

Background: DDT is a novel member of the MIF cytokine family with overlapping functions.
Results: DDT is a direct HIF target gene that is expressed widely in renal carcinoma and contributes to tumorigenesis.
Conclusion: DDT has greater tumor-promoting properties than MIF and may compensate for MIF inhibition.
Significance: Efforts to inhibit MIF signaling in cancer need to target DDT as well.
Keywords: Cancer Biology, Gene Regulation, Hypoxia, Hypoxia-inducible Factor (HIF), Kidney, DDT, MIF, Clear Cell Renal Cell Carcinoma, VHL, von Hippel-Lindau
Abstract
Clear cell renal cell carcinomas (ccRCCs) are characterized by biallelic loss of the von Hippel-Lindau tumor suppressor and subsequent constitutive activation of the hypoxia-inducible factors, whose transcriptional programs dictate major phenotypic attributes of kidney tumors. We recently described a role for the macrophage migration inhibitory factor (MIF) in ccRCC as an autocrine-signaling molecule with elevated expression in tumor tissues and in the circulation of patients that has potent tumor cell survival effects. MIF is a pleiotropic cytokine implicated in a variety of diseases and cancers and is the target of both small molecule and antibody-based therapies currently in clinical trials. Recent work by others has described D-dopachrome tautomerase (DDT) as a functional homologue of MIF with a similar genomic structure and expression patterns. Thus, we sought to determine a role for DDT in renal cancer. We find that DDT expression mirrors MIF expression in ccRCC tumor sections with high correlation and that, mechanistically, DDT is a novel hypoxia-inducible gene and direct target of HIF1α and HIF2α. Functionally, DDT and MIF demonstrate a significant overlap in controlling cell survival, tumor formation, and tumor and endothelial cell migration. However, DDT inhibition consistently displayed more severe effects on most phenotypes. Accordingly, although dual inhibition of DDT and MIF demonstrated additive effects in vitro, DDT plays a dominant role in tumor growth in vivo. Together, our findings identify DDT as a functionally redundant but more potent cytokine to MIF in cancer and suggest that current attempts to inhibit MIF signaling may fail because of DDT compensation.
Introduction
Renal cell carcinoma (RCC)2 accounts for 3–5% of all malignancies in adults, and ∼70,000 new cases of renal cancers are diagnosed every year in the United States (1). The vast majority (∼75%) of renal cancers is classified as clear cell renal cell carcinoma (ccRCC) because of their vacuolated appearance on histological sections that results from dissolution of vast lipid and cholesterol deposits (2, 3). ccRCC exhibits resistance to chemotherapy and radiotherapy, and advanced ccRCC leads to death within 5 years for nearly 90% of the affected individuals because of poor responses to conventional therapies (4). Understanding the molecular mechanisms underlying the growth and progression of ccRCC is, thus, of great interest.
ccRCCs are characterized by loss of function of the tumor suppressor gene von Hippel-Lindau (VHL) (5). The VHL protein is part of an E3 ubiquitin ligase complex that is critical for targeting the α subunits of the heterodimeric hypoxia-inducible factors (HIF-1 and HIF-2) by priming them for proteasomal degradation under normoxic conditions (6). Under hypoxic conditions, the HIFα subunits are not degraded, translocate to the nucleus, dimerize with the constitutive HIF1β subunit (also known as the aryl hydrocarbon nuclear translocator or ARNT), and transactivate a myriad of genes controlling multiple cell functions, such as angiogenesis, metabolism, cell proliferation, and survival (7). In ccRCC, impaired VHL function ablates the proteolytic regulation of HIFα subunits, leading to the constitutive activation of hypoxia pathways. The central roles of HIF1 and HIF2 in numerous pathways responsible for tumorigenesis are well established, and, as a result, HIF and HIF target genes have emerged as potential therapeutic targets in a variety of cancers, including ccRCC (8, 9).
The macrophage migration inhibitory factor (MIF) is a 115-amino acid, secreted cytokine that is normally involved in inflammation but has been implicated in a number of pathologies, including autoimmunity, obesity, and cancer (10). MIF is regulated by hypoxia and is a direct transcriptional target of HIF1α (11, 12). MIF binds to its cell surface receptor CD74, but signaling additionally requires the recruitment of coreceptors such as CD44 or CXCR2 and CXCR4 (13, 14). MIF is directly associated with the growth of several types of carcinomas, and anti-MIF therapy with immunoglobulins and antisense oligonucleotides has been shown to have antitumorigenic effects (15, 16). Prior studies from our laboratory demonstrated MIF to be a protumorigenic signaling molecule that functions in an autocrine fashion to promote ccRCC tumor growth (17). We observed the expression of MIF in the vast majority of ccRCC tumor samples and found MIF to be elevated in the blood plasma of patients with renal tumors compared with healthy controls. Thus, our studies identified MIF as a potential therapeutic target in ccRCC.
A recent study demonstrated that D-dopachrome tautomerase (DDT) is a functional homologue of MIF (18). DDT shares 34% amino acid identity with MIF and is located within 0.1 kb of MIF in both mouse and human genomes. DDT has also been shown to bind to the MIF receptor CD74 (18), although extensive studies on other receptors for DDT have yet to be reported. DDT and MIF have similar structural and enzymatic properties. Both DDT and MIF show a remarkable similarity in tertiary structure, and both have tautomerase activity (18). Coleman et al. (19) reported that DDT functionally cooperates with, and compensates for, MIF in regulating the angiogenic potential of non-small cell lung carcinoma by the additive induction of CXCL8 and VEGF expression and secretion.
Although the protumorigenic function of MIF in ccRCC has recently been established by our group, the role of DDT in ccRCC remains unexplored. In this study, we sought to investigate the role of the only known structural and functional homolog of MIF in ccRCC to determine whether DDT functions cooperatively with MIF in survival signaling. Our results indicate that DDT is a protumorigenic signaling molecule that promotes renal cell carcinoma. We find that DDT expression is controlled by the VHL/HIF axis, leading to overexpression in ccRCC tumors, and that there is a functional overlap between DDT and MIF in ccRCC signaling and tumor growth and in promoting the migration of both tumor cells and vascular endothelial cells. Strikingly, the inhibition of DDT appears to have more dramatic effects than MIF inhibition. With the observation that DDT and MIF demonstrate functional redundancy, our data demonstrate that targeting approaches that can neutralize both DDT and MIF have a greater potential for benefit.
EXPERIMENTAL PROCEDURES
Reagents
rMIF was obtained from ProSpec (East Brunswick, NJ). D-luciferin was obtained from Biosynth International, Inc. (Staad, Switzerland). A stock solution of concentration 12.5 mg/ml was prepared in 1× PBS.
