

Abstract
The cell cycle checkpoint kinases play central roles in the genome maintenance of eukaryotes. Activation of the yeast checkpoint kinase Rad53 involves Rad9 or Mrc1 adaptor-mediated phospho-priming by Mec1 kinase, followed by auto-activating phosphorylation within its activation loop. However, the mechanisms by which these adaptors regulate priming phosphorylation of specific sites and how this then leads to Rad53 activation remain poorly understood. Here we used quantitative mass spectrometry to delineate the stepwise phosphorylation events in the activation of endogenous Rad53 in response to S phase alkylation DNA damage, and we show that the two Rad9 and Mrc1 adaptors, the four N-terminal Mec1-target TQ sites of Rad53 (Rad53-SCD1), and Rad53-FHA2 coordinate intimately for optimal priming phosphorylation to support substantial Rad53 auto-activation. Rad9 or Mrc1 alone can mediate surprisingly similar Mec1 target site phosphorylation patterns of Rad53, including previously undetected tri- and tetraphosphorylation of Rad53-SCD1. Reducing the number of TQ motifs turns the SCD1 into a proportionally poorer Mec1 target, which then requires the presence of both Mrc1 and Rad9 for sufficient priming and auto-activation. The phosphothreonine-interacting Rad53-FHA domains, particularly FHA2, regulate phospho-priming by interacting with the checkpoint mediators but do not seem to play a major role in the phospho-SCD1-dependent auto-activation step. Finally, mutation of all four SCD1 TQ motifs greatly reduces Rad53 activation but does not eliminate it, and residual Rad53 activity in this mutant is dependent on Rad9 but not Mrc1. Altogether, our results provide a paradigm for how phosphorylation site clusters and checkpoint mediators can be involved in the regulation of signaling relay in protein kinase cascades in vivo and elucidate an SCD1-independent Rad53 auto-activation mechanism through the Rad9 pathway. The work also demonstrates the power of mass spectrometry for in-depth analyses of molecular mechanisms in cellular signaling in vivo.
Eukaryotic cells are most vulnerable to exogenous DNA-damaging agents during the S phase of the cell cycle, when unprogrammed DNA lesions interfere with the tightly choreographed DNA replication process. DNA damage during this phase leads to the activation of two overlapping checkpoint pathways in Saccharomyces cerevisiae, the DNA replication checkpoint and the intra-S-phase DNA damage checkpoint (1, 2). Phospho-priming for auto-activation of the central checkpoint kinase Rad53 by the upstream kinase Mec1/Tel1 depends on Mrc1 as an adaptor in the DNA replication checkpoint pathway and Rad9 as an adaptor in the DNA damage checkpoint pathway (3–10). Rad53, a well-accepted model system for studying the function and regulation of Chk2-like kinases, contains two forkhead-associated (FHA)1 domains (FHA1 and -2) and two SQ/TQ cluster domains (SCD1 and -2) enriched in Mec1/Tel1-target phosphorylation sites (11–13).
Mrc1 normally is a replisome component that functionally couples DNA Pol ε with Cdc45 and MCM helicase during replication fork progression (14, 15). As the replication forks are stalled by replication stress, the recruited checkpoint sensor kinase Mec1 phosphorylates the SCD of Mrc1, which abolishes its N-terminal interaction with Pol ε and enables Mrc1 to recruit Rad53 and promote Rad53 phosphorylation by Mec1 as an initial step in the activation of Rad53 in the Mrc1 branch (6, 14, 16). Alanine substitution of all Mec1 target sites of Mrc1 (designated the mrc1-AQ allele) has been shown to selectively disable its checkpoint function for Rad53 activation without affecting its DNA replication functions (4). In response to DNA damage, Rad9 is able to associate with damaged chromatin via its BRCT and Tudor domains, which tether it to Ser129-phosphorylated histone H2A (γH2A) and Lys79-methylated histone H3, respectively (17, 18). Alternatively, the recruitment of Rad9 onto damaged DNA could also be facilitated by its phosphorylation by CDK1, which enables the specific interaction of Rad9 with Dpb11, allowing the formation of the ternary complex of Dpb11, Mec1, and Rad9 (19, 20). Similar to Mrc1, Mec1 activates the adaptor function of Rad9 by phosphorylation of its SCD, which then binds to the Rad53-FHA domains to promote Rad53 phosphorylation by Mec1 (3, 5, 10).
Beyond serving as scaffolds to recruit Rad53, Mrc1 and Rad9 have been shown to promote Rad53 phosphorylation by Mec1 in a dose-dependent manner in vitro (3, 16), underlining their adaptor role to enhance the enzyme–substrate (Mec1–Rad53) interaction. However, how they can specifically regulate the priming phosphorylation at specific sites and how this then leads to Rad53 activation remains poorly understood. Finally, hyperphosphorylated Rad9 has also been shown to catalyze the auto-phosphorylation of recombinant Rad53 (21), but it remains to be examined whether and how this occurs in vivo.
The activation of SCD-FHA containing kinases such as human Chk2 and fission yeast Cds1 has been suggested to involve a two-step phosphorylation process: first, SCD phosphorylation by an ATM/ATR-like kinase leads to intermolecular binding to the FHA domain of another Chk2/Cds1 monomer, which then results in dimerization/oligomerization-dependent auto-phosphorylation within the kinase activation loop (22–26). In addition to the characteristic N-terminal SCD-FHA module of Chk2-like kinases, Rad53 contains another SCD2-FHA2 module C-terminal to its kinase domain. Similar to its orthologues, Rad53 activation has been proposed to depend on SCD1 phosphorylation (but not SCD2 phosphorylation) and partially redundant functions of the two FHA domains (9, 27–29). However, although Rad53-FHA1 can interact with SCD1 in a phospho-threonine (pT)-dependent manner in vitro (9, 28), it appears to be required for Rad53 activation only in G2/M-arrested cells (27, 29). In contrast, the FHA2 domain, which seems to be more important overall for Rad53 activation, does not appreciably bind phospho-SCD1 peptides in vitro (27, 28). Thus, the mechanisms by which Mrc1, Rad9, SCD1 phosphorylation, and FHA domains interact during checkpoint-dependent Rad53 priming and auto-activation remain to be elucidated.
Quantitative mass spectrometric analysis has revolutionized the functional analysis of cellular signaling pathways, including site-specific phosphorylation events of key signaling molecules (30–33), but an important caveat is that MS studies often involve protein tags or nonphysiological expression levels that can interfere with normal protein functions. For example, the integration of a triple HA tag into the endogenous RAD53 gene locus has been shown to reduce Rad53 protein levels, resulting in significantly altered checkpoint activity (34). In this study we used quantitative MS analyses to dissect the stepwise phosphorylation events of endogenous, untagged Rad53 in response to MMS-induced alkylation DNA damage and replication stress during the S phase. Together with functional analyses, our results delineate how the two Mec1 adaptors Rad9 and Mrc1 can coordinate with the four SCD1 priming sites (T5, T8, T12, and T15) to regulate the phospho-priming of Rad53 by Mec1. In addition, an SCD1-priming independent Rad53 auto-activation mechanism and the specific roles of the FHA domains during Rad53 hyperphosphorylation are also elucidated in this work.
EXPERIMENTAL PROCEDURES
Yeast Strains
All yeast strains were in the W303–1A background (with corrected RAD5) and contained sml1::HIS3. Point mutations were generated in the endogenous RAD53 locus without residual markers or protein tags, and double mutant strains were generated via mating and tetrad dissection of spores as described in supplemental Table S1. In order to specifically detect S phase DNA damage activation of Rad53 without indirect effects from dNTP fluctuations during normal S phase (35), all experiments in this study were performed in sml1Δ strains.
