

Background: Insufficient expression of survival motor neuron (SMN), a protein involved in small nuclear ribonucleoprotein (snRNP) biogenesis, causes spinal muscular atrophy (SMA).
Results: The U1A protein binds to and inhibits polyadenylation and cleavage of the SMN pre-mRNA.
Conclusion: The U1A protein is a critical regulator of SMN expression.
Significance: Understanding of the regulation of SMN expression is fundamental for developing treatments for SMA.
Keywords: mRNA, Neurodegenerative Diseases, Polyadenylation, RNA Processing, RNA-binding Proteins, SMA, SMN, Spinal Muscular Atrophy, Survival Motor Neuron, U1A
Abstract
Insufficient expression of the survival motor neuron (SMN) protein causes spinal muscular atrophy, a neurodegenerative disease characterized by loss of motor neurons. Despite the importance of maintaining adequate SMN levels, little is known about factors that control SMN expression, particularly 3′ end processing of the SMN pre-mRNA. In this study, we identify the U1A protein as a key regulator of SMN expression. U1A, a component of the U1 snRNP, is known to inhibit polyadenylation upon direct binding to mRNA. We show that U1A binds directly and with high affinity and specificity to the SMN 3′-UTR adjacent to the polyadenylation site, independent of the U1 snRNP (U1 small nuclear ribonucleoprotein). Binding of U1A inhibits polyadenylation of the SMN pre-mRNA by specifically inhibiting 3′ cleavage by the cleavage and polyadenylation specificity factor. Expression of U1A in excess of U1 snRNA causes inhibition of SMN polyadenylation and decreases SMN protein levels. This work reveals a new mechanism for regulating SMN levels and provides new insight into the roles of U1A in 3′ processing of mRNAs.
Introduction
The survival of motor neuron (SMN)2 protein, the product of the spinal muscular atrophy (SMA) disease gene, is a ubiquitously expressed protein that functions in the biogenesis of spliceosomal small nuclear ribonucleoproteins (snRNPs) (1–8). In the cytoplasm of metazoan cells, SMN binds to the seven Sm proteins (Sm B/B′, D1, D2, D3, E, F, and G) and assembles them into a heptameric ring on snRNAs (9–11). Following this Sm core assembly, the new snRNPs are imported into the nucleus and are targeted to Cajal bodies where they assemble with several additional snRNP-specific proteins (12). After further maturation, the snRNPs can then function in pre-mRNA splicing.
SMA is caused by the reduced functional expression of the SMN protein through mutation or deletion of the SMN1 gene (13). SMA is characterized by degeneration of motor neurons and subsequent atrophy of muscle (14). SMA has a broad range of clinical severity, classified as types 0–IV (15–18), and the severity of these phenotypes is tightly correlated with SMN levels in patients (19, 20). A very slight increase in SMN levels correlates with a significant lessening of severity, with milder type III patients often expressing as little as 20% more SMN than much more severe type I patients (21). Similarly, increasing SMN expression by as little as 20% in the spinal cord of mouse SMA models via delivery of scAAV9 SMN results in rescue of the phenotype (22).
Despite the obvious importance of raising SMN levels, very little is currently known about the mechanisms that regulate SMN expression in any tissue or cell line. Most of the effort to date has been in understanding the regulation of the aberrant splicing of exon 7 from the SMN2 gene, a disease modifier with a single nucleotide change that results in mis-splicing of the majority of the transcripts (23–27). Conversely, nothing is currently known about 3′ processing of the SMN pre-mRNA in the nucleus. In most mRNAs, polyadenylation is signaled by three sequences found in the 3′-UTR that interact with the basal polyadenylation machinery: an AAUAAA sequence, a CA dinucleotide at the site of 3′ cleavage and polyadenylation, and a downstream U- or GU-rich sequence (28). The AAUAAA sequence is bound by the cleavage and polyadenylation specificity factor (CPSF), a four-subunit protein complex that contains the CPSF73 endonuclease (29–31). The downstream sequence binds the cleavage stimulation factor (CstF), another multiprotein complex (32). Once both complexes are bound, additional proteins are recruited, and CPSF73 cleaves the RNA after the CA dinucleotide (30, 31), after which, poly(A) polymerase adds an adenosine tail to the cleaved 3′ end (33, 34). The SMN 3′-UTR has easily recognizable canonical CPSF and CstF binding sites but contains a UA dinucleotide instead of the canonical CA at the 3′ cleavage site. This is an inefficient site for cleavage by CPSF73, suggesting that it may be a target for regulated polyadenylation.
U1A is a dual function protein in the SMN-dependent snRNP biogenesis pathway that is known to regulate polyadenylation (35–37). U1A primarily functions as a component of the U1 snRNP. U1A, a 32-kDa RNA-binding protein, binds directly to stem-loop 2 of the U1 snRNP, where it is required for pre-mRNA splicing (38–40). U1 snRNP biogenesis is coordinated by the SMN complex, which assembles the Sm ring onto the snRNA (41–43). Changes in SMN levels, as seen in SMA, cause defects in U1 snRNP assembly and alter both the amounts of U1 snRNA and presumably the amount of U1A associated with the U1 snRNP (7, 44–46). When U1A is not associated with the U1 snRNP, it functions as a modulator of polyadenylation (35–37). snRNP-free U1A binds to tandem sites in its own mRNA, called the polyadenylation inhibition element (PIE) (35–37). Binding of U1A to the PIE inhibits polyadenylation of its own message and serves as part of a feedback mechanism to decrease U1A until it reaches proper levels.
Here, we undertake a study of the SMN 3′-UTR to identify regulatory factors that control 3′ processing of the SMN transcript. We find that U1A binds directly to sequences flanking the polyadenylation site in the 3′-UTR of the SMN pre-mRNA. In addition to being a component of the U1 snRNP, U1A is also known to bind to several mRNAs and regulates their polyadenylation (35, 47, 48). We show here that binding of U1A inhibits polyadenylation of the SMN pre-mRNA by specifically blocking 3′ cleavage of the transcript by the CPSF73 endonuclease. Further, increasing the snRNP-free levels of U1A causes a corresponding decrease in SMN protein levels. This work reveals a new mechanism regulating SMN expression and allows future investigation of this pathway in SMA.
EXPERIMENTAL PROCEDURES
Biotinylated RNA Pulldowns
pUC19 plasmids containing T7 promoters and SMN RNA sequences were linearized with BsaI and in vitro transcribed for 2 h at 37 °C in the presence of biotin-16-UTP. HeLa total extract was prepared by resuspending HeLa cell pellets in lysis buffer (40 mm Tris, pH 7.9, 100 mm KCl, 0.2 mm EDTA, 1 mm DTT, 10% glycerol). The lysate was sonicated three times for 10 s and centrifuged at maximum speed for 20 min. The supernatant was then precleared with streptavidin beads (Pierce) for 20 min at 4 °C with rotation. Ten pmol of in vitro transcribed RNA was then incubated with HeLa extract in a 25-μl reaction containing RNaseOUT (Invitrogen) and 5 μg of tRNA (Invitrogen) for 30 min at 30 °C to allow binding to occur. Afterward, prewashed streptavidin beads were added to the reactions and were tumbled at 4 °C for 1 h to capture the RNP complexes on the streptavidin beads. Beads were washed extensively with 1× RSB-150 (10 mm Tris, pH 7.4, 150 mm NaCl, 2.5 mm MgCl2) with 0.1% Nonidet P-40 and eluted into SDS-PAGE loading buffer. Samples were then run out on 4–12% NuPAGE (Invitrogen) gels and stained with Coomassie Blue or silver stain or probed with antibodies by Western. Westerns were imaged on the Li-Cor Odyssey and quantitated using the Li-Cor Image Studio 2.0 software.
