

Background: Vaccinia virus encodes several anti-inflammatory proteins that interfere with host signaling pathways.
Results: The protein A46 has a similar fold to other vaccinia virus anti-inflammatory proteins but uses a different dimer interface.
Conclusion: Variations in quaternary structure can affect the specificity of interactions of anti-inflammatory proteins.
Significance: The structure illustrates how viruses can generate versatility of function from the same protein fold.
Keywords: Bcl-2 Family Proteins, Inflammation, MyD88, Protein-Protein Interactions, Viral Immunology
Abstract
Successful vaccinia virus (VACV) replication in the host requires expression of viral proteins that interfere with host immunity, such as antagonists of the activation of the proinflammatory transcription factor NF-κB. Two such VACV proteins are A46 and A52. A46 interacts with the Toll-like receptor/interleukin-1R (TIR) domain of Toll-like receptors and intracellular adaptors such as MAL (MyD88 adapter-like), TRAM (TIR domain-containing adapter-inducing interferon-β (TRIF)-related adaptor molecule), TRIF, and MyD88, whereas A52 binds to the downstream signaling components TRAF6 and IRAK2. Here, we characterize A46 biochemically, determine by microscale thermophoresis binding constants for the interaction of A46 with the TIR domains of MyD88 and MAL, and present the 2.0 Å resolution crystal structure of A46 residues 87–229. Full-length A46 behaves as a tetramer; variants lacking the N-terminal 80 residues are dimeric. Nevertheless, both bind to the Toll-like receptor domains of MAL and MyD88 with KD values in the low μm range. Like A52, A46 also shows a Bcl-2-like fold but with biologically relevant differences from that of A52. Thus, A46 uses helices α4 and α6 to dimerize, compared with the α1-α6 face used by A52 and other Bcl-2 like VACV proteins. Furthermore, the loop between A46 helices α4-α5 is flexible and shorter than in A52; there is also evidence for an intramolecular disulfide bridge between consecutive cysteine residues. We used molecular docking to propose how A46 interacts with the BB loop of the TRAM TIR domain. Comparisons of A46 and A52 exemplify how subtle changes in viral proteins with the same fold lead to crucial differences in biological activity.
Introduction
Mammalian cells respond to the presence of pathogens by activating the innate and adaptive immune systems (1). Detection of pathogens by the innate immune system is performed by molecules such as TLRs,2 retinoic acid-inducible gene 1 (RIG-1)-like helicases and NOD (nucleotide binding domain)-like receptors (2). This recognition instigates a series of intricate signaling cascades involving a plethora of protein-protein interactions that lead to the transcription of genes encoding proteins directed toward defeating the pathogen (3). In face of this host response, pathogens have evolved systems to interfere with pathogen detection and signal transduction, leading to suppression of the immune response and enhancement of pathogen replication (4, 5).
TLRs, which are present on the plasma membrane and in the endosomes, consist of an ectodomain with leucine-rich repeats for recognition of pathogen-associated molecular patterns, a single transmembrane spanning domain, and an intracellular TIR domain (6, 7). Binding of TLRs to a ligand results in the clustering of the TLR ectodomain. As a consequence, the TLR intracellular domains dimerize and provide a platform for the binding of intracellular adaptor proteins such as MyD88 (MYD88 myeloid differentiation primary response 88), MAL/TIRAP (MyD88 adapter-like/TIR domain-containing adaptor protein), TRAM (TRIF-related adaptor molecule), and TRIF (TIR-domain-containing adapter-inducing interferon-β) (8, 9); these adaptor proteins also contain a TIR domain (10). All human TLRs except TLR3 bind to the adaptor protein MyD88; TLR3 requires instead the adapter TRIF. However, certain TLRs do not interact directly with MyD88 but instead bind first an adaptor protein that in turn binds to MyD88 (6). Thus, TLR4 binds to MAL, which then recruits MyD88 (11). In addition, TLR4 can also signal independently of MyD88 via binding to the adaptor protein TRAM, which then recruits TRIF (12). Independent of whether the signaling is performed by MyD88 or TRIF, the result is the activation of TRAF6 (TNF receptor-associated factor 6), which leads to the activation of the proinflammatory transcription factors NF-κB and the mitogen-activated protein kinase (13). MyD88 interacts with members of the IRAK (interleukin-1 receptor-associated kinase) family to activate TRAF6 whereas TRIF can bind directly to TRAF6 (14, 15). In addition, TRIF can also activate interferon regulatory factors, leading to induction of inflammatory cytokines (16).
Poxviruses, a family of viruses with a large DNA genome, modulate pathogen detection and associated signaling systems to replicate successfully. Investigation of VACV, the virus employed as the vaccine to eradicate smallpox virus in the third-quarter of the twentieth century (17), has led to the identification and characterization of virally encoded proteins that interact with various components of the signal cascade in the host immune response (18). Two anti-inflammatory proteins encoded by VACV that interfere with TLR signaling are A46 and A52 (19, 20). A46 binds to TIR domain-containing proteins such as TLR4 and the adaptor proteins MAL, MyD88, TRAM, and TRAF (21). A recent report showed that A46 specifically interferes with the interaction of the adaptor proteins with TLR4 and not with the dimerization of TLR4 or with the interaction of the adaptors with each other (22). In contrast, A52 does not interact with the TIR domain-containing adaptor proteins. Instead, it acts downstream by binding to the death domain-containing protein IRAK2 and to TRAF6 (19, 23, 24).
Structurally, A52 has been shown to possess a Bcl-2-like fold (a central α-helix surrounded by five or six other α-helices; Ref. 25) even though it has no amino acid similarity to Bcl-2 or related proteins (26). A similar Bcl-2 fold has been also observed in two further VACV anti-inflammatory proteins B14 and K7 (18, 25–27) and by three anti-apoptotic VACV proteins, F1, M11, and N1 (27). The majority of these proteins form homodimers; however, the orientation of the monomers in the dimer can vary (25).
No structure is, however, at present available for the 240-amino acid anti-inflammatory protein A46. Initially, as it binds to TIR domain-containing proteins, A46 was also proposed to possess a TIR-like domain fold (19, 21). However, based on bioinformatics comparisons of A46, A52, N1, and K7, it was later predicted to have a Bcl-2-fold (26). Furthermore, biophysical measurements confirmed a high α-helical content (28), indicative of a Bcl-2 fold. A recent report provides evidence that A46 has at least two separate binding sites, one for TRAM and one for MAL or MyD88 (22), as an oligopeptide termed VIPER corresponding to the amino acids 88–98 was capable of inhibiting the binding of A46 to TRAM but not MAL or MyD88 (29). Sequence comparisons indicate that the VIPER sequence is not conserved in A52, which would explain why A52 does not bind to TRAM. In addition, the region corresponding to the VIPER sequence partially overlaps with the dimerization domain of A52 in helix α1, suggesting there may be differences in these regions between A46 and A52 (27). Last, A46 and A52 have N-terminal extensions of 80 and 55 amino acids, respectively; their functions are at present not known.
We report here the examination of the interactions of full-length and truncated variants of A46 with its cellular binding partners MyD88 and MAL as well the structure of residues 87–229 of this VACV TLR domain-binding protein.
EXPERIMENTAL PROCEDURES
Plasmids
The A46R gene sequence of the VACV Western Reserve strain (NCBI gene ID:3707702) was synthesized by GeneArt (Regensburg, Germany). For full-size A46 expression, the full-length or truncated gene was amplified with primer set 1 (Table 1) for the complete C terminus (A46-(1–240)) or primer set 2 for the fragment containing an 11-amino acid residue deletion at C terminus (A46-(1–229)). The amplified DNA was digested with BamHI and EcoRI and ligated into the plasmid pSF15 cleaved with the same enzymes. Plasmid pSF15 is derived from a plasmid from the pET series (Novagen, Darmstadt, Germany) containing the kanamycin resistance gene. It adds the MBP sequence, 10 histidine residues, and the TEV protease recognition site to the N-terminal part of a fusion protein under the control of the T7 promoter. For the construction of the N-terminally deleted form of A46-(87–229), a primer set 3 with a His6-TEV sequence encoded by the forward primer was used. The amplified insert was treated with NcoI and BamHI and cloned into the pET11d vector (Novagen).
TABLE 1.