Cells, Cell Culture, and Constructs
The RCC4 and 786-O ccRCC cell lines were obtained from the ATCC. HUVECs were provided by Dr. Qing Wang (Lerner Research Institute, Cleveland Clinic). The ccRCC cell lines were maintained in DMEM (Thermo Scientific, Logan, UT) supplemented with 10% FBS (Invitrogen) and 50 μg/ml gentamicin (Invitrogen). HUVECs were maintained in MCDB 105 medium (Sigma-Aldrich) and supplemented with 10% FBS and bovine brain extracts (Lonza). All cell lines were maintained in a humidified incubator containing 5% CO2 at 37 °C. shRNAs were from Open Biosystems (Thermo Scientific): shDDT-1, TRCN0000178842; shDDT-2, TRCN0000377557; and shMIF, TRCN0000056818.
Immunohistochemistry
Tumor tissue microarrays comprising 38 ccRCC tumor sections were created as described previously (14). Microarrays were stained with an anti-MIF antibody (catalog no. sc-20121, Santa Cruz Biotechnology, Santa Cruz, CA) or anti-DDT antibody (catalog no. sc-86406, Santa Cruz Biotechnology) at a 1:100 dilution with no antigen retrieval and biotin/avidin amplification following standard procedures. Tumor xenograft sections were stained similarly. CD31 antibody (catalog no. sc-1506) was from Santa Cruz Biotechnology. Quantification of vessels was performed on 10 random high power fields (20×) of 2–3 tumors/group.
Colony Formation Assay
Colony survival assays of RCC4 and 786-O cells were performed after knockdown of DDT, MIF, and MIF/DDT expression with shRNA constructs versus a control construct (shGFP). 300–1000 cells/6-cm plate were plated and stained after 14 days with 0.1% crystal violet and quantified. All assays were done at least three times with individual samples in triplicate.
Cell Proliferation Assay
Cell proliferation assays of RCC4 and 786-O cells were performed after knockdown of DDT, MIF, and MIF/DDT expression with shRNA constructs versus a control construct (shGFP). Assays were performed by plating 20,000 cells in 12-well plates, trypsinizing, quantifying after 3–4 days, and diluting back to starting densities for subsequent time points. All assays were done at least three times with individual samples in triplicate.
Quantitative Real-time PCR Analysis and Chromatin Immunoprecipitation
qRT-PCR was performed using standard procedures and normalized to β-actin. Primer sequences are available upon request. ChIP was performed as described previously on RCC4VHL cells treated with 0.5% oxygen for 24 h (20). The primers used were as follows: upstream, CACTGAAAGGCCGACAGAGT and CTCTCCCATGCCTCCTCATA; HREs1 and 2, GAGACAGGGTGGGTCCACTA and CAGCAACCTGGCTTCTCATT; and HRE3, agctctgactttccgtgctc and tgaaaagttttgcccgaagt. The HIF1α antibody was from Santa Cruz Biotechnology (catalog no. sc-53546), and HIF2α was from Novus (catalog no. NB100-122)
Western Blotting
Protein lysates were made using 9 m urea, 0.075 m Tris buffer (pH 7.6), quantified with a BCA assay, and run on SDS-PAGE using standard methods. The antibodies used were anti-MIF (1:2500, catalog no. sc-20121, Santa Cruz Biotechnology), anti-DDT (1:2500, catalog no. sc-86406, Santa Cruz Biotechnology), anti-HIF1α (1:5000, catalog no. BD610958), anti-HIF2α (1:2000, catalog no. NB100-122, Novus), anti-β-actin (1:50,000), anti-GAPDH (1:25,000, catalog no. cs5174, Cell Signaling Technology), anti-phospho-ERK (1:2000, catalog no. cs9101, Cell Signaling Technology), anti-total ERK (1:10,000, catalog no. cs-9102, Cell Signaling Technology), and anti-p27 (1:20,000, catalog no. sc-1641, Cell Signaling Technology).
Wound Healing Assay
DDT-, MIF-, and DDT/MIF-depleted RCC4 cells grown to a confluent monolayer in 6-well plates were wounded horizontally and vertically with a 200-μl standard pipette tip. shGFP knockdown RCC4 cells were used as controls. The growth medium was then aspirated, and wells were washed twice with 1× PBS to remove cell debris. Six fields of cell-free wounds for each cell type were recorded at ×100 magnification immediately after the scratch and at the indicated time points. To quantify the migration of cells, the images were analyzed by TScratch software (17). The presented images are representatives of triplicate experiments with a similar outcome. In endothelial cell migration assays with rMIF, rMIF was added to the medium at a concentration of 100 ng/ml. In endothelial cell migration assays with conditioned medium, endothelial cell growth medium was conditioned overnight with DDT, MIF, DDT/MIF, and GFP knockdown RCC4 cells.
Time-lapse Video Microscopy
DDT-, MIF-, and DDT/MIF-deficient RCC4 cells were seeded in 8-well Lab-Tek chamber slides (Nalge Nunc International, Rochester, NY). GFP knockdown RCC4 cells were used as controls. Images were acquired at multiple positions within each chamber every 10 min for 24 h. Time-lapse data were analyzed with an automated two-dimensional cell tracking program in Metamorph 7.7.7.0 (MDS Analytical Technologies, Sunnyvale, CA). Assays were performed twice with individual samples in triplicate. At least 27 cells were tracked for each cell line per assay.
Mouse Tumor Assays
Six- to eight-week-old athymic BALB/c nude mice were obtained from an in-house colony. Subcutaneous mouse tumor assays were performed with DDT-depleted 786-O cells. shGFP knockdown 786-O cells were used as controls. 1 × 106 786-O cells in 100 μl of saline were injected subcutaneously in the flanks of nude mice. Tumor growth was monitored and measured twice weekly with calipers. Tumor volume was calculated by the formula V = 1/2 L × W2. Orthotopic renal tumor implantation was performed with DDT-, MIF-, and DDT/MIF-depleted luciferase-expressing 786-O cells. shGFP knockdown luciferase-expressing 786-O cells were used as controls. 1 × 105 786-O cells were injected into the subcapsular space of the left kidneys of nude mice. Nude mice were anesthetized, and a 1-cm incision was made into the left flank through which the kidney was accessed. A 5-μl mixture of 1 × 105 786-O cells and basement membrane mixture (Matrigel, BD Biosciences) was injected below the renal capsule using a Hamilton syringe and a 23-gauge needle. The needle was held in place until the Matrigel solidified, and the incisions were closed up with sutures. Tumor growth was assessed twice weekly by in vivo bioluminescence imaging using the IVISTM system (Xenogen Corp., Alameda, CA). Animals were anesthetized and injected with 200 μl of D-luciferin. Signal intensity was quantified as the sum of all detected photon counts within a region of interest using the LivingImageTM software package.
Statistical Analyses
Results are expressed as means ± S.D. Analyses were performed with GraphPad Prism 6.01. Unpaired two-tailed Student's t tests were performed to determine significance. p values ≤ 0.05 were considered significant.