MS2-MBP Pulldowns
MS2-MBP fusion protein in pMALc (49) was expressed in Escherichia coli and purified on an amylose column according to the manufacturer's specifications (New England BioLabs) with an added heparin purification step as described in Ref. 50 to remove any E. coli nucleic acids bound to the MS2 coat protein. A construct containing the SMN 3′-UTR was cloned into the 3′ end of the luciferase gene in the pmirGLO vector at PmeI (Promega). Six tandem MS2 stem-loops were cloned out of pSL-MS2–6X (AddGene 27118) by PCR and inserted into the PmeI site between the 3′ end of the luciferase coding sequence and the beginning of the 192 nucleotide SMN 3′-UTR fragment. HeLa cells were transfected with the SMN 3′-UTR construct along with a pcDNA3 plasmid containing the coding region of the U1A gene using Lipofectamine 2000 (Invitrogen). Mock transfected cells served as the negative control. The cells were incubated for 24 h before harvesting. Cells were lysed with 1× RSB-100 buffer (10 mm Tris, pH 7.4, 100 mm NaCl, 2.5 mm MgCl2) with 0.01% Nonidet P-40, protease inhibitors (Roche Applied Science) and RNaseOUT (Invitrogen). Cells were sonicated three times for 10 s on ice and then centrifuged at maximum speed for 10 min at 4 °C. Supernatant was cleared by filtering through a 0.22-μm filter and quantitated by Bradford (Bio-Rad). 8 μl of amylose beads (New England BioLabs) per reaction were washed three times with 1× PBS with 0.1% Nonidet P-40 and three times with 1× RSB-100 with 0.1% Nonidet P-40. 20 μg of MS2-MBP fusion protein per reaction was added to the washed beads and allowed to bind for 1 h before washing again with 1× RSB-100 with 0.1% Nonidet P-40. 5–8 mg of HeLa lysate was added to the immobilized MS2-MBP beads and rotated for 1 h at 4 °C. The beads were then washed extensively with 1× RSB-100 with 0.01% Nonidet P-40 before resuspending the beads in SDS-PAGE loading buffer. Samples were heated and loaded onto 4–12% NuPAGE gels (Invitrogen). Western blots were probed with antibodies against U1–70K, U1C, U1A, and tubulin.
Antibodies
The antibodies used were mouse monoclonal anti-Gemin5 (Millipore), mouse monoclonal anti-SNRPA (Santa Cruz), rabbit polyclonal anti-HuR (Abcam), mouse monoclonal anti-β-tubulin (Sigma), rabbit polyclonal anti-CstF64 (Bethyl Laboratories), rabbit polyclonal anti-DHX9 (Abcam), mouse monoclonal anti-hnRNP U (Abcam), rabbit polyclonal anti-hnRNP D (Millipore), mouse monoclonal anti-U1–70K (Millipore), rabbit polyclonal U1C (Bethyl Laboratories), rabbit polyclonal anti-FUS (Bethyl Laboratories), and mouse monoclonal anti-FLAG (Sigma). Li-Cor secondary antibodies used were donkey anti-rabbit IRDye 700DX and goat anti-mouse IRDye 800 (Rockland).
Mass Spectrometry
Mass spectrometry was performed on streptavidin pulldowns as above except that 20 individual pulldowns were pooled together and precipitated with TCA before loading on an SDS-PAGE gel. Bands were excised from Coomassie-stained gels (Bio-Rad Coomassie stain) and submitted for mass spectrometry analysis at the Fred Hutchinson Cancer Research Center Proteomics Facility. The samples were run on the Orbitrap Elite (ThermoFisher), and data were searched against the IPI Human 3.87 database using X!Tandem. The results were filtered to include only proteins with Peptide Prophet scores of less than or equal to 5% (5% error rate).
EMSA
RNA was in vitro transcribed and radiolabeled with [32P]UTP. 10,000 cpm RNA was incubated with varying concentrations of recombinant U1A protein at room temperature for 1 h in the presence of binding buffer (25 mm Tris, pH 7.4, 60 mm NaCl, 5 mm DTT, RNaseOUT, 1 mm MgCl2, 0.05% Triton X-100, 50 ng/μl tRNA, 10% glycerol). NaCl concentration, MgCl2 concentration, and SMN RNA concentrations were varied, and affinity was constant with time, verifying equilibrium conditions. The reaction was then loaded directly onto 6% native polyacrylamide gels and exposed to autoradiography film or imaged with a Phosphor screen (GE Healthcare) and quantitated on the Storm 860 (GE Healthcare) using ImageQuant 5.2 software. The data were graphed using SigmaPlot software.
In Vitro Cleavage and Polyadenylation Assays
Cleavage and polyadenylation were performed as described in Refs. 36 and 51. Briefly, in vitro cleavage was performed in 20-μl reactions containing 1 μg of tRNA, 20 mm creatine phosphate, 1 mm MgCl2, 80 units of RNAseOUT (Invitrogen), 2 mm DTT, 1 mm ATP, 0.8 mm cordycepin, 2% polyvinyl alcohol 50,000 cpm RNA, 7 μl of HeLa nuclear extract (6–8 mg/ml), and varying concentrations of recombinant U1A protein. Reactions were incubated at 30 °C for 1 h. Reactions were then stopped by the addition of proteinase K (Invitrogen) and incubated at 65 °C for 20 min, after which time the samples were extracted with TRIzol reagent (Invitrogen) and precipitated with isopropanol. The reactions were resuspended and loaded onto 6% polyacrylamide denaturing urea gels and exposed to autoradiography film. The specific polyadenylation reaction proceeded much the same way except that cordycepin 5′-triphosphate was omitted.
Recombinant Proteins
The coding sequence of U1A was cloned into pET28b(+) (Novagen) yielding a C-His-tagged U1A construct. His-tagged recombinant U1A was overexpressed in BL21 Star (DE3) cells (Invitrogen) at 30 °C. Cells were pelleted and resuspended in lysis buffer (25 mm Tris, pH 8, 300 mm NaCl, 10 mm imidazole, 0.01% Nonidet P-40, and protease inhibitors). Cells were sonicated six times for 10 s, and then the lysate was pelleted. The supernatant was loaded onto a HisTrap HP column (GE Healthcare), washed, and eluted with an imidazole gradient using the NGC Quest Chromatography system (Bio-Rad). The His tag was removed by incubation with AcTEV protease (Invitrogen).
Dual-Luciferase Assay
SMN 3′-UTR sequences were cloned into the pmirGLO (Promega) vector 3′ of the luciferase gene in between the PmeI and XbaI sites. The vector backbone was modified to remove the bovine growth hormone polyadenylation signal sequence tract. The plasmid also contains the Renilla gene as a transfection control, whereas efficiency was monitored with GFP. Plasmids were transfected into mouse 3T3 or HeLa cells with or without a U1A pcDNA3 expression plasmid using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Cells were incubated for 24 h before harvesting and assaying for luciferase activity using the Dual-Luciferase reporter assay (Promega) according to the manufacturer's protocol. The reporter assay data were collected using the Tecan Infinite F200.
