

Background: The structure and function of GPR35 are not understood.
Results: Using a GPR35 activated state molecular model, we identified crucial amino acid residues required for ligand activation using β-arrestin trafficking, ERK1/2 activation, and calcium imaging.
Conclusion: Arginines in TMH3-4-5-6 affected agonist signaling.
Significance: Identification of residues for GPR35 agonist signaling is critical for the design of ligands with improved potency.
Keywords: Arrestin, G Protein-coupled Receptors (GPCR), MAP Kinases (MAPKs), Molecular Modeling, Trafficking
Abstract
GPR35 is a G protein-coupled receptor expressed in the immune, gastrointestinal, and nervous systems in gastric carcinomas and is implicated in heart failure and pain perception. We investigated residues in GPR35 responsible for ligand activation and the receptor structure in the active state. GPR35 contains numerous positively charged amino acids that face into the binding pocket that cluster in two distinct receptor regions, TMH3-4-5-6 and TMH1-2-7. Computer modeling implicated TMH3-4-5-6 for activation by the GPR35 agonists zaprinast and pamoic acid. Mutation results for the TMH1-2-7 region of GPR35 showed no change in ligand efficacies at the K1.32A, R2.65A, R7.33A, and K7.40A mutants. However, mutation of arginine residues in the TMH3-4-5-6 region (R4.60, R6.58, R3.36, R(164), and R(167) in the EC2 loop) had effects on signaling for one or both agonists tested. R4.60A resulted in a total ablation of agonist-induced activation in both the β-arrestin trafficking and ERK1/2 activation assays. R6.58A increased the potency of zaprinast 30-fold in the pERK assay. The R(167)A mutant decreased the potency of pamoic acid in the β-arrestin trafficking assay. The R(164)A and R(164)L mutants decreased potencies of both agonists. Similar trends for R6.58A and R(167)A were observed in calcium responses. Computer modeling showed that the R6.58A mutant has additional interactions with zaprinast. R3.36A did not express on the cell surface but was trapped in the cytoplasm. The lack of surface expression of R3.36A was rescued by a GPR35 antagonist, CID2745687. These results clearly show that R4.60, R(164), R(167), and R6.58 play crucial roles in the agonist initiated activation of GPR35.
Introduction
G protein-coupled receptors (GPCRs)4 represent the largest target for current and future therapeutic agents. GPR35 is a class A GPCR that is expressed in the immune and gastrointestinal systems, dorsal root ganglia, cerebellum, and brain (Fig. 1) (1–4). GPR35 regulation appears to have profound physiological and pathophysiological implications (5–10); thus, great efforts have been made to identify compounds that regulate GPR35 (4, 5, 11–14). Kyneurinic acid (15) and lysophosphatidic acid (14), both anions, have been reported to be GPR35 endogenous ligands. Pamoic acid (5), zaprinast (4), 5-nitro-2-(3-phenylpropylamino) benzoic acid (11), nedocromil sodium (Tilade) (13), and cromolyn sodium (Intal) (13) have been reported to be GPR35 agonists, whereas methyl-5-[(tert-butylcarbamothioylhydrazinyl-idene)methyl]-1-(2,4-difluorophenyl)pyrazole-4-carboxylate (CID2745687) has been reported to be an antagonist at GPR35 (5). With the exception of zaprinast, the GPR35 agonist ligands reported to date are mono- or dicarboxylic acids that would be ionized at physiological pH. Perhaps, not surprisingly then, the GPR35 sequence contains numerous positively charged amino acids that face into the binding pocket. These residues cluster in two distinct receptor regions, the TMH3-4-5-6 region and the TMH1-2-7 region of GPR35 (Fig. 1). In work reported here, we have used a combination of modeling and mutation studies to probe the importance of these positively charged residues to the binding of two GPR35 agonists, zaprinast and pamoic acid (see drawings in Fig. 2). These studies suggest that residues in the TMH3-4-5-6 region of GPR35 play crucial roles in the agonist-binding initiated activation of this receptor.
FIGURE 1.

Schematic representation of human GPR35 receptor structure and its seven transmembrane domain regions. The TMH residue highlighted in blue in each TMH is the most highly conserved residue among class A GPCRs in that helix.
FIGURE 2.
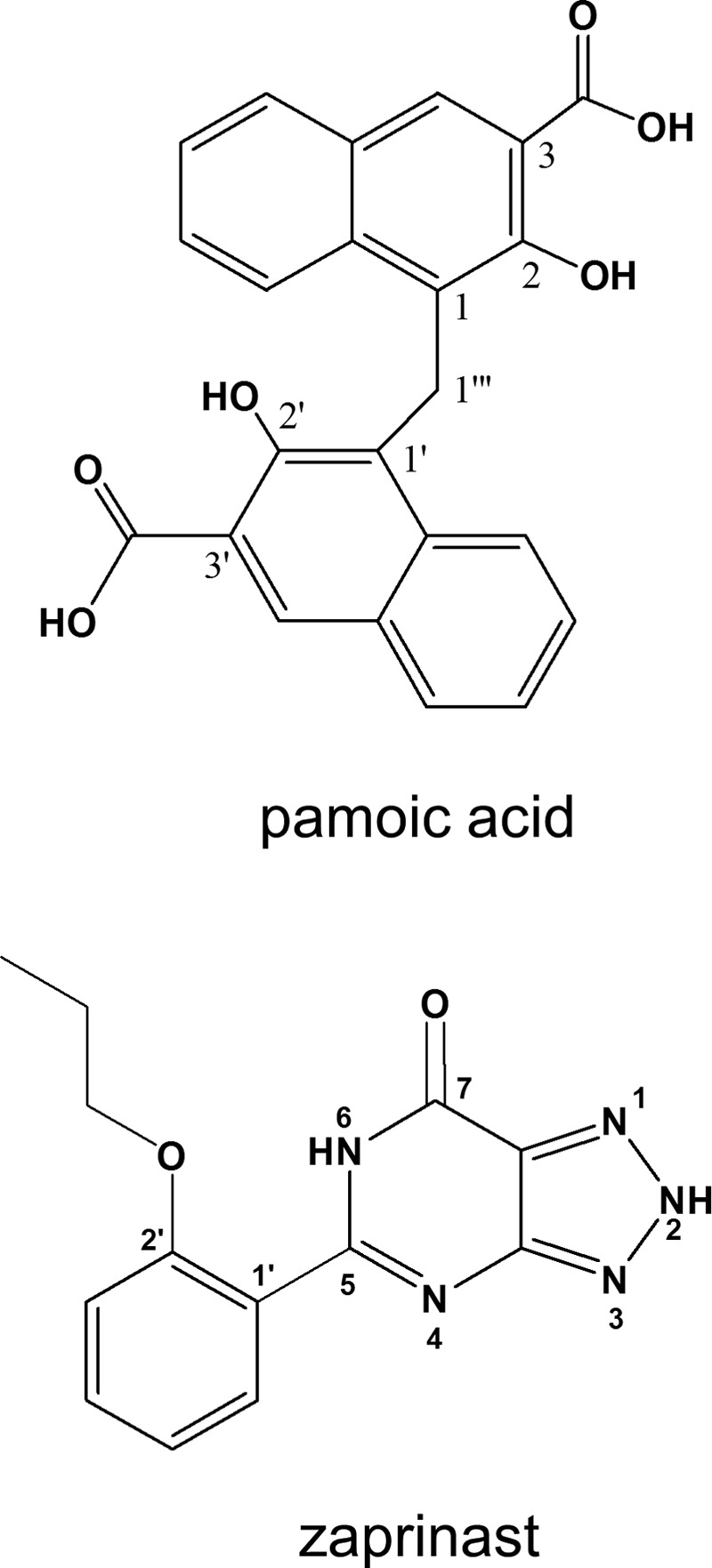
Structures of pamoic acid and zaprinast.
EXPERIMENTAL PROCEDURES
Materials
DMEM, Hanks' balanced salt solution and FBS were purchased from Cellgro, Mediatech, Inc., and Hyclone. Pamoic acid and poly-d-lysine were purchased from Sigma. Zaprinast was purchased from Tocris. Anti-phospho ERK antibodies were purchased from Cell Signaling. Anti-HA mouse monoclonal antibody was purchased from Covance. Alexa 568 goat anti-mouse antibody, Zeocin, and Lipofectamine 2000 were purchased from Invitrogen. IRDye 800-conjugated anti-mouse IgG was from LI-COR. HA-GPR35a plasmid and cell line were provided by the Duke University GPCR Assay Bank. All other reagents were obtained from Sigma or other standard sources.
Plasmids, Mutagenesis, Transfection, and Cell Culture
β-Arrestin2-green fluorescent protein (βarr2-GFP) (Renilla) is described previously (5, 16). The R3.36A, A4.59G, R4.60A, R6.58A, R(164)A/L, and K7.40A mutants were constructed using the QuikChange site-directed mutagenesis kit (Stratagene) based on the 3× HA-tagged human GPR35a in the pcDNA3.1 vector. Synthetic oligonucleotide mutagenic primers were typically 25–35 base pairs long (with 12–17 base pairs on either side of the desired mismatch region). DNA sequencing was subsequently used to ascertain the presence of the designed mutation. Stably transfected U2OS cell lines were created by transfection with wild-type or mutant human GPR35 together with βarr2-GFP plasmid by using the Lipofectamine 2000 reagent (Invitrogen) following the manufacturer's protocol. Clones were selected in growth medium containing G418 (0.4 mg/ml) and Zeocin (0.2 mg/ml) (Invitrogen) as described previously (17). Stable cell lines were then maintained in G418 (0.2 mg/ml) and Zeocin (0.1 mg/ml). U2OS and HEK293 cells transiently transfected with R3.36A were used for experiments 2 days after transfection.
Immunocytochemistry and Microscopy
The expression of wild-type and mutant GPR35 receptors was examined by immunostaining of cells with HA monoclonal antibodies. To achieve surface receptor staining, cells grown on coverslips were incubated over ice for 40 min with a 1:100 dilution of mouse monoclonal anti-HA antibody in blocking buffer (3% BSA in PBS). This was followed by appropriate washes and 40-min incubation with a 1:1500 dilution of Alexa Fluor 568 goat anti-mouse secondary antibody. Cells were then fixed with 4% paraformaldehyde for 20 min at room temperature followed by three washes with PBS. The total receptor immunostaining was done by fixing then permeabilizing cells with 0.2% Triton X-100 before the primary HA antibodies (1:500) application. Following the staining, glass coverslips were mounted on slides and were imaged on a (Nikon E800) fluorescence microscope using a 40× oil objective and 488-nm excitation for GFP. Co-immunostaining of R3.36A and calreticulin (18) was done by co-applying mouse anti-HA antibodies (1:500) and rabbit anti-calreticulin antibodies (Cell Signaling; 1:200) followed by Alexa Fluor 568 goat anti-mouse secondary antibody (1:1500) and Alexa Fluor 488 goat anti-rabbit secondary antibody (1:1000).
β-Arrestin Assay for Determining Receptor Responsiveness
The β-arrestin assay was described previously (5). Briefly, stable cell lines were plated onto poly-d-lysine-coated coverslips in 24-well plates. Cells were maintained at 37 °C in 5% CO2 until ready for experiments (80–85% confluent) and washed once with Hanks' balanced salt solution before drug application, and experiments were performed in Hanks' balanced salt solution. Agonist-stimulated redistribution of βarr2-GFP was assessed following drug treatment for 40 min at room temperature. Cells were fixed and imaged as described above. Quantification of βarr2-GFP aggregates was described previously (5). Concentration-effect curves for agonist-mediated receptor activation were analyzed by nonlinear regression techniques using GraphPad Prism 5.0 software (GraphPad), and data were fitted to sigmoidal dose-response curves to obtain EC50 or IC50 values. Statistical analysis was performed using one-way analysis of variance followed by Dunnett's post-test or two-tailed unpaired Student's t test. p values of <0.05 are considered significant.