RESULTS
DDT Is Highly Expressed in ccRCC Tissues
To determine the expression levels of DDT in ccRCC tumor samples, we stained multitumor tissue microarrays consisting of multiple (2–5) sections of 38 pathologically confirmed clear cell tumors with a DDT-specific antibody. The stained tumors were scored as one of four categories: negative, low, moderate, or high staining (numerically scored as 0–3). Averages scores were computed for staining of each sample. Examples of the three types of positive DDT staining are shown in Fig. 1A. The majority of the tumor samples showed positive DDT staining (36 of 38, 95%). 21% of the samples (8 of 38) demonstrated high staining, 45% demonstrated moderate staining (17 of 38), and only 5% were negative (2 of 38) (Fig. 1B). Associated “normal” sections for some tumors also existed on the array and generally displayed staining in the low range of the scale. We stained a serial tissue microarray slide with a MIF-specific antibody and scored the staining levels similarly. Similar results were observed: positive staining in 97% (37 of 38), high staining in 26% (10 of 38), moderate staining in 50% (19 of 38), and negative staining in 3% (1 of 38). Consistent with the roles as secreted factors and signaling through a receptor-mediated mechanism, DDT and MIF staining showed a prominent localization to the cell membrane (Fig. 1A). In tumors expressing high levels of DDT and MIF, both cytoplasmic and nuclear staining are also evident, suggesting potential intracellular roles as well. To assess whether DDT and MIF demonstrate a correlation in staining, the staining results were quantified (low = 1, moderate = 2, high = 3), multiple sections of the same tumors were averaged, and Spearman's correlation coefficient ρ was used to measure the association between DDT and MIF expression on the arrays. A positive correlation (R2 = 0.3686; p = 0.0001) was observed (Fig. 1C). Similar results were also observed when we queried the Oncomine database and found a correlated expression of DDT and MIF mRNA in the Yusenko and Beroukhim data sets (Fig. 1, D and 1E). Together, these results demonstrate that DDT is highly expressed in the majority of ccRCC tumors and that the expression levels of DDT correlate with MIF expression.
FIGURE 1.

DDT and MIF expression are strongly correlated in ccRCC. A, representative images showing high, moderate, and low immunohistochemical staining for DDT and MIF in serial ccRCC tumor tissues. A normal kidney control is also shown on the array. Scale bar = 25 μm. B, quantitation of staining. DDT, like MIF, is expressed in the vast majority of ccRCC tissues. Mod, moderate; Neg., negative. C, correlation between DDT staining and MIF staining on the ccRCC tissue microarray shown by Pearson correlation coefficient (R2 = 0.3686, p = 0.0001. D and E, significant correlation of DDT and MIF gene expression in two ccRCC studies in Oncomine. The axes display log2-transformed, median-centered expression values.
DDT Is a Hypoxia-regulated, HIFα Direct Target Gene
To determine the mechanism of regulation of DDT in ccRCC, we assessed the dependence of DDT transcriptional expression on VHL status and on hypoxia in vitro. The expression levels of DDT were determined by quantitative RT-PCR in the VHL-deficient ccRCC cell line RCC4 and compared with RCC4 cells that have reconstituted VHL expression. As shown in Fig. 2A, there was a significant decrease in expression of DDT upon reconstitution of VHL, suggesting that DDT, like MIF, is regulated by the VHL/HIF pathway and overexpressed because of the loss of VHL. There was more than a 4-fold decrease in MIF expression and more than a 3-fold decrease in DDT. As expected, on Western blot analyses, there was a concomitant decrease in the expression levels of HIF1α and DDT upon reconstitution of VHL in RCC4 cells and of HIF2α and DDT in the 786-O VHL-deficient ccRCC line, which do not express HIF1α (Fig. 2B). We next tested the hypoxic induction of DDT expression in RCC4VHL cells. Like the canonical HIF target GLUT1, both DDT and MIF demonstrated significant hypoxia inducibility after 24-h exposure to 0.5% oxygen. Similar to the RCC4 and RCC4VHL comparison, hypoxic treatment of RCC4VHL cells led to a 3- to 5-fold transcriptional induction of DDT and MIF (Fig. 2C). To test the effects of hypoxia in a non-ccRCC cell type, 293T cells were treated with either 2% or 0.5% O2 and compared with cells in normoxia (21% O2) on Western blot analyses for DDT expression. DDT demonstrated an oxygen-dependent increase in expression in concordance with HIF1α stabilization (Fig. 2D). Although HIF1α appears to be maximally stabilized at 2%, it is known that asparagine hydroxylation by factor inhibiting HIF (FIH) continues to antagonize HIF1α until more severe hypoxia is achieved and HIF1 activity reaches a maximum (near 0.5% O2) (21). Finally, to determine whether DDT is a HIF1α- or HIF2α-dependent gene, we employed shRNA to HIF1α and HIF2α and determined the effects on DDT expression in RCC4 cells. With shRNAs that demonstrated an efficient knockdown of HIF1α and HIF2α (Fig. 2E), we observed that the expression of DDT is regulated by both HIF1α and HIF2α. In contrast, GLUT4 proved to be more dependent on HIF1α, whereas VEGF is more dependent HIF2α, in agreement with published findings (22).
FIGURE 2.

DDT is a hypoxia-inducible HIF target gene. A, qRT-PCR of DDT and MIF expression in the presence or absence of VHL in RCC4 cells. B, Western blot analyses of HIF1α or HIF2α and DDT in the presence or absence of VHL in RCC4 cells (left panel) and 786-O cells (right panel). C, qRT-PCR of RCC4VHL cells under 21% oxygen or hypoxia (0.5% O2) for 24 h. D, Western blot analysis of HIF1α and DDT proteins in 293T cells under 21% oxygen or hypoxia (2% O2 and 0.5% O2) for 24 h. E, Western blot analysis of HIF1α or HIF2α after knockdown by shRNAs in RCC4 cells. F, qRT-PCR of GLUT4, VEGF, and DDT in RCC4VHL after HIF1α or HIF2α knockdown.
To determine whether DDT is a direct target of HIF, we then assessed whether HIF1α or HIF2α can associate with the DDT promoter under hypoxia by ChIP. As shown in Fig. 3A, the proximal DDT promoter contains at least three putative HREs (hypoxia response elements) by computational analysis (MatInspector). We designed primers to an upstream region as a negative control and surrounding the three HREs (HRE1 and 2 together and HRE3). Following ChIP on RCC4VHL cells exposed to either 21 or 0.5% O2 for 24 h, we found that HIF1α- and HIF2α-specific antibodies could pull down protein-DNA complexes that include HRE3 but not HRE1 and 2 (Fig. 3, B–D). Hypoxia induced a 2.5-fold enrichment of the HRE3 region of the DDT promoter compared with normoxia and to control IgG pull-down for both HIF1α of HIF2α. Collectively, the results imply that DDT is regulated by the VHL/HIF pathway and is a HIF1α and HIF2α direct target gene.
FIGURE 3.