Real Time PCR
Cells expressing U1A and control cells were harvested with TRIzol (Invitrogen), and the RNA was extracted. cDNA was made from the RNA using SMART Moloney murine leukemia virus reverse transcriptase (Clontech) according to the manufacturer's protocol. SMN and U1 snRNA RNA levels were quantitated using primers: SMN forward, ACGGTTGCATTTACCCAGCTA; SMN reverse, CAGATTTTGCTCCTCTCTATTTCCA; U1 forward, CCATGATCACGAAGGTGGTTT; and U1 reverse, ATGCAGTCGAGTTTCCCACAT. PCRs were set up using SYBR Green PCR Master Mix (Applied Biosystems) according to the manufacturer's protocol. PCR conditions were 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The real time PCR was performed on the Applied Biosystems 7300 and quantitated using the comparative Ct method. Samples were normalized to 18 S rRNA. Primers were 18 S forward (CCTTTAACGAGGATCCATTGGA) and 18 S reverse (CGAGCTTTTTAACTGCAGCAACT).
Statistics
All data are presented as the means ± the standard error from at least three independent experiments. Statistical significance between two groups was determined by using the two-tailed Student's t test. Resulting p values less than 0.05 were considered significant.
RESULTS
Identification of Proteins That Bind the Polyadenylation Site of the SMN 3′-UTR
To identify proteins that function in the polyadenylation of the SMN transcript, mass spectrometry was performed on biotinylated RNA/streptavidin pulldown samples. We designed a 192-nucleotide construct spanning the sequences ∼93 nucleotides upstream of the CPSF binding site to 74 nucleotides downstream of the cleavage site. This fragment contains the polyadenylation site and all nearby polyadenylation signal sequences and binding sites for the general polyadenylation machinery. As a control, we also generated a fragment of RNA that lacks all the polyadenylation signal sequences. In this way, we could verify that our pre-mRNA fragment is assembling the polyadenylation complex. The biotinylated RNA constructs were incubated with HeLa extract, and associated proteins were isolated using streptavidin beads. HeLa total extract was used so we could also potentially identify proteins involved in regulated RNA decay. The proteins were separated by SDS-PAGE, and a segment of the gel representing the full lane (molecular mass, 15–250 kDa) was submitted for mass spectrometry analysis and compared with a corresponding segment from the control pulldown. Table 1 is a summary of top mRNA processing-related proteins identified with Peptide Prophet scores of 5% or less that were found to bind to the SMN 3′-UTR, but not the control RNA (see supplemental Table S1 for complete data). Common contaminating proteins such as keratins and ribosomal components were discarded from our final analysis.
TABLE 1.
Summary of SMN 3′-UTR interacting proteins identified by mass spectrometry
The table shows a summary of selected SMN 3′UTR- interacting proteins that were identified by mass spectrometry and potentially function in pre-mRNA processing. Shown are the gene names of proteins identified with Peptide Prophet scores of less than or equal to 5%. Contaminating proteins identified in a control streptavidin pulldown were subtracted. Bold text is the SNRPA gene encoding the U1 snRNP-protein U1A.
| Category | Gene name |
|---|---|
| SMN complex | GEMIN5 |
| Cleavage and polyadenylation | CPSF2 |
| CPSF4 | |
| CSTF1 | |
| CSTF2 | |
| FIP1L1 | |
| NONO | |
| NUDT21 | |
| PTBP1 | |
| SNRPA | |
| Other mRNA processing proteins | CCAR2 |
| COIL | |
| CUGBP1 | |
| DDX21 | |
| DHX30 | |
| ELAVL2 | |
| FUS/TLS | |
| HNRNPC | |
| HNRNPCL1 | |
| HNRNPD | |
| HNRNPL | |
| HNRNPR | |
| HNRNPU | |
| IGF2BP2 | |
| KHDRBS1 | |
| LARP5 | |
| LSM12 | |
| LSM14A | |
| NCBP1 | |
| PABPC4 | |
| PAIP1 | |
| PRPF31 | |
| PUM1 | |
| RBM5 | |
| RBM8A | |
| RBM25 | |
| RBM39 | |
| SART1 | |
| SFPQ | |
| SFRS11 | |
| SNRNP200 | |
| SNRNPD1 | |
| SNRNPD3 | |
| SNRPE | |
| STAU1 | |
| STRAP | |
| STRBP | |
| SYF2 | |
| THRAP3 | |
| TIAL1 | |
| U2AF2 | |
| ZNF326 |
We found 52 unique proteins that are known to function in RNA processing, including several members of the general polyadenylation machinery, such as cleavage and polyadenylation factors CPSF2, CPSF4, CPSF5 (NUDT21), CstF1, and CstF2, confirming that our construct assembles polyadenylation complexes. Interestingly, we also identified SNRPA (U1A), a bifunctional protein primarily involved in pre-mRNA splicing but also shown to function in regulated polyadenylation. We additionally identified NONO (p54nrb), SFPQ (PSF), and PTBP1 (PTB), three factors known to bind snRNP-free U1A and function with it in polyadenylation (48, 52, 53). In addition to factors involved in polyadenylation, we identified Gemin5, the RNA-binding protein of the SMN complex (54). Additionally, ELAVL1 (HuR) and HNRNPD (hnRNP D/Auf1), two factors involved in regulated mRNA decay (55–58), were found to be enriched in the SMN sample. HuR has been previously shown to bind and regulate turnover of the SMN mRNA, further verifying our experimental approach (59).
We confirmed the mass spectrometry results using Western blot analysis of the proteins isolated with the SMN 3′-UTR (Fig. 1A). Gemin5, hnRNP U, HuR, CstF64, U1A, and hnRNP D (Auf1) were positively identified in our samples. Gemin5, hnRNP U, CstF64, and U1A were unique hits by mass spectrometry and are present only in the SMN pulldown sample. HuR and hnRNP D were enriched, but not unique, in the mass spectrometry results; however, both were confirmed by Western blot. DHX9 was present in both control and SMN 3′-UTR mass spectrometry samples, so it was used as a nonspecific control for Western blot, whereas tubulin was used as a negative control. FUS/TLS, although identified in the mass spectrometry results, was only very slightly enriched by Western blot.
FIGURE 1.
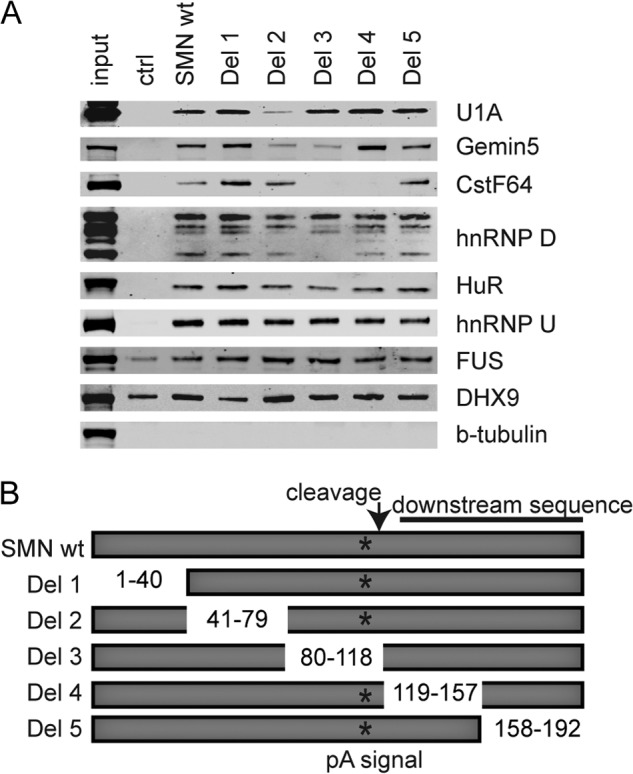
Confirmation of mass spectrometry results. A, Western blots of streptavidin pulldowns of biotinylated SMN 3′-UTR RNAs were probed with indicated antibodies. B, diagram depicting the SMN deletion constructs used in A. ctrl, control; Del, deletion.