Cell Synchronization, MMS Treatment, and Preparation of Spiked-in SILAC Standard
Yeast cells were grown in yeast peptone dextrose at 30 °C. Log phase cultures (optical density at 600 nm ≈ 0.4) were arrest in G1 phase by two pulses of α-factor treatment (9 μg/ml) for 75 min each. Cells were then washed extensively and resuspended in prewarmed yeast peptone dextrose with or without 0.05% MMS. The lysates were prepared from cells released from α-factor arrest into yeast peptone dextrose (45 min for collecting S phase Rad53) or yeast peptone dextrose containing 0.05% MMS (45 min for collecting maximal activated Rad53 in S phase). The SILAC isogenic strain (CAN1 (plasma membrane Arg permease), arg4::KAN lys2::NAT) was grown in minimal dropout media supplemented with l-lysine-13C6,15N2 and l-arginine-13C6,15N4 (ISOTEC, Sigma, St. Louis, Missouri), and lysates were prepared from mid-log phase cells (OD 600 nm ∼ 0.8 to 1) after 0.05% MMS treatment for 2 h.
Western Blotting and Immunoprecipitation
For Western blots, lysates were prepared using bead beating at 4 °C in lysis buffer (50 mm Tris-HCl, pH 7.5, 0.5% Nonidet P-40, 0.5% Triton-X 100, 150 mm NaCl, 5 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, EDTA-free Protease Inhibitor Mixture (Roche), 5 mm sodium pyrophosphate, 50 mm sodium fluoride, 10 mm β-glycerophosphate, and Phosphatase Inhibitor Mixture (Sigma)) and separated by 8% standard SDS-PAGE. The rabbit polyclonal anti-Rad53 antibody was raised and affinity-purified using recombinant Rad53-FHA2 domain and then used for monitoring Rad53 characteristic gel shifts. Mouse monoclonal antibody EL7E1 was used for Rad53 immunoprecipitation and detection. Anti-Rad9 was raised and affinity-purified using recombinant Rad9-BRCT domain, anti-γH2A from Abcam, Cambridge, Massachusetts (ab15083), and anti-Clb5 from Santa Cruz Biotechnology, Dallas, Texas (sc6704). For Rad53 immunoprecipitation from large culture for MS analysis, ∼150 to 200 mg of the lysate protein was collected as described above and adjusted to a protein concentration of about 10 mg/ml. The endogenous Rad53 was then immunoprecipitated using EL7E1 and protein-G Mag Sepharose Xtra beads from GE Healthcare (28–9670-70) at 4 °C overnight. After extensive washing of the beads with the lysis buffer and PBS with 0.1% Tween-20, the bound Rad53 and associated proteins were eluted with the SDS sample buffer. After spiking in “heavy” EL7E1 immunopurified Rad53 metabolically labeled with l-lysine-13C6,15N2 and l-arginine-13C6,15N4 (ISOTEC), the mixture was separated by 9.5% standard SDS-PAGE followed by in-gel trypsin digestion for LC-MS/MS analysis.
Enrichment of Phosphopeptides with TiO2 Beads
The resulting tryptic fragments of Rad53 and co-purified proteins were incubated with 2 mg of TiO2 beads (GL Science, Torrance, CA) or 50 μl of TiO2 Mag Sepharose (GE Healthcare) in 1 m glycolic acid (Sigma) prepared in 80% acetonitrile with 5% (v/v) trifluoroacetic acid in water. After incubation for 30 min with vigorous shaking, the beads were washed with the above binding buffer and then with a solution of 80% acetonitrile with 1% (v/v) trifluoroacetic acid twice. The bound phosphopeptides were then eluted from the beads with 5% NH3 twice. After acidification with formic acid, the eluted phosphopeptides were desalted by C18 StageTips (Proxeon Biosystems, Thermo, Waltham, Massachusetts, USA).
LC-MS/MS and Data Analysis
The resulting peptides were generated from in-gel trypsin digestion, which cleaves at the C terminus of lysine and arginine residues. The peptide nanoflow LC-MS/MS experiments were performed using a reverse-phase C18 capillary column and an LTQ-Orbitrap hybrid mass spectrometer (Thermo) equipped with a nanoelectrospray ion source (New Objective, Woburn, Massachusetts) and an ultrahigh pressure LC (Waters Corporation, Milford, Massachusetts, USA) or HPLC pump (Agilent Technology, Santa Clara, CA). The scan cycle was initiated with a full-scan survey MS (m/z 320–1600) experiment in the Orbitrap followed by data-dependent MS/MS experiments in the linear ion trap on the 12 or 20 most abundant ions detected in the full MS scan. The raw MS and MS/MS spectra were processed by MaxQuant (1.2.2.5 or 1.3.0.5) with the Andromeda search engine for protein and peptide identification in a target-decoy Saccharomyces cerevisiae protein database (yeast orf trans all 05-Jan-2010.fasta; 6754 entries) (36). The precursor and fragment mass tolerance were set to 6 ppm and 0.5 Da, respectively, with up to two missed cleavages. Cysteine carbamidomethylation was used as a fixed modification, and the variable modifications were methionine oxidation; protein N-terminal acetylation; asparagine deamidation; and serine, threonine, and tyrosine phosphorylation. The false discovery rate was set at 1% for peptides, proteins, and sites, and the minimum peptide length allowed was seven amino acids. In order to precisely quantitate the changes in the Rad53 phosphorylation and in the association of Mrc1 and Rad9 with Rad53, the abundance of the unlabeled and SILAC-labeled tryptic fragments was also manually examined and calculated based on the peak areas in the extracted ion chromatogram for their top three isotopic peptide precursor ions with 25 ppm mass tolerance using Xcalibur (Thermo). The results for the quantitation of Rad53 phosphorylation were obtained from two to six independent biological repeats for all of the yeast strains used in this study. The lists of all detected Rad53 tryptic fragments and Rad53 and Mrc1 phosphorylation site identifications are organized in supplemental Tables S2–S4. All annotated, mass-labeled spectra for supplemental Tables S2–S4 can be found in supplemental Table S5.
RESULTS
Quantitative MS Analysis of Endogenous Rad53 Phosphorylation in Response to S Phase DNA Damage
Whereas the Rad9-dependent DNA damage checkpoint pathway functions during all cell cycle phases, the Mrc1 pathway is only active during S phase (2, 37). Thus, in order to be able to assess the contributions of both adaptors to Rad53 activation under comparable conditions, we chose to monitor Rad53 phosphorylation in response to S phase MMS treatment. To investigate Rad53 phosphorylation under entirely physiological conditions, we took advantage of the recently developed monoclonal antibody EL7E1 (38), which can efficiently immunoprecipitate endogenous Rad53 with or without MMS (supplemental Fig. S1A). We employed a variant of the SILAC-based quantitative MS approach, “spike-in SILAC” (39), to evaluate the effects of different mutations on the site-specific phosphorylation states of Rad53 under ideal growth conditions. As shown in Fig. 1A, phosphorylation-dependent electrophoretic mobility shifts of Rad53 were maximal after the G1-arrested yeast cells had been released for 45 min into S phase in the presence of 0.05% MMS. To avoid measuring secondary responses, a 45-min MMS treatment was chosen as the standard condition in this study. As indicated in Fig. 1B, all unlabeled EL7E1 affinity-purified “light Rad53” experimental samples were internally standardized by spiking with the same amount of “heavy Rad53” reference sample prepared via DNA damage treatment of an isogenic WT strain (except for lys2Δ arg4Δ CAN1 to ensure even labeling) grown in 13C/15N-Lys/Arg containing minimal medium. The mixture of the heavy (H) and light (L) Rad53 samples was then separated by SDS-PAGE (supplemental Fig. S1B), followed by in-gel tryptic digestion and LC-MS/MS analysis using an LTQ-Orbitrap hybrid MS analyzer. The resulting peptide identification covered around 90% of the Rad53 protein sequence (supplemental Table S2). Two types of phosphopeptide analysis were performed: the relative levels of phosphorylation (the WT data were set to be 100%) using the normalized heavy phosphopeptide as the reference, and the estimated phosphostoichiometry (i.e. percent site occupancy) based on depletion of the corresponding unphosphorylated peptide counterpart. The detailed algorithms are further elaborated below.