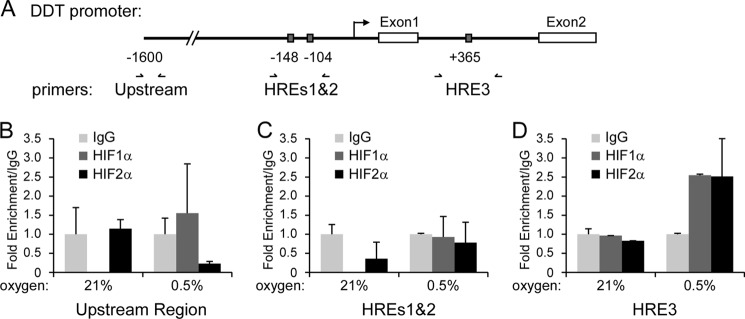
HIF1α and HIF2α bind to the DDT promoter. A, diagram of the proximal promoter region of DDT, identified putative HREs, and ChIP primers. Shown is a ChIP analysis of RCC4VHL cells exposed to 21% of 0.5% oxygen for 24 h and amplified with primers to an upstream region (B), HREs1 and 2 (C), and HRE3 (D) following pull-down with IgG, HIF1α, or HIF2α antibodies.
DDT Knockdown Reduces Cell Survival and Growth Factor Signaling in ccRCC Cells
To assess the functional significance of DDT in ccRCC, DDT expression was stably inhibited by two independent shRNAs in RCC4 cells via lentivirus-mediated transduction, and functional assays were performed. Upon knockdown, cells displayed significant decrease in DDT expression compared with shGFP control cells, as verified by Western blot analysis (Fig. 4A). When progrowth signaling was assessed, DDT-deficient cells showed decreased ERK phosphorylation and a concurrent increase in p27, the cell cycle-dependent kinase inhibitor (Fig. 4A), much like what we have found previously for MIF knockdown (17). We then assessed the growth characteristics of ccRCC cells that lack DDT expression by proliferation assays and by clonogenic survival assays. DDT knockdown reduced monolayer cell proliferation by roughly 50% (Fig. 4B) and clonogenic survival by 70–85% (C). The reduction in colonies occurred in both the number as well as the sizes of the colonies. Interestingly, the reduced cell and colony numbers occurred in the absence of induction of cell death, as measured by trypan blue staining and flow cytometry (data not shown). MIF has been linked to induction of apoptosis in a number of other systems (23, 24). However, our group has previously noted the absence of death in renal carcinoma cells upon MIF inhibition (17). Together, these data suggest that DDT promotes growth and clonogenic survival signaling of ccRCC cells.
FIGURE 4.
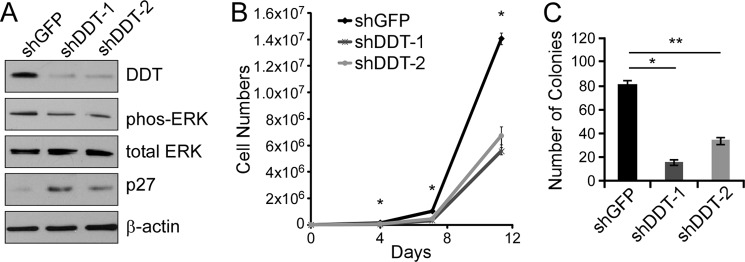
DDT knockdown impairs growth and survival of RCC4 cells in vitro. A, Western blot analysis of control (shGFP) or DDT knockdown (shDDT-1 and shDDT-2) in RCC4 cell lysates probed with DDT, phospho-ERK (phos-ERK), total ERK, p27, and β-actin antibodies. B, cell proliferation assay of control or DDT knockdown RCC4 cells. *, p < 0.00015 for both shDDTs versus shGFP at all points measured. C, colony formation assay of control or DDT knockdown RCC4 cells. *, p = 0.0002; **, p = 0.0008.
DDT Knockdown Impairs ccRCC Tumor Growth
To expand our observations to an additional ccRCC cell line and to assess the effects of DDT knockdown on tumorigenic potential, we next utilized the tumorigenic 786-O ccRCC line. 786-O shGFP or shDDT cells were produced and assessed for knockdown by qRT-PCR (Fig. 5A). The cells were then subjected to colony survival assays as before, and we observed that DDT inhibition led to a 63% diminution of clonogenicity (Fig. 5B). The cells were also injected subcutaneously into the flanks of nude mice and followed over a period of 6 weeks to monitor tumor growth. Control 786-O cells generated 1.0-cm3 tumors in just over 5 weeks and were sacrificed because of the large tumor burden. In contrast, DDT knockdown cells demonstrated a significant decrease in tumor growth rate (Fig. 5C), resulting in a 61.3% decrease in tumor volume at the study end point. Thus, in both RCC4 and 786-O cells, the DDT signaling pathway displays a significant role in tumorigenic potential.
FIGURE 5.
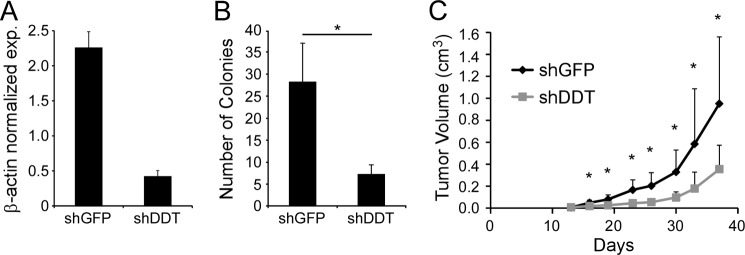
DDT knockdown impairs growth and survival of 786-O cells both in vitro and in vivo. A, qRT-PCR of DDT expression following knockdown with shDDT in 786-O cells. exp., expression. B, colony formation assay of control (shGFP) or DDT knockdown (shDDT) in 786-O cells. *, p = 0.015. C, xenograft tumor assay of 786-O cells with DDT knockdown (n = 10) and control (n = 10). *, p < 0.033 for all data points beyond day 16.
DDT/MIF Cooperate in Clonogenic Survival and Tumor Growth
Because of the structural and enzymatic similarities between DDT and MIF (18) and their roles in tumorigenic processes in ccRCC described here, we hypothesized that DDT may act in concert with MIF. To test the effect of inhibition of both DDT and MIF in ccRCC cells, we next performed double knockdown experiments in both RCC4 and 786-O cells. Verification of single and double shRNA knockdown of DDT and MIF expression in RCC4 cells was obtained by Western blot analysis (Fig. 6A) and by qRT-PCR for 786-O cells (C). We first assessed signaling in the RCC4 cells by measuring Akt and ERK phosphorylation. Both MIF inhibition and DDT inhibition individually decreased phospho-ERK expression, whereas dual inhibition of DDT and MIF showed a further significant decrease in phospho-ERK (Fig. 6A). Interestingly, Akt phosphorylation was unaffected by single knockdown of either DDT or MIF, consistent with our previous findings (17). However, dual knockdown led to significant decreases in Akt phosphorylation. Finally, we measured p27 and observed that either knockdown was sufficient to lead to dramatic increases in p27 levels. The growth characteristics of ccRCC cells were next measured by clonogenic survival. Both DDT inhibition and MIF inhibition individually decreased the clonogenicity of ccRCC cells, whereas dual inhibition of DDT and MIF showed an additional decrease in the clonogenic capacity of ccRCC cells (Fig. 6, B and D). Collectively, these results show that DDT and MIF have similar contributions in ccRCC growth and survival and that DDT and MIF may work in an additive or redundant manner in ccRCC.