To confirm that the binding of these proteins to the SMN 3′-UTR was indeed specific to the SMN sequence and also to delineate the binding site for these proteins, we performed streptavidin pulldowns with a series of biotinylated SMN 3′-UTR deletions. We generated a series of 35–40-nucleotide deletion mutants of the 192-nucleotide fragment of the SMN 3′-UTR (Fig. 1, A and B). We found that deletion 2 bound less U1A, indicating that a U1A binding motif resides in the region of nucleotides 41–79 of the 3′-UTR construct. Deletion 2 and deletion 3 bound less Gemin5, indicating that the sequence flanking the polyadenylation sequence may contain a binding site for Gemin5. The same was true for some isoforms of hnRNP D. CstF64 bound less to deletion 3 and deletion 4, which is expected because those two deletions remove the polyadenylation and cleavage sequences. The other proteins did not show preference for any particular region of this SMN 3′-UTR fragment and may bind indirectly or nonspecifically to the SMN 3′-UTR.
U1A Specifically Binds Sequences Flanking the Polyadenylation Site
The U1 snRNP protein, U1A, is already known to function as a regulator of polyadenylation (35–37). Further, U1 snRNP is assembled by the SMN complex (41–43). Therefore, we chose to investigate this interaction further as a likely regulatory element for SMN expression. We hypothesized that U1A functions to inhibit polyadenylation of the SMN pre-mRNA much as it does for its own pre-mRNA.
One possibility was that U1A might be associating with the SMN 3′-UTR as part of the U1 snRNP. However, we did not detect either U1–70K or U1C, the other two proteins specific to the U1 snRNP, by mass spectrometry. To confirm this, streptavidin pulldowns were performed again and Western blotted for U1–70K and U1C (Fig. 2A). We did not detect either U1–70K or U1C bound to the UTR in Western blots despite the fact that U1A is readily detectable, indicating that the U1 snRNP is not present, and U1A binds independently to the SMN 3′-UTR.
FIGURE 2.
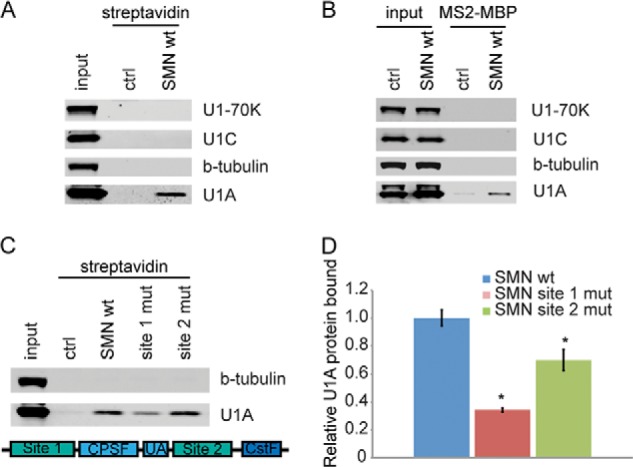
Sequence-specific binding of snRNP-free U1A to the SMN 3′-UTR. A, Western blot of streptavidin pulldowns probed with antibodies against U1 snRNP proteins. B, Western blot of MS2-MBP pulldown. C, top, Western blot of streptavidin pulldowns with mutant SMN RNA. Bottom, diagram of SMN depicting the locations of site 1 and site 2 mutations. D, quantitation of U1A in pulldowns shown in B. *, p < 0.05. ctrl, control; mut, mutation.
We also performed pulldowns of SMN 3′-UTR-containing mRNAs tagged with MS2 hairpins to confirm that the interaction occurs in cells undergoing co-transcriptional mRNA processing (Fig. 2B). HeLa cells were transfected with a luciferase reporter containing the MS2 hairpin-tagged SMN 3′-UTR attached to an intronless luciferase gene. The MS2 hairpin-SMN construct was co-transfected with the U1A cDNA. The cells were incubated for 24 h before isolating MS2-hairpin mRNP complexes with immobilized MS2-MBP fusion protein on amylose beads. Western blots of the proteins bound to the MS2-reporter constructs showed that U1A protein binds to the SMN 3′-UTR construct and is not found in the mock transfected control. Furthermore, U1–70K and U1C, components of the U1 snRNP, do not associate with the SMN 3′-UTR. Tubulin was used as the negative control that does not bind to the control or to the SMN reporter. This result shows that U1A can bind to the SMN 3′-UTR in vivo as well as in vitro and is independent of the U1 snRNP.
U1A contains two RRM domains, one of which binds directly to mRNAs and the U1 snRNA by recognition of an AUUGYAY heptanucleotide consensus sequence (38, 60). U1A binds to its own mRNA by recognizing a PIE element containing tandem U1A binding sites in its own 3′-UTR (35–37). We examined the sequence of the SMN 3′-UTR and identified tandem consensus U1A binding sequences flanking the polyadenylation site (Fig. 2C, bottom). The first site, 36 nucleotides upstream of the CPSF binding site, has the sequence AUUGUAC and is predicted to be a high affinity U1A site. The second site, AUUGUAU, found 17 nucleotides downstream of the cleavage site is predicted to bind U1A with lower affinity. This arrangement of combining one higher and one lower affinity sites is similar to what is found in a canonical PIE, where U1A typically dimerizes on the RNA across the two sites (61).
To verify that U1A binds to the predicted consensus U1A binding sites, we created SMN 3′-UTR constructs with mutations to each of the predicted U1A binding sequences. These RNAs were added to HeLa extract, and association with U1A was monitored by streptavidin pulldown and Western blot. We found that mutation of either of these sites reduces association of U1A with the SMN 3′-UTR in cell extract. Fig. 2C shows the Western blot for U1A, and Fig. 2D shows the quantitation. Mutation of the first site reduces U1A binding by 65.73 ± 1.5% of that of the wild type SMN sequence. Mutation of the second site also significantly reduces U1A binding to SMN 3′-UTR, but to a lesser extent than for the first site, by 30.05 ± 7.5%. This difference indicates weaker binding at this site than at the first site, as predicted. These results demonstrate that U1A specifically recognizes binding sites flanking the polyadenylation site on the SMN pre-mRNA.
U1A Directly Binds the SMN 3′-UTR
To show that U1A binds directly and with high specificity to the SMN 3′-UTR, we measured direct binding with recombinant U1A protein and in vitro transcribed SMN 3′-UTR RNA. Upon addition of increasing amounts of U1A protein, we observed binding of first one and then a second molecule of U1A to the SMN 3′-UTR (Fig. 3A). Mutation of the upstream, higher affinity site (site 1) resulted in a loss of the monomer species, although U1A eventually dimerized at higher protein concentrations on the downstream, low affinity site (site 2) (Fig. 3B). To verify that the band represented binding of U1A to site 2 and not simply low affinity binding to the mutant site 1, we also mutated the second site, causing complete loss of U1A binding in the absence of both sites (Fig. 3C). This is reminiscent of what occurs when U1A binds its own 3′-UTR (35, 61). In that case, U1A first recognizes the high affinity site (35, 61). Following binding to RNA, U1A dimerizes through extensive contacts between the N-terminal RRM domains as well as a peptide adjacent to the RRM (62, 63). Once dimerized, the RRM from the second U1A contacts the lower affinity RNA site (61–63).
FIGURE 3.
