296W | L234Y + K290Y + Y296W | 40.7 | 36.92 |
| Anti-Her2 IgG1 (W202) | Homodimer | A330F + K334V + L234Y + K290Y + Y296W | A330F + K 334V + L234Y + K290Y + Y296W | 77.23 | 50.55 |
| anti-Her2 IgG1 (W188) | Heterodimer | K392D + K409D + S239D + A330F + K334V | E356K + D399K + L234Y + K290Y + Y296W | 5.38 | 3.26 |
| Anti-Her2 IgG1 (W203) | Homodimer | S239D + A330F + K334V | S239D + A330F + K334V | 16.35 | 9.08 |
| Anti-Her2 IgG1 (W211) | Homodimer | L234Y + K290Y + Y296W | L234Y + K290Y + Y296W | 40.7 | 36.92 |
| Anti-Her2 IgG1 (W204) | Homodimer | S239D + A330F + K334V + L234Y + K290Y + Y296W | S239D + A330F + K334V + L234Y + K290Y + Y296W | 11.03 | 5.9 |
S239D significantly improves the binding of Fc to FcγRIIIA (13). On top of anti-Her2 IgG1 variant W144, S239D was added in the Fc chain containing A330F + K334V to make variant W188, so that each Fc chain of W188 has three different ADCC-enhancing mutations in addition to the heterodimerization charge pair mutations. Similarly, homodimeric anti-Her2 IgG1 variants W203, W211, and W204 were made for comparisons (Table 4). Heterodimeric variant W188 is more effective than all homodimeric variants for competing the binding of human IgG1 to FcγRIIIA Val-158 and Phe-158 allotypes (Fig. 6 and Table 4). These results suggested that asymmetrical Fc engineering could work more effectively than conventional (symmetrical) Fc engineering to improve the binding of Fc to FcγRIIIA.
Asymmetrically Engineered IgG1 Variants Confer High CH2 Stability
To determine whether the ADCC enhancement alterations may destabilize and aggregate the antibody, we tested the domain stability of anti-Her2 IgG1 variants by DSC. As shown in Fig. 7A, the wild type anti-Her2 IgG1 (M01) had a Tm for Fab/CH3 domains at 82 °C and for CH2 domain at 72 °C. The homodimeric variants M02 (S239D/I332E) and M03 (S239D/I332E/A330L) had the same Tm for the Fab/CH3 domain at 82 °C as wild type IgG1; however, their Tm for the CH2 domain decreased more than 20 °C to 52 and 51, respectively. Asymmetrically engineered anti-Her2 IgG1 variants (M04, W23, W141, W144, W157, W165, W168, W187, and B50) all had the same Tm for the Fab region at 82 °C, their Tm for the CH2/CH3 domains decreased slightly from 72 to ∼68 °C when comparing with homodimeric anti-Her2 wild type IgG1 (M01), indicating that the asymmetrical Fc engineering causes only a small change on the CH2 domain stability of the modified antibodies. Both anti-Her2 IgG1 variants afuco-W117 and afuco-W125 have similar DSC profile as variants W117 and W125, respectively (Fig. 7B), implicating that afucosylation does not impact the CH2 stability of heterodimeric IgG1 antibodies.
FIGURE 7.