Oligonucleotides used in this work
F, forward; R, reverse.
| Set | Residues | Oligonucleotide |
|---|---|---|
| 1 | A46-(1–240) | F, 5′-CGCAAGGGATCCCAGCAAATGGCGTTTGATATATC-3′ |
| R, 5′-CCCGAATTCTTATACATCCGTTTCCCTG-3′ | ||
| 2 | A46-(1–229) | F, 5′-CGCAAGGGATCCCAGCAAATGGCGTTTGATATATC-3′ |
| R, 5′-GCCCGAATTCTTATGTAGACGAATCATCGTC-3′ | ||
| 3 | A46-(89–229) | F, 5′-GCAAGCCATGGCACATCACCATCACCATCACGAAAACCTGTATTTTCAGGGAAATAAGTATAGTTTTAAAC-3′ |
| R, 5′-CCCGGGATCCTTATGTAGACGAATCATCGTC-3′ | ||
| 4 | TIR/MyD88 | F, 5′-CCACGGATCCATGGGCATCACCACCCTTG-3′ |
| R, 5′-CCCGGAATTCTCAGGGCAGGGACAAAGC3′ | ||
| 5 | A46-(89–229) | F, 5′-GGATATGATAAGTTGTGATTCTAG-3′ |
| C155S | R, 5′-CTAGAATCACAACTTATCATATCC-3′ | |
| 6 | A46-(89–229) | F, 5′-GTATAGGAGTATGGCATTAGCC-3′ |
| C181S | R, 5′-GGCTAATGCCATACTCCTATAC-3′ | |
| 7 | A46-(89–229) | F, 5′-CAATCTAAGTAAGTATCTACGCG-3′ |
| C205S | R, 5′-CGCGTAGATACTTACTTAGATTG-3′ | |
| 8 | Myc-A46-(1–240)-FLAG | F, 5′-GCCCGAATTCCGAGAATGGAGCAGAAACTCATCTCTGAAGAGGATCTG GCGTTTGATATATC-3′ |
| R, 5′-CCGCTCGAGTTACTTATCGTCGTCATCCTTGTAATCTACATCCGTTTCCC-3′ | ||
| 9 | A46-(87–229)-FLAG | F, 5′-GCCCGAATTCCGAGAATGGGAAATAAGTATAGTTTTAAAC-3′ |
| R, 5′-CCGCTCGAGTTACTTATCGTCGTCATCCTTGTAATCTGTAGACGAATCATCG-3′ |
For expression in mammalian cells, full-length and truncated versions of A46 protein were amplified with primer set 8 (1–240 amino acid residues) or primer set 9 (residues 87–229) from the full-length A46 plasmid synthesized by GeneArt. The resulting DNA sequences coded for the full-length A46 protein containing N-terminal myc and C-terminal FLAG tags and A46-(87–229) with C-terminal FLAG tag, respectively. The amplified DNA fragments were digested with XhoI and EcoRI restriction enzymes and ligated into the pCAGGS vector (30). PCR mutagenesis was used to insert the indicated point mutations into A46 with the primer sets 5 to 7 (see Table 1).
The nucleotide sequence of murine MyD88 protein encoding for amino acid residues 146 to 296 was obtained by PCR from cDNA isolated from RAW 264.7 mouse leukemic monocyte macrophage cells with primer set 4. The isolated DNA was digested with BamHI and EcoRI and cloned into the pGEX4T1 vector cleaved with the same enzymes (GE Healthcare). The plasmid encoding a GST fusion of the TIR domain of human MAL (amino acid residues 75–235) was a kind gift from Dr. H. Tochio (31).
Expression and Purification of Full-length A46
Escherichia coli BL21 (DE3) cells were transformed with the pSF15 plasmids carrying the A46-(1–240) or A46-(1–229) genes. Overnight precultures grown in 5 ml of LB containing kanamycin (50 mg/liter) were inoculated into 2 liters of fresh medium and grown at 37 °C until mid-log phase (A600 = 0.6). Cultures were cooled to 23 °C, and expression was induced with 0.25 mm isopropyl 1-thio-β-d-galactopyranoside. After 5 h of expression at 23 °C, cells were pelleted and resuspended in a buffer containing 20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 10 mm β-mercaptoethanol, 5% glycerol. Cells were lysed in an EmulsiFlex-C3 homogenizer (Avestin, Ottawa) and cleared by centrifugation at 18,000 rpm for 30 min. The cleared lysate was loaded onto amylose resin (New England Biolabs, Ipswich, MA) pre-equilibrated with lysis buffer. After washing with 10 column volumes of lysis buffer, recombinant TEV protease in TEV protease cleavage buffer (20 mm Tris-HCl, pH 8.0, 200 mm NaCl, 15 mm β-mercaptoethanol) was loaded on the amylose column for the overnight digestion of the fusion protein at 4 °C. After overnight incubation, the cleaved A46 protein was eluted from column with lysis buffer. Unprocessed fusion protein and MBP were subsequently eluted in buffer with 10 mm maltose. Further purification by SEC was performed on a HiLoad 26/60 Superdex 200 (GE Healthcare) gel-filtration column in a buffer containing 20 mm Tris-HCl, pH 8.5, and 10 mm DTT.
Expression and Purification of the N-terminal-deleted A46
E. coli BL21 (DE3) LysS competent cells were transformed with the pET11d vector carrying the A46-(87–229) sequence. Overnight precultures grown in 5 ml of LB containing ampicillin (100 mg/liter) and chloramphenicol (34 mg/liter) were inoculated into 2 liters of fresh antibiotic-containing media and grown at 37 °C until mid-log phase (A600 = 0.6). Cultures were cold-shocked on ice for 20 min, and protein expression was started with the addition of 0.3 mm isopropyl 1-thio-β-d-galactopyranoside and continued at 16 °C overnight. Cells were harvested by centrifugation and resuspended in 20 mm Tris-HCl, pH 8.5, 100 mm NaCl, 25 mm imidazole, 10 mm β-mercaptoethanol. For lysis, cells were freeze-thawed and sonicated on ice. The soluble fraction was clarified by centrifugation at 18,000 rpm for 30 min and subsequent filtering. For the initial purification, the lysate was loaded on the 5 ml of Ni-NTA-agarose (5 Prime), washed with 10 column volumes of lysis buffer, and eluted in a buffer containing 20 mm Tris-HCl, pH 8.5, 200 mm NaCl, 200 mm imidazole, and 10 mm β-mercaptoethanol. For His-tag removal, recombinant TEV protease was added, and the protein solution was dialyzed overnight at 4 °C against 20 mm Tris-HCl, pH 8.5, 150 mm NaCl, 25 mm imidazole, and 15 mm β-mercaptoethanol. For the removal of uncleaved protein, the protein fraction was loaded four times on a 5-ml Ni-NTA gravity column. The final purification was achieved by SEC on a HiLoad 16/60 Superdex 75 column equilibrated with 20 mm Tris-HCl, pH 8.5, and 10 mm DTT.
For the production of selenomethionine (Se-Met)-labeled A46-(87–229) protein, M9 medium containing Se-Met was prepared (Molecular Dimensions, Suffolk, UK), and expression was performed according to the manufacturer's protocol. Purification was as described for the unlabeled protein.
Expression and Purification of TIR/MyD88 and TIR/MAL
Expression and purification of the TIR domain of MyD88 (TIR/MyD88) as a GST fusion protein was performed in E. coli BL21 (DE3) as described in Ohnishi et al. (32). The TIR domain of MAL (TIR/MAL) was expressed and purified as a GST fusion as TIR/MyD88 except for the removal of the GST tag. For the GST-TIR/MyD88 construct, the tag was removed by thrombin cleavage; for GST-TIR/MAL, it was cleaved with Factor Xa. The tags were removed by SEC on a 26/60 Hiload Superdex column equilibrated with 20 mm Tris-HCl, pH 8.5, 100 mm NaCl, 5% glycerol, and 1 mm Tris(2-carboxyethyl)phosphine for TIR/MAL and 20 mm potassium phosphate, pH 6.5, 100 mm NaCl, and 10 mm DTT (32) for TIR/MyD88.
Size Exclusion by FPLC followed by Multiangle Laser Light Scattering
For the assessment of the oligomeric state and molecular weight of MBP-A46-(1–240), A46-(1–240), methylated A46-(1–229), and A46-(87–229), a Superdex 200 10/300 exclusion column, equilibrated with 20 mm Tris-HCl 8.0, 20 mm NaCl, 5% glycerol, 5 mm β-mercaptoethanol, was connected to a miniDAWN Tristar light scattering instrument (Wyatt Technologies, Santa Barbara, CA). Data analysis was performed using the manufacturer's software ASTRA.
Studying Protein-Protein Interactions by Microscale Thermophoresis
Microscale thermophoresis protein-protein interaction studies were performed on the Monolith NT.115 (Nanotemper Technologies, Munich, Germany) using fluorescently labeled proteins as described (33, 34). For the TIR/MAL and TIR/MyD88 protein labeling, the standard labeling kit for the fluorescent dye Alexa Fluor 647 from Nanotemper was used. Solutions of unlabeled A46-(1–240), A46-(1–229) C205S, A46-(87–229), A46-(87–229) C205S, TIR/MAL, TIR/MyD88 R196A/R288A were serially diluted from 150–450 μm to 8–20 nm in the presence of 30–70 nm concentrations of one of the labeled TIR/MAL, TIR/MyD88, or TIR/MyD88 R196A/R288A proteins. Measurements were performed at 25 °C in 20 mm Tris-HCl, pH 8.5, 100 mm NaCl, 5% glycerol, 1 mm Tris(2-carboxyethyl)phosphine, 1 mm EDTA, 0.05% Tween 20 using 50% LED power and 80% IR-laser power. Data analysis was performed with Nanotemper analysis software, v.1.2.101.