Fig. 1.

Rad53 phosphorylation site mapping in EL7E1-immuno-purified endogenous, untagged Rad53 after S phase MMS treatment. A, Western blot analysis for phosphorylation-dependent electrophoretic mobility shifts of Rad53. Samples were collected during α-factor G1 arrest (0 min) and at the indicated time points after release into the S phase with 0.05% MMS. B, conditions and workflow of the spike-in SILAC approach to quantitate the site-specific phosphorylation level and the estimated phosphostoichiometry of Rad53. C, precursor mass spectral scans for an unmodified Rad53670–679 fragment FLLQDGDEIK (a) and the unphosphorylated (b) and mono-, di-, tri-, and tetraphosphorylated (c–f, respectively) SCD1 Rad531–17 fragment MENITQPTQQSTQATQR (the N-terminal Met was N-acetylated and S-oxidized) in unlabeled light (L1–L3) experimental samples relative to the SILAC-labeled heavy (H) DNA-damaged WT reference sample. Mass-to-charge ratio (m/z) of the monoisotopic precursor is labeled at each top panel. Mec1/Tel1 consensus (SQ/TQ) phosphorylation sites (D) and other sites (E) identified in this study are also presented.
As illustrated in row I of Fig. 1B, the L/H ratios of Rad53 tryptic fragments that did not contain phosphorylation or other post-translational modification sites were used to normalize the input levels of unlabeled Rad53. It was then possible to compare the relative phosphorylation levels of specific sites (or multisites) between different Rad53 samples (row II in Fig. 1B), including the mrc1-AQ rad9Δ control strain in which both mediator pathways are disabled, to allow identification of the checkpoint-dependent phosphorylation events. Examples of the raw precursor MS scan are shown in Fig. 1C for an unmodified Rad53670–679 fragment as the input control (a) and the unphosphorylated (b) and mono-, di-, tri-, and tetraphosphorylated (c–f, respectively) Rad53-SCD1 fragments (Rad531–17, MENITQPTQQSTQATQR) in unlabeled light (L1–L3) experimental samples relative to the SILAC-labeled heavy (H) DNA-damaged WT reference sample. Comparison of the MMS-treated WT sample (L2) with the untreated sample (L1) and the mrc1-AQ rad9Δ control (L3) indicated that phosphorylation of the four TQ motifs (c–f) was checkpoint- and DNA-damage-dependent.
Furthermore, as phosphorylation at a given site results in lower levels of the corresponding unphosphorylated peptides (row III of Fig. 1B), quantitation of the relative depletion of unphosphorylated peptides compared with that in the unperturbed S phase sample allowed us to estimate the changes of phosphostoichiometry in response to DNA damage, with the caveat that in tryptic peptides with multiple phosphorylation sites (e.g. SCD1 or SCD2), this depletion analysis can only estimate the total fraction of phosphorylated peptides without resolution of individual sites or specific multiphosphorylation states. For example, note that as a consequence of substantial SCD1 phosphorylation after S phase checkpoint activation (Fig. 1C, L2, c–f), the level of the corresponding unphosphorylated SCD1 fragment Rad531–17 (L2, b) was considerably reduced relative to the unperturbed WT (L1, b) or the MMS-treated mrc1-AQ rad9Δ (L3, b).
New Phosphorylation Sites, Multisites, and Estimated Stoichiometry
Altogether, for endogenous, untagged Rad53 in response to S phase MMS treatment, we detected significantly elevated in vivo phosphorylation at 14 potential Mec1/Tel1-target motifs (SQ/TQs) (Figs. 1D and 2A; supplemental Fig. S2A; supplemental Table S3), including 6 new sites (T12, T15, S53, S473, S480, and T731) not identified in previous reports (3, 28, 40–42). We also detected phosphorylation at 41 mostly DNA damage–inducible non-SQ/TQ sites (Figs. 1E and 2B; supplemental Fig. S2B; supplemental Table S3), including 15 newly identified in vivo phosphorylation sites. Interestingly, all four TQs in SCD1 and all four SQs in SCD2 were found to be phosphorylatable and simultaneously phosphorylatable (Fig. 1C; supplemental Fig. S2C; supplemental Table S2). The MS2 spectral assignments for identification of the tetraphosphorylated SCD1 and SCD2 peptides are shown in supplemental Figs. S2D and S2E, respectively.
Fig. 2.

Quantitative mass spectrometric analyses of Rad53 phosphorylation upon S phase DNA damage. A and B, bar graphs showing the relative phosphorylation levels of the DNA-damage-inducible Mec1/Tel1-target (A) and autophosphorylation (B) sites in the indicated strains after S phase MMS treatment. The asterisk (*) marks the crucial autoactivating residue T354. C, bar graphs showing the estimated phosphostoichiometries of selected Mec1/Tel1-target sites (a) and autophosphorylation sites (a) based on the relative depletion ratios of the corresponding unphosphorylated peptides. Note that “S11” presented in a small font is not a Mec1/Tel1 target site.
It is also important to note that the phosphostoichiometry varied substantially between different sites for the DNA-damage-induced Rad53 hyperphosphorylation (Fig. 2C, supplemental Fig. S2F). The closely spaced Mec1/Tel1 phosphorylation site clusters SCD1 (T5/T8/T12/T15/S24) and SCD2 (S473/S480/S485/S489) generally showed high phosphorylation stoichiometry (>50%) in response to S phase DNA damage, and the estimated phosphostoichiometry of the critical activation loop residue T354 was about 50% (Fig. 2C). Taken together, the large number of phosphorylation sites and multisites and the high phosphostoichiometry of many of these sites provide a molecular explanation for the extensive electrophoretic mobility shifts of Rad53 that have been well established previously as shown in Fig. 1A.
Identification of Kinase-dependent Rad53 Phosphorylation Events with the Kinase-deficient rad53-K227A Allele
To functionally characterize these Rad53 phosphorylation events, we performed MS analyses in MMS-treated, otherwise isogenic strain containing the kinase-deficient rad53-K227A allele (43) (this section) or checkpoint mediator-deficient mrc1-AQ, rad9Δ, and double mutant mrc1-AQ rad9Δ strains (next section) for comparison with WT.
As shown by the relative levels of MMS-inducible phosphorylation sites (Figs. 2A and 2B; supplemental Figs. S2A and S2B) and the relative depletion of the corresponding unphosphorylated peptides (Fig. 2C; supplemental Fig. S2F), defective Rad53 kinase activity caused little or no decrease in its SQ/TQ phosphorylation states. In contrast, the relative phosphorylation levels of several non-SQ/TQ sites, including that of the key activation loop residue T354 (38, 44), were greatly diminished in the kinase-defective strain (Fig. 2B), indicating that these are auto-phosphorylation sites. However, some of these autokinase sites (S185, S547, S560, T563, and S635) retained minor phosphorylation levels in the kinase-defective strain, indicative of low-level transphosphorylation by other kinases (Fig. 2B). Another group of phosphorylation sites (supplemental Fig. S2B; S198, S373, S750, S766, and S789) were partially reduced in the absence of Rad53 kinase activity but still occurred relatively efficiently, suggesting that they are transphosphorylated by other kinases whose activity is directly or indirectly modulated by Rad53 activity (e.g. Polo kinases involved in down-regulating checkpoint activity) (40, 45, 46).