In-Cell Western Assay for ERK Activity
The assay was described previously (5). Briefly, cells were grown to confluence in 96-well plates and serum-starved overnight prior to assay. Following the 20-min agonist treatment at room temperature, the medium was removed, and 4% paraformaldehyde in PBS was added to fix cells for 1 h at room temperature. Cells were then permeabilized by 0.1% Triton X-100 in PBS for five washes, 10 min per wash. LI-COR blocking buffer was added, and samples were shaken on a rotator for 1 h. Primary antibodies against phosphor-ERK1/2 (Cell Signaling Technology; 1:100) were applied for 2 h, and then secondary antibodies goat anti-rabbit 800CW (1:800) were applied overnight in a cold room. Sapphire700 (LI-COR, 1:1000) and DRAQ5 (Biostatus Limited, 1:2000) were added together with the secondary antibodies for normalization. The plate was dried and then scanned using a LI-COR Odyssey Infrared Imager set at 169 μm resolution, 3 focus offset, and 4.5–6 intensity. Data were analyzed using Excel and Prism 5.0 software.
Calcium Imaging
Intracellular Ca2+ measurements were performed as described previously (19). Briefly, GPR35 and mutant stable U2OS cells were loaded with fura-2/AM and mounted in an open bath chamber (RP-40LP; Warner Instruments, Hamden, CT) on the stage of an inverted microscope Nikon Eclipse TiE (Optical Apparatus Co., Ardmore, PA). Fura-2/AM fluorescence (emission = 510 nm) following alternate excitation at 340 and 380 nm was acquired at a frequency of 0.25 Hz. Images were acquired and analyzed using Nikon NIS-Elements AR 3.1 software (Optical Apparatus Co.). The ratio of the fluorescence signals (340/380 nm) was converted to Ca2+ concentrations.
Quantification of Receptor Surface Expression
We used average fluorescent signal intensity analysis of surface receptor staining on the whole cell surface to measure total expression levels (20). Within the confines of the demarcation of the cell area in each image, we measured the average fluorescent signal to sample receptor expression level by using ImageJ software (1.47v; National Institutes of Health). Sampling the protein expression level of receptors in the easily definable cell area in the images provides an estimate of the total receptor expression that can be used to compare WT and mutant expression levels.
Modeling of GPR35 Using GPCR Crystallization Data
During the development of this model, several GPCRs have been crystallized, including rhodopsin (21–23), opsin (24) the β2-adrenergic receptor (β2-AR) (25–27), the β1-adrenergic receptor (28), the A2-adenosine receptor (29), and the CXCR4 receptor (30). The model of the activated form of GPR35 is based on the 2.4 Å resolution crystal structures of the β2-AR (25). The initial, canonical comparison of GPR35 with other class A GPCRs was based on a sequence comparison. The GPR35 sequence was aligned with the sequences of rhodopsin, β2-AR, A2-adenosine, and the cannabinoid CB1 and CB2 receptors, using highly conserved residues as alignment guides as done previously for the CB1 (31) and CB2 (32) receptors. GPR35 contains many of the highly conserved class A residue patterns in TMHs 1, 2, 3, 4, 5 (N1.50, D2.50, and (E)DRY motif in TMH3, C3.25, W4.50, and P5.50). However, there are some notable sequence variations that may alter the geometry of one or more helices in GPR35 as described below.
Conformational Memories (CM) Technique for Calculating TMH Conformation
For TMH 1, 2, 4, 5, 6, and 7, a series of possible, sequence-dictated conformations that deviate from the β2-AR template were calculated using the CM Monte Carlo/simulated annealing technique (33, 34). The CM technique uses multiple Monte Carlo/simulated annealing random walks employing the CHARMM all atom force field (35) in a distance-dependent dielectric at 310 K. With this technique, nearly all dihedrals and bond angles are defined as variable, with various restrictions. Bond angle variation ranges were defined as either ±8° for default bond angles or ±15° for side chain angles involving polar hydrogens (i.e., C-O-H: Ser, Thr, Tyr) or the flexible C-S-C bond angle in methionine. The backbone dihedrals Φ, Ψ, and ω had standard dihedral variation ranges of ±10°, ±10°, and ±20°, respectively, and the side chain dihedrals had full range of motion. If the sequence contained a known helix deforming residue such as proline, serine (36), threonine (36), or glycine, a larger variation range of up to ±50° on the Φ and Ψ was implemented to allow for increased flexibility. An ideal helix (Φ = −62.9° and Ψ = −41.6°) with the GPR35 sequence for each TMH was built, and a minimum of 105 conformers were generated for each GPR35 helix independently.
TMH1 in GPR35 has three nonconsecutive glycines located at positions 1.34, 1.39, and 1.46. An ideal helix with the GPR35 sequence was built, and three independent calculations were performed with increased flexibility of the backbone (Φ/Ψ of ± 50°) at the residues mentioned above.
TMH2 in GPR35 has a proline that is shifted by one residue intracellularly as compared with the β2-AR sequence: P2.59 in the β2AR and P2.58 in GPR35. An ideal helix with the GPR35 sequence was built, and a flexible region (Φ/Ψ ± 50°) was implemented into the P2.58 to L2.54 region to evaluate the flexibility at this proline position.
TMH4 in GPR35 lacks the conserved proline located at the extracellular end of the β2-AR TMH4, but it contains a potential helix deforming motif, GS (G4.55–S4.56). Two independent conformational searches were performed from an ideal helix starting structure to determine the impact of this motif. The first explored only the increased flexibility of the backbone at G4.55 (Φ/Ψ of ± 50°), and the second explored both backbone flexibility and χ1 of S4.56 restrained to g(−) (36).
TMH5 in GPR35 has two proline residues; the conserved proline at position 5.50 and an additional proline at 5.43. The flexibly of the proline kink of the conserved proline (G5.45 to P5.50) was restricted to empirically based backbone dihedral ranges, and the proline kink of nonconserved proline (S5.39 to P5.42) was given increased backbone flexibility (Φ/Ψ of ± 50°).
TMH6 in GPR35 lacks the highly conserved CWXP motif but has the conservative substitution, CFXP. The importance of exploring the possible conformational space of TMH6 lies in the documented changes in TMH6 conformation that occur when a class A GPCR is activated (37). In β2-AR and rhodopsin, the most dramatic structural change associated with activation is the conformational change in TMH6 about the highly conserved CWXP hinge region (38–45). Two independent conformational searches were performed with an identical, ideal helix starting structure to be representative of either an R or R* state. The flexibility of the backbone of the conserved proline kink region (V6.46 to P6.50) was restricted to the empirically based ranges of the antagonist bound GPCR crystal structures for the R state and allowed full flexibility (Φ/Ψ of ± 50°) for the R* state. G6.54 was also considered flexible ((Φ/Ψ of ± 50°) in both calculations.
TMH7 in GPR35 lacks the conserved P7.50 in the NPXXY motif and has the nonconservative substitution of DAXXY. To determine whether the GPR35 TMH7 sequence may suggest a flexible region within the same area as the conserved proline kink region, an increased flexibility (Φ/Ψ ± 50°) was introduced in the region of C7.46 to A7.50. The TMH7 conformation chosen for bundle creation was one that allowed D7.43 to hydrogen-bond to Y3.32. This interaction is found in the CXCR4 crystal structure (30).
Construction of the GPR35 Inactive State TMH Bundle
The conformers generated by the CM calculations were used to construct the TMH region of the receptor. The receptor was assembled by aligning the intracellular regions of TMHs 1, 2, 4, and 5–7 with the β2-AR template. In the R model, R3.50 and T6.30 form a hydrogen bond that mimics the R3.50/D or E6.30 ionic lock common in class A GPCRs. The bundle was then pulled apart 3 Å and energy-minimized for 1500 iterations utilizing the OPLS2005 all atom force field in Macromodel 9.1 (Schrodinger Inc., Portland, OR) employing a distance-dependent dielectric with extended cutoffs (nonbonded, 8.0 Å; electrostatic, 20.0 Å; and hydrogen bonding, 4.0 Å) and the Polak-Ribiere conjugate minimization scheme to pack the TMHs and relieve side chain clashes. A harmonic constraint was placed on the backbone (ϕ, ψ, and ω) of 500 kj/mol to maintain helicity. For the inactive (R) bundle, several highly conserved residue side chain dihedrals were restrained to crystal structure ranges.
Modeling of the Intra/Extracellular Loops and Receptor Termini
Modeler 8.2 (46, 47) was used to generate the N and C termini (Met-1 to Pro-16 and Pro-293 to Ala-309, respectively) and the intra- and extracellular loops (IC1: Arg-48 to Glu-54, IC2: Pro-121 to Ser-129, IC3: Ala-199 to Thr-212, EC1: Asp-82 to Thr-86, EC2: Leu-153 to His-168, EC3: Gly-244 to Ala-247). A restrictive distance was implemented between Cys-(162), located on the EC2 loop, and Cys-3.25(89) to emulate the class A highly conserved disulfide bridge between residues at these positions. A restricted distance was also implemented between C(8) in the N terminus and C7.25(248) at the EC end of TMH7 to emulate the disulfide bridge seen in the CXCR4 crystal structure (30). An EC-2 loop conformation was selected that had the second residue past the disulfide bridge (R(164)) pointing into the bundle as this is seen in the β2-AR (25–27) and other x-ray crystal structures. With the TMHs frozen, the final loops were energy-minimized to a gradient of 0.01 kcal/mol in a high dielectric of 80 using the same force field, cutoffs, and conjugate method described earlier.
Modeling of the Human GPR35 R* Receptor
To create an activated GPR35 model (R*), The EC2 and IC3 loops were removed, and a second TMH6 conformer was chosen that was straighter than TMH6 in the R model. This conformer was chosen so that no hydrogen bond was broken. With the loops and termini frozen, the TMH region was energy-minimized for 500 iterations in a distance-dependent dielectric utilizing the same force field, cutoffs, and conjugate method described earlier, with a harmonic constraint was placed on the backbone (ϕ, ψ, and ω) of 1000 kj/mol to maintain helicity. The EC2 and IC3 loops were then modeled as described above, including the distance restraint in the EC2 loop to mimic the conserved disulfide bridge. The final loops were then energy-minimized, as described above, to a gradient of 0.01 kcal/mol in a high dielectric of 80.
Modeling of the Human R6.58A Mutant GPR35 R* Receptor
To create the R6.58A R* receptor model, residue 6.58 in the WT R* bundle was mutated to Ala, and the bundle was reminimized using the same protocol as described above.
Ligand Conformational Analysis
Complete conformational analyses were performed on pamoic acid and zaprinast. Because pamoic acid (pKa1 = 2.51, pKa2 = 3.1) is a dianion at physiological pH (48), conformational analyses were performed on the dianionic form. Zaprinast (Fig. 2) can exist in several tautomeric forms, so conformational analyses were performed for each tautomer.
Hartree-Fock ab initio calculations at the 6–31+G* level as encoded by Spartan '08 (Wavefunction, Inc., Irvine, CA) were performed for each compound. For each comformational search, local energy minima were identified by the rotation of subject dihedrals 360° in 60° increments (6-fold search), followed by a Hartree-Fock 6–31+G* energy minimization of each rotamer generated. For this reason, the energy minimum of each tautomeric form was assessed at the 6–31+G* level. Pamoic acid conformational analyses were performed in vacuo and using a SM8 water solvation model as implemented in Spartan '08 (Wavefunction, Inc.).