Protein Crystallization and Data Collection
Plate-shaped crystals of A46-(87–229)C205S were initially obtained a protein concentration of 3.5 mg/ml in 20 mm Tris-HCl, pH 8.5, and 10 mm DTT with the PACT Premier (Molecular Dimensions, Suffolk, UK) G2 solution (0.2 m NaBr, 0.1 m Bistris propane, pH 7.5, and 20% PEG 3350) using the sitting-drop vapor diffusion technique and a nanodrop-dispensing robot (Phoenix RE; Rigaku Europe, Kent, UK). The crystals were then optimized to 100 mm Bistris propane, pH 7.5, 100 mm NaBr, 18% (w/v) PEG 3350 using the hanging drop vapor diffusion method at 22 °C. Crystals with selenium methionine labeled A46-(87–229)C205S were obtained in 100 mm Bistris propane, pH 7.5, 150 mm NaBr, 18% (w/v) PEG 3350. For data collection, crystals were mounted in the loop, soaked in the reservoir solution supplemented with 20% glycerol, and flash-frozen in liquid nitrogen. The diffraction data set was collected at 100 K at the peak of selenium at λ = 0.979 Å at the beamline ID-29 at the European Synchrotron Radiation Facility (Grenoble, France) to 1.99 Å resolution, processed using the XDS package (35), and converted to mtz format using the program AIMLESS (36). Crystals belonged to the space group C2221 (a = 107.12 Å, b = 107.64 Å, c = 137.9 Å).
Structure Determination and Refinement
The structure was solved by single-wavelength anomalous diffraction. 12 possible selenium sites, which belong to four molecules of A46, were located by SHELXD (37) followed by phasing and density modification by SHELXE (37). Autobuilding was carried out using the program ARP/wARP (38) and resulted in tracing 93% of the model. The structure was refined using the program REFMAC (39) and Phenix Refine (40), and rebuilding was done using the program Coot (41). Stereochemistry and structure quality were checked using the program MolProbity (42). Data collection and refinement statistics are reported in Table 2.
TABLE 2.
Data collection and refinement statistics

SAXS Analysis
SAXS experiments were performed at 0.99 Å wavelength ESRF at BioSAXS beamline BM29 (Grenoble, France) equipped with PILATUS 1 m. The detector distance was set up at 2.867 m. The data were collected using protein concentration at 2.5 and 3.5 mg/ml and at 0.028 nm−1 < q < 4.5 nm−1 range of scattering vector. The samples were in the buffer containing 20 mm Tris-HCl, pH 8.5, 10 mm DTT, and the measurements were performed at 20 °C. The data were processed and analyzed using the ATSAS program package (43) and PRIMUS (44). The radius of gyration Rg and forward scattering I(0) were calculated by Guinier approximation. The maximum particle dimension Dmax and P(r) function were evaluated using the program GNOM (45) The analysis of SAXS data by Guinier approximation shows no concentration dependence effect, indicating the samples are homogeneous and free of aggregation. The ab initio modeling was simulated using DAMMIF software (46).
Cell Culture
Human embryonic kidney 293T (HEK293T) cells were a kind gift of Dr. G. Versteeg. Cells were maintained in Dulbecco's modified Eagle's medium containing 10% (v/v) FCS, 100 μg/ml streptomycin, and 100 units/ml penicillin.
Reporter Gene Assay
HEK293T cells were seeded (1–2 × 105 per well) in 24-well plates and transfected with plasmids carrying either full-length myc-A46-(1–240)-FLAG or truncated variant A46-(87–229)-FLAG at the indicated concentrations together with 70 ng of NF-κB-luc reporter plasmid (Firefly luciferase) and 30 ng of pRL-TK (Renilla luciferase) internal control. For transfections TransIT-LT1 transfection reagent (Mirus, Madison, WI) was used according to the manufacturer's instructions. The total amount of DNA (500 ng) was kept constant by the addition of pCAGGS empty vector. After 40 h of incubation, transfected cells were stimulated by 0.32 ng/ml of IL-1β for 6 h. Subsequently, cells were lysed in Passive Lysis Buffer (Promega, Madison), and whole cell lysates were analyzed for luciferase activity using Dual-Luciferase Reporter assay (Promega). Firefly luciferase activity was normalized by Renilla luciferase activity.
RESULTS
Functional Analysis of A46 Protein Variants
To obtain sufficient material to investigate the properties of VACV A46 protein, we first constructed a plasmid expressing A46 with an N-terminal MBP tag (MBP-A46; Fig. 1A). The linker region between the MBP tag and the A46 sequence contained an oligo-histidine tag as well as the cleavage site for the TEV protease. Cleavage with TEV proteinase should result in a protein containing a 16-amino acid extension (GHMASMTGGQQMGRGS) at the N terminus. Expression of this fusion protein led to soluble protein that could be purified by affinity chromatography on an amylose column. Subsequent incubation with TEV proteinase liberated the A46 protein, which could then be purified to homogeneity by SEC. Yields were between 0.5 and 1 mg/liter of culture. In order not to have to rely on standard proteins and an assumption of their conformation, we coupled SEC to multi-angle laser light scattering analysis to enable absolute molecular masses to be calculated (47). Such measurements revealed that the full-length A46 had an apparent molecular mass of 110 kDa (Fig. 2A), suggesting that the protein is a tetramer. In contrast, the MBP-A46 fusion protein behaved as a dimer (150 kDa) (Fig. 2A). We also examined the mobilities of the tetrameric and dimeric proteins by non-reducing gels PAGE. No difference was found to those observed in the presence of β-mercaptoethanol (data not shown). This indicates that the oligomerization of A46 does not involve cysteine residues.
FIGURE 1.
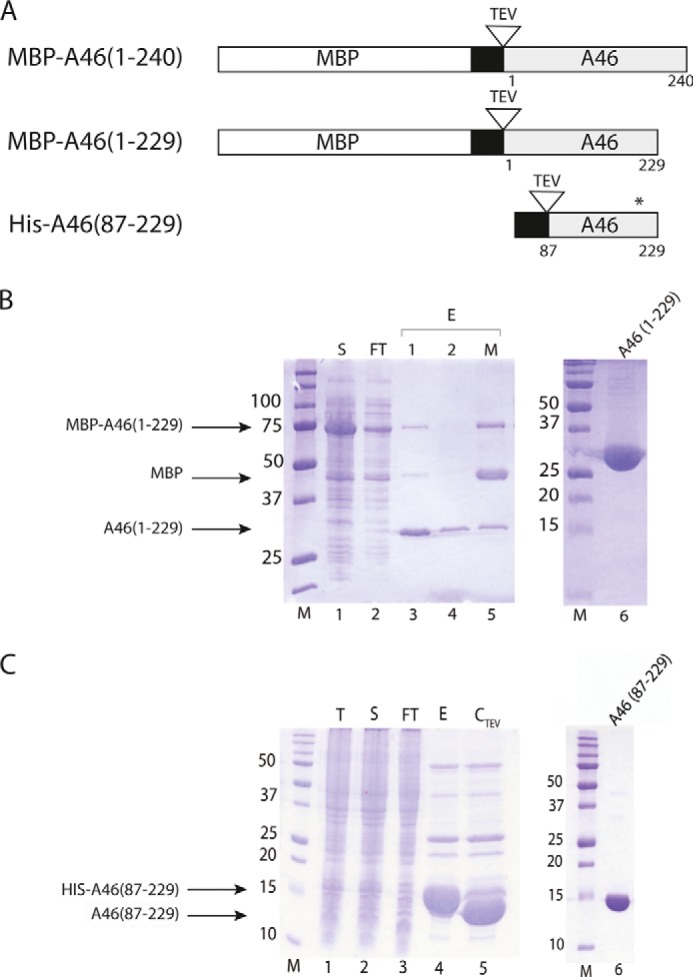
Expression and purification of A46 variants. A, schematic representation of A46 variants used in this work. The MBP tag is indicated by the open bar, the His tag is indicated by the black bar, the A46 is indicated by the gray bar, and the TEV site is indicated by an open triangle. The position of the modified Cys-205 residue is marked by an asterisk. B, SDS-PAGE analysis of the purification of MBP-A46-(1–229). Samples of total protein from each of the indicated fractions were applied to gels containing 12.5% (except for lane 6, 15%) acrylamide. Lane 1, soluble protein loaded onto amylose column (S); lane 2, amylose column flow-through (FT); lanes 3 and 4, eluate from the amylose column after TEV cleavage (E1, E2); lane 5, eluate from the amylose column after TEV cleavage in the presence of maltose. Lane 6, 15 μg of concentrated pooled fraction after SEC (note 15% acrylamide percentage). M, molecular mass markers. C, SDS-PAGE analysis on gels containing 15% acrylamide of the purification of His-A46-(87–229). Lane 1, crude cell extract (T); lane 2, total soluble protein in crude cell extract loaded on to Ni-NTA beads (S); lane 3, flow-through from Ni-NTA beads (FT); lane 4, eluate from Ni-NTA beads with imidazole (E); lane 5, eluate after incubation with TEV protease (CTEV); lane 6, 7.5 μg concentrated pooled fraction after SEC. Proteins were visualized with Coomassie Brilliant Blue R250. Numbers on the left indicate apparent molecular sizes (in kDa).
FIGURE 2.