Mrc1 and Rad9 Function Redundantly in Rad53 Phosphorylation Events
In the mrc1-AQ rad9Δ strain, in which both mediator pathways were disabled, all of these DNA-damage-inducible phosphorylation levels were dramatically reduced (Fig. 2; supplemental Figs. S2A–S2C and S2F), except for residually detectable low phosphorylation levels in the SCD2 domain (Figs. 2Ab and 2Ca) and the C-terminal SQSQ motif (S793/S795; supplemental Figs. S2A and S2Fa). Whereas disabling both mediators was detrimental to Rad53 hyperphosphorylation and activation, the single mutants mrc1-AQ and rad9Δ had surprisingly negligible effects on the phosphorylation levels at most SQ/TQ sites and the Rad53 autophosphorylation sites including the autoactivation site T354, as shown in the same set of figures. Thus, in response to alkylating DNA damage during DNA replication, the Mrc1 and Rad9 pathways appear to function virtually redundantly without any notable preference for directing Mec1 phosphorylation to particular Rad53 residues.
Because the lack of identifiable pathway-specific phospho-fingerprints was surprising, we tested whether the absence of one mediator pathway leads to an increased association of Rad53 with the other adaptor for S phase DNA damage signaling. Tryptic Mrc1 phosphopeptides, mostly derived from its SCD domain, were readily detectable (supplemental Table S4) and quantifiable (supplemental Fig. S3A) in Rad53 immune complexes by MS analysis after TiO2 enrichment (47). Relative to levels in the MMS-treated WT, the level of Rad53-associated Mrc1 increased by ∼3-fold in the rad9Δ strain (Fig. 3Aa; supplemental Fig. S3A). Conversely, the level of Rad9 association in similar analyses of the mrc1-AQ strain increased by only ∼30% relative to the MMS-treated WT (Fig. 3Ab; supplemental Fig. S3B). Thus, compensation for the absence of Rad9 may involve relative hyperactivation of the Mrc1 branch, whereas the Rad9 branch may already be highly engaged and competent to deal with S phase DNA damage without the need for significant hyperactivation in the absence of the Mrc1 checkpoint branch.
Fig. 3.

Effects of rad9Δ, mrc1-AQ, and rad53-4AQ on Rad53 hyperphosphorylation. A, relative association levels of Mrc1 (a) and Rad9 (b) with Rad53 during MMS-perturbed S phase in the indicated strains. See supplemental Figs. S3A and S3B for raw MS spectra used for quantitation. B, effects of SCD1, Rad9, and Mrc1 mutations on the relative phosphorylation levels (a) and the phosphostoichiometries based on depletion ratios of corresponding unphosphorylated peptides (b) for selected high-stoichiometric SQ/TQ phosphorylation sites. C, effects of SCD1, Rad9, and Mrc1 mutations on the relative phosphorylation levels of S350 and T354 within the activation loop after 45 min (a) and 135 min (b) of MMS treatment in S phase. See supplemental Fig. S3C for additional MS quantitation of Rad53 autophosphorylation. D, Western blot analysis for phosphorylation-dependent electrophoretic mobility shifts of Rad53. Clb5 and γH2A are shown as S phase and DNA damage markers, respectively. Samples were collected during α-factor G1 arrest (0 min) and at the indicated time points after release into the S phase with 0.05% MMS.
SCD1 Phospho-priming Is a Key Regulator but Not Sole Determinant of Rad53 Autoactivation
The integrity of the SCD1 TQ cluster has previously been reported to be required for efficient Rad53 activation (9, 28), and our MS results showed that all four TQs were indeed phosphorylatable during S phase DNA damage responses (Fig. 1C; supplemental Fig. S2D). To directly assess the importance of SCD1 for Rad53 trans- and autophosphorylation, we analyzed phospho-patterns in the rad53-4AQ strain, in which these four threonine residues are replaced by alanine. The 4AQ mutation had little effect on the phosphorylation of other SQ/TQ motifs, except S24 in the same SQ/TQ cluster (Fig. 3B). In contrast, it led to a profound reduction in the phosphorylation of all autophosphorylation sites (supplemental Fig. S3Ca), including the key autoactivation site pT354 as well as pS350 in the kinase activation loop, which were reduced to 25.8% ± 0.5% and 27% ± 10% of WT levels, respectively (Fig. 3Ca). This was also supported by Western blot analysis of phosphorylation-dependent electrophoretic Rad53 mobility shifts in response to S phase MMS treatment, which were considerably reduced in rad53-4AQ at 45 min, when the WT showed maximal shifts (Fig. 3D, lanes 2 and 6). In addition, the activation of Rad534AQ was considerably slower than that of the WT (lanes 1–2 and 5–8), and this strain accumulated higher γH2A levels (Fig. 3D, lower panel, lanes 5–8), suggesting increased replicative DNA damage (48). Together, these results confirm the importance of SCD1 phosphorylation for efficient Rad53 activation, but they also indicate that autoactivation can be accomplished via an alternative, though less efficient, mechanism without phospho-primed SCD1.
Rad9 but Not Mrc1 Also Functions as a Scaffold for SCD1-independent Rad53 Autoactivation
The finding that Rad53 was unaffected by single mrc1-AQ or rad9Δ mutations prompted us to test whether either of these pathways was involved in the residual Rad53 autoactivation in the rad53-4AQ strain. Interestingly, the combined absence of SCD1 phosphorylation sites and Rad9 in rad53-4AQ rad9Δ almost completely abolished the activating Rad53 autophosphorylation at T354, whereas this effect was considerably more modest in the rad53-4AQ mrc1-AQ double mutant (Fig. 3C; supplemental Fig. S3C). In line with the results of autophosphorylation, MMS-induced Rad53 mobility shifts were further reduced relative to rad53-4AQ in both double mutants at 45 min (Fig. 3D, lanes 6, 10, and 14), but the autophosphorylation and the mobility shifts increased at later time points in rad53-4AQ mrc1-AQ but not rad53-4AQ rad9Δ (Fig. 3D, lanes 9–16; supplemental Fig. S3Cb). These results suggest that Rad9 is essential for residual Rad53 activation in the absence of SCD1 phosphorylation, whereas the Mrc1 branch plays only a relatively minor role in the SCD1-independent pathway. As neither mrc1-AQ nor rad9Δ had substantial effects on the phosphorylation of other Rad53 SQ/TQ sites in rad53-4AQ (Fig. 3B), Rad9 cannot fulfill this SCD1-independent function by increasing phosphorylation of other SQ/TQ sites during the priming step. Collectively, these results suggest that, in addition to mediating phospho-priming, Rad9 is able to directly contribute to the activating autophosphorylation step as a scaffold catalyst at physiological protein levels in vivo, as proposed based on biochemical reconstitution assays in vitro (21).
We next analyzed the relative associations of Mrc1 and Rad9 with immunoprecipitated Rad53 under these conditions. Based on MS (Fig. 3Ab; supplemental Fig. S3B) and immunoprecipitation/Western blot analyses (supplemental Fig. S3D), Rad9 association with Rad53 after 45 min of S phase MMS treatment increased by ∼2.5-fold in rad53-4AQ as well as in rad53-4AQ mrc1-AQ strains relative to that in the WT strain. In contrast, under the same conditions, Mrc1 association with Rad53 increased by ∼10-fold relative to the WT in rad53-4AQ, and by almost 30-fold in rad53-4AQ rad9Δ (Fig. 3Aa; supplemental Fig. S3A). The finding that despite highly increased Mrc1 association Rad53 cannot be activated in rad53-4AQ rad9Δ cells demonstrates that Mrc1 cannot substitute for Rad9 in the residual SCD1-independent autoactivation mechanism. Together, these results indicate that Rad9 can contribute to both steps of Rad53 activation in vivo, whereas Mrc1 regulates only the priming step in response to S phase MMS damage.