Docking of Ligands
The global minimum energy conformer of pamoic acid (dianion) and the global minimum energy conformer of the 2,6-dihydro tautomeric state of zaprinast were used for docking studies (see numbering system in Fig. 2). Because of recent experimental data (49), Y3.32(96) was used as the primary interaction site for zaprinast, and two arginine residues located near the binding pocket (R3.36(100) and R4.60(151)) were used as primary interactions for the pamoic acid (dianion). Initially, each compound was manually docked in the binding site of GPR35 R* (or R6.58A R*) model.
Ligand/Receptor Minimization
The ligand-receptor complexes were minimized using an OPLS2005 all atom force in Macromodel 9.9 (Schrodinger Inc.). An extended cutoff (nonbonded, 8.0 Å; electrostatic, 20.0 Å; hydrogen bonding, 4.0 Å) was used in each stage of the calculation. The minimization was broken into two stages: 1) the minimization of the ligand and the TM regions. The first step consisted of 1250 steps of conjugate gradient minimization using a distance-dependent dielectric. Harmonic constraints of 1000 kcal/mol were placed on select dihedrals of the ligand to maintain conformational similarity to the low energy conformer calculated at the Hartree-Fock 6–31+G* level of theory as the receptor. The ligand restraints were released for the last 250 steps to allow the ligand to adapt to the optimized binding pocket. The EC and IC loop regions were frozen during this stage. 2) The extracellular and intracellular loops were minimized for 500 steps in a generalized born/surface area continuum solvation model for water, whereas the TM regions and the ligand atoms were held frozen.
Glide Docking Studies
The automatic docking program, Glide (Schrodinger Inc.) was then used to first score the manual docks and then explore other possible binding conformations or receptor site interactions with flexible docking. Glide was used to generate a grid based on the centroid of the ligand in the binding site (from the manual docks). Any hydrophobic region defined in the grid generation that contacted the ligand was selected as important to the flexible docking procedure. The box for flexible docking was defined large enough to encompass Y3.32(96) as the primary interaction site for zaprinast and R3.36(100) and R4.60(151) as primary interactions for pamoic acid. Standard precision (SP) was selected, and flexible docking was invoked unless scoring a manual dock (50).
RESULTS
GPR35 Receptor Model Development
A homology model of the GPR35 inactive state was developed using the β2-AR crystal structure (Protein Data Bank code 2RH1 (25)) as the template. This initial model was refined based on sequence dictated differences between GPR35 and the β2-AR crystal structure. The key differences between these two TMH bundles are described below.
TMH1
Despite the presence of three glycines (G1.34, G1.30, and G1.46) in TMH1, conformational memories calculations revealed that TMH1 in GPR35 is similar to TMH1 in β2-AR, which leans away from the TMH bundle on the extracellular side.
TMH2
Like the CXCR4 receptor (30), GPR35 contains the XX(C/S/T)LP motif in TMH2, with proline position at 2.58. This motif creates a tightly wound proline kink where the C/S/T residues in the proline kink region are in g(−), supporting the tightly wound kink structure. The β2-AR has a relaxed broad turn in the 2.59 proline kink region. The different turn ratio between the β2-AR and the GPR35 TMH2 proline kink region supports TMH2 fitting into the general seven-TMH GPCR structure, but with a different set of residues facing into the binding site. For example, residue V2.60 faces into the binding pocket of GPR35, whereas residue 2.60 in the β2-AR structure (25) is in the TMH2–3 interface and not available from within the binding pocket. Another consequence of the Pro at 2.58 in GPR35 is that the extracellular portion of TMH2 leans toward TMH3. It is important to note that had the GPR35 TMH2 retained the β2-AR TMH2 conformation, the GPR35 EC1 loop would not have been able to join the EC ends of TMH2 and TMH3, because the GPR35 EC1 loop is only two amino acids long (SD). In contrast, the EC1 loop of the β2-AR is six amino acids in length (KMWTFG).
TMH4
GPR35 lacks the proline at 4.60 found in the β2-AR. CM results suggest that the lack of this proline at 4.60 causes a local change at the extracellular end of TMH4, which leans toward TMH5 compared with the β2-AR crystal structure (Fig. 3).
FIGURE 3.
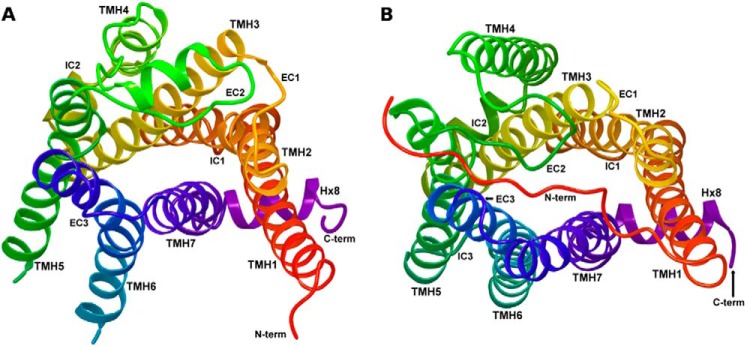
A comparison of the activated β2-AR crystal structure (37) (A) and the GPR35 R* (active) state model (B) is illustrated here. The receptors are each shown from an extracellular view.
TMH5
TMH5 in GPR35 has a proline at 5.50 and at 5.43. Because the β2-AR TMH5 has the conserved P5.50 as does GPR35 TMH5, the backbone torsions for that Pro kink were preserved and only the region around P5.43 in GPR35 TMH5 was explored via CM. These calculations suggested that P5.43 leans the extracellular end of TMH5 away from the binding crevice in GPR35. This change is accommodated in GPR35 because the GPR35 EC2 loop is longer than β2-AR (GPR35 11 amino acids versus 5 amino acids in β2-AR).
TMH6
The CM output gives helices that bend inward toward the binding crevice and also helices that bend toward TMH7. From the helices generated by CM, we picked two helices, one that would maintain the R3.50/T6.30 hydrogen bond TMH6 R (C-α to C-α distance = 6.15 Å) and one for which this interaction would be broken. TMH6 R had the following bend, wobble, and face shift angles: 32.8°, −62.1°, and 129.5°. TMH6 R* had the following bend, wobble, and face shift angles: 32.3°, −71.2°, and 83.3°. The differences in these two conformers are in their wobble and face shift angles. These changes result in the intracellular end of TMH6 in R* pivoting away from the IC end of TMH3.
TMH7
GPR35 TMH7 lacks the highly conserved class A GPCR NPXXY motif, having instead DAICY. The lack of a proline residue in TMH7 resulted in CM output that did not have the usual proline induced bend and deformation seen in crystal structures of other class A GPCRs. However, GPR35 has a charged residue, K7.40, that faces toward TMH2 in the binding pocket that should draw water into the TMH1-2-7 region. We have shown previously for GPR55 that such charged residues (D7.43 and D7.49 for GPR55) in TMH7 become hydrated, and this water further perturbs the backbone structure on TMH7 by hydrogen bonding to backbone carbonyls. The net result is a region of 3–10 helix from 7.43 to 7.50.
GPR35 Model Activated Form (R*)
Fig. 3 illustrates a comparison of the activated β2-AR crystal structure (37) (Fig. 3A) and the GPR35 active state model (Fig. 3B). Here the view is from the extracellular side of each receptor. One hallmark of GPCR activation is the breaking of the ionic lock between R3.50 and E/D6.30 (T6.30 in GPR35), which allows TMH6 to change conformation. The result is an intracellular opening of the receptor, exposing residues that can interact with the C terminus of Gα (51). The active form of the GPR35 receptor model differs from the inactive form mainly in changes at the intracellular side of the receptor. The intracellular end of TMH6 has changed conformation and moved its IC end, breaking the hydrogen bond between R3.50 and T6.30. The distance between the Cα carbons of R3.50 and T6.30 is 11.8 Å in the GPR35 R* model, indicating loss of interaction between the IC ends of TMH3 and TMH6. This distance is comparable to the opening formed in the rhodopsin meta II (activated state) crystal structure (R3.50–E6.30 Cα distance = 14.7 Å) (52) in which the C-terminal fragment of G-α of transducin is inserted. The size of this same opening in the nanobody stabilized β2-AR crystal structure (Fig. 3A), is larger (R3.50 and E6.30 Cα distance = 17.2 Å) (37), because of the physical insertion of the nanobody higher than the C-terminal fragment of G-α of transducin, which causes the IC opening to enlarge.
GPR35 R6.58A Model Activated Form (R*)
To create the R6.58A mutant, residue 6.58 in the WT R* bundle was mutated to Ala, and the bundle was reminimized. This mutation did not change the overall conformation of TMH6. The mutant TMH6 had the following bend, wobble, and face shift angles: 32.7°, −74.4°, and 83.1°. The distance between the Cα carbons of R3.50 and T6.30 in the R6.58A mutant R* model was 11.9 Å, indicating loss of interaction between the IC ends of TMH3 and 6.
Ligand Conformational Analysis: Pamoic Acid Dianion
Pamoic acid (pKa1 = 2.51, pKa2 = 3.1) (53) is a dianion at physiological pH. For this reason, pamoic acid was docked as a dianion here. Conformational analyses were performed in vacuo and in a SM8 water solvation model as implemented in Spartan '08. Both analyses yielded the same global minimum energy conformer. In this conformation, the ring systems are nearly perpendicular to each other (C2-C1-C1‴-C1′ = 118.5°; C2′-C1′-C1‴-C1 = 118.3°) with the carboxyl (and hydroxyl) groups pointing away from each other (for numbering system, see Fig. 2).
Docking Studies: Pamoic Acid
Pamoic Acid/Wild-type Human GPR35 R*
We used our GPR35 activated state model to probe the nature of the interactions of pamoic acid dianion within the GPR35-binding pocket. Pamoic acid dianion is both charged and highly aromatic. Docking studies suggested that the pamoic acid-binding pocket is in the TMH3-4-5-6 region of GPR35. Fig. 4A illustrates the final docked pose of pamoic acid in the GPR35 R* model (Glide score SP 5.0, −8.81 kcal/mol). In this docked position, pamoic acid is able to form several highly favorable polar interactions with Arg residues, including several salt bridges and cation-π interactions (residues colored magenta). The ligand also forms hydrogen bonds (residues colored orange) and predominantly van der Waals interactions with other residues (residues colored yellow). The geometries of these interactions are discussed below. Pamoic acid establishes a salt bridge, as well as a hydrogen bond with the EC2 loop residue, R(164). The N–O distances are 2.68 and 2.82 Å, and the N–H—O angles are 166 and 162°, respectively. An additional EC2 loop residue, R(167), forms a salt bridge with the other acid moiety of the ligand. The N–O distance is 2.71 Å, and the N–H—O angle is 161°. Pamoic acid also forms a salt bridge with R3.36. The N–O distance is 2.63 Å, and the N–H—O angle is 164°. Q3.29 hydrogen bonds with this same carboxyl group having a N–O distance of 2.80Å and a N–H—O angle of 177°. R4.60 forms a parallel and a tilted T cation-π interaction with the two sets of fused rings of pamoic acid. The distance of the central carbon of the R4.60 guanidinium group and the centroid of the ring that is parallel to the R4.60 is 4.19 Å. The distance and angle of the central carbon of the R4.60 guanidinium group and the centroid of the ring that forms the tilted T cation-π interaction are 3.94 Å and 60°, respectively. The TMH residue, S5.39, hydrogen-bonds with the other acid moiety of pamoic acid. The hydrogen bond O–O distance is 2.70 Å, and the O–H—O angle is 165°. The residues T166, L6.55, and L3.33 have van der Waals interactions with pamoic acid dianion. Finally, R6.58 lies above the pamoic acid-binding pocket and has no direct interactions with pamoic acid (colored gray).