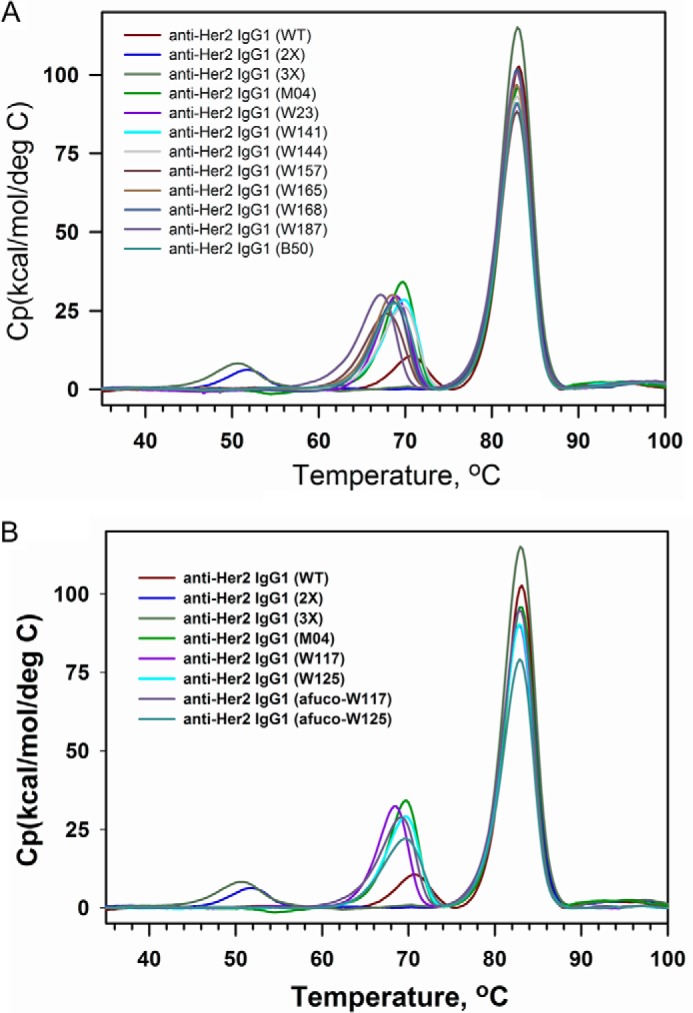
DSC profile of full-length IgG1 antibodies containing variant Fc regions. A, anti-Her2 IgG1 containing the wild type Fc (WT); S239D/I332E mutations (2X); S239D/I332E/A330L mutations (3X); heterodimerization mutations K392D and K409D in one Fc chain and E356K and D399K in the other Fc chain (M04); ADCC-enhanced variants W23, W141, W144, W157, W165, W168, W187, and B50. B, anti-Her2 IgG1 containing the wild type Fc (WT), S239D/I332E mutations (2X), S239D/I332E/A330L mutations (3X), heterodimerization mutations K392D and K409D in one Fc chain and E356K and D399K in the other Fc chain (M04); ADCC-enhanced variants W117, W125, afucosylated W117, and afucosylated W125. The various anti-Her2 human IgG1 antibodies used in these assays are indicated by alias in the graph, and the substitutions contained in each antibody are shown in Table 3. The transition peak at the lowest temperature for each profile typically corresponds to CH2 domain.
Asymmetrically Engineered IgG1 Variants Induce Potent ADCC Killing against Tumor Cells in Vitro
Cell lines expressing high (tumor cell line SK-BR-3) and medium (tumor cell line JIMT-1) levels of Her2 expression were used as target cells for the assessment of anti-Her2 human IgG1 ADCC activity (human NK cells as effector cells). Killing of SK-BR-3 cells was antibody concentration-dependent (Fig. 8, A and B). Control antibodies M01 (having a wild type Fc region) and M04 (having a Fc region containing only heterodimerization alterations) exhibited 60–75% maximal killing at the highest concentration of antibody tested (2,667 pm). Cell killing dropped off steeply at lower antibody concentrations for M01 and M04. However, antibodies containing variant Fc regions, including W23, W117, W125, W141, W144, W165, W168, and W187, exhibited increased ADCC maximal response and potency as compared with M01 or M04. The variant W187 exhibited the greatest ADCC potency at EC50 of 0.0123 pm in this assay; it also bound with the highest affinity to human FcγRIIIA (Table 3).
FIGURE 8.

ADCC activity of anti-Her2 IgG1 antibodies containing Fc variants. The graph shows the percent of cells killed in an assay for antibody-dependent cellular cytotoxicity (% specific lysis) versus log10 of antibody concentration. A, percent of SK-BR-3 cells killed with anti-Her2 IgG1 containing the Fc variants of M01, M04, W23, W117, W125, and W141. B, percent of SK-BR-3 cells killed with anti-Her2 IgG1 containing the Fc variants of. M01, M04, W144, W165, W168, and W187. C, percent of JIMT-1 cells killed with anti-Her2 IgG1 containing the wild type Fc (wt), afucosylated wild type Fc (afuco-wt), variant W117 and afucosylated W117 (afuco-W117). D, percent of JIMT-1 cells killed with anti-Her2 IgG1 containing the wild type Fc (wt), afucosylated wild type Fc (afuco-wt), and variant W125 and afucosylated W125 (afuco-W125). The designation afuco preceding an alias means that the antibody lacks fucose on the glycan at Asn-297 of Fc region. The various human IgG1 antibodies used in these assays are indicated by aliases in the graph, and the substitutions contained in each antibody are indicated in Table 3. Effect concentrations at 50% of maximal killing (EC50, pm) are shown beside the designated Fc variants in the figure.