Both Mrc1 and Rad9 Adaptors Are Required for Sufficient SCD1 Priming Phosphorylation when Fewer SCD1-TQ Motifs Are Preserved
It has been suggested previously that a single threonine in Rad53-SCD1, with preference for T8, is largely sufficient to prime Rad53 for activation (28). This was confirmed here, as rad53-T8-3AQ maintained 70% ± 10% of WT T354 autophosphorylation, compared with 25.8% ± 0.5% in rad53-4AQ (Fig. 4A; supplemental Fig. S4Aa). However, this was no longer true in the absence of Rad9, where the corresponding figures were 13.7% ± 0.3% in rad53-T8-3AQ rad9Δ and 3.5% ± 0.4% in rad53-4AQ rad9Δ (Fig. 4A; supplemental Fig. S4Ab). We thus quantified the phosphorylation state of SCD1 in these strains. Interestingly, the relative level of T8 phosphorylation in rad53-T8-3AQ was reduced to ∼40% when RAD9 was deleted (Fig. 4B; supplemental Fig. S4B). This result is in contrast to the WT, in which the deletion of RAD9 did not significantly affect the level of SCD1 phosphorylation (Fig. 2Aa). These results suggest that reducing the number of TQ motifs could render SCD1 a poorer Mec1 target in the absence of Rad9, leading to impaired phospho-priming.
Fig. 4.
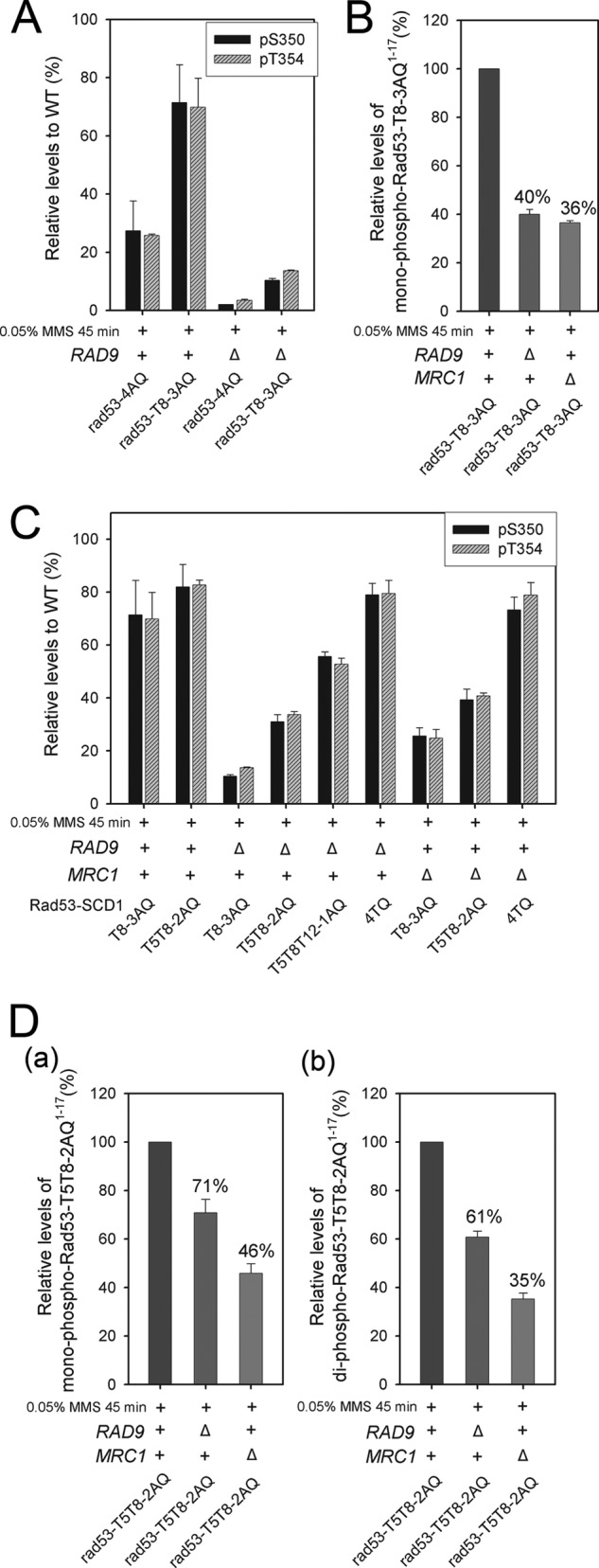
Multiple SCD1 TQ motifs are required for efficient Rad53 priming and autoactivation when either Mec1 adaptor is abolished. A, C, relative S350 and T354 phosphorylation levels in the indicated strains. B, effect of rad9Δ or mrc1Δ on the relative levels of monophosphorylated rad53-T8-3AQ1–17 fragment MENIAQPTQQSAQAAQR in Rad53T8-3AQ. See supplemental Figs. S4B and S4C for the raw extracted ion chromatogram data used for quantitation. D, effect of rad9Δ or mrc1Δ on the relative levels of mono- (a) and diphosphorylated (b) rad53-T5T8-2AQ1–17 fragment MENITQPTQQSAQAAQR in Rad53T5T8-2AQ. See supplemental Fig. S4D for the raw extracted ion chromatogram data used for quantitation.
We then examined the effect of mrc1Δ on the levels of pT8 and activation loop phosphorylation in rad53-T8-3AQ. As indicated in Fig. 4B and supplemental Fig. S4C, the induction of pT8 in rad53-T8-3AQ was also reduced to ∼36% when MRC1 was deleted. In accordance with the impaired phospho-priming, the corresponding pT354 was reduced to 25% ± 3% of WT levels in rad53-T8-3AQ mrc1Δ (Fig. 4C). Taken together, the results suggest that the presence of both mediators is required for sufficient phosphorylation of T8 in the absence of other N-terminal TQ motifs in rad53-T8-3AQ.
We next examined whether increasing the number of SCD1 TQ motifs improved Rad53 activation in the absence of Rad9 or Mrc1 by increasing the overall likelihood of SCD1 phosphorylation. In rad53-T5T8-2AQ rad9Δ or rad53-T5T8-2AQ mrc1Δ, although the relative levels of mono- and diphosphorylated SCD1 fragments were still lower than those in rad53-T5T8-2AQ with WT RAD9 and MRC1 (Fig. 4D; supplemental Fig. S4D), the availability of two TQs not only led to certain levels of diphosphorylated species (Fig. 4Db) but also alleviated the reduction of the relative levels of SCD1 monophosphorylation (Fig. 4Da) compared with the effect of rad9Δ or mrc1Δ on the rad53-T8-3AQ mutant in Fig. 4B. Furthermore, when all four SCD1 TQs were available, we observed minimal effects on SCD1 phosphorylation in rad9Δ and mrc1-AQ (Figs. 2A and 2C) or mrc1Δ (supplemental Fig. S4E). These results suggest that the presence of multiple TQs enables the efficient phosphorylation of SCD1 in the presence of a single Mec1 adaptor. In light of the effects in promoting the overall levels of priming SCD1 phosphorylation, the preservation of more TQs in SCD1 proportionally improved the autoactivating T354 phosphorylation when either Rad9 or Mrc1 was absent (Fig. 4C; supplemental Figs. S4Ab and S4Ac). Similar results were obtained for pS350 within the activation loop (Fig. 4C), as well as for most of the other high-occupancy autophosphorylation sites (data not shown). This correlation between the level of priming phosphorylation and the phosphorylation at the activation loop demonstrates that the autoactivation of Rad53 is highly regulated by the priming efficiency. Altogether, whereas an intact TQ cluster supports efficient priming phosphorylation of SCD1 when only one of the Mec1 adaptors is present, reducing the number of TQ motifs turns SCD1 into a proportionally poorer Mec1 target, which then requires both Mrc1 and Rad9 for sufficient priming phosphorylation to support substantial Rad53 autoactivation.