FIGURE 4.
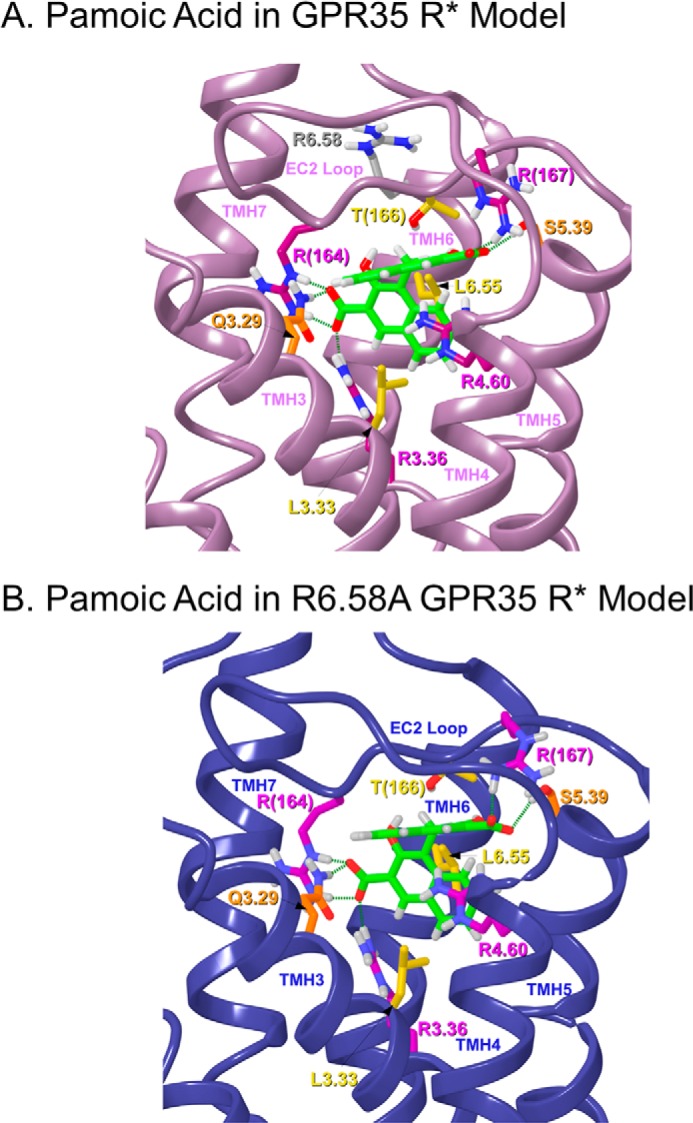
This figure illustrates the final docked poses of pamoic acid dianion in the human WT GPR35 R* model (A) and in the R6.58A GPR35 R* model (B). Residues that form salt bridge and cation-π interactions are colored magenta, whereas residues that form hydrogen bonds are colored orange, and residues that contribute mainly van der Waals interactions are colored yellow. Residues that are not part of the ligand-binding pocket are colored gray.
Pamoic Acid/Human R6.58A Mutant GPR35 R*
Despite the fact that pamoic acid is predicted to dock in the arginine rich TMH3-4-5-6 region of GPR35, not all of these arginines appear to be necessary for ligand binding. A case in point is R6.58. Our modeling studies (see above) suggest that this residue does not interact with pamoic acid. We created an R6.58A GPR35 R* model to examine possible changes occurring upon mutation. Fig. 4B illustrates the final docked pose of pamoic acid in the GPR35 R* model (Glide score SP 5.0, −8.95 kcal/mol). In this docked position, pamoic acid is able to form several highly favorable polar interactions with Arg residues, including several salt bridges and cation-π interactions (residues colored magenta). The ligand also forms hydrogen bonds (residues colored orange) and predominantly van der Waals interactions with other residues (residues colored yellow). The interactions that pamoic acid has in the R6.58A mutant are identical to WT GPR35. R(164) has a salt bridge and a hydrogen bond with one of the acid moieties of pamoic acid. The N–O distances are 2.65 and 2.92 Å, and the N–H—O angles are 165 and 169°, respectively. Pamoic acid also forms a salt bridge with R3.36. The N–O distance is 2.84 Å, and the N–H—O angle is 169°. Also, Q3.29 hydrogen-bonds with this same carboxyl group having a N–O distance of 2.78 Å and a N–H—O angle of 161°. Residues S5.39 and R(167) have a hydrogen bond and a salt bridge with the other acid moiety of pamoic acid. The hydrogen bond O–O and salt bridge N–O distances are 3.03 and 2.63 Å, whereas the O–H—O and N–H—O angles are 162 and 165°, respectively. R4.60 forms a parallel and a tilted T cation-π interaction with the two sets of fused rings of pamoic acid. The distance of the central carbon of the guanidinium of R4.60 and the centroid of the ring that is parallel to the R4.60 is 3.96 Å. The distance and angle of the central carbon of the guanidinium of R4.60 and the centroid of the ring that forms the tilted T cation-π interaction are 3.87 Å and 63°. Other amino acids that have predominantly van der Waals interaction energies with pamoic acid are T166, L6.55, and L3.33.
Ligand Conformational Analysis: Zaprinast
The zaprinast tautomer used for docking, 5-(2-propoxy-phenyl)-2,6-dihydro[1,2,3]-triazolo-[4,5-d]pyrimidin-7-one) was the global minimum energy conformer for the 2,6-dihydro tautomeric state (see numbering system in Fig. 2). This tautomer was 1.55 kcal/mol above the lowest energy tautomer global min as calculated by Hartree-Fock, which was the 3,6-dihydro form. Although this tautomer choice costs energy, the location of a proton at N2 facilitates a key interaction with Y3.32, a residue found to be important for zaprinast action at GPR35 (49) (see “Discussion”). The global minimum energy conformer for this tautomeric form of zaprinast has the two ring systems nearly in plane with each other (C2′-C1′-C5-N6 = 16.1°) with the 2-propoxy group near N6 of the bicyclic ring (see Fig. 2 for numbering system).
Docking Studies: Zaprinast
Zaprinast-GPR35 R* Complex
The EC50 of zaprinast is considerably higher than that of pamoic acid. Docking studies suggested that like pamoic acid, the binding pocket for zaprinast is in the TMH3-4-5-6 region of GPR35. Fig. 5A illustrates the final docked pose of zaprinast in the GPR35 R* model (Glide score SP 5.0, −4.14 kcal/mol). Unlike pamoic acid, zaprinast is not charged and therefore cannot form salt bridges like pamoic acid. In its docked position, zaprinast is able to form hydrogen bonds (residues colored orange) and predominantly van der Waals interactions with other residues (residues colored yellow). The C=O oxygen of the zaprinast fused ring system can form a hydrogen bond with R(164). The N–O distance is 2.64 Å, and the N–H—O angle is 168°. Q3.29 is too far to hydrogen-bond with zaprinast, but this residue has significant van der Waals interactions with zaprinast. R4.60 (colored cyan) forms a boundary of the zaprinast-binding pocket but does not have a significant direct interaction with zaprinast. Several other amino acids have predominantly van der Waals interactions with zaprinast, including L3.33, L6.51, and L6.55. Y3.32 has a tilted T aromatic stack with the heterocyclic fused ring of zaprinast. The distance is 4.14 and 5.65 Å for the five- and six-member rings, respectively, and the angle is 49°. R6.58 and R(167) (colored gray) are above the zaprinast-binding pocket and have no direct interaction with zaprinast.
FIGURE 5.
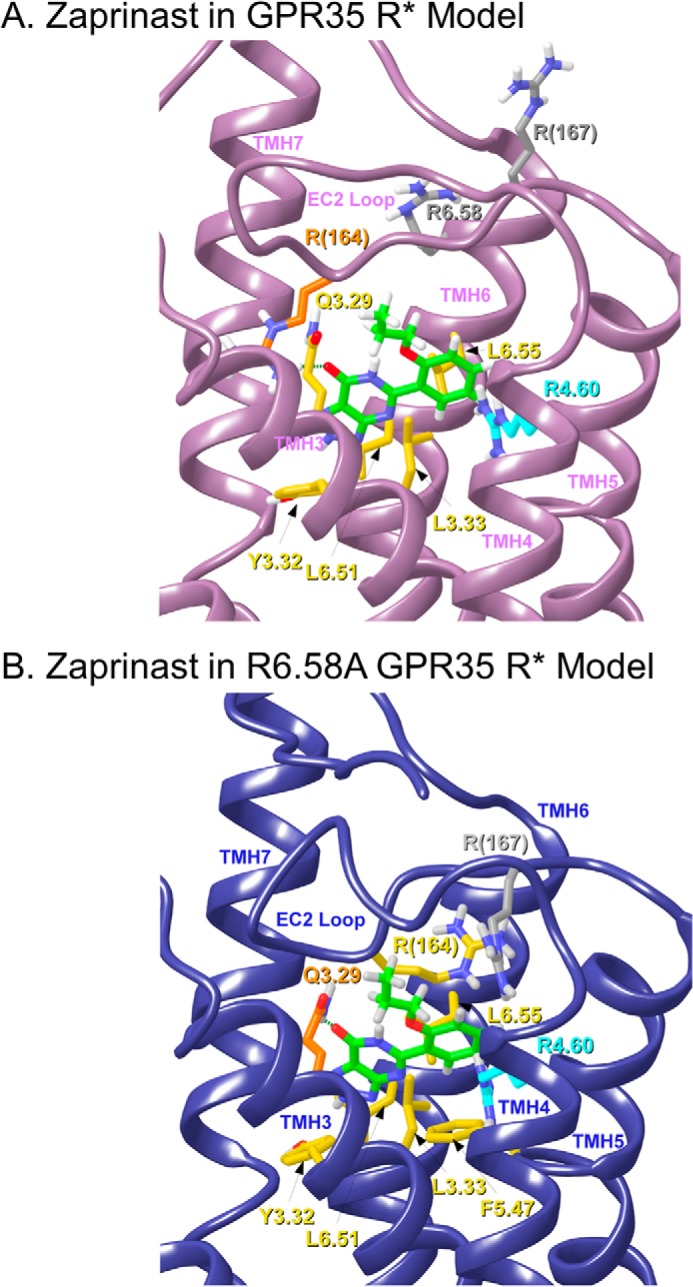
This figure illustrates the final docked poses of zaprinast in the human WT GPR35 R* model (A) and in the R6.58A GPR35 R* model (B). Residues that form hydrogen bonds are colored orange, and residues that contribute mainly van der Waals interactions are colored yellow. Residues that are not part of the ligand-binding pocket are colored gray, whereas residues that may form a boundary of the binding pocket are colored cyan.
Zaprinast-GPR35 R6.58A R* Complex
Fig. 5B illustrates the final docked pose of zaprinast in the R6.58A GPR35 R* model (Glide score SP 5.0, −5.38 kcal/mol). Our modeling studies suggest that R6.58 in WT GPR35 does not interact with zaprinast but produces crowding in the binding region. Removal of R6.58 via the R6.58A mutation is actually favorable for zaprinast because it acquires additional interactions compared with WT. In its docked position, zaprinast is able to form hydrogen bonds (residues colored orange) as well as predominantly van der Waals interactions with other residues (residues colored yellow). Zaprinast can form a hydrogen bond between Q3.29 and the C=O oxygen of its fused ring system. The N–O distance is 2.84 Å, and N–H—O angle is 143.5°. Additional contributors to the zaprinast interaction energy come from residues that form predominantly van der Waals interactions with the ligand. These include L6.55, R(164), L3.33, and L6.51. Zaprinast also has two aromatic stacking interactions that add to its receptor interaction, one with F5.47 and one with Y3.32. F5.47 forms a tilted T aromatic stack with the propoxy-benzyl ring of zaprinast. The ring centroid to centroid distance is 6.22 Å, and the angle of the planes of the two rings is 50°. The fused ring system also stacks with F5.47. The ring centroid to centroid distances of the five- and six-member rings are 5.61 and 5.90 Å, respectively, and the plane to plane angle is 59°. Y3.32 forms a tilted T interaction with the fused ring system of zaprinast. The centroid to centroid distance of the two rings is 4.77 Å, and the plane to plane angle is 72°. R4.60 (colored cyan) forms a boundary of the zaprinast-binding pocket but does not have a significant interaction with zaprinast. R(167) (colored gray) is above the zaprinast-binding pocket and has no direct interaction with zaprinast.