As observed with SK-BR-3 cells, ADCC mediated by anti-Her2 antibody variants against JIMT-1 cells was also concentration-dependent. Anti-Her2 wild type IgG1 antibody achieved only about 64% maximal cell lysis at the highest antibody concentration tested and the EC50 at 98.3 pm. An afucosylated preparation of wild type antibody (afuco-WT) achieved 86% maximal specific cell lysis at the highest concentration tested and showed >100-fold increase in potency (EC50, 0.274 pm). The asymmetrically engineered IgG1 variant W117 exhibited enhanced ADCC killing compared with wild type IgG1 antibody, reaching similar maximal killing to that of afucosylated wild type IgG1 (Fig. 8C) and exhibiting an 85-fold improvement in potency (EC50, 1.15 pm) as compared with wild type IgG1 antibody. An afucosylated preparation of the same antibody (afuco-W117) showed even greater killing activity (EC50, 0.015 pm), a >6,500-fold improvement than that of wild type IgG1. A similar type of increase in ADCC potency was observed with an afuco-W125 variant (EC50, 0.061 pm) as compared with its fucosylated counterpart W125 (EC50, 3.992 pm in Fig. 8D). These results demonstrate a further improvement in ADCC activity when afucosylation is combined with amino acid changes that increase Fc binding affinity to FcγRIIIA.
The results validated that all 14 of the antibody variants described here display potent ADCC-mediated killing of targeted tumor cells. Each demonstrated a potency of EC50 <1 pm, which was far greater than the potency of the wild type antibody or an antibody containing only charge pair mutations. Furthermore, lower EC50 values for ADCC generally correlated with lower EC50 values for FcγRIIIA binding, which would be expected because binding to and clustering of FcγRIIIA are the prerequisite for antibodies to mediate ADCC activity.
Asymmetrically Engineered IgG1s That Show Enhanced ADCC Activity Inhibit the Tumor Growth in Xenograft Tumor Models for Gastric/Ovarian and Breast Cancers
To answer the question whether IgG1 variants that exhibit enhanced ADCC activity through an asymmetrical engineering approach could trigger potent tumor killing in vivo, their ability to inhibit tumor growth in two different mouse xenograft tumor models was assessed. Based on the improved binding affinity to mouse FcγRIV (Fig. 5C and Table 3), we carried out the xenograft studies in CB-17/SCID mice. Gastric NCI-N87 tumor cells express Her2 at about 450,000 sites per cell, and they express a specific tumor antigen at about 200,000 sites per cell. As shown in Fig. 9A, tumors in NCI-N87-bearing mice treated with isotype control anti-SAv IgG1 antibody grew steadily over the course of the study; however, NCI-N87 tumor growth was significantly inhibited in mice treated with anti-TuAg IgG1 S239D/I332E/A330L variant (p < 0.05). The anti-TuAg IgG1 asymmetrical variant W165 inhibited NCI-N87 tumor growth to a greater extent than the S239D/I332E/A330L variant (p < 0.05). These results showed that in CB-17/SCID mice the ADCC-enhanced IgG1s inhibited tumor growth when the targeted tumor antigen was expressed at lower levels. Anti-Her2 wild type IgG1 or ADCC-enhanced IgG1 variants (W165 and afuco-W117) all significantly inhibited (p < 0.001) NCI-N87 tumor growth, with the afuco-W117 showing a trend of being the most active one. At the relatively high antibody dose (10 mg/kg) and dosing frequency tested, any differences, which might exist in the in vivo anti-tumor activity for the different anti-Her2 human IgG1s, could not be teased out in xenografted tumors expressing a high level of antigen Her2. As a further study (Fig. 9B), the effect of the anti-Her2 antibody variants was tested in a JIMT-1 xenograft, which expresses lower Her2 levels (180,000 sites per cell on average) than NCI-N87 cells. Significant inhibition of the JIMT-1 tumor growth was observed in mice treated with heterodimeric anti-Her2 IgG1 variants. The tumor growth inhibition for all variants appeared to be greater than that mediated by anti-Her2 wild type IgG1. The percent tumor growth inhibition observed for the treatment groups was 71.6, 80.2, and 61.2% for anti-Her2 IgG1 variant W165, anti-Her2 IgG1 variant afuco-W117, and anti-Her2 wild type IgG1, respectively, compared with the anti-SAv human IgG1 isotype control (p = 0.0004, 0.0002, and 0.0959, respectively). Furthermore, a greater reduction of the tumor size was achieved by both anti-Her2 IgG1 variant W165 and anti-Her2 IgG1 variant afuco-W117, compared with anti-Her2 wild type IgG1. A significant difference was observed in the treatments with anti-Her2 IgG1 variant afuco-W117 (p = 0.0481), whereas no significant difference was found in the treatments with anti-Her2 IgG1 variant W165 (p = 0.0745) when compared with anti-Her2 wild type IgG1. The studies suggested that in mice there is a benefit when glyco-engineering and protein-engineering technologies are combined as described in the anti-Her2 IgG1 variant afuco-W117. The high affinity of afuco-W117 human IgG1 for mouse FcγRIV may help to explain the increased anti-tumor activity in this model (Table 3).