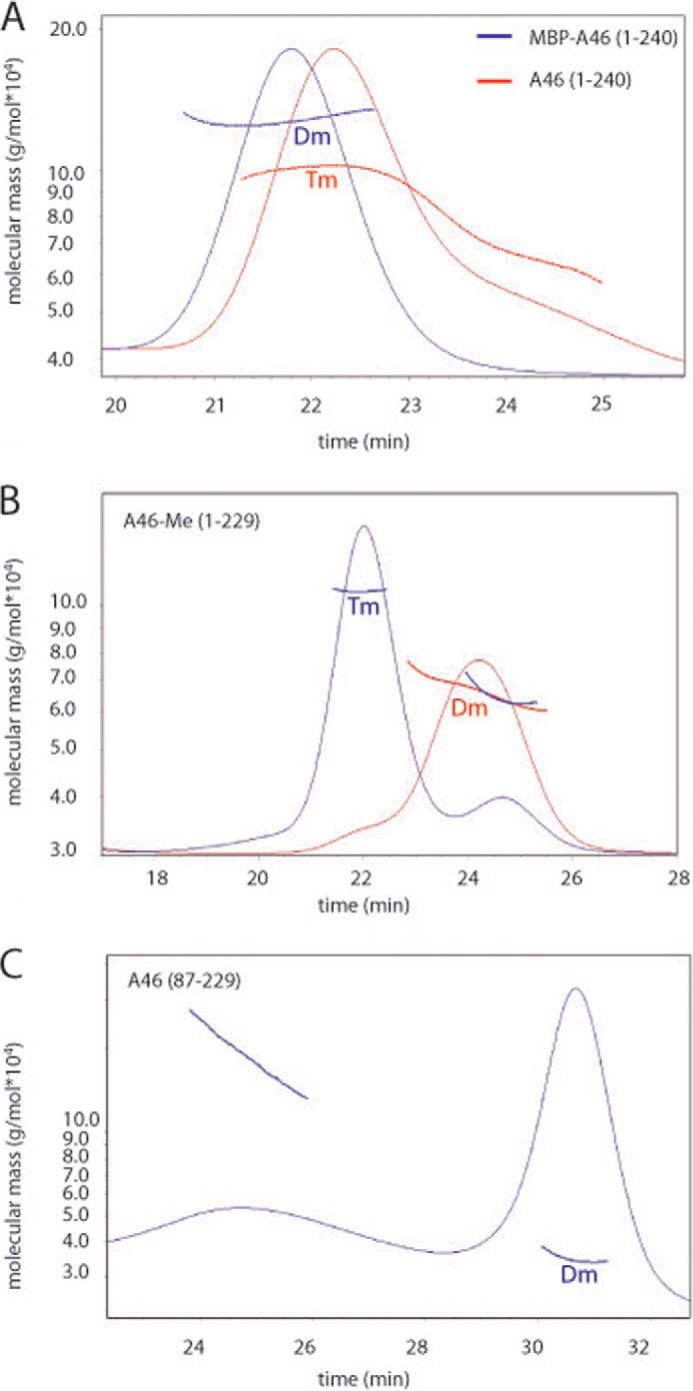
Examination of the solution oligomeric states of indicated A46 variants by SEC-multiangle laser light scattering. A, wild-type A46 behaves as a tetramer (Tm), whereas MBP tagged full-length A46 behaves a dimer (Dm). B, methylated A46-(1–229) was separated into tetramer and dimer populations by size-exclusion chromatography, and the resulting fractions were subjected to static light scattering. C, The N- and C-terminally truncated A46-(87–229) variant that was successfully crystallized behaves as a dimer on static light scattering. 100–200 μg of the indicated proteins was applied to a Superdex 10/300 column, and the light scattering was measured using a miniDAWN Tristar light scattering instrument. Molecular masses were calculated directly from the light scattering measurements.
To investigate the functionality of this protein, we first employed isothermal calorimetry. However, under the conditions employed, we were unable to obtain reproducible measurements because of the precipitation of one of the interaction partners. Consequently, we turned to the method of microscale thermophoresis to examine the binding of the full-length A46 to the recombinant fluorescently labeled TIR domains of MyD88 and MAL. Microscale thermophoresis effectively measures changes in the hydration shell, charge, or size of macromolecules (33, 34). The interaction of A46-(1–240) with TIR/MyD88 showed a KD of 0.79 μm (Table 3), based on the assumption that A46 is a tetramer, as indicated by SEC-multiangle laser light scattering measurements. Values in parentheses in Table 3 indicate the KD values should the tetramer of A46 actually bind two molecules of TIR/MyD88. The amount of protein that could be obtained from the A46-(1–240) construct was limited; therefore, we also investigated the binding behavior of the variant A46-(1–229)C205S (described below); this variant was made in attempts to make the protein more soluble and amenable to crystallography. A representative binding curve for A46 with TIR/MyD88 is shown in Fig. 3A. With this material, we found a reproducible binding constant (0.5 μm, Table 3) that was close to that (0.79 μm) obtained with A46-(1–240). A46-(1–229)C205S could also bind to a variant of MyD88 bearing the double mutation R196A/R288A that had previously been shown to inhibit binding of MyD88 to MAL (Table 3). This would suggest that A46 binds to a site on MyD88 that is different to that bound by the TIR/MAL (31).
TABLE 3.
Properties and binding constants (KD, μm) of A46 variants
Measurements of binding constants by microscale thermophoresis were limited by the amounts of purified proteins available. Thus, the absolute values for each interaction are given, as recommended by Vaux (63). Tm, tetramer; Dm, dimer; M, monomer; ND, not determined.
| Crystals | Quaternary structure | Labeled protein |
|||
|---|---|---|---|---|---|
| TIR/MAL | TIR/MyD88 | TIR/MyD88 R196A/R288A | |||
| A46-(1–240) | N | Tm | No saturation | 0.79 (1.58) | ND |
| A46-(1–229) C205S | N | Tm | 1.44, 1.56 (2.88, 3.12) | 0.46, 0.52 (0.96, 1.04) | 0.52 (1.04) |
| A46-(87–229) | Poor | Dm | 2.24, 2.30 | 0.89, 1.24 | ND |
| A46-(87–229) C205S | Y | Dm | 3.22 | 1.09 | 1.26 |
| TIR/MAL | M | 3.40, 4.08, 4.56, 5.21 | No saturation | ||
| TIR/MyD88 R196A/R288A | M | 10.60, 11.90, 12.80 | |||
FIGURE 3.
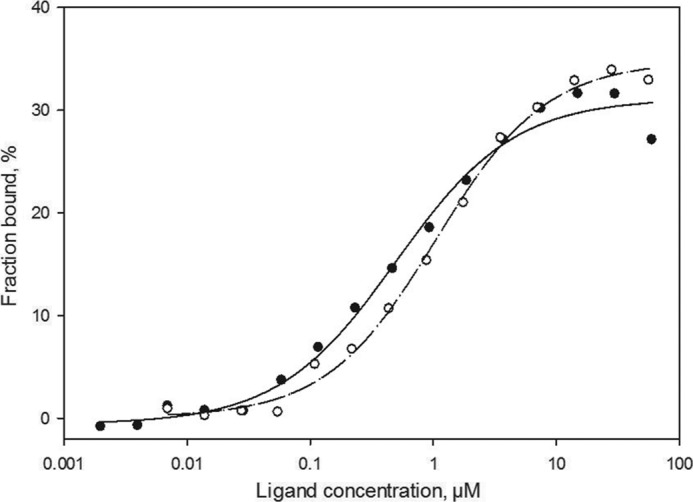
Analysis of A46 interactions with microscale thermophoresis. Unlabeled full-length A46C205 (closed circles, 1.8 nm to 30 μm) or A46-(87–229) (open circles, 5.2 nm to 170 μm) were titrated into a fixed concentration of labeled TIR/MyD88 (30–70 nm). Both A46 variants demonstrated similar binding properties with apparent KD values of 0.5 and about 1.0 μm, respectively. Values are an average of the two measurements presented in Table 3.
An N-terminal Truncation of A46 Is Amenable to Crystallography
Having shown that recombinant A46 is functional, both the MBP-A46 fusion protein and the A46 protein from which the MBP had been removed were then used in crystallization trials. Small crystals were observed with the full-length A46 protein alone; however, they failed to grow, and their generation was irreproducible.
The inability to obtain crystals from a protein often reflects the presence of unstructured regions or flexible domains. We used a multi-construct approach to optimize soluble protein expression and increase the number of variants for crystallization. These data showed that the variant MBP-A46-(1–229) containing four extra amino acids at the N terminus (GSQQ) was expressed at high levels and had good solubility properties. The purification is shown in Fig. 1B. This protein also behaved as a tetramer. However, we were not able to obtain crystals of this variant or of any other variants of A46-(1–229) (e.g. C205S).
We then subjected A46-(1–229) containing the four additional residues at the N terminus to reductive lysine methylation in an attempt to improve crystal formation (48). Examination of the modified protein by SEC showed that ∼50% was still behaving as a tetramer, whereas the remaining 50% eluted at a volume corresponding to a dimer. Separation of the tetramer (molecular mass 110 kDa) and dimer molecular mass 60 kDa) fractions and analysis of the molecular mass (Fig. 2B) confirmed the nature of the quaternary structure. Samples of both tetramer and dimer fractions were placed into crystallization trials, again, however, without success. Examination of the modified protein by mass spectrometry revealed that residues Lys-11, Lys-25, and Lys-45 were most consistently methylated. We, therefore, mutated all of these three residues to alanine in A46-(1–229) to generate a protein that would be more homogeneous than that produced by reductive methylation. This protein also showed a 50% distribution in the population of tetramers and dimers and was also refractory to crystallization.