FIGURE 6.

DDT and MIF additively regulate ccRCC growth and survival. A, Western blot analysis of control (shGFP/shGFP), DDT knockdown (shGFP/shDDT), MIF knockdown (shMIF/shGFP), and dual DDT/MIF knockdown (shDDT/shMIF) RCC4 cell lysates probed with MIF, DDT, phospho-ERK (p-ERK), total ERK (t-ERK), phospho-Akt (p-Akt), total Akt (t-Akt), p27, and β-actin antibodies. B, colony formation assay of RCC4 cells with DDT, MIF, or dual knockdown. *, p = 0.002; **, p = 0.005; ***, p = 0.001, ****, p = 0.0007, *****, p = 0.00001, ******, p = 0.003. C, qRT-PCR of 786-O cells with DDT, MIF, or dual knockdown. D, colony formation assay of 786-O cells with DDT, MIF, or dual knockdown. *, p = 0.001; **, p = 0.004; ***, p = 0.0007, ****, p = 0.006, *****, p = 0.0008, ******, p = 0.006. E, in vivo bioluminescence imaging of orthotopically implanted, luciferase-expressing 786-O cells with DDT, MIF, or dual knockdown. Differences between shGFP/shGFP (n = 5) and shMIF/shGFP (n = 6) are statistically significant at day 14 (p = 0.04). shGFP/shDDT and shMIF/shDDT differences from shGFP/shGFP are statistically significant at all points after day 10 (p < 0.025).
To assess the effects of dual inhibition of DDT and MIF on tumor growth, we used an orthotopic tumor model by injecting luciferase-expressing 786-O cells into the subcapsular space of the kidneys of nude mice. 105 shGFP/shGFP, shGFP/shDDT, shMIF/shGFP, or shMIF/shDDT cells were implanted, and tumor growth was monitored by bioluminescent imaging twice per week over a 6-week period. DDT and MIF knockdown cells demonstrated decreases in the tumor growth rate compared with the control cells. Interestingly, however, the DDT knockdown was significantly more effective in limiting tumor growth than the MIF knockdown, despite similar levels of knockdown (Fig. 6C). Tumors from dual DDT and MIF knockdown cells, in fact, showed no further decrease in the tumor growth rate than the DDT knockdown alone (Fig. 6E).
We next analyzed the histology of the tumors after harvest to determine whether biochemical changes observed in vitro were recapitulated in vivo. We stained serial sections for DDT, MIF, and p27 (Fig. 7). As expected, control tumors stained strongly for both DDT and MIF, whereas the knockdowns displayed little or no expression of the appropriate proteins. Likewise, as seen in vitro, knockdown of either DDT or MIF was sufficient to induce a dramatic increase in expression of p27. Together, the in vitro and in vivo results not only demonstrate that DDT and MIF have a functional overlap and cooperative roles in ccRCC tumorigenesis but also suggest a potentially greater dependence on DDT expression in vivo.
FIGURE 7.

DDT and MIF knockdown tumors display increased p27 staining and decreased vascular density in xenograft orthotopic 786-O tumors. shGFP, shDDT, shMIF, and shDDT/MIF tumors were sectioned and stained for MIF, DDT, p27, and CD31. Decreases in CD31-positive vessels (arrows) averaged from 10 high power fields are significant: shGFP versus shDDT, p = 0.0017; shGFP versus shMIF, p = 0.0003; shGFP versus shDDT/MIF, p = 5.3 × 10−9; shDDT versus shDDT/MIF, p = 0.003; and shMIF versus shDDT/MIF, p = 0.027. Representative sections are shown. Scale bar = 50 μm.
DDT and MIF Promote Tumor Cell and Endothelial Cell Migration
Increasing cell migration is a tumor growth-promoting property of MIF (10). To determine whether there is a role for DDT in tumor cell and endothelial cell migration, scratch wound healing assays were performed. Confluent RCC4 cells with shDDT, shMIF, shMIF/DDT, or shGFP were wounded and photographed over an 18-h period. After 18 h, shGFP cells had almost completely repaired the wound site, leaving only 7% of the wound open (Fig. 8, A and B). In contrast, inhibition of either DDT or MIF significantly decreased cell migration, as seen by retarded wound healing ability. MIF-deficient cells left 22% of the wound open, and DDT-deficient cells left 30% of the wound open. Dual inhibition of DDT and MIF showed the most dramatic decrease in wound healing migration, with only 50% of the wound filled (Fig. 8, A and B). Although DDT and MIF knockdown cells demonstrated decreased growth rates compared with control cells (Fig. 4B), the wound healing assays were quantified after relatively short (≤18 h) healing periods, arguing against cell growth being a significant confounding factor in decreased would healing. In fact, we calculated the proliferation rates of the cells from cell growth assays and found that all of the cells had doubling times over 45 h (shGFP/shGFP = 45 h, shGFP/shDDT = 52 h, shMIF/shGFP = 55 h, and shMIF/shDDT = 68 h).
FIGURE 8.

DDT and MIF regulate cell migration. A, wound healing assay of control (shGFP), shDDT, shMIF, and shDDT/MIF RCC4 cells. B, quantitation of the percentage of unrepaired wound in A. *, p = 0.0334; **, p = 0.0069; ***, p = 0.0063; ****, p = 0.0343. C, mean velocity (micrometers/second) of GFP-, DDT-, MIF-, and DDT/MIF-depleted cells in time-lapse video microscopy. *, p = 6.92 × 10−7; **, p = 5.34 × 10−7; ***, p = 1.57 × 10−10; ****, p = 2.52 × 10−3; *****, p = 1.52 × 10−3. D, wound healing assay of HUVECs in media with and without rMIF (100 ng/ml). E, quantitation of the percentage of unrepaired wound in D (*, p = 0.0053) and the percentage of HUVEC-unrepaired wound in the presence of conditioned media from control, shDDT, shMIF, and shDDT/MIF RCC4 cells (**, p = 0.0059; ***, p = 0.0176; ****, p = 0.0190).