FIGURE 9.

Antitumor activity of anti-TuAg and anti-Her2 HuIgG1 antibodies containing variant Fc regions in NCI-N87 and JIMT-1 xenograft human tumor models. A, NCI N87-tumor bearing CB-17/SCID mice were treated with the indicated antibodies at a dose of 10 mg/kg twice per week for 3 weeks via intraperitoneal administration with anti-SAv huIgG1, anti-TuAg clone H158 huIgG1 (S239D/I332E/A330L), anti-TuAg clone H158 huIgG1 (W165), anti-Her2 huIgG1 (WT), anti-Her2 huIgG1 (W165), and anti-Her2 huIgG1 (afuco-W117). The results are represented as mean tumor volume (mm3) (n = 10 mice/group); bars, ± S.E. The graph shows tumor volume measurement over time in days post-tumor implantation. At study end, 3/10, 5/10, and 5/10 mice did not exhibit measureable tumor following treatment with anti-Her2 IgG1 W165, wild type (WT), afuco-W117, respectively. B, JIMT-1-tumor xenograft bearing CB-17/SCID mice were treated once per week for 4 weeks via intraperitoneal administration with 10 mg/kg human IgG1 isotype control antibody, anti-Her2 huIgG1 (WT), anti-Her2 huIgG1 (W165), and anti-Her2 huIgG1 (afuco-W117). The various human IgG1 antibodies used in these assays are indicated by the alias in the graph, and the substitutions contained in each antibody are indicated in Table 3.
DISCUSSION
Based on the nature of asymmetric interaction between the Fc region of IgG and FcγRIII (15), we hypothesized that an optimal way to improve Fc binding to FcγRIII is to make asymmetrical changes in the two Fc chains so that each Fc chain can be individually optimized to interact with the FcγRIII and thus enhance the ADCC activity of engineered antibodies. We made heterodimeric IgG1 by separately introducing two charge pair residues (K392D + K409D and E356K + D399K) in the CH3 domain of each Fc chain (21). We constructed Fc libraries in the scFv-Fc fragment or dummy Fc, and we then expressed heterodimeric scFv-Fc/Fc from mammalian HEK-293 cells. We screened more than 9,000 individual clones using a high throughput competition AlphaLISA® assay and identified 71 single mutants with improved binding to FcγRIIIA (Phe-158). These single mutants were grouped based on their positions and further combined to seek synergy in heterodimeric IgG1 format. To this end, we have identified a panel of novel Fc heterodimer variants with significant improvement binding to FcγRIIIA (both Phe-158 and Val-158 allotypes), high CH2 melting temperature, increased ADCC activity in tumor cell killing assays, and strong anti-tumor activity in mice xenograft tumor models. We found that afucosylation of Fc variants further increases the affinity of Fc to FcγRIIIA, leading to much higher ADCC activity. Some novel Fc variants impart higher binding to FcγRIIA and unchanged binding to FcγRIIB.