We next used limited proteolysis to characterize domains of A46 for stability and flexibility. SDS-PAGE analysis of limited digests of A46-(1–229) with elastase, trypsin, chymotrypsin, and thermolysin (data not shown) showed that cleavage was only obtained at high concentrations of proteinase and that only two fragments of ∼10 and 15 kDa were generated. Mass spectrometric analysis revealed that the smaller fragment comprised N-terminal residues 1–63, whereas the larger fragment comprised residues 92–229. Consequently, we expressed a number of fragments (as His-Tag fusions) derived from various regions of the larger fragment. Proteins expressed from the fragments Ser-81–Thr-229 and Asn-87–Thr-229 were soluble and could be purified (Fig. 1C) and eluted mostly at the position corresponding to a dimer (Fig. 2C), although a minor fraction did appear to be undergoing aggregation (Fig. 2C).
The fragment Asn-87–Thr-229 (termed A46-(87–229)) bound the TIR domains of MAL and MyD88 with binding constants of 2.3 and 1 μm, respectively, assuming that it binds as a dimer (Fig. 3B and Table 3). In addition, it also gave rise to micro-crystals. After buffer optimization, micro-crystals were again obtained but in a wider range of conditions. All attempts to optimize crystal growth remained, however, unsuccessful. We noted that hyperactive cysteine residues can often hamper further growth of protein crystals (see for example Nickerson and Wong (49)). There are four cysteine residues (Cys-155, -156, -181, and -205) present in A46-(87–229). We, therefore, attempted to express and purify four modified A46-(87–229) proteins, each bearing a particular cysteine to serine mutation. Three of the mutated proteins (C155S, C181S, and C205S) could be expressed and purified. Diffraction quality crystals were obtained with the A46-(87–229) protein bearing the C205S mutant. This variant retained the ability to bind to the TIR domains of MAL and wild-type MyD88 with similar KD values as unmodified A46-(87–229) (3.22 μm versus 2.3 μm and 1.09 μm versus 1.24 μm, respectively; Table 3), indicating that the mutation had not affected the binding properties of the molecule (Table 3).
A46-(87–229)C205S crystals diffracted on an in-house x-ray source to 2.8 Å. However, the phases could not be solved using the structure of A52. Consequently, this mutant was then labeled with selenium methionine. The labeled protein yielded crystals in essentially the same conditions that diffracted to 2 Å in the European Synchrotron Radiation Facility (Grenoble, France) ID-29 beam-line (Table 2), allowing the phases to be solved using the anomalous selenium signal. All three selenium methionine residues per subunit could be located. A full description of buffer optimization and crystal generation will be presented elsewhere.3
Structure of VACV A46 and Relationship to Other bcl-2-like Vaccinia Proteins
The structure of A46-(87–229) is shown in Figs. 4 and 5. Good electron density for the four molecules in the asymmetric unit was observed for all atoms except for those at the N and C termini. Thus, no density was observed for the first three N-terminal residues (87–89) in all four molecules. At the C terminus, the number of residues for which density was visible varied with the chain, with chain A lacking the least (two) and chain C lacking the most (eight) residues. The orientation of the C termini also varied between the molecules, indicating some degree of flexibility in this region. However, overall the structure of the four chains was very similar; superimposition of chains B, C, and D onto chain A gave r.m.s.d. values ranging between 0.28 and 0.47 Å over 132 superimposed Cα atoms.
FIGURE 4.

Amino acid alignment of A46 and A52 showing secondary structure alignments. A sequence alignment was first generated using the T-coffee algorithm (59) and then modified by hand. Residues identified by Franklin and Khan (27) as forming the hydrophobic core of vaccinia proteins with a Bcl-2 fold are shown in reverse, as are the conserved arginine and aspartic acid residues in α1 and α2, respectively. Residues 90–98 that make up part of the VIPER peptide are shown in bold.
FIGURE 5.

Overall structure of A46. A, view of the overall structure of A46 colored as a spectrum from the N terminus to the C terminus. The side chains of the nine residues of the 11-mer VIPER peptide visible in the structure are shown as sticks as are those of Cys-155 and Cys-156. Carbon atoms are colored in light yellow, oxygen atoms are in red, nitrogen atoms are in blue, and sulfur atoms are in dark yellow. B, 2Fo − Fc electron density map contoured at 1.2δ of the minor confirmation of C156 observed in molecule A, illustrating the disulfide bridge to C155. C and D, electrostatic surface potentials of A46, blue being electropositive and red being electronegative. In C the orientation of the molecule is that in A, in D it is rotated 180°. Drawings were made with PyMOL (60) and APBS (61).
As predicted by González and Esteban (26) and Oda et al. (28), the A46-(87–229) protein shows a typical Bcl-2 fold (27) comprising a central α-helix (α5) surrounded by six further α-helices (Fig. 5A). Unusually, helix α4 is kinked at its C terminus, bringing it close to helix α5. In addition, this region has a high B factor and the electron density in molecule A in the asymmetric unit indicates presence of more than one conformation of the side chains of Cys-155 and Cys-156. Two alternative conformations could be modeled showing ∼65 and 35% occupancy in refinement. The minor conformation is involved in an intramolecular disulfide bridge formed between residues Cys-155 and Cys-156 (Fig. 5A); the electron density for the disulfide bridge conformation is shown in Fig. 5B. This feature may be absent in the other three molecules in the asymmetric unit because of the oxidation of the reducing compound remaining from the protein storage buffer during crystal formation. Whatever the reason, these residues of helix α5 appear to be flexible and can exist in at least two conformations. Last, the completely α-helical structure of A46 bears no relationship to the globular mixed α-helix, β-sheet TIR-domain (see Fig. 7) fold as had initially been proposed (19, 21).
FIGURE 7.
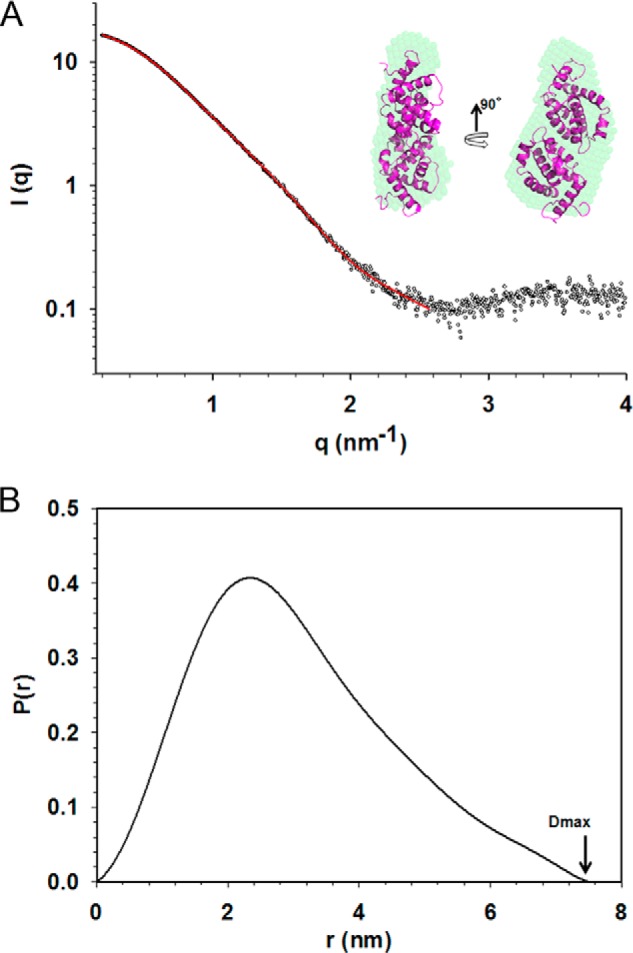
Analysis of scattering data of A46. A, average ab initio bead model of A46 (green) fits the dimer from crystal structure (magenta). The experimental SAXS data are represented as an open sphere, whereas the ab initio fit is in red. The data were fit at resolution of about 25 Å. B, distance distribution analysis calculated using SAXS data.
A peptide mimic termed VIPER comprising 11 A46 residues (88–98) was shown to inhibit the interaction of A46 with the binding partner TRAM but not with MyD88 or MAL (29). Nine (90–98) of these residues are visible in the structure of A46 (shown in bold in Fig. 4 and as sticks in Fig. 5A). All except the first two residues lie in helix α1, with Leu-93 pointing out toward the solvent; the surface potential of this region (Fig. 5C) has a general positive charge, as suggested earlier in a model of A46 by Lysakova-Devine et al. (29). Examination of the surface potential of other parts of the protein revealed a negatively charged stripe on the opposite face to that of VIPER (Fig. 5D) that could interact with positively charged surfaces found on MyD88 and MAL.
Of the poxviral proteins adopting a Bcl-2-like fold, A46 shows the highest sequence identity (16%) to A52. This close relationship between A46 and A52 is further strengthened when the two proteins are aligned (Fig. 4) and superimposed (Fig. 6A); the average r.m.s.d. is 1.71 Å over 111 superimposed Cα atoms. Superimposition of the A46 structure to those of the other related Bcl-2-like proteins such as B14 and N1 gives r.m.s.d. values of 2.45 Å over 92 superimposed Cα atoms and 2.35 Å over 105 superimposed Cα atoms, respectively, supporting the notion that the A52 protein is the closest relative of A46 (data not shown).