To assess migration on an individual cell basis, we next performed time-lapse photography on RCC4 DDT- and MIF-depleted cells. At least 27 individual cells on subconfluent plates were photographed every 10 min over a 24-h period, and mean velocities of migration were calculated. DDT and MIF depletion individually decreased the mean velocity of RCC4 cells (37.9% and 41.9%, respectively) (Fig. 8C). Dual DDT- and MIF-depleted cells showed the most prominent decreased mean velocity (56.7%). Thus, DDT and MIF appear to promote the migration of tumor cells in vitro in an additive fashion. Thus, we conclude that both DDT and MIF promote migration in ccRCC cells.
MIF has also been implicated in promoting the migration of endothelial cells, which play a major role in ccRCC tumor growth (25). Therefore, we next investigated the role of DDT and MIF in endothelial cell migration. Recombinant human MIF (rMIF) promoted efficient HUVEC cell migration in wound healing assays compared with normal HUVEC growth medium, reducing the remaining wound after 8 h from 20% to less than 5% (Fig. 8, D and E). HUVECs incubated with conditioned medium from RCC4 cells also showed improved wound healing compared with normal medium, leaving 11% of the wound unrepaired. Media collected from MIF-deficient and DDT-deficient RCC4 cells each showed a decreased ability to induce cell migration. Conditioned medium from double knockdown cells showed a similar effect. Together, these results demonstrate that DDT functionally cooperates with MIF in promoting endothelial cell migration, whereas the lack of a combined effect of double knockdown may reveal a threshold level of MIF/DDT signaling. Thus, in the renal model, our data suggest that DDT and MIF have clear roles in both tumor cell autonomous functions as well as in programing aspects of the microenvironment.
Finally, to determine whether MIF and DDT affect endothelial cells and blood vessels in vivo, we stained the knockdown tumors described above for CD31, an endothelial cell marker. In agreement with the in vitro studies, shGFP tumors had significantly greater numbers of CD31-positive cells and vessels throughout their tumors than the shDDT and shMIF tumors (Fig. 7, bottom row). From 10 high power fields (portions of which are shown in Fig. 7), we quantified the average number of vessels and found statistically significant decreases between shGFP (16.7 ± 3.5) and shDDT (8.5 ± 4.2), shGFP and shMIF (7.7 ± 5.2), and shGFP and shDDT/shMIF (3.5 ± 2.0). Double knockdown cells also displayed statistically significantly decreases compared with the single knockdowns. It is unclear whether MIF and DDT only affect migration in this context or proliferation as well. Nonetheless, the data suggest that DDT and MIF can affect renal tumors via both autocrine and paracrine effects.
DISCUSSION
Recent work from our laboratory has established the important role of MIF in the development of ccRCC for both therapeutic and diagnostic perspectives (17). In this study, we demonstrate that DDT and MIF have significantly overlapping and/or redundant functions in promoting ccRCC. DDT and MIF have strongly correlated expression patterns in ccRCC tumor samples because of commonalities in hypoxia-dependent signaling of the two genes that, as we have now demonstrated, are both direct targets of the HIFα transcription factors. Furthermore, like MIF, DDT inhibition leads to decreased cell growth, decreased colony survival, and decreased tumor growth in xenograft tumors, although DDT appears to be a more potent molecule in promoting tumor growth. We further find that both DDT and MIF contribute to tumor cell and endothelial cell migration. Finally, an important conclusion of our study is that dual inhibition of DDT and MIF leads to the most significant phenotypes, suggesting that therapeutic strategies aimed at inhibiting MIF signaling need to be expanded to target DDT as well.
Current therapeutic strategies for ccRCC rely on restricting tumor-associated angiogenesis using anti-VEGF compounds such as monoclonal antibodies against VEGF-A (bevacizumab) or VEGFR tyrosine kinase inhibitors (sorafenib, sunitinib, etc.) (26). However, variable efficacies and tumor resistance to these treatments have emerged as major problems. Identification of novel therapeutic targets that can be used in conjunction with VEGF inhibition is an important research effort. Because HIF deregulation via VHL loss of function is an almost universal event in the development of ccRCC, elucidation of the roles of HIF targets in renal tumors that are aberrantly expressed and are “druggable” is a promising approach. DDT and MIF are attractive molecules for targeting because of the fact that they are secreted cytokines and can be inhibited extracellularly. Although immunohistochemical measurement of expression of DDT and MIF did not correlate with the tumor stage in our cohort on our tumor microarray, HIF targets are not typically markers of disease progression in ccRCC because of the fact that HIF deregulation is an early event present throughout the tumor (27), in contrast to other solid tumors where heterogeneous hypoxia drives HIF stabilization and target gene expression and correlates with a poorer outcome (28). The systemic measurement of DDT and MIF may, however, have a diagnostic or prognostic significance as a measure of tumor burden, therapeutic response, or tumor recurrences.
As proinflammatory cytokines, the roles for DDT and MIF in tumor immunology and inflammation are important considerations not described in this study. Indeed, although using human xenograft cell lines allows the investigation of DDT and MIF in human cancer lines, it excludes the opportunity to determine the roles in the immunological aspects of their functions. Recently, Simpson et al. (29) demonstrated that, in the murine breast cancer cell line 4T1, MIF expression plays a critical role in the recruitment of myeloid-derived suppressor cells in a syngeneic orthotopic tumor model, thereby promoting tumor growth by restricting immune function. In contrast, MIF depletion did not affect cell growth in vitro or tumor growth in immunocompromised animals. It is clear, then, that different tumor systems display different requirements for MIF expression. Studies in human glioblastoma, colon, prostate, and renal cancer systems (among others) have found significant phenotypes following MIF inhibition (17, 30–32). Importantly, our current studies would suggest that, in cases where MIF expression is dispensable for tumorigenesis, it is possible that DDT expression is sufficient to overcome MIF deficiency. Furthermore, in immunocompetent systems, it is possible that MIF functions are further magnified or complemented by the presence of DDT. Together, the cooperative roles of DDT and MIF in both tumor cell autonomous and in microenvironmental functions argue for dual inhibitory approaches in the treatment of renal carcinoma.
The structural and functional similarities of DDT and MIF presented here and described recently by others (18, 33, 34), in conjunction with our findings of correlated expression patterns in ccRCC tumors, suggest a possible cooperative role of the two chemokines in ccRCC. Tumor studies in mice, however, demonstrated a distinctly more significant effect of DDT inhibition than of MIF inhibition. Several possibilities exist to explain these observations. First, it is known that human cells can respond to mouse MIF (35), leading to the potential mitigation of MIF knockdown in tumor cells by systemic mouse MIF. Although likely, it is as yet unclear whether systemic mouse DDT can replace depleted human DDT in tumor cell signaling. Second, the functions of DDT and MIF may be overlapping but not fully redundant. Indeed, Merk et al. (18) demonstrated that both DDT and MIF can catalyze the tautomerization of p-hydroxyphenylpyruvate in vitro, but noted ∼10-fold less activity of DDT for the substrate, likely because of structural differences in the active sites of the molecules. In our own studies, we consistently found DDT depletion to be more detrimental to renal cancer cells than MIF depletion, although it is difficult to specifically control for absolute levels of knockdown of different genes. Thus, it is possible that DDT is functioning in additional pathways critical to tumor cell proliferation. Finally, the more dramatic effects of MIF knockdown on ccRCC cells in vitro than on tumors in vivo (which does not occur for DDT) present the possibility that the tumor microenvironment in vivo can compensate for MIF loss in a way that it cannot compensate for DDT loss. The variety of cells that interact with tumor cells, including inflammatory cells, vascular endothelial cells, stromal cells, etc., may promote tumor cell survival to compensate for MIF loss but not for DDT inhibition. Again, different functions of DDT and MIF may be revealed by our data. Further studies are clearly needed to elucidate the range of functions of DDT in cancer.