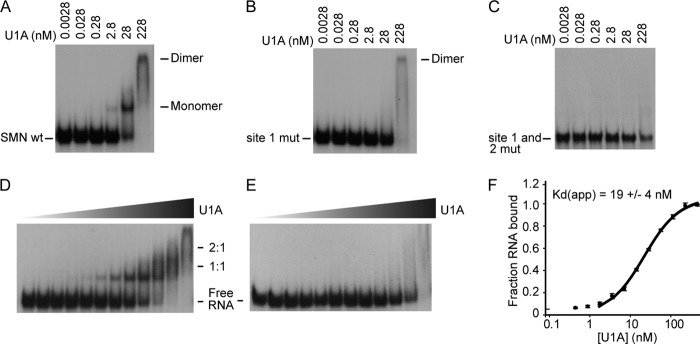
U1A binds directly to the SMN 3′-UTR with high affinity and specificity. Gel shift assays of radiolabeled SMN RNA incubated with recombinant U1A protein were performed. A, 10-fold titrations of U1A protein with SMN wild type 3′-UTR. B, 10-fold dilutions of U1A protein with SMN site 1 mutant. C, 10-fold dilutions of U1A with the SMN site 1 and site 2 mutants. D, 2-fold dilutions of U1A protein with SMN wild type 3′-UTR under equilibrium conditions. E, 2-fold dilutions of U1A protein with SMN site 1 mutant under equilibrium conditions. F, binding curve of experiment in D with Kd.
We determined the affinity of the interaction of U1A with the SMN 3′-UTR using EMSAs under equilibrium conditions. U1A binds to the SMN 3′-UTR with an apparent Kd of 19 ± 4 nm (Fig. 3, D and F) for binding of the first molecule of U1A to the RNA. Mutation of the first U1A binding site greatly reduces U1A protein binding to the SMN 3′-UTR, as shown in Fig. 3E, demonstrating high specificity of U1A for the SMN 3′-UTR. Taken together, these data show that U1A directly binds the SMN 3′-UTR with high affinity and specificity.
U1A Specifically Inhibits 3′ Processing of the SMN Pre-mRNA
Because U1A binds near the polyadenylation site of the SMN 3′-UTR, and U1A is known to function as an inhibitor of polyadenylation, we investigated the function of U1A in 3′ processing of the SMN pre-mRNA. We first performed in vitro polyadenylation assays with [32P]UTP-labeled SMN 3′-UTR constructs that contain the polyadenylation signal sequences and 3′ cleavage site. Labeled RNA was incubated with HeLa nuclear extract under conditions that favor polyadenylation and cleavage, allowing endogenous CPSF, CstF, and PAP present in the nuclear extract to cleave and polyadenylate the substrate RNA. Polyadenylation of the labeled RNAs causes the appearance of a smear of larger species on a denaturing polyacrylamide gel. Fig. 4 (A–C) shows that the addition of recombinant U1A protein inhibits the formation of poly(A) tails on the RNA. Cordycepin is included in the reaction as a control, because it is an ATP analog that will not allow 3′ extension by PAP, verifying that the signal on the gel is due to polyadenylation. U1A has been shown to bind and directly inhibit PAP (36). Recombinant U1A readily inhibited PAP in solution, so the inhibition we observed was not RNA-specific. Also, polyadenylation of the SMN transcript was particularly inefficient, possibly in part because of the UA dinucleotide at the 3′ cleavage site and/or other possible negative regulatory elements, allowing spurious polyadenylation at the 3′ end of the construct. We therefore directly examined the 3′ cleavage step that is independent of PAP and that occurs immediately prior to the poly(A) tail addition. Cleavage assays were performed much like the polyadenylation assay, except that cordycepin 3′ triphosphate was added. Under these conditions, cleavage will still occur, but poly(A) addition is blocked, allowing detection of the 118-nucleotide cleaved intermediate (Fig. 4D). Addition of U1A to the wild type SMN pre-mRNA construct in nuclear extract effectively inhibited 3′ cleavage of mRNA (43.8 ± 10.7% decrease; Fig. 4E). Mutation of either of the U1A sites (site 1 and site 2 mutations) blocked the inhibition by U1A, restoring proper cleavage of the pre-mRNA. These results demonstrate that binding of U1A to the SMN 3′-UTR specifically blocks 3′ cleavage of the SMN pre-mRNA prior to the poly(A) addition step and that both U1A binding sites are required for inhibition of cleavage.
FIGURE 4.

U1A inhibits polyadenylation and cleavage of the SMN 3′-UTR. A, polyadenylation of the U1A 3′-UTR in the presence or absence of U1A protein. p(A) indicates the polyadenosine tails seen upon polyadenylation of a substrate pre-mRNA. B, polyadenylation of the SMN 3′-UTR in the presence or absence of U1A protein. C, polyadenylation assay in the presence of increasing amounts of U1A protein. Cordycepin is shown as an inhibitor of polyadenylation. D, 3′ cleavage assay in the presence of increasing amounts of U1A protein. pA scramble indicates a mutant pre-mRNA construct with a disrupted CPSF binding site, which prohibits polyadenylation. E, cleavage of SMN 3′-UTR mutants in the presence or absence of U1A protein. FL, full length; mut, mutation.
U1A Overexpression Reduces SMN Levels in Cell Culture
Because U1A inhibits 3′ processing of the SMN pre-mRNA in vitro and in cell extract, we next tested whether U1A affects SMN expression in living cells. Under normal growth conditions, most or all of U1A is bound to the U1 snRNA as a component of the U1 snRNP (64). Regulation of polyadenylation by U1A has been observed under conditions where U1A is overexpressed relative to the U1 snRNA, resulting in a significant pool of snRNP-free U1A (35, 47, 65). Under these conditions, excess U1A binds to its own mRNA and inhibits its own expression through decreased polyadenylation as part of an autoregulatory loop to restore U1A to proper levels. We wanted to investigate whether these same conditions also resulted in down-regulation of SMN. A construct containing the U1A coding sequence under the control of the CMV promoter was transfected into HeLa cells. Twenty-four hours later, the cells were harvested and analyzed for protein and RNA expression. Fig. 5A shows a Western blot of cells transfected with either 2 or 4 μg of U1A plasmid. U1A expression increased with increasing amounts of plasmid, with transfection efficiencies of ∼75–80% for the 2-μg samples and 85–90% for the 4-μg. Transfection of 4 μg of plasmid resulted in an ∼2-fold increase in U1A protein relative to mock transfected controls, whereas U1 snRNA was only slightly increased, verifying that we had generated snRNP-free U1A (Fig. 5B). Under these conditions, we observe a dose-dependent decrease in endogenous SMN protein and mRNA expression that is inversely correlated with U1A expression (Fig. 5C). Because U1A also functions as part of the pre-mRNA splicing machinery, it was possible that overexpression of U1A could affect SMN expression by perturbing splicing of the endogenous mRNA. To eliminate this possibility, we generated luciferase reporter constructs that contain the luciferase open reading frame with either the luciferase 3′-UTR, the U1A 3′-UTR, or the SMN 3′-UTR. These constructs were transfected into mouse 3T3 cells or HeLa cells along with a construct for overexpression of human U1A. A separate cell line overexpressing FLAG-tagged U1A was used to verify that U1A could target its own endogenous expression (Fig. 5D). After 24 h, cells were assayed for U1A protein levels as well as luciferase activity. Fig. 5E show that co-transfection of U1A results in a significant decrease in luciferase from reporters tagged with either the U1A or the SMN 3′-UTRs, whereas a luciferase construct lacking a 3′-UTR was used as a negative control, and expression was not significantly different in the presence or absence of U1A overexpression.
FIGURE 5.