Multiple SCD1 Threonines Are Required for RAD53-dependent DNA Damage Survival in the Absence of the Mrc1 or Rad9 Adaptor
Because Rad53 activity plays a key role in DNA damage survival, we performed DNA damage survival assays to assess the biological significance of these findings. Similar to T354 phosphorylation, relative to results in rad53-4AQ, preserving a single SCD1 threonine in rad53-T8-3AQ (or rad53-T5-3AQ) improved MMS survival to near the WT level in the presence of both mediators (Fig. 5A). In accordance with the effects on pT354, when combined with rad9Δ, a single threonine in SCD1 (rad53-T5-3AQ rad9Δ or rad53-T8-3AQ rad9Δ) only modestly improved the extreme MMS hypersensitivity relative to rad53-4AQ rad9Δ, with further gradual survival improvements with the preservation of two (rad53-T5T8-2AQ rad9Δ) or three threonines (rad53-T5T8T12-1AQ rad9Δ or rad53-T5T8T15-1AQ rad9Δ) (Fig. 5A). Similar results were also observed for the effect of mrc1Δ on fewer TQ preserved SCD1 mutants, though with less attenuation than in the effect by rad9Δ (Fig. 5B). This is likely because Mrc1 carries out the adaptor function in response to replication stress mainly during S phase (6), whereas Rad9 is required throughout the cell cycle in response to DNA damage (2, 4, 37). Like in the regulation for Rad53 autoactivation, multiple SCD1 priming sites are required for sustaining Rad53-dependent DNA damage survival in the presence of a single Mec1 adaptor.
Fig. 5.

Multiple SCD1 TQ motifs are required for sustaining RAD53-dependent DNA damage survival in the presence of a single Mec1 adaptor. Clonal survival assays of the indicated SCD1 mutants in the presence and absence of Rad9 (A) or Mrc1 (B). Unsynchronized cultures were treated with the indicated doses of MMS for 3 h and plated on yeast peptone dextrose medium. Survival was calculated from the fraction of colonies formed relative to the untreated control.
The FHA2 Domain Plays a Major Role in SCD1 Priming Phosphorylation
The FHA-mediated binding of Rad53 to Rad9 and Mrc1 has been shown to be involved in its phospho-priming by Mec1 (3, 5, 8, 10, 16). We therefore examined the contribution of Rad53-FHA domains to the phosphorylation of Rad53. We analyzed the phospho-pattern in rad53-R70A, rad53-R605A, and rad53-R70A-R605A strains upon S phase DNA damage, when the pT binding ability of FHA1 and/or FHA2 is specifically impaired by alanine substitution of the respective critical arginine residue (27, 29, 49–51). As indicated by the Western blot analysis of Rad53 mobility shifts (Fig. 6A), hyperphosphorylation of Rad53 depends mainly on the pT binding activity of FHA2, and FHA1 is required only for residual Rad53 phosphorylation in the absence of a functional FHA2 domain.
Fig. 6.

FHA domain functions in the regulation of Rad53 hyperphosphorylation. A, Western blot analysis for phosphorylation-dependent electrophoretic mobility shifts of Rad53 in the indicated strains. Samples were collected after 45 min of 0.05% MMS treatment in S phase. B, effects of the indicated FHA mutations on the depletion ratios of the corresponding unphosphorylated Rad531–17 fragment. C, monoisotopic peak extracted ion chromatograms (with 25 ppm tolerance) for an unmodified Rad53670–679 fragment (a) and the unphosphorylated Rad531–17 fragment (b) in the indicated, unlabeled light strains relative to the normalized spiked-in heavy DNA-damaged WT references. Note that L1 and L2 peaks in (a) and (b) here correspond to those in Figs. 1Ca and 1Cb, except that only the monoisotopic peak is outputted here in an expanded view. D, effects of the indicated FHA, SCD1, and Rad9 mutations on the relative phosphorylation levels of selected high-stoichiometric autophosphorylation sites.
We then used MS-based relative depletion analysis to evaluate the effects of FHA mutations on the total phosphorylation within the SCD1 priming site cluster. As shown in Figs. 6B and 6C, the phosphorylation-dependent depletion of unphosphorylated Rad531–17 (MENITQPTQQSTQATQR) was significantly reduced in rad53-R605A, but not in rad53-R70A, suggesting that the priming phosphorylation of Rad53 by Mec1 is predominantly mediated by FHA2. Similar to the results of Rad53 mobility shifts (Fig. 6A), SCD1 priming was further reduced in rad53-R70A-R605A (Figs. 6B and 6C), indicative of a low level of redundancy between the two FHA domains in Rad53 priming. In accordance with the decreased levels of SCD1 priming phosphorylation in rad53-R605A and rad53-R70A-R605A, the relative phosphorylation levels of the autoactivating T354, as well as other high-stoichiometric autophosphorylation sites, also decreased correspondingly (Fig. 6Da). Together, these results suggest that the phospho-priming of Rad53 is predominantly contributed by the pT binding activity of FHA2, which is crucial for sustaining the hyperphosphorylation of Rad53 upon S phase DNA damage.
FHA1-pSCD1 Binding Is Not the Major Regulator for Rad53 Autoactivation
Relative to Rad53-FHA2, Rad53-FHA1 has been linked to a much wider range of interaction partners (9, 10, 28, 52–56), in particular the phosphorylated Rad53-SCD1 since the intermolecular interaction of phospho-SCD/FHA is widely believed to be a paradigm for the autoactivation mechanism of Chk2-like kinases (22–26). However, the finding that rad53-R70A did not significantly impair Rad53 autophosphorylation (Fig. 6Da), while on the other hand rad53-4AQ showed substantial impairment of Rad53 autophosphorylation (Fig. 3C; supplemental Fig. S3C), raised the possibility that FHA1-pSCD1 binding might not play a major role in the autoactivation of Rad53. To test this possibility, we studied the pattern of Rad53 autophosphorylation in the absence of Rad9 (rad53-4AQ rad9Δ and rad53-R70A rad9Δ) that allows for selective assessment of the SCD1-specific autoactivation pathway. As indicated in Fig. 6Db, whereas the SCD1 mutation almost completely abolished the total levels of pT354 and other autophosphorylation sites in rad9Δ, the FHA1 mutation caused only a modest effect when combined with rad9Δ. These results suggest that, in contrast to other Chk2-like kinases, FHA1-pSCD1 interactions cannot be the major mechanism in the pSCD1-specific Rad53 autoactivation step in response to S phase alkylation DNA damage.
Relative to rad53-R70A rad9Δ, rad53-R605A rad9Δ led to a more pronounced defect in Rad53 autophosphorylation (Fig. 6Db). This result, however, does not suggest a major role of FHA2 in the pSCD1-specific Rad53 autoactivation either, because it can be attributed to the noticeably impaired phospho-priming in the SCD1 domain in rad53-R605A rad9Δ (but not in rad53-R70A rad9Δ) (supplemental Fig. S5). The results in supplemental Fig. S5 also suggest that in the absence of Rad9, FHA2 still plays a major role in the priming of SCD1, presumably via the Mrc1 pathway.