A significant amount of effort has been put into the development of inhibitory strategies to target MIF. Small molecules from a variety of compound screens, as well as structure-aided design strategies, have produced a number of compounds with anti-MIF activities (36–38). In addition, fully human monoclonal antibodies that target MIF are being developed for therapeutic purposes (39). A recent study demonstrated the effectiveness of anti-MIF antibodies in inhibiting human prostate cancer growth (32). With the novel observation that DDT and MIF demonstrate functional redundancy in ccRCC, however, a targeting approach that neutralizes both DDT and MIF has a greater potential for benefit in ccRCC with reduced chances of complications than strategies that target one factor alone. It is unclear at this point whether DDT and MIF demonstrate a significant overlap in expression in other cancers, but the likelihood is high because of the prevalence of hypoxia in almost all solid tumors. Therefore, in conclusion, our studies define a novel mechanism of HIF-dependent tumorigenesis that relies on functionally similar members of the MIF cytokine family, supporting investigations into approaches for therapeutic intervention that will inactivate both DDT and MIF signaling.
Acknowledgments
We thank the core facilities of the Case Comprehensive Cancer Center (supported by P30CA43703) (Cytometry and Microscopy Core, Small Animal Imaging Core) and the Lerner Research Institute of the Cleveland Clinic Foundation (LRI Imaging Core).
This work was supported by American Cancer Society Grant 121762-RSG-12-097-01-CCG) and by a grant from the Concern Foundation for Cancer Research.
- RCC
- renal cell carcinoma
- ccRCC
- clear cell renal cell carcinoma
- VHL
- von Hippel-Lindau
- HIF
- hypoxia-inducible factor
- MIF
- macrophage migration inhibitory factor
- DDT
- D-dopachrome tautomerase
- HUVEC
- human umbilical vein endothelial cell
- HRE
- hypoxia response element
- qRT-PCR
- quantitative real-time PCR
- rMIF
- recombinant human macrophage migration inhibitory factor.
REFERENCES
- 1. Siegel R., Naishadham D., Jemal A. (2013) Cancer statistics, 2013. CA-Cancer J. Clin. 63, 11–30 [DOI] [PubMed] [Google Scholar]
- 2. Drabkin H. A., Gemmill R. M. (2012) Cholesterol and the development of clear-cell renal carcinoma. Curr. Opin. Pharmacol. 12, 742–750 [DOI] [PubMed] [Google Scholar]
- 3. Ericsson J. L., Seljelid R., Orrenius S. (1966) Comparative light and electron microscopic observations of the cytoplasmic matrix in renal carcinomas. Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 341, 204–223 [DOI] [PubMed] [Google Scholar]
- 4. (2012) ACS Cancer Facts and Figures. Cancer Facts and Figures. American Cancer Society. Cancer Society, Atlanta, GA [Google Scholar]
- 5. Rini B. I., Campbell S. C., Escudier B. (2009) Renal cell carcinoma. Lancet 373, 1119–1132 [DOI] [PubMed] [Google Scholar]
- 6. Kaelin W. G., Jr. (2008) The von Hippel-Lindau tumour suppressor protein. O2 sensing and cancer. Nat. Rev. Cancer 8, 865–873 [DOI] [PubMed] [Google Scholar]
- 7. Semenza G. L. (2012) Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rankin E. B., Giaccia A. J. (2008) The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 15, 678–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patiar S., Harris A. L. (2006) Role of hypoxia-inducible factor-1α as a cancer therapy target. Endocr. Relat. Cancer 13, S61–75 [DOI] [PubMed] [Google Scholar]
- 10. Bach J. P., Rinn B., Meyer B., Dodel R., Bacher M. (2008) Role of MIF in inflammation and tumorigenesis. Oncology 75, 127–133 [DOI] [PubMed] [Google Scholar]
- 11. Baugh J. A., Gantier M., Li L., Byrne A., Buckley A., Donnelly S. C. (2006) Dual regulation of macrophage migration inhibitory factor (MIF) expression in hypoxia by CREB and HIF-1. Biochem. Biophys. Res. Commun. 347, 895–903 [DOI] [PubMed] [Google Scholar]
- 12. Welford S. M., Bedogni B., Gradin K., Poellinger L., Broome Powell M., Giaccia A. J. (2006) HIF1α delays premature senescence through the activation of MIF. Genes Dev. 20, 3366–3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shi X., Leng L., Wang T., Wang W., Du X., Li J., McDonald C., Chen Z., Murphy J. W., Lolis E., Noble P., Knudson W., Bucala R. (2006) CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 25, 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bernhagen J., Krohn R., Lue H., Gregory J. L., Zernecke A., Koenen R. R., Dewor M., Georgiev I., Schober A., Leng L., Kooistra T., Fingerle-Rowson G., Ghezzi P., Kleemann R., McColl S. R., Bucala R., Hickey M. J., Weber C. (2007) MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 13, 587–596 [DOI] [PubMed] [Google Scholar]
- 15. Chesney J., Metz C., Bacher M., Peng T., Meinhardt A., Bucala R. (1999) An essential role for macrophage migration inhibitory factor (MIF) in angiogenesis and the growth of a murine lymphoma. Mol. Med. 5, 181–191 [PMC free article] [PubMed] [Google Scholar]
- 16. Meyer-Siegler K. L., Leifheit E. C., Vera P. L. (2004) Inhibition of macrophage migration inhibitory factor decreases proliferation and cytokine expression in bladder cancer cells. BMC Cancer 4, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Du W., Wright B. M., Li X., Finke J., Rini B. I., Zhou M., He H., Lal P., Welford S. M. (2013) Tumor-derived macrophage migration inhibitory factor promotes an autocrine loop that enhances renal cell carcinoma. Oncogene 32, 1469–1474 [DOI] [PubMed] [Google Scholar]