Agonist-induced β-arr2-GFP Trafficking in Mutant GPR35 Cells
Stable U2OS cell lines expressing both β-arr2-GFP and each of the mutants as well as WT were made to assess agonist-induced receptor activation of β-arr2-GFP trafficking. Cell surface expression of the receptor was confirmed by HA immunostaining for WT, A4.59G, R4.60A, R6.58A, R(164)A/L, K7.40A (Fig. 6), K1.32A, R2.65A, and R(167)A cell lines (data not shown). Application of pamoic acid (1 μm) or zaprinast (10 μm) induced the typical β-arr2-GFP aggregates in WT, A4.59G, R(164)A/L, and K7.40A cell lines (Fig. 7) and K1.32A, R2.65A, and R(167)A cell lines (data not shown). The R4.60A mutant did not respond to treatment with pamoic acid or zaprinast. Although R6.58A cells also showed β-arr2-GFP aggregates upon pamoic acid or zaprinast treatment (Fig. 7), an abnormal morphological change was observed. Treated R6.58A cells rounded up, this made the β-arr2-GFP trafficking assay unreliable for this cell line.
FIGURE 6.
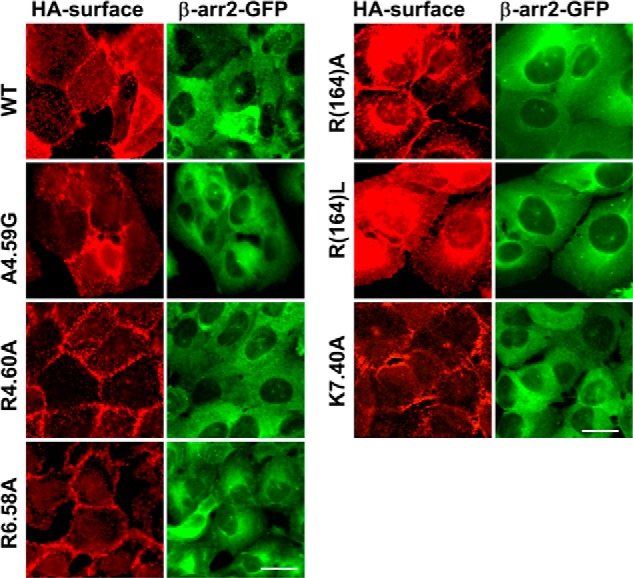
Immunofluorescent confirmation of surface expressing of GPR35 mutant receptors and cytosolic βarr2-GFP. Wild-type GPR35, A4.59G, R4.60A, R6.58A, R(164)A, R(164)L, and K7.40A showed typical surface receptor staining pattern using HA monoclonal antibody. All these mutant cell lines showed βarr2-GFP expression similar to wild-type GPR35 cells. Scale bars, 20 μm.
FIGURE 7.
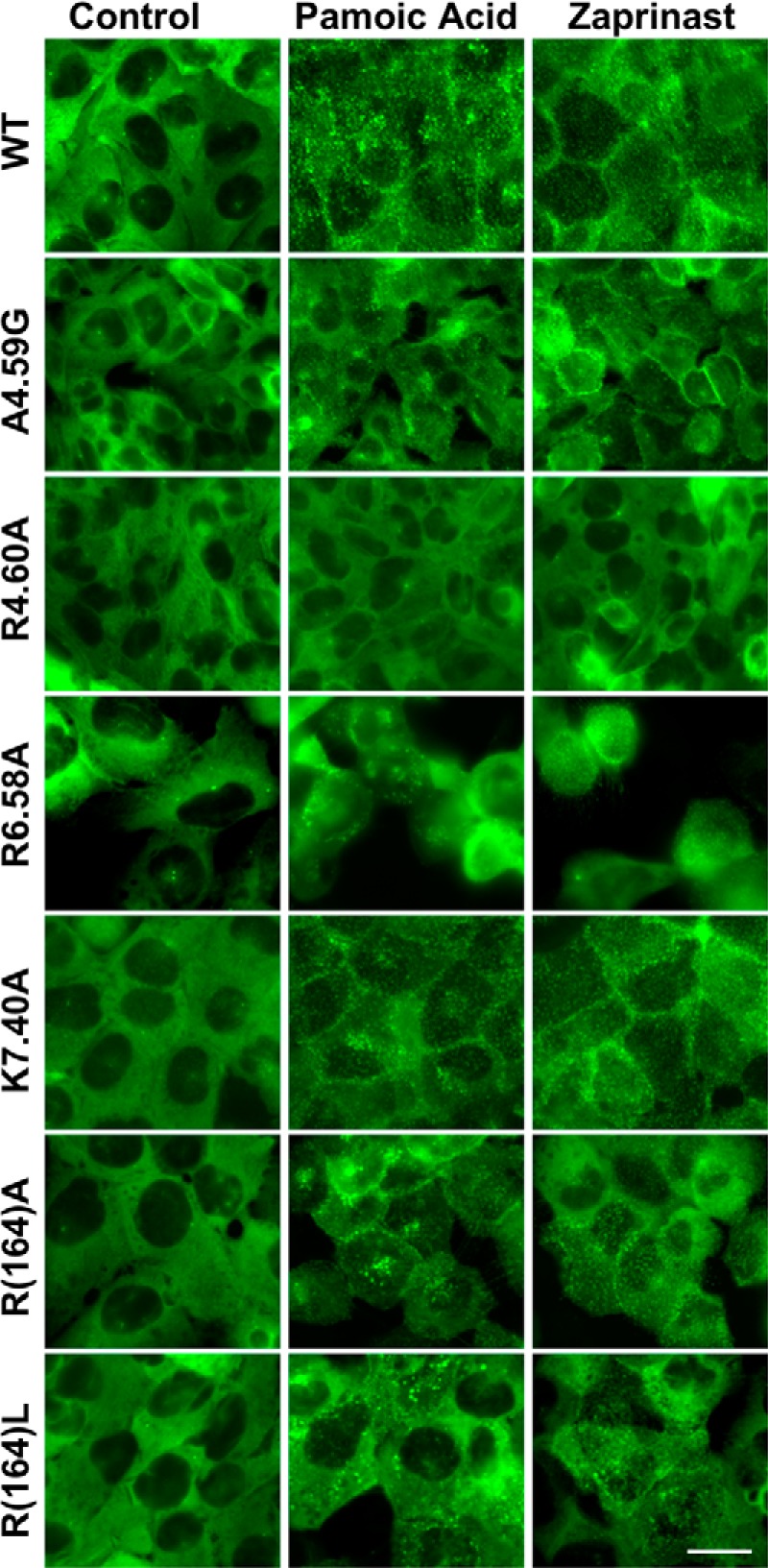
Agonist-mediated βarr2-GFP response in mutant GPR35 cells. Stable U2OS cell lines co-expressing β-arr2-GFP and each of the GPR35 mutants were made to examine the receptor activity. Representative images showed cells in vehicle control, pamoic acid (1 μm), and zaprinast (10 μm). No β-arr2-GFP trafficking was observed at control condition for all the cell lines. WT, A4.59G, R6.58A, R(164)A, R(164)L, and K7.40A showed β-arr2-GFP aggregates upon both pamoic acid and zaprinast application. R6.58A cells rounded up after both pamoic acid and zaprinast treatment. R4.60A did not show βarr2-GFP response in both pamoic acid and zaprinast treatments. Scale bar, 20 μm.
HA immunostaining of the R3.36A and R7.33A (data not shown) mutant U2OS cell lines showed no cell surface expression (Fig. 8A, left panels), but instead showed strong cytosolic expression in permeabilized cells (Fig. 8A, second panels from left). Immunostaining of HEK293 cells transiently transfected with R3.36A plasmid also showed no surface expression but strong cytosolic expression (Fig. 8A, right two panels). No β-arr2-GFP response was observed in R3.36A mutant cells upon pamoic acid or zaprinast treatment (Fig. 8B). To localize the cytosolic HA-tagged R3.36A mutant, U2OS cells expressing R3.36A alone were permeabilized and immunostained with HA and the ER-resident protein calrecticulin (18). High magnification of confocal images reveals a perinuclear labeling that is characteristic of ER staining (Fig. 8C). Co-localization of the ER protein calreticulin with the R3.36A GPR35 suggests that the R3.36A mutant is retained intracellularly at the ER. This is consistent with the lack of cell surface receptor immunoreactivity for R3.36A (Fig. 8A). Treatment of R3.36A U2OS cells with GPR35 antagonist CID2745687 (5) for 16 h rescued the surface expression of R3.36A, suggesting a chaperone function by this antagonist.
FIGURE 8.
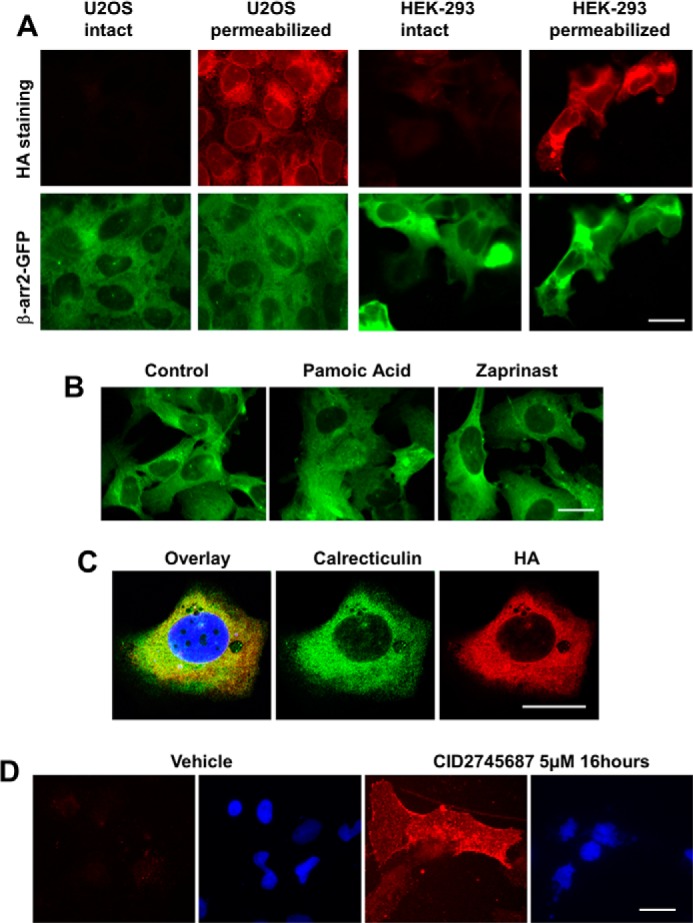
Expression of GPR35 mutant R3.36A in U2OS and HEK293 cells. A, U2OS and HEK293 cells are co-transfected with HA-tagged R3.36A and βarr2-GFP. Cell surface staining of HA-tagged R3.36A did not show specific signal in both U2OS and HEK-293 cells. HA staining of permeabilized cells showed strong HA antibody signal in both U2OS and HEK-293. βarr2-GFP was expressed in both U2OS and HEK-293 cells. B, transiently transfected U2OS cells expressing both R3.36A and β-arr2 GFP were treated with vehicle control, pamoic acid (10 μm), and zaprinast (10 μm). No difference between drug-treated and control cells was observed in terms of β-arr2 GFP trafficking. C, co-localization of HA and calreticulin in permeabilized U2OS cells transiently transfected with HA-tagged R3.36A. D, CID2745687 rescued the surface expression of R3.36A. HA staining is shown in red; DAPI staining is shown in blue. Scale bar, 20 μm.