DISCUSSION
In this study, we combined advanced mass spectrometric quantification of phosphorylation site occupancy and mutational analyses to delineate the specific roles of Rad9, Mrc1, and Rad53 SCD1 and FHA1/2 domains in the activation of endogenous Rad53 in response to S phase DNA damage treatment in vivo. The studies led to four major findings regarding the phospho-priming step: (i) As a Mec1 adaptor, Mrc1 and Rad9 can function redundantly (one is sufficient) when all four TQs are present. (ii) Reciprocally, the TQ motifs can function redundantly (one is sufficient) when both adaptors are present. (iii) However, the priming is impaired when neither the two adaptors nor the four TQ sites are intact. Under these circumstances, the priming efficiency is proportional to the number of TQ motifs. (iv) FHA2 plays a major role in the interaction of Rad53 with Rad9 or Mrc1 for Rad53 priming by Mec1. These features are incorporated into a model shown in Fig. 7A.
Fig. 7.
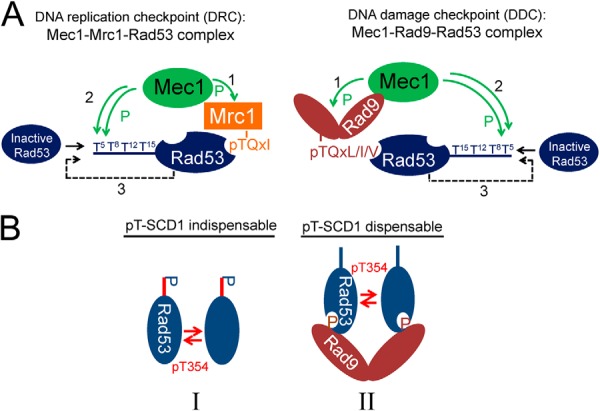
Proposed models for the priming and autoactivation mechanisms of Rad53 upon S phase alkylation DNA damage. A, proposed models for the formation of Rad53 priming complexes in Mrc1-mediated DNA replication checkpoint and Rad9-mediated DNA damage checkpoint pathways. Mrc1 and Rad9 phosphorylation by Mec1 (step 1) enable the recruitment of Rad53 for priming by Mec1 via binding to Rad53-FHA2 (step 2). The re-priming of Rad53 may occur to ensure a sufficient level of priming phosphorylation (step 3), which promotes substantial Rad53 autoactivation. B, proposed models of possible intermediate complexes I and II involved in the in vivo autoactivation of Rad53.
The studies also identified three significant mechanistic features for the autoactivation step: (i) The level of autophosphorylation is correlated with the priming efficiency (i.e. the overall phosphorylation stoichiometry of the TQ cluster). (ii) However, in contrast to the other Chk2-like kinases, the priming-mediated Rad53 activation does not seem to involve direct binding between the FHA1 domain and phosphorylated SCD1 as the major mechanism, though a minor role cannot be ruled out. (iii) In the absence of TQ sites, Rad9 but not Mrc1 can mediate an SCD1-independent activation mechanism of Rad53, though with less efficiency.
Possible Molecular Basis for the Redundant Functions of Mrc1 and Rad9 in the Phospho-priming Step
Although our finding that Rad9 and Mrc1 function redundantly during the Rad53 priming step is consistent with previous observations (6), it should be stressed that there was not a single transphosphorylation site “sentinel” specific for the Mrc1- or Rad9-associated checkpoint pathway during MMS-damaged S phase (Fig. 2A; supplemental Fig. S2A), which suggests that both scaffolds recruit Rad53 in a structurally very similar manner for priming by Mec1, as illustrated by the model shown in Fig. 7A. Interaction of Mec1-phosphorylated pTQxL/I/V in Rad9 with the Rad53-FHA2 domain has previously been proposed to be important for Rad53 priming by Mec1 (5). Interestingly, our MS analysis of Rad53-interacting protein tryptic fragments revealed several different Mrc1 peptides containing phosphorylated pTQxI motifs (supplemental Table S4), which fits with the preferential phospho-ligand specificity of Rad53-FHA2 (49). Based on this, together with our finding that the pT-binding activity of FHA2 is critical for Rad53 priming (Figs. 6A–6C), it seems reasonable to speculate that the interaction of Rad53-FHA2 with Mrc1-pTQxI or Rad9-pTQxL/I/V could likely be the major structural basis for the redundant functions of Mrc1 and Rad9 in the phospho-priming step of Rad53 activation. In contrast to the ability of Rad9 to dimerize or oligomerize in vivo (20, 21, 57), Mrc1 monomers are believed to be stably tethered to Polε and the replisome with only its Mec1-phosphorylated N-terminal region available for Rad53 binding (14), which could explain its inability to dimerize Rad53 for the autoactivation step.
Coordination of Mec1 Adaptors and Mec1 Target Sites for Optimal Priming of Rad53 by Mec1
Although the redundancy of four individual priming sites in Rad53 activation has been suggested previously based on the study of single TQ-preserved SCD1 (9, 28), here we found that both mediators, Mrc1 and Rad9, are required for sufficient phospho-priming through a single priming site in order to facilitate efficient Rad53 autoactivation (Fig. 4). Likewise, although the potential redundant roles of Mrc1 and Rad9 in S phase Rad53 activation have been reported previously (6), we found that they were no longer redundant when the number of TQ sites was limited. Taken together, our results demonstrate for the first time the coordination between the multiple adaptors and the multiple TQ sites—when the adaptor is only minimally available, the priming sites need to be maximally available, and vice versa.
Possible Intermediate Complexes of Rad53 Autoactivation
The detailed phosphorylation results obtained with various mutants allow us to propose two alternative Rad53 autoactivation intermediates for the in vivo autoactivation of Rad53 (Fig. 7B). In intermediate I, the inactive Rad53 could possibly be dimerized for activation-loop phosphorylation in a pT-SCD1-dependent manner. Note that in addition to mono-pT-SCD1-Rad53, we also observed di-, tri-, and tetra-pT-SCD1-Rad53 (Figs. 1C and 2Aa; supplemental Table S2). It remains to be established, however, whether multiphosphorylated SCD1 is also involved or even favored in this mechanism. In II, the inactive Rad53 with or without SCD1 phosphorylation can be dimerized via Rad9 scaffolds, mainly by tethering to the Mec1-phosphorylated Rad9-SCD involving its FHA domains (3, 5, 8, 10). Because SCD1 is highly phosphorylated in the WT upon MMS-induced DNA damage during S phase (Fig. 2C), and because ablation of the four SCD1 TQ motifs in rad53-4AQ has a much greater effect than deletion of Rad9 on the autoactivation of Rad53 (Figs. 2B and 3C), we believe that the pT-SCD1-indispensable intermediate I is the favorable route in sustaining Rad53 autoactivation.
Given that direct FHA1-pT-SCD1 binding does not seem to be a major mechanism in Rad53 autoactivation and that FHA2 (which is key to the priming step) does not bind pSCD1 with appreciable affinity (9, 28), we propose the possible involvement of an unknown scaffold protein in intermediate I, which either could be able to dimerize or may contain at least two separate pT-binding regions to facilitate dimerization of SCD1-phospho-primed Rad53. Potential candidates that fulfill this requirement include the dimeric 14-3-3 proteins Bmh1 and Bmh2, which have previously been shown to bind Rad53 and affect its activation (58, 59), and the multi-BRCT domain containing proteins Esc4/Rtt107 and Dbp11, which are associated with damage sites and have also been linked to multiple roles in checkpoint regulation (19, 60–62). These candidates, however, were not detected in our preliminary proteomic analyses of binding partners in rad53-4AQ and the WT (only Dun1 was shown to be stably associated with WT Rad53 but not rad534AQ), which nonetheless does not rule out their involvement in Rad53 activation, as formation of the activation complex could be very transient.