FcγRIIIA is co-expressed with other FcγRs on many mononuclear phagocytes but is solely expressed by NK cells. FcγRIIIA bridges the NK cells and antibodies. Perforin, granzyme, and others released from NK cells strongly kill tumor cells that generally have down-regulated expression of MHC class I molecules. Early studies argued that NK cells display impaired functionality and capability to infiltrate tumors of cancer patients, and limited numbers of infiltrating NK cells are unlikely to greatly contribute to the elimination of tumor cells (22, 23). However, anti-Her2 trastuzumab-treated patients with complete remission or partial remission were found to have a higher in situ infiltration of leukocytes with a higher capability to mediate in vitro ADCC activity (24). Osteosarcoma patients treated with anti-EGF receptor cetuximab have substantially increased NK cells; NK cells from treated patients were as efficient in inducing ADCC activity against autologous tumor cells as NK cells from healthy donors (25). The afucosylated anti-CD20 antibody (GA101) when combined with chemotherapy has improved overall survival than the combination of anti-CD20 (Rituximab) regular antibody and chemotherapy in patients with chronic lymphocytic leukemia in phase 3 trials. ADCC is a very important mechanism of action for antibodies to mediate killing to tumor cells.
Compared with the conventional Fc engineering approach in which the same mutations are present in both Fc chains, the asymmetrical engineering strategy allows us to utilize residue changes that may not be compatible in the context of the Fc homodimer. For example, several residues in the lower hinge region of Fc are involved in direct interaction with FcγRIIIA at both Fc sides; therefore, it is difficult to introduce the same amino acid change that can enhance interaction in the lower hinge region of both Fc chains. In fact, most of the mutations reported in the lower hinge region have led to decreased binding to Fc receptors, e.g. L234A/L235A mutation (26). Using the asymmetrical approach, we were able to identify several mutations in the lower hinge region (e.g. L234Y, L235S, and G236Y in Table 2) that can enhance FcγRIIIA binding when introduced into either only one Fc chain such as the W165 variant or both Fc chains such as the B50 variant (Table 3). Similarly, many of the Fc mutations identified in this work showed enhanced binding to FcγRIIIA significantly better than those in conventional Fc homodimer format (Table 4).
Asymmetrical Fc engineering allowed us to identify Fc variants that can increase FcγRIIIA binding but without other liabilities such as decreased stability. For example, heterodimeric IgG1 variants described in this work all showed high CH2 melting temperature with only 4 °C less than that of wild type IgG1 and good manufacturability (data not shown). This is in contrast to some of the Fc variants in homodimeric form, e.g. S239D/I332E (2X) and S239D/I332E/A330L (3X), which both have a decreased Tm with more than 20 °C for the CH2 domain (Figs. 2B and 7). Therefore, the asymmetrically engineered antibodies would be predicted to be less likely to have production and stability issues than the symmetrically (conventionally) engineered antibodies. By using different sets of mutations on two Fc chains, the asymmetrical approach may allow the fine-tuning of the two binding interfaces with FcγRIIIA. As shown in Table 3, all the Fc heterodimer variants exhibit higher binding affinity to both Phe-158 and Val-158 allotypes of FcγRIIIA than wild type IgG1. Interestingly, when compared with engineered homodimeric IgG1, e.g. S239D/I332E/A330L (3X) variant, some of our Fc heterodimers showed a similar degree of overall affinity improvement to FcγRIIIA as measured by Biacore, but they typically have a slower off-rate. It remains to be explored whether the slower off-rate to FcγRIIIA, which may allow longer engagement of NK cells to tumor cells, could be translated into a better in vivo efficacy.
The asymmetrical engineering approach may also allow a better discrimination of highly homologous Fc receptors such as activating receptor FcγRIIA and inhibitory receptor FcγRIIB. As shown in Fig. 5, our anti-Her2 heterodimeric IgG1 variants except W187 exhibit enhanced binding to FcγRIIA (His-131) but not to FcγRIIA (Arg-131) or FcγRIIB. In contrast, the homodimeric IgG1 S239D/I332E variant containing S239D/I332E mutations in both Fc chains showed significantly enhanced binding to FcγRIIB. It could be predicted that the asymmetrically engineered IgG1 variants may potentiate stronger ADCP activity in addition to the enhanced ADCC activity. When the engagement of the inhibitory FcγRIIB is preferred, e.g. for the treatment of autoimmune diseases or for antibodies to target the TNF receptor family members (27, 28), asymmetrical Fc engineering could also be applied in identifying Fc mutants with stronger binding to FcγRIIB only.