To compare the potencies of these mutants, β-arr2-GFP response was quantified as previously described (5). The WT GPR35 has EC50 values for pamoic acid and zaprinast-induced activation of 52 nm and 1.0 μm, respectively (Table 1). The substitution of Ala4.59 with a glycine did not significantly change the potency for β-arr2-GFP response (Fig. 9 and Table 1). Although modest rightward shifts in the concentration response curves were observed with the K7.40A mutant receptor for pamoic acid and zaprinast (Fig. 9, A and B), they were not significantly different from the WT GPR35 receptor (Table 1). Both R(164)A and the R(164)L mutants showed a robust rightward shift in the concentration response curves for pamoic acid and zaprinast (Fig. 9, C and D, and Table 1). There was no β-arr2-GFP response in the R3.36A and R4.60A mutants (Table 1). The EC50 value of R6.58A was not obtained because the morphological changes made the quantification of aggregates unreliable.
TABLE 1.
βarr2-GFP trafficking activities of wild-type and mutant cell lines
ND, not detected; NT, not tested.
| Cell line | EC50 (CI) |
|
|---|---|---|
| Pamoic acid | Zaprinast | |
| WT | 52.0 nm (33.8–80.0) | 1.0 μm (0.7–1.3) |
| R3.36A | ND | ND |
| A4.59G | 55.7 nm (35.7–83.6) | 0.9 μm (0.5–1.6) |
| R4.60A | ND | ND |
| R6.58A | NT | NT |
| K7.40A | 111.3 nm (65.4–189.3) | 1.4 μm (1.0–1.9) |
| R(164)A | 783 nm (214–2859)a | 5.2 μm (3.0–9.0)a |
| R(164)L | 944 nm (391–2277)a | 6.1 μm (1.6–23)a |
| K1.32A | 29.7 nm (6.8–130.4) | 1.4 μm (0.5–4.4) |
| R2.65A | 43.2 nm (20.2–92.4) | 0.5 μm (0.2–1.2) |
| R(167)A | 200.5 nm (87.5–459.8)a | 1.6 μm (0.9–2.7) |
| R7.33A | ND | ND |
a p < 0.05. EC50 values and 95% confidence intervals are shown from n > 3 experiments.
FIGURE 9.
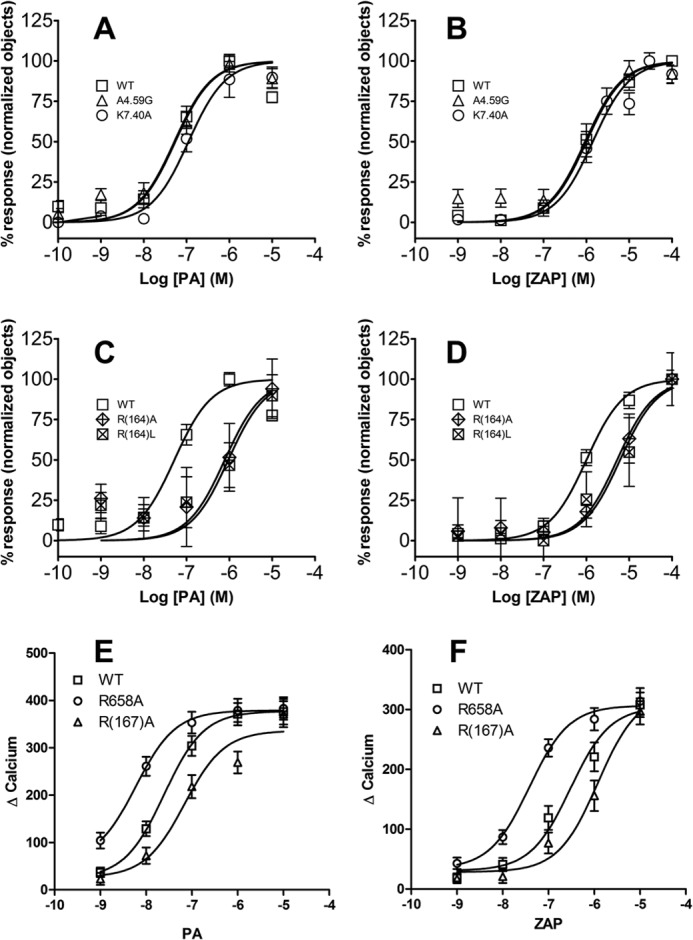
β-Arrestin2 and calcium responses of wild-type and mutant GPR35 receptors. A, β-arrestin2 response of WT, A4.59G, and K7.40A to the agonist pamoic acid. B, β-arrestin2 response of WT, A4.59G, and K7.40A to the agonist zaprinast. C, β-arrestin2 response of WT, R(164)A, and R(164)L to the agonist pamoic acid. D, β-arrestin2 response of WT, R(164)A, and R(164)L to the agonist zaprinast. E, calcium response of WT, R(167)A, and R6.58A to the agonist pamoic acid. F, calcium response of WT, R(167)A, and R6.58A to the agonist zaprinast. Each data point represents the mean ± S.E. of at least three independent experiments performed in quadruplicate.
Agonist-induced ERK1/2 Activation in Mutant GPR35 Cells
In addition to the β-arr2-GFP trafficking assay, agonist-induced ERK1/2 phosphorylation was used to examine the functional effects of GPR35 mutations. A downstream effector of GPR35 activation is the G protein-dependent ERK1/2 phosphorylation. It has been shown to be independent of the β-arrestin pathway (5). Table 2 summarizes the activities of ERK1/2 by pamoic acid and zaprinast from WT and mutant GPR35 receptors. The EC50 values of agonists, pamoic acid, and zaprinast on the mutants A4.59G and K7.40A were comparable and not statistically different from the WT receptor. In contrast, a ∼30-fold increase in the potency of zaprinast was observed in the R6.58A mutant receptor (83 nm) compared with the wild-type GPR35 receptor (2.8 μm). A modest increase in potency of pamoic acid in R6.58A (16 nm) was observed as well, compared with WT (62.3 nm).
TABLE 2.
The effects of amino acid mutations of recombinant GPR35 on the ERK1/2 activation induced by pamoic acid and zaprinast
ND, not detected; NT, not tested.
| Cell line | EC50 (CI) |
|
|---|---|---|
| Pamoic acid | Zaprinast | |
| WT | 62.3 nm (30.5–127) | 2.8 μm (0.5–15) |
| R3.36A | NT | NT |
| A4.59G | 45.6 nm (6.7–309) | 0.5 μm (0.1–2.7) |
| R4.60A | ND | ND |
| R6.58A | 16.0 nm (6.6–38) | 0.08 μm (0.02–0.3)a |
| K7.40A | 70.7 nm (23.2–215) | 0.5 μm (0.22–1.1) |
a p < 0.05. EC50 values and 95% confidence intervals are shown from n > 3 experiments performed in triplicate.
Calcium Responses in Wild-type and Mutant GPR35 Cells to Agonists
Because GPR35 is a Gi/o-coupled receptor, calcium is a downstream effector. Calcium response curves of mutant R6.58A shifted to the left compared with WT (Fig. 9, E and F). With zaprinast, the EC50 values were 0.3 μm (0.25–0.35 CI) for WT and 0.038 μm (0.034–0.044 CI) for R6.58A. With pamoic acid, the EC50 values were 25 nm (21–28 CI) for WT and 6.0 nm (5.1–6.9 CI) for R6.58A. On the contrary, the mutant R(167)A showed right shifted curves, indicating lower potencies for agonists. The EC50 values were 69 nm (56–84 CI) for pamoic acid and 1.2 μm (1.0–1.4) for zaprinast in the R(167)A mutant. These shifts of potencies in the mutants are consistent with the β-arr2-GFP response (Table 1) and ERK1/2 activation (Table 2) results. Quantification of receptor expression levels of WT and mutants (Table 3) showed no significant difference between them, indicating that the expression level is not a contributing factor for the shifts of response curves of β-arr2-GFP, ERK1/2, and calcium.
TABLE 3.
Quantification of receptor surface expression level of GPR35 and mutants by HA antibody immunostaining
n ≥ 4 images in each group.
| Cell line | Immunostaining intensity (mean ± S.E.) |
|---|---|
| WT | 12.29 ± 1.13 |
| R(164)A | 11.65 ± 0.91 |
| R(167)A | 13.22 ± 1.04 |
| R6.58A | 9.60 ± 1.25 |
DISCUSSION
To date, there is no x-ray crystal structure information available for the human GPR35 receptor; thus, in the present study, we have used computational modeling together with site-directed mutagenesis to explore the structural features of human GPR35 involved in agonist-induced receptor activation. The GPR35 sequence contains numerous positively charged amino acids that face into the binding pocket. These residues cluster in two distinct receptor regions: the TMH3-4-5-6 region and the TMH1-2-7 region of GPR35 (Fig. 1).
The most prominent positively charged amino acid in the TMH 1-2-7 region of the GPR35-binding pocket is K7.40, whereas other positively charged residues that face into the binding pocket but are more extracellular include K1.32, R2.65, and R7.33. Mutation of each of these residues to Ala resulted in the retention of WT EC50 for pamoic acid and zaprinast. Because many of the ligands for GPR35 are anions, a lack of change in potency for any of these TMH1-2-7 residues suggests that the ligand-binding pocket of GPR35 may not lie in the TMH 1-2-7 region of GPR35.
The majority of the positively charged residues that face into the binding pocket are located in the TMH 3-4-5-6 region. These residues include R3.36, R4.60, and R6.58 and the EC-2 loop residues R(164) and R(167). In contrast to results for the TMH 1-2-7-binding pocket region, we found that mutation of many of the positively charged binding site residues in the TMH 3-4-5-6 region affected both pamoic acid and zaprinast signaling. Fig. 10 illustrates the relative positions of pamoic acid and zaprinast in their docked positions as determined by Glide docking studies. In this view from lipid looking between TMH5 and TMH6, it is clear that both ligands bind in the same general area (TMH3-4-5-6 region); however, their positions are shifted relative to each other. Pamoic acid extends further up toward extracellular space than does zaprinast and also moves closer to TMH4.
FIGURE 10.