FIGURE 6.

Comparison of the structures of A46 and A52. A, superimposition of A46 (magenta) to residues 54–189 of A52 (cyan). Faces involved in dimer formation are shown in light pink (A46) and light blue (A52). B, the A46 dimer. Molecule C of A46 (magenta) is in the same orientation as in Fig. 6A, and molecule D is colored as a spectrum from the N terminus to the C terminus. Residues involved in dimer formation are shown as sticks. C, enlarged view of the residues in the A46 dimer interface shown in A, rotated 45° on the x axis to allow better visualization of the side chains. D, as in A, except that the second monomer is included for each protein. Drawings were made as in Fig. 5.
The most striking structural difference between A46 and A52 is in the arrangement of C terminus of helix α4 and the subsequent loop connecting it to the central helix α5 (Figs. 4 and 6A). This difference is likely of functional relevance as a peptide termed P13 derived from the residues of the α4-α5 loop and the first five residues of α5 (residues 125–135 of A52) has been shown to inhibit NF-κB activation (50, 51). In addition, the two cysteine residues of α4 in A46 that are able to form a disulfide bridge are not found in A52. A further difference between A46 and A52 is that the loop between α1 and α2 is shorter in A46. In helix α1, which contains the VIPER peptide residues, the orientations of the A52 residues are almost identical. However, the presence of residues with different properties in A52 (e.g. Thr-58 and Glu-61, equivalent to Leu-93 and Ala-96 in A46) should prevent A52 binding to TRAM. Other features of note are the rotation of helix α6 of A46 ∼15° closer to helix α1 and the C-terminal extension observed in A46.
Franklin and Khan (27) identified 17 conserved non-polar residues involved in the packing of viral Bcl-2-like proteins (depicted in reverse in Fig. 4). Only eight of these residues are identical between A46 and A52, with six of the seven conserved residues in the central helix α5 being different. In addition, the above authors also identified a conserved arginine residue in helix α1 that forms a salt bridge with a conserved aspartate residue in helix α2 (Fig. 4, both residues also in reverse). This salt bridge is also present in A46. However, it does not extend to helix α5 as it does in A52. The aspartate residue found in A52 is replaced by a lysine in A46 (Fig. 4, C terminus of α5). Lysine is also present at this position in N1 of VACV and M11 of myxoma virus (27).
A further similarity of A46 and A52 is the closed nature of the BH3 groove, found in proteins such as N1 and M11. In these proteins, this groove, encompassed by helices α2 to α5, is constitutively open, allowing them to bind BH3 peptides of pro-apoptotic Bcl-2 proteins (27, 52). Based on these data, Cooray et al. (52) termed N1 an anti-apoptotic protein; however, Postigo and Way have provided evidence that N1 does not bind pro-apoptotic Bcl-2 proteins (53). In A52, the groove is occluded by the presence of a longer α2 helix and the side chains of residues 92–94 and 117–122. In A46, helix α2 has the same number of residues as that of A52 (Fig. 4). In addition, the side chains of the residues equivalent to 92–94 and 117–122 of A52 are 122–124 and 147–152 are very similar (Fig. 4). Thus, as in A52, the BH3 groove should also be closed in A46.
As shown in Fig. 3, the A46-(87–229) protein behaves as a dimer. Does the arrangement of the molecules in the asymmetric unit shed light on its nature? Close inspection reveals that the four molecules in the asymmetric unit are arranged as two dimers. The interface between the monomers in the two dimers lies between helices α4 and α6 (termed the 4–6 face; Fig. 6B). Analysis of the crystal packing contacts with the PISA server (54) gave a complex significance score of 0.6 for this interface; the total buried surface area is around 700 Å2, comprising 10% of the total accessible area of the molecule. The side chains involved in the 4–6 face are depicted in Fig. 6C; a list of all residues involved in the dimer interface and the extent of their buried surface area are given in Table 4. In addition, the 4–6 face was observed as the dimer interface in a second crystal form of A46-(87–229)C205S, which contained only two molecules per asymmetric unit (data not shown).
TABLE 4.
Summary of residues involved in the A46 dimer interface
Bond type: H, hydrogen; S, salt bridge.
| Residue | Buried surface area | Accessible surface area | Length of bond | Bond type | Interacting residue |
|---|---|---|---|---|---|
| Å2 | Å2 | Å | |||
| Lys-92 | 9.98 | 66.46 | 3.73 | HS | Asp-140 |
| Ser-141 | 45.48 | 57.99 | 3.78 / 3.45 / 2.58 | H | Ser-191/Asp-190/His-187 |
| His-187 | 68.51 | 87.08 | 2.58 | H | Ser-141 |
| Leu-143 | 21.91 | 23.42 | |||
| Phe-144 | 127.66 | 142.7 | |||
| Val-147 | 13.23 | 22.11 | |||
| Tyr-179 | 108.29 | 139.69 | 2.84 | H | Glu-186 |
| Arg-180 | 10.51 | 10.94 | 2.96/3.92, 2.96, 3.15, 3.30 | H/S | Glu-186/Glu-186 |
| Met-182 | 32.12 | 105.27 | |||
| Ala-183 | 35.3 | 35.46 | |||
| Leu-184 | 0.67 | 0.67 | |||
| Ser-189 | 1.11 | 3.07 | |||
| Asn-190 | 35.5 | 93.21 | 3.45 | H | Ser-141 |
SAXS analysis provided further support for this interpretation. Ab initio modeling calculated by DAMMIF (46) at 25 Å resolution shows that overall shape of the models is in agreement with the dimer obtained from crystal structure (Fig. 7A). The radius of gyration (Rg) calculated by the Guinier approximation was about 2.30 ± 0.04 nm, which is in agreement with the analysis using the P(r) function (Fig. 7B).
Phe-144 and His-187, both extensively buried in the interface, were investigated by replacing the residues with alanine. We were unable to express the variant A46-(87–229)F144A. In contrast, the variant A46-(87–229)H187A could be expressed, was present as a dimer, and had KD values for binding to MyD88 and MAL very similar to those of A46-(87–229) (data not shown). Further mutagenesis studies will be necessary to determine the contributions of the 13 residues in Table 4, indicated as being part of the dimer interface.
DISCUSSION
Several hurdles had to be overcome to allow us to characterize and determine the structure of the VACV A46 protein. Production of soluble full-length protein was achieved only when the protein was extended by four amino acids at the N terminus. The full-length protein behaved essentially as a tetramer, although static light scattering indicated the presence of lower molecular weight material that could correspond to the dimeric species (Fig. 2A). The importance of tetramer formation in full-length A46 and the oligomerization state in the infected cell remains to be investigated. Removal of the N-terminal 86 residues of the protein gave rise to a species that was essentially dimeric. Both the tetrameric and dimeric forms of A46 were capable of binding to the TIR domains of the interaction partners MyD88 and MAL with KD values in the low micromolar range. Full-length A46 has been shown to inhibit IL-1β induced activation of transcription from the NF-κB promoter (19). We also confirmed that the dimeric A46-(87–229) inhibits IL-1β induced NF-κB activation (Fig. 8).
FIGURE 8.

Inhibition of IL-1β stimulation of NF-κβ transcription by A46. HEK293T cells were transfected with the indicated amounts of plasmids. After 40 h of incubation, the cells were stimulated with 0.32 ng/ml of IL-1β and incubated for a further 6 h. NF-κβ reporter gene activity was then measured. Data are expressed as the relative stimulation from a representative experiment from a minimum three separate experiments, each performed in triplicate. EV, empty vector
Despite the thorough characterization, diffraction quality crystals could, however, not be obtained from these forms; this was only achieved by introducing the mutation C205S into A46-(87–229). Examination of the structure showed that residue 205 is involved in crystal packing interactions with helix α3; these may have been hampered by the oxidation of the side chain of Cys-205.
The structure of A46 shows a Bcl-2 fold that is clearly related to those of A52, B14, K7, and N1, as predicted by González and Esteban (26) and Oda et al. (28). Superimposition of the A46 structure showed that the most closely related structure is that of A52 (111 residues, r.m.s.d. over 111 superimposed Cα atoms 1.71 Å), as also suggested by sequence alignments. The protein interaction partners of A46 and A52 are however different, and there does not appear to be any overlap. Which differences between the structures could account for these differences in their protein-protein interactions? For A52, the loop region of five residues between α4 and α5 termed P13 has been proposed to be part of interaction site for the host protein IRAK2 (25, 51). This loop has only two residues in A46 and is preceded by the two adjacent cysteine residues Cys-155 and Cys-156 at the end of the kinked helix α4 that can exist in at least two conformations. Together, these differences should prevent A46 from interacting with IRAK2 or any other ligand that is recognized by this sequence in A52. At the same time, given the unusual aspects of α4-α5 loop in A46, one can speculate that it may also be a site for ligand interaction in this protein.