- 18. Merk M., Zierow S., Leng L., Das R., Du X., Schulte W., Fan J., Lue H., Chen Y., Xiong H., Chagnon F., Bernhagen J., Lolis E., Mor G., Lesur O., Bucala R. (2011) The D-dopachrome tautomerase (DDT) gene product is a cytokine and functional homolog of macrophage migration inhibitory factor (MIF). Proc. Natl. Acad. Sci. U.S.A. 108, E577–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Coleman A. M., Rendon B. E., Zhao M., Qian M. W., Bucala R., Xin D., Mitchell R. A. (2008) Cooperative regulation of non-small cell lung carcinoma angiogenic potential by macrophage migration inhibitory factor and its homolog, D-dopachrome tautomerase. J. Immunol. 181, 2330–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krieg A. J., Rankin E. B., Chan D., Razorenova O., Fernandez S., Giaccia A. J. (2010) Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 α enhances hypoxic gene expression and tumor growth. Mol. Cell Biol. 30, 344–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lisy K., Peet D. J. (2008) Turn me on. Regulating HIF transcriptional activity. Cell Death Differ. 15, 642–649 [DOI] [PubMed] [Google Scholar]
- 22. Carroll V. A., Ashcroft M. (2006) Role of hypoxia-inducible factor (HIF)-1α versus HIF-2α in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function. Implications for targeting the HIF pathway. Cancer Res. 66, 6264–6270 [DOI] [PubMed] [Google Scholar]
- 23. Lue H., Thiele M., Franz J., Dahl E., Speckgens S., Leng L., Fingerle-Rowson G., Bucala R., Lüscher B., Bernhagen J. (2007) Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene 26, 5046–5059 [DOI] [PubMed] [Google Scholar]
- 24. Nguyen M. T., Lue H., Kleemann R., Thiele M., Tolle G., Finkelmeier D., Wagner E., Braun A., Bernhagen J. (2003) The cytokine macrophage migration inhibitory factor reduces pro-oxidative stress-induced apoptosis. J. Immunol. 170, 3337–3347 [DOI] [PubMed] [Google Scholar]
- 25. Huang D., Ding Y., Li Y., Luo W. M., Zhang Z. F., Snider J., Vandenbeldt K., Qian C. N., Teh B. T. (2010) Sunitinib acts primarily on tumor endothelium rather than tumor cells to inhibit the growth of renal cell carcinoma. Cancer Res. 70, 1053–1062 [DOI] [PubMed] [Google Scholar]
- 26. Najjar Y. G., Rini B. I. (2012) Novel agents in renal carcinoma. A reality check. Ther. Adv. Med. Oncol. 4, 183–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mandriota S. J., Turner K. J., Davies D. R., Murray P. G., Morgan N. V., Sowter H. M., Wykoff C. C., Maher E. R., Harris A. L., Ratcliffe P. J., Maxwell P. H. (2002) HIF activation identifies early lesions in VHL kidneys. Evidence for site-specific tumor suppressor function in the nephron. Cancer Cell 1, 459–468 [DOI] [PubMed] [Google Scholar]
- 28. Vaupel P., Mayer A. (2007) Hypoxia in cancer. Significance and impact on clinical outcome. Cancer Metastasis Rev. 26, 225–239 [DOI] [PubMed] [Google Scholar]
- 29. Simpson K. D., Templeton D. J., Cross J. V. (2012) Macrophage migration inhibitory factor promotes tumor growth and metastasis by inducing myeloid-derived suppressor cells in the tumor microenvironment. J. Immunol. 189, 5533–5540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baron N., Deuster O., Noelker C., Stüer C., Strik H., Schaller C., Dodel R., Meyer B., Bacher M. (2011) Role of macrophage migration inhibitory factor in primary glioblastoma multiforme cells. J. Neurosci. Res. 89, 711–717 [DOI] [PubMed] [Google Scholar]
- 31. Dessein A. F., Stechly L., Jonckheere N., Dumont P., Monté D., Leteurtre E., Truant S., Pruvot F. R., Figeac M., Hebbar M., Lecellier C. H., Lesuffleur T., Dessein R., Grard G., Dejonghe M. J., de Launoit Y., Furuichi Y., Prévost G., Porchet N., Gespach C., Huet G. (2010) Autocrine induction of invasive and metastatic phenotypes by the MIF-CXCR4 axis in drug-resistant human colon cancer cells. Cancer Res. 70, 4644–4654 [DOI] [PubMed] [Google Scholar]
- 32. Hussain F., Freissmuth M., Völkel D., Thiele M., Douillard P., Antoine G., Thurner P., Ehrlich H., Schwarz H. P., Scheiflinger F., Kerschbaumer R. J. (2013) Human anti-macrophage migration inhibitory factor antibodies inhibit growth of human prostate cancer cells in vitro and in vivo. Mol. Cancer Ther. 12, 1223–1234 [DOI] [PubMed] [Google Scholar]
- 33. Brock S. E., Rendon B. E., Yaddanapudi K., Mitchell R. A. (2012) Negative regulation of AMP-activated protein kinase (AMPK) activity by macrophage migration inhibitory factor (MIF) family members in non-small cell lung carcinomas. J. Biol. Chem. 287, 37917–37925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xin D., Rendon B. E., Zhao M., Winner M., McGhee Coleman A., Mitchell R. A. (2010) The MIF homologue D-dopachrome tautomerase promotes COX-2 expression through β-catenin-dependent and -independent mechanisms. Mol. Cancer Res. 8, 1601–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bernhagen J., Mitchell R. A., Calandra T., Voelter W., Cerami A., Bucala R. (1994) Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF). Biochemistry 33, 14144–14155 [DOI] [PubMed] [Google Scholar]
- 36. Jorgensen W. L., Gandavadi S., Du X., Hare A. A., Trofimov A., Leng L., Bucala R. (2010) Receptor agonists of macrophage migration inhibitory factor. Bioorg. Med. Chem. Lett. 20, 7033–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Al-Abed Y., Dabideen D., Aljabari B., Valster A., Messmer D., Ochani M., Tanovic M., Ochani K., Bacher M., Nicoletti F., Metz C., Pavlov V. A., Miller E. J., Tracey K. J. (2005) ISO-1 binding to the tautomerase active site of MIF inhibits its pro-inflammatory activity and increases survival in severe sepsis. J. Biol. Chem. 280, 36541–36544 [DOI] [PubMed] [Google Scholar]
- 38. Winner M., Meier J., Zierow S., Rendon B. E., Crichlow G. V., Riggs R., Bucala R., Leng L., Smith N., Lolis E., Trent J. O., Mitchell R. A. (2008) A novel, macrophage migration inhibitory factor suicide substrate inhibits motility and growth of lung cancer cells. Cancer Res. 68, 7253–7257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kerschbaumer R. J., Rieger M., Völkel D., Le Roy D., Roger T., Garbaraviciene J., Boehncke W. H., Müllberg J., Hoet R. M., Wood C. R., Antoine G., Thiele M., Savidis-Dacho H., Dockal M., Ehrlich H., Calandra T., Scheiflinger F. (2012) Neutralization of macrophage migration inhibitory factor (MIF) by fully human antibodies correlates with their specificity for the β-sheet structure of MIF. J. Biol. Chem. 287, 7446–7455 [DOI] [PMC free article] [PubMed] [Google Scholar]