In this report, we demonstrated that some of the anti-HER2 heterodimeric IgG variants such as W117 and W125, when produced as afucosylated form, can further increase binding to FcγRIIIA. This was achieved by increased on-rate and decreased off-rate to both Val-158 and Phe-158 allotypes of FcγRIIIA, suggesting that a further binding improvement to FcγRIIIA can be achieved by combining glyco-engineering with an asymmetric protein engineering approach. This is consistent with earlier reports on engineered variants of anti-CD20 antibodies (29) or scFv-Fc proteins (30) where additive or synergistic binding improvements were observed when both glyco-engineering and protein engineering techniques were applied. The afucosylated W117 and W125 variants can bind to FcγRIIIA with even higher affinity than the S239D/I332E/A330L (3X) variant (Fig. 5 and Table 3), the most potent Fc variant reported as regular IgG1 homodimer so far.
The Fc variants described here can potentially be applied to many asymmetrical types of Fc fusions that are based on Fc heterodimers such as bispecific antibodies (bsAbs). bsAbs can simultaneously recognize two different antigens or two different epitopes of the same antigen. bsAbs have long been considered as an attractive approach to combine the additive or potentially synergistic effects exhibited by combinations of two monoclonal antibodies. For example, bsAbs have been used to neutralize different pathogenic mediators to alleviate the symptom of chronic diseases, to inhibit tumor growth by targeting different signaling pathways or different molecules in the same pathway, and to redirect cellular activity by recruiting different types of cells (31, 32). Over the past 2 decades, more than 45 different formats of bsAb have been developed for different biological applications (33). Among the many different forms of bispecific antibodies, bispecific IgG based on the Fc heterodimer is a promising format because it maintains the overall size and structure of the regular IgG with good bioavailability and pharmacokinetic profiles (34). It may be desirable to develop bsAbs with enhanced ADCC activity in the future for the treatment of cancers and infectious diseases. The asymmetrically engineered Fc variants described in this report could be integrated in the heterodimeric bispecific IgG1 to make best-in-class therapeutic antibodies.
Acknowledgments
We thank Paul Kodama and Kenneth Prentice for their mass spectrometry analysis and Michael Wittekind for support.
Addendum
During the preparation of this manuscript, Mimoto et al. (36) published their work on the identification of Fc variants with increased FcγR binding affinity by using a similar asymmetrical engineering strategy, further validating the feasibility of this approach.
The concept of asymmetrical Fc engineering and partial results were presented in PEPTALK at Hotel Del Coronado, San Diego, CA, on January 13, 2012. A provisional patent application was filed in the United States Patent Office on March 16, 2011, which was published as WO 2012/125850 on September 20, 2012. All authors were or are employed at Amgen, Inc.
- FcγRs
- Fc receptors for IgG
- ADCC
- antibody-dependent cellular cytotoxicity
- ADCP
- antibody-dependent cell-mediated phagocytosis
- scFv
- single chain fragment of variable region
- PEI
- polyethylenimine
- TuAg
- Tumor Antigen
- EGAD
- a genetic algorithm for protein design
- DSC
- differential scanning calorimetry
- SAv
- streptavidin
- bsAbs
- bispecific antibodies
- hu
- human.