A46 is an intracellular protein. Is the finding of the presence of a disulfide bridge between Cys-155 and Cys-156 in one of the molecules in the asymmetric unit of biological significance? It is noteworthy that VACV possesses a system of three oxidoreductases for intracellular disulfide bridge formation (55). Although A46 is not one of the nine known target proteins, it will be of interest to examine whether this protein requires the disulfide bridge for its biological properties. However, the removal of the potential to form the disulfide bridge by introduction of the mutation C155S did not interfere with the binding of A46 to MyD88 or MAL, suggesting that, in vitro at least, these interactions do not require the disulfide bridge.
Full-length A46 behaves as tetramer, whereas the N-terminal truncated form is a dimer, suggesting that in the full-length form two dimers are held together by the N-terminal 80 residues that are absent from the structure. Analysis with the PISA algorithm identified the α4-α6 helices as the dimer interface found in A46-(87–229)C205S. SAXS data also supported the 4–6 face being involved in dimer formation (Fig. 7). This is a further difference to A52, B14, and N1L, all of which use the α1-α6 face for the dimer interface. The position of the dimer interaction faces are depicted on the superimposition of A46 and A52 in Fig. 6A. The net result of the different interfaces is that the non-superimposed chain of the A46 dimer is rotated about 90° that of the corresponding A52 chain (Fig. 6D). It is noteworthy that the three helices of A46 and A52 involved in dimer formation show some of the greatest structural differences between the two proteins (Figs. 4 and 6A). Furthermore, it is not surprising that helix α1 is not involved in A46 dimer formation, as six residues of this helix in A46 are part of the VIPER peptide that has been shown to inhibit A46 binding to TRAM (29). A46-(87–229) still dimerized when His-187 residue was substituted by alanine and bound the adaptor proteins with similar KD values to the wild type, suggesting that the dimer possess a high stability (data not shown). Nevertheless, we cannot at present rule out similar behavior with that of the VACV protein B14 that uses the same face to bind to the IκB kinase β (IKKβ) subunit of the IKK complex as it does to dimerize (56).
The interaction of the full-length and truncated versions of A46 with the TIR domains of TIR/MyD88 and TIR/MAL was investigated and measured with microscale thermophoresis. KD values for both proteins were in the sub- to low micromolar range. Values obtained for the interaction with TIR/MAL were in good agreement of that previously measured by Oda et al. (1.7 μm; Ref. 28). Interestingly, the KD values of A46 with either TIR/MyD88 or TIR/MAL were about 2-fold lower than those measured for the interaction of TIR/MyD88 with TIR/MAL, suggesting that A46 could prevent the host proteins from interacting with each other.
The location of the binding site (or sites) on A46 for TIR/MAL and TIR/MyD88 remains an open question. Stack and Bowie (22) demonstrated that A46 possesses separate binding sites for TRAM on the one hand and MAL and MyD88 on the other. To search for possible interaction sites, we attempted docking studies with the known structures of MAL (57, 58) and MyD88 (31) using the ClusPro server. However, these studies were inconclusive. For MAL at least, this probably arose because in the two independent MAL structures published, the so-called BB loop of these TIR domain-containing proteins that is required for the interaction (22) shows no density and is, therefore, disordered (57, 58).
In contrast, the binding site of TRAM has been determined to include residues 87–98 of A46. With this information as a support for interpretation, we again used the protein-protein interaction server ClusPro and a model of the TIR domain of TRAM (29), as the structure of TRAM has not been solved. One result predicts an interaction of the BB loop of TRAM, containing the characteristic proline residue Pro-116 (13) and residues of α1 contained in the VIPER peptide (Fig. 9). Importantly, the A46 residue Leu-93 is very close to Phe-112 of TRAM. This interaction might explain why Leu-93 was the only A46 residue whose substitution led to the loss of inhibition by the VIPER peptide (29). Nevertheless, one must remember that this docking simulation is based on a model of TRAM and that only the structure determination of A46/TRAM complex will resolve this question.
FIGURE 9.

Docking model of A46 with TRAM. The coordinates of A46 and the model of TRAM generated by (29) were submitted to the ClusPro protein-protein docking server (62). One docking result that showed an interaction of the VIPER peptide with the BB-loop of TRAM was chosen and visualized. A46 is colored as a spectrum from the N terminus to the C terminus, the VIPER residues are shown as sticks (carbon atoms are colored in light yellow, oxygen atoms are in red, and nitrogen atoms are in blue) as are residues Phe-144 and His-187 to mark the 4–6 dimer interface. TRAM is colored in cyan, with the residues of the BB loop are shown in sticks. The inset shows an enlargement of the VIPER sequence of A46 and the BB loop of TRAM.
In summary, we have characterized biochemically and determined the structure of residues 87–229 of VACV protein A46. The structures illustrate how viruses can generate versatility of function from the same fold by using different faces for dimerization. In addition, this work provides the basis for experiments to determine the nature and location of the binding sites on A46 for its battery of interaction partners.
Acknowledgments
We thank Kristina Djinović-Carugo, Gang Dong, Julius Kostan, Thomas Leonard, and Ekaterina Shimanovskaya for support in suggesting approaches to protein crystallization as well as data collection and interpretation, Flavia Leite for support with VACV biology, Vitaly Sedlyarov for murine macrophage cDNA, Gijs Versteeg and Stefan Benke for help with NF-κβ promoter assays, and Dr. H. Tochio for the MAL plasmid and Dr T. Monie for the coordinates of the TRAM model. We thank Kristina Djinović-Carugo, Thomas Leonard, and Gijs Versteeg for critical reading. We acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facilities, and we thank Barbara Machado.
This work was supported by Austrian Science Fund Grant W1221, “DK: Structure and Interaction of Biological Macromolecules,” and the Medical University of Vienna.
The atomic coordinates and structure factors (code 4LQK) have been deposited in the Protein Data Bank (http://wwpdb.org/).
S. Fedosyuk and T. Skern, manuscript submitted.
- TLR
- Toll-like receptor
- TIR
- Toll-like receptor/interleukin-1 receptor
- MyD88
- MYD88 myeloid differentiation primary response 88
- MAL/TIRAP
- MyD88 adapter-like/TIR domain-containing adaptor protein
- TRAM
- TRIF-related adaptor molecule
- TRIF
- TIR-domain-containing adapter-inducing interferon-β
- TRAF
- TNF receptor-associated factor
- NF-κB
- nuclear factor κ light chain-enhancer of activated B cell
- IRAK
- interleukin-1 receptor-associated kinase
- VACV
- vaccinia virus
- VIPER
- viral inhibitory peptide of TLR4
- MBP
- maltose-binding protein
- TEV
- tobacco etch virus
- GST
- glutathione S-transferase
- SEC
- size exclusion chromatography
- SAXS
- small-angle x-ray scattering
- Ni-NTA
- nickel-nitrilotriacetic acid
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Medzhitov R., Janeway C., Jr. (2000) Innate immunity. N. Engl. J. Med. 343, 338–344 [DOI] [PubMed] [Google Scholar]
- 2. Aoshi T., Koyama S., Kobiyama K., Akira S., Ishii K. J. (2011) Innate and adaptive immune responses to viral infection and vaccination. Curr. Opin. Virol. 1, 226–232 [DOI] [PubMed] [Google Scholar]
- 3. O'Neill L. A. (2006) How Toll-like receptors signal. What we know and what we don't know. Curr. Opin. Immunol. 18, 3–9 [DOI] [PubMed] [Google Scholar]
- 4. Bowie A. G., Unterholzner L. (2008) Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 8, 911–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kotwal G. J., Hatch S., Marshall W. L. (2012) Viral infection. An evolving insight into the signal transduction pathways responsible for the innate immune response. Adv. Virol. 2012, 131457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kawai T., Akira S. (2007) Signaling to NF-κB by Toll-like receptors. Trends Mol. Med. 13, 460–469 [DOI] [PubMed] [Google Scholar]
- 7. Botos I., Davies D. R. (2010) Toll-like receptors-Structure and Signalling. In Handbook of Cell Signalling (Bradshaw R. A., Dennis E. A., eds) pp. 139–143, 2nd Ed., Elsevier, San Diego, CA [Google Scholar]
- 8. Akira S., Takeda K. (2004) Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511 [DOI] [PubMed] [Google Scholar]
- 9. West A. P., Koblansky A. A., Ghosh S. (2006) Recognition and signaling by toll-like receptors. Annu. Rev. Cell Dev. Biol. 22, 409–437 [DOI] [PubMed] [Google Scholar]
- 10. O'Neill L. A., Bowie A. G. (2007) The family of five. TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7, 353–364 [DOI] [PubMed] [Google Scholar]
- 11. Kagan J. C., Medzhitov R. (2006) Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 125, 943–955 [DOI] [PubMed] [Google Scholar]
- 12. Kagan J. C., Su T., Horng T., Chow A., Akira S., Medzhitov R. (2008) TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 9, 361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Núñez Miguel R., Wong J., Westoll J. F., Brooks H. J., O'Neill L. A., Gay N. J., Bryant C. E., Monie T. P. (2007) A dimer of the Toll-like receptor 4 cytoplasmic domain provides a specific scaffold for the recruitment of signalling adaptor proteins. PLoS ONE 2, e788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Napetschnig J., Wu H. (2013) Molecular basis of NF-κB signaling. Annu. Rev. Biophys. 42, 443–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sato S., Sugiyama M., Yamamoto M., Watanabe Y., Kawai T., Takeda K., Akira S. (2003) Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-κ B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 171, 4304–4310 [DOI] [PubMed] [Google Scholar]