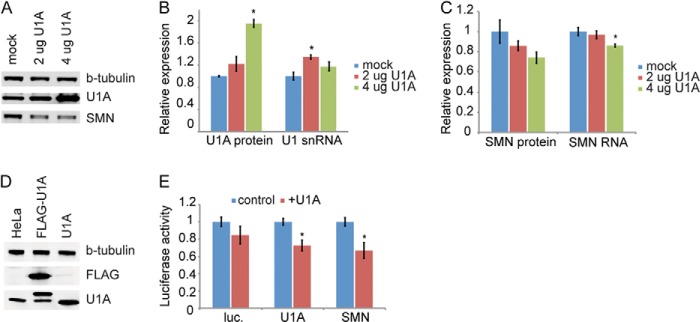
U1A overexpression decreases SMN levels. A, Western blot of HeLa cells overexpressing U1A. B, quantitation of U1A protein levels and U1 snRNA levels. C, quantitation of SMN protein and RNA levels in cells overexpressing U1A protein. D, Western blot showing overexpression of U1A and a FLAG-tagged U1A in HeLa cells. E, quantitation of luciferase protein produced with or without overexpression of U1A as measured by the Dual-Luciferase assay. *, p < 0.05.
DISCUSSION
The results described above demonstrate that U1A binds to the SMN 3′-UTR both in vitro and in vivo. Binding of U1A to the SMN 3′-UTR specifically inhibits 3′ cleavage of the mRNA, causing a decrease in polyadenylation and a corresponding decrease in SMN protein expression. This is the first example of any protein regulating SMN levels through 3′ processing of the SMN transcript.
The primary U1A binding site is immediately upstream of the CPSF binding site and 3′ cleavage site. U1A binds to this site with high affinity and specificity both in vitro and in cell extracts. We find a second potential, albeit much weaker, U1A binding site immediately downstream of the cleavage site, reminiscent of the PIE arrangement. This site is likely only bound when U1A dimerizes following recruitment through the higher affinity upstream site. Although we could not detect significant direct binding to this site independent from the first site, mutation of this second site does cause a small but significant decrease in the association of U1A with the SMN 3′-UTR in cell extracts, as well as a loss of binding in vitro when the first site has already been mutated, and the complete loss of U1A-dependent inhibition of 3′ cleavage. An alternative explanation is that U1A primarily functions through the higher affinity upstream site, either alone or in conjunction with other, as yet unidentified, proteins, and that the second site mutant is affecting the local secondary structure. Further structural and biophysical work will be necessary to determine exactly how U1A interacts with this downstream site.
In the best understood system, U1A inhibits polyadenylation of its own mRNA through dimerization on a PIE in its own 3′-UTR (35–37). In that case, the two U1A binding sites are both upstream of the polyadenylation site, and binding of U1A inhibits poly(A) tail addition by poly(A) polymerase (PAP). In the case of the SMN transcript, the higher affinity site is upstream of the cleavage and polyadenylation site, but the second site is 17 nucleotides downstream of the cleavage site. In this case, we find that U1A blocks polyadenylation by specifically inhibiting the 3′ cleavage step immediately prior to poly(A) tail addition by PAP. One explanation for this difference in mechanism may be that the SMN 3′-UTR contains a noncanonical, inefficient UA dinucleotide at the cleavage site, leaving cleavage more amenable to inhibition. Further, U1A dimerization across the cleavage site could potentially affect the geometry of the cleavage site. Indeed, U1A has been shown to inhibit both 3′ cleavage and polyadenylation of the IgM heavy chain mRNA during B cell differentiation, and, similarly to the SMN system, U1A binds to the IgM heavy chain pre-mRNA both upstream and downstream of the regulated cleavage site. U1A may be functioning on the SMN mRNA in a manner similar to the IgM heavy chain system (47, 66, 67). Identifying the molecular details of the inhibitory mechanism presents an interesting avenue for further investigation.
U1A is also a component of the U1 snRNP, which itself functions in polyadenylation. Recent work has identified a global role for the U1 snRNP in regulation of alternative polyadenylation (68), and U1 snRNP sites have been shown to work synergistically with direct U1A binding sites on mRNAs (69, 70). We did not detect U1 snRNP bound to the SMN 3′-UTR either in vitro or in vivo, nor did we identify any U1 snRNP binding sequences in the SMN 3′-UTR. Rather, in this case, U1A appears to be acting independent of the U1 snRNP.
U1A inhibits its own polyadenylation as part of a feedback mechanism. When U1A levels are too high, U1A binds to its own mRNA to inhibit its own expression until U1A levels return to normal. We now extend this feedback to SMN, the protein responsible for the biogenesis of the U1 snRNP. High levels of U1A not only signal to reduce U1A expression, but also to reduce SMN expression, and presumably U1 snRNP production, until levels of U1A are brought back to normal. This work demonstrates that regulation of expression of the basal splicing machinery is more complex and tightly controlled than previously thought.
There is much interest in understanding how SMN levels are controlled in cells. Small increases in SMN have the potential to have a significant impact on SMA pathogenesis. Increasing SMN levels to restore function in SMA has largely focused on replacement of the defective SMN1 gene, increasing transcription, and correction of aberrant splicing of the SMN2 mRNA transcript. Because low levels of SMN cause SMA, any regulatory pathway that could be exploited to restore SMN expression to improve function of motor neurons will be critical. Here, we describe a new mechanism of controlling SMN expression through 3′ processing of its transcript. Whether the repression of SMN by U1A is active in motor neurons remains unknown, although U1 snRNP levels and activity are tightly coupled to SMN expression. Reduction of SMN, as seen in SMA, causes tissue-specific defects in the expression of U1 snRNP, potentially changing the balance between snRNP-bound and snRNP-free U1A. This work allows investigation of this pathway in SMA.
Acknowledgments
We acknowledge Aalapi Patel for technical assistance; Xiaoyan Guan, Owen Branson, and Dr. Michael Freitas for assistance in preparing samples for mass spectrometry; Dr. Daniel Kiss for assistance in data analysis; and the Fred Hutchinson Cancer Research Center Proteomics Core for mass spectrometry analysis.
This work was supported, in whole or in part, by National Institutes of Health Grant R01NS077010 (to D. B.).

This article contains supplemental Table S1.
- SMN
- survival motor neuron
- SMA
- spinal muscular atrophy
- snRNP
- small nuclear ribonucleoprotein
- CPSF
- cleavage and polyadenylation factor
- CstF
- cleavage stimulation factor
- PIE
- polyadenylation inhibition element
- PAP
- poly(A) polymerase.