REFERENCES
- 1. Reichert J. M., Valge-Archer V. E. (2007) Development trends for monoclonal antibody cancer therapeutics. Nat. Rev. Drug Discov. 6, 349–356 [DOI] [PubMed] [Google Scholar]
- 2. Nimmerjahn F., Ravetch J. V. (2008) Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 8, 34–47 [DOI] [PubMed] [Google Scholar]
- 3. Gessner J. E., Heiken H., Tamm A., Schmidt R. E. (1998) The IgG Fc receptor family. Ann. Hematol. 76, 231–248 [DOI] [PubMed] [Google Scholar]
- 4. Cartron G., Dacheux L., Salles G., Solal-Celigny P., Bardos P., Colombat P., Watier H. (2002) Therapeutic activity of humanized ant-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood 99, 754–758 [DOI] [PubMed] [Google Scholar]
- 5. Musolino A., Naldi N., Bortesi B., Pezzuolo D., Capelletti M., Missale G., Laccabue D., Zerbini A., Camisa R., Bisagni G., Neri T. M., Ardizzoni A. (2008) Immunoglobin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with Her-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 26, 1789–1796 [DOI] [PubMed] [Google Scholar]
- 6. Weng W. K., Levy R. (2003) Two immunoglobulin G Fc receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 21, 3940–3947 [DOI] [PubMed] [Google Scholar]
- 7. Clynes R. A., Towers T. L., Presta L. G., Ravetch J. V. (2000) Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 6, 443–446 [DOI] [PubMed] [Google Scholar]
- 8. Green S. K., Karlsson M. C., Ravetch J. V., Kerbel R. S. (2002) Disruption of cell-cell adhesion enhances antibody-dependent cellular cytotoxicity: implications for antibody-based therapeutics of cancer. Cancer Res. 62, 6891–6900 [PubMed] [Google Scholar]
- 9. Nimmerjahn F., Ravetch J. V. (2005) Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science 310, 1510–1512 [DOI] [PubMed] [Google Scholar]
- 10. Shinkawa T., Nakamura K., Yamane N., Shoji-Hosaka E., Kanda Y., Sakurada M., Uchida K., Anazawa H., Satoh M., Yamasaki M., Hanai N., Shitara K. (2003) The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 278, 3466–3473 [DOI] [PubMed] [Google Scholar]
- 11. Ferrara C., Brünker P., Suter T., Moser S., Püntener U., Umaña P. (2006) Modulation of therapeutic antibody effector functions by glycosylation engineering: influence of Golgi enzyme localization domain and co-expression of heterologous β1,4-N-acetylglucosaminyltransferase III and Golgi α-mannosidase II. Biotechnol. Bioeng. 93, 851–861 [DOI] [PubMed] [Google Scholar]
- 12. Shields R. L., Namenuk A. K., Hong K., Meng Y. G., Rae J., Briggs J., Xie D., Lai J., Stadlen A., Li B., Fox J. A., Presta L. G. (2001) High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J. Biol. Chem. 276, 6591–6604 [DOI] [PubMed] [Google Scholar]
- 13. Lazar G. A., Dang W., Karki S., Vafa O., Peng J. S., Hyun L., Chan C., Chung H. S., Eivazi A., Yoder S. C., Vielmetter J., Carmichael D. F., Hayes R. J., Dahiyat B. I. (2006) Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. U.S.A. 103, 4005–4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stavenhagen J. B., Gorlatov S., Tuaillon N., Rankin C. T., Li H., Burke S., Huang L., Vijh S., Johnson S., Bonvini E., Koenig S. (2007) Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low affinity activating Fcγ receptors. Cancer Res. 67, 8882–8890 [DOI] [PubMed] [Google Scholar]
- 15. Sondermann P., Huber R., Oosthuizen V., Jacob U. (2000) The 3.2-Å crystal structure of the human IgG1 Fc fragment-FcgRIII complex. Nature 406, 267–273 [DOI] [PubMed] [Google Scholar]
- 16. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 17. Chenna R., Sugawara H., Koike T., Lopez R., Gibson T. J., Higgins D. G., Thompson J. D. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31, 3497–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pokala N., Handel T. M. (2005) Energy functions for protein design: adjustment with protein-protein complex affinities, models for the unfolded state, and negative design of solubility and specificity. J. Mol. Biol. 347, 203–227 [DOI] [PubMed] [Google Scholar]
- 19. Zhang J., Liu X., Bell A., To R., Baral T. N., Azizi A., Li J., Cass B., Durocher Y. (2009) Transient expression and purification of chimeric heavy chain antibodies. Protein Expr. Purif. 65, 77–82 [DOI] [PubMed] [Google Scholar]
- 20. Warrens A. N., Jones M. D., Lechler R. I. (1997) Splicing by overlap extension by PCR using asymmetric amplification: an improved technique for the generation of hybrid proteins of immunological interest. Gene 186, 29–35 [DOI] [PubMed] [Google Scholar]