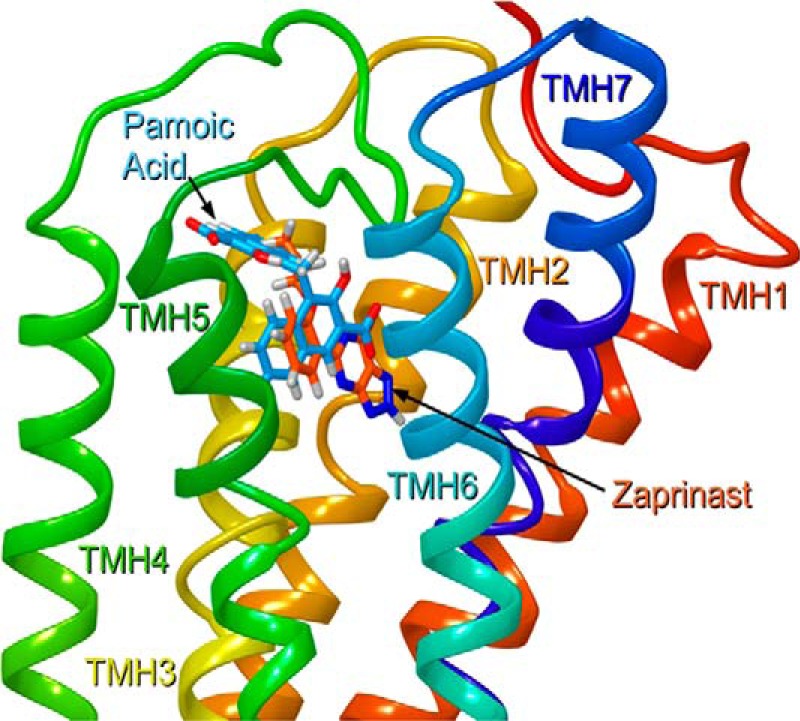
The relative positions of pamoic acid (blue) and zaprinast (orange) in their docked conformations as determined by Glide docking studies. In this view from lipid looking between TMH5 and TMH6, it is clear that both ligands bind in the same general area (TMH3-4-5-6 region); however, their positions are shifted relative to each other. Pamoic acid extends further up toward extracellular space than does zaprinast and pamoic acid also moves closer to TMH4.
Many of the interactions identified by docking studies, particularly for pamoic acid, are salt bridge and cation-π interactions, which are strong interactions. It is important to emphasize, however, that the presence of water will have a dampening effect on such electrostatic interactions. This would be particularly true for residues in loop regions but is likely true even for transmembrane positively charged residues here, because the high concentration of positively charged residues in the GPR35-binding pocket likely draws waters down into the binding pocket. One therefore cannot expect as profound a change upon mutation and loss of a salt bridge in GPR35 compared with other receptors that may have an analogous salt bridge shielded from water.
Many of the mutations performed here had very striking effects. The EC-2 loop residue R(164) is shown here by mutation and modeling studies to be a key interaction site for pamoic acid, with a significant increase in EC50 values (R164A 15-fold; R164L, 18-fold) upon mutation to an uncharged residue. R(164) forms a hydrogen bond, rather than a salt bridge, with zaprinast. Mutation of R(164) to an uncharged amino acid would be expected, therefore, to have a less profound effect on the EC50 of zaprinast when compared with the effect of this mutation on pamoic acid. The 5-fold increase in EC50 for zaprinast at the R(164)A and R(164)L mutants, therefore, is consistent with this result.
R(167) in the EC2 loop is also shown here by modeling and mutation studies to be a key interaction for pamoic acid, but not for zaprinast. R(167) forms a salt bridge with the part of pamoic acid that is located near the extracellular end of TMH5. This interaction is with the carboxyl group with which R(164) does not interact. The R(167) interaction with pamoic acid is shown to be weaker by mutation studies than the R(164) interaction but still significant with an increase in EC50 of almost 4-fold for pamoic acid. Zaprinast does not interact with R(167), as shown by modeling, because zaprinast is located further from TMH5. Because the position of zaprinast does not shift toward TMH5 in the R6.58A mutant, no interaction with R(167) is present there as well.
Our modeling studies suggest that at the pamoic acid binding site, R4.60 forms a parallel and a tilted T cation-π interaction with both fused rings of pamoic acid. Although R4.60 does not form a very high energy interaction with zaprinast, this residue forms the boundary of the zaprinast-binding pocket and therefore could be anticipated to play an important structural role in the zaprinast-GPR35 R*-binding pocket (Fig. 5). We show here that mutation of Arg4.60 to alanine abolished the agonist-induced receptor activation for both pamoic acid and zaprinast and in two independent assays of β-arr2-GFP trafficking and ERK1/2 phosphorylation. This suggests that R4.60(151) is a key residue in the GPR35 R*-binding pocket that may serve to initially align the ligand in the binding pocket either through direct coulombic interactions via a cation-π interaction (pamoic acid) or by defining one edge of the binding pocket (zaprinast).
R6.58 is located near the extracellular end of TMH6 and points toward the TMH3-4-5-6 region of GPR35. Our modeling studies suggest that this residue does not directly interact with pamoic acid and that the interactions that pamoic acid has in the R6.58A mutant receptor are almost identical, with the exception of the exchange of a direct interaction with Q(157) for R(167). The R6.58A mutation does not cause a large drop in the overall interaction energy of pamoic acid, because interaction with other residues (principally T(166)) improves in the absence of R6.58. Thus, modeling would predict that the R6.58A mutation should have little effect on the efficacy of pamoic acid.
Our modeling studies suggest that R6.58 does not interact with zaprinast either but produces crowding in the binding region. Removal of R6.58 via the R6.58A mutation is actually favorable for zaprinast. The improved receptor interaction results from multiple stronger interactions at L6.55, R(164), L3.33, and L6.51. In each case, the gains are largely due to improved van der Waals interactions. Thus, modeling would predict that zaprinast should have a lower EC50 in the R6.58A mutant.
Mutation studies showed that R6.58A increases the potency of zaprinast in the pERK assay. On the other hand, it causes a roundup of cells upon agonist treatment in the β-arr2-GFP trafficking assay. Because of these morphological changes upon agonist application, a reliable quantification of arrestin trafficking could not be obtained for this mutant. This morphological change upon agonist application was not observed in WT and other mutant cell lines. Interestingly, the calcium response assay showed increased potencies for both zaprinast and pamoic acid in the 6.58A mutant, similar to what was observed in the pERK assay.
Modeling studies suggest that R3.36 is a major contributor to the interaction energy of pamoic acid at GPR35 R*. This residue also interacts with zaprinast, but in a lower energy interaction. An R3.36A mutation can be expected to impact both ligands, but pamoic acid more significantly. Mutation of Arg3.36 to alanine prevented GPR35 receptor expression on the cell surface; it was trapped in the endoplasmic reticulum. This suggests that R3.36A mutation may cause a structural misfolding during the protein maturation process. This is consistent with the ability of the GPR35 antagonist to rescue cell surface expression of R3.36A (Fig. 8D). A similar chaperone function by ligands has been previously documented for V2 vasopressin receptor (18). In the functional assay, there was no βarr2-GFP trafficking response for this mutant upon pamoic acid and zaprinast application. Lack of βarr2 recruitment response for the mutant R3.36A was reported in another study as well (49). The total loss of βarr2 response for R3.36A may be due to the lack of surface expression. Milligan and co-workers (49) have reported that the R3.36A mutation abolished βarr2 recruitment in response to several GPR35 agonists. A recent mutant and modeling study by MacKenzie et al. (54) also indicated the contributions of R4.60, R(164), and R6.58 to ligand binding.
The A4.59G mutant is a random mutation created as a control. This residue actually faces lipid in the GPR35 R* model. Mutation studies show here that this mutation did not change the EC50 of pamoic acid or zaprinast relative to WT.
The Milligan group has reported that Y3.32 is also an important residue for the action of zaprinast (49). Modeling studies reported here suggest that Y3.32 is a part of both the pamoic acid- and zaprinast-binding pockets at GPR35 R*, with its interaction being stronger for zaprinast. In the present study, however, function in response to pamoic acid or zaprinast could not be detected at a human Y3.32L mutant.
Our data strongly support that direct interactions of the ligands with the mutated residues is responsible for the observed functional effects. However, in the absence of radioligand binding experiments, direct interactions are implied and not proven at this point. A recent mutant and modeling study by MacKenzie et al. (54) also indicated the contributions of R4.60, R(164), and R6.58 to ligand binding using functional assays.
Recently it was reported that zaprinast is 38-fold more potent for rat GPR35 than human GPR35 (55). Kynurenic acid also has a higher potency for rat than human GPR35 (15). These findings imply that the ligand-binding site in human versus rat might be distinct. The origins of this difference are currently under investigation.
Acknowledgment
We thank Dr. Lawrence Barak (Duke University) for advice regarding the antagonist experiments.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 DA023204 (to M. E. A.), KO5 DA021358 (to P. H. R.), and P30 DA013429.
- GPCR
- G protein-coupled receptor
- βarr2-GFP
- β-arrestin2-green fluorescent protein
- β2-AR
- β2-adrenergic receptor
- CID2745687
- methyl-5-[(tert-butylcarbamothioylhydrazinyl-idene)methyl]-1-(2,4-difluorophenyl)pyrazole-4-carboxylate
- CM
- conformational memories
- TMH
- transmembrane helix
- CI
- confidence interval
- SP
- standard precision
- IC
- intracellular
- EC
- extracellular
- ER
- endoplasmic reticulum.