REFERENCES
- 1. Eggert C., Chari A., Laggerbauer B., Fischer U. (2006) Spinal muscular atrophy. The RNP connection. Trends Mol. Med. 12, 113–121 [DOI] [PubMed] [Google Scholar]
- 2. Fischer U., Englbrecht C., Chari A. (2011) Biogenesis of spliceosomal small nuclear ribonucleoproteins. Wiley Interdiscip. Rev. RNA 2, 718–731 [DOI] [PubMed] [Google Scholar]
- 3. Liu Q., Fischer U., Wang F., Dreyfuss G. (1997) The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell 90, 1013–1021 [DOI] [PubMed] [Google Scholar]
- 4. Meister G., Eggert C., Fischer U. (2002) SMN-mediated assembly of RNPs. A complex story. Trends Cell Biol. 12, 472–478 [DOI] [PubMed] [Google Scholar]
- 5. Paushkin S., Gubitz A. K., Massenet S., Dreyfuss G. (2002) The SMN complex, an assemblyosome of ribonucleoproteins. Curr. Opin. Cell Biol. 14, 305–312 [DOI] [PubMed] [Google Scholar]
- 6. Simic G. (2008) Pathogenesis of proximal autosomal recessive spinal muscular atrophy. Acta Neuropathol. 116, 223–234 [DOI] [PubMed] [Google Scholar]
- 7. Wan L., Battle D. J., Yong J., Gubitz A. K., Kolb S. J., Wang J., Dreyfuss G. (2005) The survival of motor neurons protein determines the capacity for snRNP assembly. Biochemical deficiency in spinal muscular atrophy. Mol. Cell. Biol. 25, 5543–5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Workman E., Kolb S. J., Battle D. J. (2012) Spliceosomal small nuclear ribonucleoprotein biogenesis defects and motor neuron selectivity in spinal muscular atrophy. Brain Res. 1462, 93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Achsel T., Stark H., Lührmann R. (2001) The Sm domain is an ancient RNA-binding motif with oligo(U) specificity. Proc. Natl. Acad. Sci. U.S.A. 98, 3685–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kambach C., Walke S., Nagai K. (1999) Structure and assembly of the spliceosomal small nuclear ribonucleoprotein particles. Curr. Opin. Struct. Biol. 9, 222–230 [DOI] [PubMed] [Google Scholar]
- 11. Stark H., Dube P., Lührmann R., Kastner B. (2001) Arrangement of RNA and proteins in the spliceosomal U1 small nuclear ribonucleoprotein particle. Nature 409, 539–542 [DOI] [PubMed] [Google Scholar]
- 12. Hebert M. D., Szymczyk P. W., Shpargel K. B., Matera A. G. (2001) Coilin forms the bridge between Cajal bodies and SMN, the spinal muscular atrophy protein. Genes Dev. 15, 2720–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lefebvre S., Bürglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M., Le Paslier D., Frezal J., Cohen D., Weissenbach J., Munnich A., Melki J. (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165 [DOI] [PubMed] [Google Scholar]
- 14. Crawford T. O., Pardo C. A. (1996) The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 3, 97–110 [DOI] [PubMed] [Google Scholar]
- 15. Munsat T. L. (1991) Workshop Report. International SMA Collaboration. Neuromuscul. Disord. 1, 81 [Google Scholar]
- 16. Munsat T. L., Davies K. E. (1992) International SMA consortium meeting (26–28 June 1992, Bonn, Germany). Neuromuscul. Disord. 2, 423–428 [DOI] [PubMed] [Google Scholar]
- 17. Dubowitz V. (1999) Very severe spinal muscular atrophy (SMA type 0). An expanding clinical phenotype. Eur. J. Paediatr. Neurol. 3, 49–51 [DOI] [PubMed] [Google Scholar]
- 18. Zerres K., Rudnik-Schoneborn S., Forkert R., Wirth B. (1995) Genetic basis of adult-onset spinal muscular atrophy. Lancet 346, 1162. [DOI] [PubMed] [Google Scholar]
- 19. McAndrew P. E., Parsons D. W., Simard L. R., Rochette C., Ray P. N., Mendell J. R., Prior T. W., Burghes A. H. (1997) Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 60, 1411–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parsons D. W., McAndrew P. E., Iannaccone S. T., Mendell J. R., Burghes A. H., Prior T. W. (1998) Intragenic telSMN mutations. Frequency, distribution, evidence of a founder effect, and modification of the spinal muscular atrophy phenotype by cenSMN copy number. Am. J. Hum. Genet. 63, 1712–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arnold W. D., Burghes A. H. (2013) Spinal muscular atrophy. Development and implementation of potential treatments. Ann. Neurol. 74, 348–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Foust K. D., Wang X., McGovern V. L., Braun L., Bevan A. K., Haidet A. M., Le T. T., Morales P. R., Rich M. M., Burghes A. H., Kaspar B. K. (2010) Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 28, 271–274 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23. Cartegni L., Krainer A. R. (2002) Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 30, 377–384 [DOI] [PubMed] [Google Scholar]
- 24. Gennarelli M., Lucarelli M., Capon F., Pizzuti A., Merlini L., Angelini C., Novelli G., Dallapiccola B. (1995) Survival motor neuron gene transcript analysis in muscles from spinal muscular atrophy patients. Biochem. Biophys. Res. Commun. 213, 342–348 [DOI] [PubMed] [Google Scholar]
- 25. Kashima T., Manley J. L. (2003) A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 34, 460–463 [DOI] [PubMed] [Google Scholar]
- 26. Lorson C. L., Hahnen E., Androphy E. J., Wirth B. (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U.S.A. 96, 6307–6311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Monani U. R., Lorson C. L., Parsons D. W., Prior T. W., Androphy E. J., Burghes A. H., McPherson J. D. (1999) A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183 [DOI] [PubMed] [Google Scholar]
- 28. Tian B., Graber J. H. (2012) Signals for pre-mRNA cleavage and polyadenylation. Wiley Interdiscip. Rev. RNA 3, 385–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jenny A., Hauri H. P., Keller W. (1994) Characterization of cleavage and polyadenylation specificity factor and cloning of its 100-kilodalton subunit. Mol. Cell. Biol. 14, 8183–8190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ryan K., Calvo O., Manley J. L. (2004) Evidence that polyadenylation factor CPSF-73 is the mRNA 3′ processing endonuclease. RNA 10, 565–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mandel C. R., Kaneko S., Zhang H., Gebauer D., Vethantham V., Manley J. L., Tong L. (2006) Polyadenylation factor CPSF-73 is the pre-mRNA 3′-end-processing endonuclease. Nature 444, 953–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takagaki Y., Manley J. L., MacDonald C. C., Wilusz J., Shenk T. (1990) A multisubunit factor, CstF, is required for polyadenylation of mammalian pre-mRNAs. Genes Dev. 4, 2112–2120 [DOI] [PubMed] [Google Scholar]
- 33. Wahle E. (1991) A novel poly(A)-binding protein acts as a specificity factor in the second phase of messenger RNA polyadenylation. Cell 66, 759–768 [DOI] [PubMed] [Google Scholar]
- 34. Wahle E. (1995) Poly(A) tail length control is caused by termination of processive synthesis. J. Biol. Chem. 270, 2800–2808 [DOI] [PubMed] [Google Scholar]
- 35. Boelens W. C., Jansen E. J., van Venrooij W. J., Stripecke R., Mattaj I. W., Gunderson S. I. (1993) The human U1 snRNP-specific U1A protein inhibits polyadenylation of its own pre-mRNA. Cell 72, 881–892 [DOI] [PubMed] [Google Scholar]
- 36. Gunderson S. I., Beyer K., Martin G., Keller W., Boelens W. C., Mattaj L. W. (1994) The human U1A snRNP protein regulates polyadenylation via a direct interaction with poly(A) polymerase. Cell 76, 531–541 [DOI] [PubMed] [Google Scholar]
- 37. Gunderson S. I., Vagner S., Polycarpou-Schwarz M., Mattaj I. W. (1997) Involvement of the carboxyl terminus of vertebrate poly(A) polymerase in U1A autoregulation and in the coupling of splicing and polyadenylation. Genes Dev. 11, 761–773 [DOI] [PubMed] [Google Scholar]