- 21. Gunasekaran K., Pentony M., Shen M., Garrett L., Forte C., Woodward A., Ng S. B., Born T., Retter M., Manchulenko K., Sweet H., Foltz I. N., Wittekind M., Yan W. (2010) Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 285, 19637–19646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Albertsson P. A., Basse P. H., Hokland M., Goldfarb R. H., Nagelkerke J. F., Nannmark U., Kuppen P. J. (2003) NK cells and the tumour microenvironment: implications for NK-cell function and anti-tumour activity. Trends Immunol. 24, 603–609 [DOI] [PubMed] [Google Scholar]
- 23. Esendagli G., Bruderek K., Goldmann T., Busche A., Branscheid D., Vollmer E., Brandau S. (2008) Malignant and nonmalignant lung tissue areas are differentially populated by natural killer cells and regulatory T cells in non-small cell lung cancer. Lung Cancer 59, 32–40 [DOI] [PubMed] [Google Scholar]
- 24. Gennari R., Menard S., Fagnoni F., Ponchio L., Scelsi M., Tagliabue E., Castiglioni F., Villani L., Magalotti C., Gibelli N., Oliviero B., Ballardini B., Da Prada G., Zambelli A., Costa A. (2004) Pilot study of the mechanism of action of preoperative trastuzumab in patients with primary operable breast tumors overexpressing HER2. Clin. Cancer Res. 10, 5650–5655 [DOI] [PubMed] [Google Scholar]
- 25. Pahl J. H., Ruslan S. E., Buddingh E. P., Santos S. J., Szuhai K., Serra M., Gelderblom H., Hogendoorn P. C., Egeler R. M., Schilham M. W., Lankester A. C. (2012) Anti-EGFR antibody cetuximab enhances the cytolytic activity of natural killer cells toward osteosarcoma. Clin. Cancer Res. 18, 432–441 [DOI] [PubMed] [Google Scholar]
- 26. Hezareh M., Hessell A. J., Jensen R. C., van de Winkel J. G., Parren P. W. (2001) Effector function activities of a panel of mutants of a broadly neutralizing antibodies against human immunodeficiency virus type 1. J. Virol. 75, 12161–12168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li F., Ravetch J. V. (2011) Inhibitory Fcγ receptors engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science 333, 1030–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li F., Ravetch J. V. (2012) Apoptotic and antitumor activity of death receptor antibodies require inhibitory Fcγ receptor engagement. Proc. Natl. Acad. Sci. U.S.A. 109, 10966–10971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Masuda K., Kubota T., Kaneko E., Iida S., Wakitani M., Kobayashi-Natsume Y., Kubota A., Shitara K., Nakamura K. (2007) Enhanced binding affinity for FcγRIIIa of fucose-negative antibody is sufficient to induce maximal antibody-dependent cellular cytotoxicity. Mol. Immunol. 44, 3122–3131 [DOI] [PubMed] [Google Scholar]
- 30. Repp R., Kellner C., Muskulus A., Staudinger M., Nodehi S. M., Glorius P., Akramiene D., Dechant M., Fey G. H., van Berkel P. H., van de Winkel J. G., Parren P. W., Valerius T., Gramatzki M., Peipp M. (2011) Combined Fc-protein- and Fc-glyco-engineering of scFv-Fc fusion proteins synergistically enhances CD16a binding but does not further enhance NK-cell mediated ADCC. J. Immunol. Methods 373, 67–78 [DOI] [PubMed] [Google Scholar]
- 31. Kufer P., Lutterbüse R., Baeuerle P. A. (2004) A revival of bispecific antibodies. Trends Biotechnol. 22, 238–244 [DOI] [PubMed] [Google Scholar]
- 32. Chames P., Baty D. (2009) Bispecific antibodies for cancer therapy: the light at the end of the tunnel? mAbs 1, 539–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kontermann R. E. (2012) Dual targeting strategies with bispecific antibodies. mAbs 4, 182–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Klein C., Sustmann C., Thomas M., Stubenrauch K., Croasdale R., Schanzer J., Brinkmann U., Kettenberger H., Regula J. T., Schaefer W. (2012) Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. mAbs 4, 653–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Edelman G. M., Cunningham B. A., Gall W. E., Gottlieb P. D., Rutishauser U., Waxdal M. J. (1969) The covalent structure of an entire γG immunoglobulin molecule. Proc. Natl. Acad. Sci. U.S.A. 63, 78–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mimoto F., Igawa T., Kuramochi T., Katada H., Kadono S., Kamikawa T., Shida-Kawazoe M., Hattori K. (2013) Novel asymmetrically engineered antibody Fc variant with superior FcγR binding affinity and specificity compared with afucosylated Fc variant. MAbs 5, 229–236 [DOI] [PMC free article] [PubMed] [Google Scholar]