REFERENCES
- 1. O'Dowd B. F., Nguyen T., Marchese A., Cheng R., Lynch K. R., Heng H. H., Kolakowski L. F., Jr., George S. R. (1998) Discovery of three novel G-protein-coupled receptor genes. Genomics 47, 310–313 [DOI] [PubMed] [Google Scholar]
- 2. Guo J., Williams D. J., Puhl H. L., 3rd, Ikeda S. R. (2008) Inhibition of N-type calcium channels by activation of GPR35, an orphan receptor, heterologously expressed in rat sympathetic neurons. J. Pharmacol. Exp. Ther. 324, 342–351 [DOI] [PubMed] [Google Scholar]
- 3. Ohshiro H., Tonai-Kachi H., Ichikawa K. (2008) GPR35 is a functional receptor in rat dorsal root ganglion neurons. Biochem. Biophys. Res. Commun. 365, 344–348 [DOI] [PubMed] [Google Scholar]
- 4. Taniguchi Y., Tonai-Kachi H., Shinjo K. (2006) Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett. 580, 5003–5008 [DOI] [PubMed] [Google Scholar]
- 5. Zhao P., Sharir H., Kapur A., Cowan A., Geller E. B., Adler M. W., Seltzman H. H., Reggio P. H., Heynen-Genel S., Sauer M., Chung T. D., Bai Y., Chen W., Caron M. G., Barak L. S., Abood M. E. (2010) Targeting of the orphan receptor GPR35 by pamoic acid. A potent activator of extracellular signal-regulated kinase and β-arrestin2 with antinociceptive activity. Mol. Pharmacol. 78, 560–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neubig R. R. (2010) Mind your salts. When the inactive constituent isn't. Mol. Pharmacol. 78, 558–559 [DOI] [PubMed] [Google Scholar]
- 7. Fallarini S., Magliulo L., Paoletti T., de Lalla C., Lombardi G. (2010) Expression of functional GPR35 in human iNKT cells. Biochem. Biophys. Res. Commun. 398, 420–425 [DOI] [PubMed] [Google Scholar]
- 8. Okumura S., Baba H., Kumada T., Nanmoku K., Nakajima H., Nakane Y., Hioki K., Ikenaka K. (2004) Cloning of a G-protein-coupled receptor that shows an activity to transform NIH3T3 cells and is expressed in gastric cancer cells. Cancer Sci. 95, 131–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shrimpton A. E., Braddock B. R., Thomson L. L., Stein C. K., Hoo J. J. (2004) Molecular delineation of deletions on 2q37.3 in three cases with an Albright hereditary osteodystrophy-like phenotype. Clin. Genet. 66, 537–544 [DOI] [PubMed] [Google Scholar]
- 10. Cosi C., Mannaioni G., Cozzi A., Carlà V., Sili M., Cavone L., Maratea D., Moroni F. (2011) G-protein coupled receptor 35 (GPR35) activation and inflammatory pain. Studies on the antinociceptive effects of kynurenic acid and zaprinast. Neuropharmacology 60, 1227–1231 [DOI] [PubMed] [Google Scholar]
- 11. Taniguchi Y., Tonai-Kachi H., Shinjo K. (2008) 5-Nitro-2-(3-phenylpropylamino)benzoic acid is a GPR35 agonist. Pharmacology 82, 245–249 [DOI] [PubMed] [Google Scholar]
- 12. Wang J., Simonavicius N., Wu X., Swaminath G., Reagan J., Tian H., Ling L. (2006) Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 281, 22021–22028 [DOI] [PubMed] [Google Scholar]
- 13. Yang Y., Lu J. Y., Wu X., Summer S., Whoriskey J., Saris C., Reagan J. D. (2010) G-protein-Coupled receptor 35 is a target of the asthma drugs cromolyn disodium and nedocromil sodium. Pharmacology 86, 1–5 [DOI] [PubMed] [Google Scholar]
- 14. Oka S., Ota R., Shima M., Yamashita A., Sugiura T. (2010) GPR35 is a novel lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 395, 232–237 [DOI] [PubMed] [Google Scholar]
- 15. Barth M. C., Ahluwalia N., Anderson T. J., Hardy G. J., Sinha S., Alvarez-Cardona J. A., Pruitt I. E., Rhee E. P., Colvin R. A., Gerszten R. E. (2009) Kynurenic acid triggers firm arrest of leukocytes to vascular endothelium under flow conditions. J. Biol. Chem. 284, 19189–19195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barak L. S., Ferguson S. S., Zhang J., Martenson C., Meyer T., Caron M. G. (1997) Internal trafficking and surface mobility of a functionally intact β2-adrenergic receptor-green fluorescent protein conjugate. Mol. Pharmacol. 51, 177–184 [DOI] [PubMed] [Google Scholar]
- 17. Tao Q., McAllister S. D., Andreassi J., Nowell K. W., Cabral G. A., Hurst D. P., Bachtel K., Ekman M. C., Reggio P. H., Abood M. E. (1999) Role of a conserved lysine residue in the peripheral cannabinoid receptor (CB2). Evidence for subtype specificity. Mol. Pharmacol. 55, 605–613 [PubMed] [Google Scholar]
- 18. Morello J. P., Salahpour A., Laperrière A., Bernier V., Arthus M. F., Lonergan M., Petäjä-Repo U., Angers S., Morin D., Bichet D. G., Bouvier M. (2000) Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J. Clin. Investig. 105, 887–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brailoiu E., Rahman T., Churamani D., Prole D. L., Brailoiu G. C., Hooper R., Taylor C. W., Patel S. (2010) An NAADP-gated two-pore channel targeted to the plasma membrane uncouples triggering from amplifying Ca2+ signals. J. Biol. Chem. 285, 38511–38516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ito T., Kwon H. Y., Zimdahl B., Congdon K. L., Blum J., Lento W. E., Zhao C., Lagoo A., Gerrard G., Foroni L., Goldman J., Goh H., Kim S. H., Kim D. W., Chuah C., Oehler V. G., Radich J. P., Jordan C. T., Reya T. (2010) Regulation of myeloid leukaemia by the cell-fate determinant Musashi. Nature 466, 765–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li J., Edwards P. C., Burghammer M., Villa C., Schertler G. F. (2004) Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol. 343, 1409–1438 [DOI] [PubMed] [Google Scholar]
- 22. Okada T., Fujiyoshi Y., Silow M., Navarro J., Landau E. M., Shichida Y. (2002) Functional role of internal water molecules in rhodopsin revealed by x-ray crystallography. Proc. Natl. Acad. Sci. U.S.A. 99, 5982–5987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Palczewski K., Kumasaka T., Hori T., Behnke C. A., Motoshima H., Fox B. A., Le Trong I., Teller D. C., Okada T., Stenkamp R. E., Yamamoto M., Miyano M. (2000) Crystal structure of rhodopsin. A G protein-coupled receptor. Science 289, 739–745 [DOI] [PubMed] [Google Scholar]
- 24. Park J. H., Scheerer P., Hofmann K. P., Choe H. W., Ernst O. P. (2008) Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454, 183–187 [DOI] [PubMed] [Google Scholar]
- 25. Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Kuhn P., Weis W. I., Kobilka B. K., Stevens R. C. (2007) High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rasmussen S. G., Choi H. J., Rosenbaum D. M., Kobilka T. S., Thian F. S., Edwards P. C., Burghammer M., Ratnala V. R., Sanishvili R., Fischetti R. F., Schertler G. F., Weis W. I., Kobilka B. K. (2007) Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 450, 383–387 [DOI] [PubMed] [Google Scholar]
- 27. Rosenbaum D. M., Cherezov V., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Yao X. J., Weis W. I., Stevens R. C., Kobilka B. K. (2007) GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science 318, 1266–1273 [DOI] [PubMed] [Google Scholar]
- 28. Warne T., Serrano-Vega M. J., Baker J. G., Moukhametzianov R., Edwards P. C., Henderson R., Leslie A. G., Tate C. G., Schertler G. F. (2008) Structure of a β1-adrenergic G-protein-coupled receptor. Nature 454, 486–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jaakola V. P., Griffith M. T., Hanson M. A., Cherezov V., Chien E. Y., Lane J. R., Ijzerman A. P., Stevens R. C. (2008) The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322, 1211–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu B., Chien E. Y., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., Stevens R. C. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McAllister S. D., Rizvi G., Anavi-Goffer S., Hurst D. P., Barnett-Norris J., Lynch D. L., Reggio P. H., Abood M. E. (2003) An aromatic microdomain at the cannabinoid CB(1) receptor constitutes an agonist/inverse agonist binding region. J. Med. Chem. 46, 5139–5152 [DOI] [PubMed] [Google Scholar]
- 32. Zhang R., Hurst D. P., Barnett-Norris J., Reggio P. H., Song Z. H. (2005) Cysteine 2.59(89) in the second transmembrane domain of human CB2 receptor is accessible within the ligand binding crevice. Evidence for possible CB2 deviation from a rhodopsin template. Mol. Pharmacol. 68, 69–83 [DOI] [PubMed] [Google Scholar]
- 33. Guarnieri F., Weinstein H. (1996) Conformational memories and the exploration of biologically relevant peptide conformations. An illustration for the gonadotropin-releasing hormone. J. Am. Chem. Soc. 118, 5580–5589 [Google Scholar]
- 34. Whitnell R. M., Hurst D. P., Reggio P. H., Guarnieri F. (2008) Conformational memories with variable bond angles. J. Comput. Chem. 29, 741–752 [DOI] [PubMed] [Google Scholar]
- 35. MacKerell A. D., Jr., Banavali N., Foloppe N. (2000) Development and current status of the CHARMM force field for nucleic acids. Biopolymers 56, 257–265 [DOI] [PubMed] [Google Scholar]
- 36. Ballesteros J. A., Deupi X., Olivella M., Haaksma E. E., Pardo L. (2000) Serine and threonine residues bend α-helices in the χ1 = g(−) conformation. Biophys. J. 79, 2754–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rasmussen S. G., Choi H.-J., Fung J. J., Pardon E., Casarosa P., Chae P. S., DeVree B. T., Rosenbaum D. M., Thian F. S., Kobilka T. S., Schnapp A., Konetzki I., Sunahara R. K., Gellman S. H., Pautsch A., Steyaert J., Weis W. I., Kobilka B. K. (2011) Structure of a nanobody-stabilized active state of the β2-adrenoreceptor. Nature 469, 175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Altenbach C., Kusnetzow A. K., Ernst O. P., Hofmann K. P., Hubbell W. L. (2008) High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc. Natl. Acad. Sci. U.S.A. 105, 7439–7444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Farrens D. L., Altenbach C., Yang K., Hubbell W. L., Khorana H. G. (1996) Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 274, 768–770 [DOI] [PubMed] [Google Scholar]
- 40. Ghanouni P., Steenhuis J. J., Farrens D. L., Kobilka B. K. (2001) Agonist-induced conformational changes in the G-protein-coupling domain of the β2 adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 98, 5997–6002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Javitch J. A., Fu D., Liapakis G., Chen J. (1997) Constitutive activation of the β2 adrenergic receptor alters the orientation of its sixth membrane-spanning segment. J. Biol. Chem. 272, 18546–18549 [DOI] [PubMed] [Google Scholar]
- 42. Jensen A. D., Guarnieri F., Rasmussen S. G., Asmar F., Ballesteros J. A., Gether U. (2001) Agonist-induced conformational changes at the cytoplasmic side of transmembrane segment 6 in the β2 adrenergic receptor mapped by site-selective fluorescent labeling. J. Biol. Chem. 276, 9279–9290 [DOI] [PubMed] [Google Scholar]
- 43. Lin S. W., Sakmar T. P. (1996) Specific tryptophan UV-absorbance changes are probes of the transition of rhodopsin to its active state. Biochemistry 35, 11149–11159 [DOI] [PubMed] [Google Scholar]
- 44. Nakanishi J., Takarada T., Yunoki S., Kikuchi Y., Maeda M. (2006) FRET-based monitoring of conformational change of the β2 adrenergic receptor in living cells. Biochem. Biophys. Res. Commun. 343, 1191–1196 [DOI] [PubMed] [Google Scholar]
- 45. Scheerer P., Park J. H., Hildebrand P. W., Kim Y. J., Krauss N., Choe H. W., Hofmann K. P., Ernst O. P. (2008) Crystal structure of opsin in its G-protein-interacting conformation. Nature 455, 497–502 [DOI] [PubMed] [Google Scholar]
- 46. Fiser A., Do R. K., Sali A. (2000) Modeling of loops in protein structures. Protein Sci. 9, 1753–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 48. Stahl H. P., Wermuth C. G. (2011) Handbook of Pharmaceutical Salts: Properties, Selection, and Use, 2nd Ed., Wiley-VCH, Hoboken, NJ [Google Scholar]
- 49. Jenkins L., Alvarez-Curto E., Campbell K., de Munnik S., Canals M., Schlyer S., Milligan G. (2011) Agonist activation of the G protein-coupled receptor GPR35 involves transmembrane domain III and is transduced via Gα and β-arrestin-2. Br. J. Pharmacol. 162, 733–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., Shenkin P. S. (2004) Glide. A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 [DOI] [PubMed] [Google Scholar]
- 51. Hamm H. E., Deretic D., Arendt A., Hargrave P. A., Koenig B., Hofmann K. P. (1988) Site of G protein binding to rhodopsin mapped with synthetic peptides from the α subunit. Science 241, 832–835 [DOI] [PubMed] [Google Scholar]
- 52. Choe H. W., Kim Y. J., Park J. H., Morizumi T., Pai E. F., Krauss N., Hofmann K. P., Scheerer P., Ernst O. P. (2011) Crystal structure of metarhodopsin II. Nature 471, 651–655 [DOI] [PubMed] [Google Scholar]
- 53. Greco K., Wright J. (2003) Aripiprazole, olanzapine and haloperidol pamoate salts. Alkermes Controlled Therapeutics Inc., Cambridge, MA [Google Scholar]
- 54. Mackenzie A. E., Caltabiano G., Kent T. C., Jenkins L., McCallum J. E., Hudson B. D., Nicklin S. A., Fawcett L., Markwick R., Charlton S. J., Milligan G. (2014) The antiallergic mast cell stabilizers lodoxamide and bufrolin as the first high and equipotent agonists of human and rat GPR35. Mol. Pharmacol. 85, 91–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jenkins L., Brea J., Smith N. J., Hudson B. D., Reilly G., Bryant N. J., Castro M., Loza M. I., Milligan G. (2010) Identification of novel species-selective agonists of the G protein-coupled receptor GPR35 that promote recruitment of β-arrestin-2 and activate Gα13. Biochem. J. 432, 451–459 [DOI] [PubMed] [Google Scholar]