- 38. Scherly D., Boelens W., van Venrooij W. J., Dathan N. A., Hamm J., Mattaj I. W. (1989) Identification of the RNA binding segment of human U1 A protein and definition of its binding site on U1 snRNA. EMBO J. 8, 4163–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lutz-Freyermuth C., Query C. C., Keene J. D. (1990) Quantitative determination that one of two potential RNA-binding domains of the A protein component of the U1 small nuclear ribonucleoprotein complex binds with high affinity to stem-loop II of U1 RNA. Proc. Natl. Acad. Sci. U.S.A. 87, 6393–6397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lutz-Freyermuth C., Keene J. D. (1989) The U1 RNA-binding site of the U1 small nuclear ribonucleoprotein (snRNP)-associated A protein suggests a similarity with U2 snRNPs. Mol. Cell. Biol. 9, 2975–2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fischer U., Liu Q., Dreyfuss G. (1997) The SMN–SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell 90, 1023–1029 [DOI] [PubMed] [Google Scholar]
- 42. Meister G., Bühler D., Pillai R., Lottspeich F., Fischer U. (2001) A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat. Cell Biol. 3, 945–949 [DOI] [PubMed] [Google Scholar]
- 43. Pellizzoni L., Yong J., Dreyfuss G. (2002) Essential role for the SMN complex in the specificity of snRNP assembly. Science 298, 1775–1779 [DOI] [PubMed] [Google Scholar]
- 44. Gabanella F., Carissimi C., Usiello A., Pellizzoni L. (2005) The activity of the spinal muscular atrophy protein is regulated during development and cellular differentiation. Hum. Mol. Genet. 14, 3629–364216236758 [Google Scholar]
- 45. Winkler C., Eggert C., Gradl D., Meister G., Giegerich M., Wedlich D., Laggerbauer B., Fischer U. (2005) Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev. 19, 2320–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Workman E., Saieva L., Carrel T. L., Crawford T. O., Liu D., Lutz C., Beattie C. E., Pellizzoni L., Burghes A. H. (2009) A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum. Mol. Genet. 18, 2215–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Phillips C., Jung S., Gunderson S. I. (2001) Regulation of nuclear poly(A) addition controls the expression of immunoglobulin M secretory mRNA. EMBO J. 20, 6443–6452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hall-Pogar T., Liang S., Hague L. K., Lutz C. S. (2007) Specific trans-acting proteins interact with auxiliary RNA polyadenylation elements in the COX-2 3′-UTR. RNA 13, 1103–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Macías S., Bragulat M., Tardiff D. F., Vilardell J. (2008) L30 binds the nascent RPL30 transcript to repress U2 snRNP recruitment. Mol. Cell 30, 732–742 [DOI] [PubMed] [Google Scholar]
- 50. Zhou Z., Reed R. (2003) Purification of functional RNA-protein complexes using MS2-MBP. Curr. Protoc. Mol. Biol. Chapter 27, Unit 27.23 [DOI] [PubMed] [Google Scholar]
- 51. Wahle M., Keller W. (1994) 3′ Processing of mRNA. In RNA Processing: A Practical Approach (Higgins S. J., Hames B. D., ed) pp. 1–34, IRL Press at Oxford University Press, New York [Google Scholar]
- 52. Lutz C. S., Cooke C., O'Connor J. P., Kobayashi R., Alwine J. C. (1998) The snRNP-free U1A (SF-A) complex(es). Identification of the largest subunit as PSF, the polypyrimidine-tract binding protein-associated splicing factor. RNA 4, 1493–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liang S., Lutz C. S. (2006) p54nrb is a component of the snRNP-free U1A (SF-A) complex that promotes pre-mRNA cleavage during polyadenylation. RNA 12, 111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Battle D. J., Lau C. K., Wan L., Deng H., Lotti F., Dreyfuss G. (2006) The Gemin5 protein of the SMN complex identifies snRNAs. Mol. Cell 23, 273–279 [DOI] [PubMed] [Google Scholar]
- 55. Ma W. J., Cheng S., Campbell C., Wright A., Furneaux H. (1996) Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J. Biol. Chem. 271, 8144–8151 [DOI] [PubMed] [Google Scholar]
- 56. Myer V. E., Fan X. C., Steitz J. A. (1997) Identification of HuR as a protein implicated in AUUUA-mediated mRNA decay. EMBO J. 16, 2130–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang W., Wagner B. J., Ehrenman K., Schaefer A. W., DeMaria C. T., Crater D., DeHaven K., Long L., Brewer G. (1993) Purification, characterization, and cDNA cloning of an AU-rich element RNA-binding protein, AUF1. Mol. Cell. Biol. 13, 7652–7665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. DeMaria C. T., Brewer G. (1996) AUF1 binding affinity to A+U-rich elements correlates with rapid mRNA degradation. J. Biol. Chem. 271, 12179–12184 [DOI] [PubMed] [Google Scholar]
- 59. Farooq F., Balabanian S., Liu X., Holcik M., MacKenzie A. (2009) p38 mitogen-activated protein kinase stabilizes SMN mRNA through RNA binding protein HuR. Hum. Mol. Genet. 18, 4035–4045 [DOI] [PubMed] [Google Scholar]
- 60. Tsai D. E., Harper D. S., Keene J. D. (1991) U1-snRNP-A protein selects a ten nucleotide consensus sequence from a degenerate RNA pool presented in various structural contexts. Nucleic Acids Res. 19, 4931–4936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. van Gelder C. W., Gunderson S. I., Jansen E. J., Boelens W. C., Polycarpou-Schwarz M., Mattaj I. W., van Venrooij W. J. (1993) A complex secondary structure in U1A pre-mRNA that binds two molecules of U1A protein is required for regulation of polyadenylation. EMBO J. 12, 5191–5200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Klein Gunnewiek J. M., Hussein R. I., van Aarssen Y., Palacios D., de Jong R., van Venrooij W. J., Gunderson S. I. (2000) Fourteen residues of the U1 snRNP-specific U1A protein are required for homodimerization, cooperative RNA binding, and inhibition of polyadenylation. Mol. Cell. Biol. 20, 2209–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Varani L., Gunderson S. I., Mattaj I. W., Kay L. E., Neuhaus D., Varani G. (2000) The NMR structure of the 38 kDa U1A protein-PIE RNA complex reveals the basis of cooperativity in regulation of polyadenylation by human U1A protein. Nat. Struct. Biol. 7, 329–335 [DOI] [PubMed] [Google Scholar]
- 64. Milcarek C., Martincic K., Chung-Ganster L. H., Lutz C. S. (2003) The snRNP-associated U1A levels change following IL-6 stimulation of human B-cells. Mol. Immunol. 39, 809–814 [DOI] [PubMed] [Google Scholar]
- 65. Guan F., Palacios D., Hussein R. I., Gunderson S. I. (2003) Determinants within an 18-amino-acid U1A autoregulatory domain that uncouple cooperative RNA binding, inhibition of polyadenylation, and homodimerization. Mol. Cell. Biol. 23, 3163–3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Phillips C., Gunderson S. (2003) Sequences adjacent to the 5′ splice site control U1A binding upstream of the IgM heavy chain secretory poly(A) site. J. Biol. Chem. 278, 22102–22111 [DOI] [PubMed] [Google Scholar]
- 67. Phillips C., Pachikara N., Gunderson S. I. (2004) U1A inhibits cleavage at the immunoglobulin M heavy-chain secretory poly(A) site by binding between the two downstream GU-rich regions. Mol. Cell. Biol. 24, 6162–6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Berg M. G., Singh L. N., Younis I., Liu Q., Pinto A. M., Kaida D., Zhang Z., Cho S., Sherrill-Mix S., Wan L., Dreyfuss G. (2012) U1 snRNP determines mRNA length and regulates isoform expression. Cell 150, 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gunderson S. I., Polycarpou-Schwarz M., Mattaj I. W. (1998) U1 snRNP inhibits pre-mRNA polyadenylation through a direct interaction between U1 70K and poly(A) polymerase. Mol. Cell 1, 255–264 [DOI] [PubMed] [Google Scholar]
- 70. Guan F., Caratozzolo R. M., Goraczniak R., Ho E. S., Gunderson S. I. (2007) A bipartite U1 site represses U1A expression by synergizing with PIE to inhibit nuclear polyadenylation. RNA 13, 2129–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]