- 16. Tatematsu M., Ishii A., Oshiumi H., Horiuchi M., Inagaki F., Seya T., Matsumoto M. (2010) A molecular mechanism for Toll-IL-1 receptor domain-containing adaptor molecule-1-mediated IRF-3 activation. J. Biol. Chem. 285, 20128–20136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walsh S. R., Dolin R. (2011) Vaccinia viruses. Vaccines against smallpox and vectors against infectious diseases and tumors. Expert. Rev. Vaccines 10, 1221–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bahar M. W., Graham S. C., Chen R. A., Cooray S., Smith G. L., Stuart D. I., Grimes J. M. (2011) How vaccinia virus has evolved to subvert the host immune response. J. Struct. Biol. 175, 127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bowie A., Kiss-Toth E., Symons J. A., Smith G. L., Dower S. K., O'Neill L. A. (2000) A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 97, 10162–10167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perdiguero B., Esteban M. (2009) The interferon system and vaccinia virus evasion mechanisms. J. Interferon Cytokine Res. 29, 581–598 [DOI] [PubMed] [Google Scholar]
- 21. Stack J., Haga I. R., Schröder M., Bartlett N. W., Maloney G., Reading P. C., Fitzgerald K. A., Smith G. L., Bowie A. G. (2005) Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J. Exp. Med. 201, 1007–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stack J., Bowie A. G. (2012) Poxviral protein A46 antagonizes Toll-like receptor 4 signaling by targeting BB loop motifs in Toll-IL-1 receptor adaptor proteins to disrupt receptor:adaptor interactions. J. Biol. Chem. 287, 22672–22682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harte M. T., Haga I. R., Maloney G., Gray P., Reading P. C., Bartlett N. W., Smith G. L., Bowie A., O'Neill L. A. (2003) The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 197, 343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Keating S. E., Maloney G. M., Moran E. M., Bowie A. G. (2007) IRAK-2 participates in multiple toll-like receptor signaling pathways to NFκB via activation of TRAF6 ubiquitination. J. Biol. Chem. 282, 33435–33443 [DOI] [PubMed] [Google Scholar]
- 25. Graham S. C., Bahar M. W., Cooray S., Chen R. A., Whalen D. M., Abrescia N. G., Alderton D., Owens R. J., Stuart D. I., Smith G. L., Grimes J. M. (2008) Vaccinia virus proteins A52 and B14 Share a Bcl-2-like fold but have evolved to inhibit NF-κB rather than apoptosis. PLoS Pathog. 4, e1000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. González J. M., Esteban M. (2010) A poxvirus Bcl-2-like gene family involved in regulation of host immune response. Sequence similarity and evolutionary history. Virol. J. 7, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Franklin E., Khan A. R. (2013) Poxvirus antagonism of innate immunity by Bcl-2 fold proteins. J. Struct. Biol. 181, 1–10 [DOI] [PubMed] [Google Scholar]
- 28. Oda S., Franklin E., Khan A. R. (2011) Poxvirus A46 protein binds to TIR domain-containing Mal/TIRAP via an α-helical sub-domain. Mol. Immunol. 48, 2144–2150 [DOI] [PubMed] [Google Scholar]
- 29. Lysakova-Devine T., Keogh B., Harrington B., Nagpal K., Halle A., Golenbock D. T., Monie T., Bowie A. G. (2010) Viral inhibitory peptide of TLR4, a peptide derived from vaccinia protein A46, specifically inhibits TLR4 by directly targeting MyD88 adaptor-like and TRIF-related adaptor molecule. J. Immunol. 185, 4261–4271 [DOI] [PubMed] [Google Scholar]
- 30. Niwa H., Yamamura K., Miyazaki J. (1991) Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–199 [DOI] [PubMed] [Google Scholar]
- 31. Ohnishi H., Tochio H., Kato Z., Orii K. E., Li A., Kimura T., Hiroaki H., Kondo N., Shirakawa M. (2009) Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 10260–10265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ohnishi H., Tochio H., Kato Z., Kimura T., Hiroaki H., Kondo N., Shirakawa M. (2010) 1H, 13C, and 15N resonance assignment of the TIR domain of human MyD88. Biomol. NMR Assign. 4, 123–125 [DOI] [PubMed] [Google Scholar]
- 33. Jerabek-Willemsen M., Wienken C. J., Braun D., Baaske P., Duhr S. (2011) Molecular interaction studies using microscale thermophoresis. Assay Drug Dev. Technol. 9, 342–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seidel S. A., Dijkman P. M., Lea W. A., van den Bogaart G., Jerabek-Willemsen M., Lazic A., Joseph J. S., Srinivasan P., Baaske P., Simeonov A., Katritch I., Melo F. A., Ladbury J. E., Schreiber G., Watts A., Braun D., Duhr S. (2013) Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59, 301–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kabsch W. (2010) Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schneider T. R., Sheldrick G. M. (2002) Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 58, 1772–1779 [DOI] [PubMed] [Google Scholar]
- 38. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 40. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 42. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Konarev P. V., Petoukhov M. V., Volkov V. V., Svergun D. I. (2006) ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 39, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS. A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 45. Svergun D. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 46. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lechauve C., Bouzhir-Sima L., Yamashita T., Marden M. C., Vos M. H., Liebl U., Kiger L. (2009) Heme ligand binding properties and intradimer interactions in the full-length sensor protein dos from Escherichia coli and its isolated heme domain. J. Biol. Chem. 284, 36146–36159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Walter T. S., Meier C., Assenberg R., Au K. F., Ren J., Verma A., Nettleship J. E., Owens R. J., Stuart D. I., Grimes J. M. (2006) Lysine methylation as a routine rescue strategy for protein crystallization. Structure 14, 1617–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nickerson D. P., Wong L. L. (1997) The dimerization of Pseudomonas putida cytochrome P450cam. Practical consequences and engineering of a monomeric enzyme. Protein Eng. 10, 1357–1361 [DOI] [PubMed] [Google Scholar]
- 50. Tsung A., McCoy S. L., Klune J. R., Geller D. A., Billiar T. R., Hefeneider S. H. (2007) A novel inhibitory peptide of Toll-like receptor signaling limits lipopolysaccharide-induced production of inflammatory mediators and enhances survival in mice. Shock 27, 364–369 [DOI] [PubMed] [Google Scholar]
- 51. McCoy S. L., Kurtz S. E., Macarthur C. J., Trune D. R., Hefeneider S. H. (2005) Identification of a peptide derived from vaccinia virus A52R protein that inhibits cytokine secretion in response to TLR-dependent signaling and reduces in vivo bacterial-induced inflammation. J. Immunol. 174, 3006–3014 [DOI] [PubMed] [Google Scholar]
- 52. Cooray S., Bahar M. W., Abrescia N. G., McVey C. E., Bartlett N. W., Chen R. A., Stuart D. I., Grimes J. M., Smith G. L. (2007) Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J. Gen. Virol. 88, 1656–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Postigo A., Way M. (2012) The vaccinia virus-encoded Bcl-2 homologues do not act as direct Bax inhibitors. J. Virol. 86, 203–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krissinel E. (2010) Crystal contacts as nature's docking solutions. J. Comput. Chem. 31, 133–143 [DOI] [PubMed] [Google Scholar]
- 55. Bisht H., Brown E., Moss B. (2010) Kinetics and intracellular location of intramolecular disulfide bond formation mediated by the cytoplasmic redox system encoded by vaccinia virus. Virology 398, 187–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Benfield C. T., Mansur D. S., McCoy L. E., Ferguson B. J., Bahar M. W., Oldring A. P., Grimes J. M., Stuart D. I., Graham S. C., Smith G. L. (2011) Mapping the IκB kinase β (IKKβ)-binding interface of the B14 protein, a vaccinia virus inhibitor of IKKβ-mediated activation of nuclear factor κB. J. Biol. Chem. 286, 20727–20735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lin Z., Lu J., Zhou W., Shen Y. (2012) Structural insights into TIR domain specificity of the bridging adaptor Mal in TLR4 signaling. PLoS ONE 7, e34202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Valkov E., Stamp A., Dimaio F., Baker D., Verstak B., Roversi P., Kellie S., Sweet M. J., Mansell A., Gay N. J., Martin J. L., Kobe B. (2011) Crystal structure of Toll-like receptor adaptor MAL/TIRAP reveals the molecular basis for signal transduction and disease protection. Proc. Natl. Acad. Sci. U.S.A. 108, 14879–14884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Notredame C., Higgins D. G., Heringa J. (2000) T-Coffee. A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 [DOI] [PubMed] [Google Scholar]
- 60. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA [Google Scholar]
- 61. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems. Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kozakov D., Hall D. R., Beglov D., Brenke R., Comeau S. R., Shen Y., Li K., Zheng J., Vakili P., Paschalidis I. Ch., Vajda S. (2010) Achieving reliability and high accuracy in automated protein docking. ClusPro, PIPER, SDU, and stability analysis in CAPRI rounds 13–19. Proteins 78, 3124–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vaux D. L. (2012) Research methods. Know when your numbers are significant. Nature 492, 180–181 [DOI] [PubMed] [Google Scholar]