Implication for the in Vivo Activation Mechanisms of Rad53 and Other Chk2-like Kinases
Here we have combined the power of yeast genetics to introduce specific point mutations without the need for residual selection markers that may indirectly affect protein levels and functions (63), a Rad53-specific monoclonal antibody to efficiently purify the endogenous untagged protein, and advanced mass spectrometry to study Rad53 activation under physiological conditions. This approach has enabled the detection of numerous novel Rad53 in vivo phosphorylation sites for which the kinases responsible may have been limiting under substrate overexpression conditions, and the results suggest that the crucial autoactivation step of Chk2-like kinases does not always need to involve direct phospho-SCD1/FHA domain interactions. Although the important finding we have uncovered for Rad53 might be unique for those fungal Chk2-like kinases that contain duplicate SCD-FHA modules, the notion that physiological analyses are crucial for the study of Rad53 activation might prompt similar analyses to revisit the role of individual phosphorylation sites in the in vivo regulation of mammalian Chk2-like kinases. Regardless of whether the mechanisms deciphered here are applicable to mammalian checkpoint responses, all proteins studied here—Rad53, Mrc1, and Rad9—are highly conserved in human pathogenic fungi such as Candida, where they are involved in regulating the switch to more invasive and drug-resistant hyphal growth forms (64, 65), and where they might represent potential drug targets to stop the spread of nosocomial fungal infections.
Supplementary Material
Acknowledgments
MS experiments were performed at the Genomics Research Center and the Core Facility for Protein Structural Analysis (CFPSA), supported by the National Science Council of Taiwan. Peptides were obtained from the Peptide Synthesis Core of the Institute of Biological Chemistry, Academia Sinica.
Footnotes
Author contributions: E.C., J.H., and M.T. designed research; E.C., N.C.H., and S.W. performed research; E.C., N.C.H., S.W., and A.P. contributed new reagents or analytic tools; E.C., N.C.H., J.H., and M.T. analyzed data; E.C., J.H., and M.T. wrote the paper.
* Work in M.D.T.'s lab was supported by Grant EX95–9508NI from the National Health Research Institute (NHRI) and an Academia Sinica Investigator Award. Work in J.H.'s lab was supported by grants and fellowships from the National Health and Medical Research Council of Australia (NHMRC), and in part by the Victorian Government's Operational Infrastructure Support Program.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- FHA
- forkhead-associated
- MMS
- methyl methanesulfonate
- pT
- phospho-threonine
- SCD
- SQ/TQ cluster domain
- SILAC
- stable isotope labeling by amino acids in cell culture
- WT
- wild type.
REFERENCES
- 1. Nyberg K. A., Michelson R. J., Putnam C. W., Weinert T. A. (2002) Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 36, 617–656 [DOI] [PubMed] [Google Scholar]
- 2. Melo J., Toczyski D. (2002) A unified view of the DNA-damage checkpoint. Curr. Opin. Cell Biol. 14, 237–245 [DOI] [PubMed] [Google Scholar]
- 3. Sweeney F. D., Yang F., Chi A., Shabanowitz J., Hunt D. F., Durocher D. (2005) Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr. Biol. 15, 1364–1375 [DOI] [PubMed] [Google Scholar]
- 4. Osborn A. J., Elledge S. J. (2003) Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 17, 1755–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schwartz M. F., Duong J. K., Sun Z., Morrow J. S., Pradhan D., Stern D. F. (2002) Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol. Cell 9, 1055–1065 [DOI] [PubMed] [Google Scholar]
- 6. Alcasabas A. A., Osborn A. J., Bachant J., Hu F., Werler P. J., Bousset K., Furuya K., Diffley J. F., Carr A. M., Elledge S. J. (2001) Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 3, 958–965 [DOI] [PubMed] [Google Scholar]
- 7. Ma J.-L., Lee S.-J., Duong J. K., Stern D. F. (2006) Activation of the checkpoint kinase Rad53 by the phosphatidyl inositol linase-like kinase Mec1. J. Biol. Chem. 281, 3954–3963 [DOI] [PubMed] [Google Scholar]
- 8. Schwartz M. F., Lee S. J., Duong J. K., Eminaga S., Stern D. F. (2003) FHA domain-mediated DNA checkpoint regulation of Rad53. Cell Cycle 2, 384–396 [PubMed] [Google Scholar]
- 9. Lee S.-J., Schwartz M. F., Duong J. K., Stern D. F. (2003) Rad53 phosphorylation site clusters are important for Rad53 regulation and signaling. Mol. Cell Biol. 23, 6300–6314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun Z., Hsiao J., Fay D. S., Stern D. F. (1998) Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science 281, 272–274 [DOI] [PubMed] [Google Scholar]
- 11. Mahajan A., Yuan C., Lee H., Chen E. S. W., Wu P.-Y., Tsai M.-D. (2008) Structure and function of the phosphothreonine-specific FHA domain. Sci. Signal. 1, re12. [DOI] [PubMed] [Google Scholar]
- 12. Antoni L., Sodha N., Collins I., Garrett M. D. (2007) CHK2 kinase: cancer susceptibility and cancer therapy—two sides of the same coin? Nat. Rev. Cancer 7, 925–936 [DOI] [PubMed] [Google Scholar]
- 13. Traven A., Heierhorst J. (2005) SQ/TQ cluster domains: concentrated ATM/ATR kinase phosphorylation site regions in DNA-damage-response proteins. Bioessays 27, 397–407 [DOI] [PubMed] [Google Scholar]
- 14. Lou H., Komata M., Katou Y., Guan Z., Reis C. C., Budd M., Shirahige K., Campbell J. L. (2008) Mrc1 and DNA polymerase [var epsilon] function together in linking DNA replication and the S phase checkpoint. Mol. Cell 32, 106–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Szyjka S. J., Viggiani C. J., Aparicio O. M. (2005) Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol. Cell 19, 691–697 [DOI] [PubMed] [Google Scholar]
- 16. Chen S.-H., Zhou H. (2009) Reconstitution of Rad53 activation by Mec1 through adaptor protein Mrc1. J. Biol. Chem. 284, 18593–18604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hammet A., Magill C., Heierhorst J., Jackson S. P. (2007) Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 8, 851–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wysocki R., Javaheri A., Allard S., Sha F., Côté J., Kron S. J. (2005) Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol. Cell. Biol. 25, 8430–8443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pfander B., Diffley J. F. X. (2011) Dpb11 coordinates Mec1 kinase activation with cell cycle-regulated Rad9 recruitment. EMBO J. 30, 4897–4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Granata M., Lazzaro F., Novarina D., Panigada D., Puddu F., Abreu C. M., Kumar R., Grenon M., Lowndes N. F., Plevani P., Muzi-Falconi M. (2010) Dynamics of Rad9 chromatin binding and checkpoint function are mediated by its dimerization and are cell cycle–regulated by CDK1 activity. PLoS Genet. 6, e1001047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gilbert C. S., Green C. M., Lowndes N. F. (2001) Budding yeast Rad9 is an ATP-dependent Rad53 activating machine. Mol. Cell 8, 129–136 [DOI] [PubMed] [Google Scholar]
- 22. Ahn J.-Y., Li X., Davis H. L., Canman C. E. (2002) Phosphorylation of threonine 68 promotes oligomerization and autophosphorylation of the Chk2 protein kinase via the forkhead-associated domain. J. Biol. Chem. 277, 19389–19395 [DOI] [PubMed] [Google Scholar]
- 23. Li J., Taylor I. A., Lloyd J., Clapperton J. A., Howell S., MacMillan D., Smerdon S. J. (2008) Chk2 oligomerization studied by phosphopeptide ligation. J. Biol. Chem. 283, 36019–36030 [DOI] [PubMed] [Google Scholar]
- 24. Oliver A. W., Paul A., Boxall K. J., Barrie S. E., Aherne G. W., Garrett M. D., Mittnacht S., Pearl L. H. (2006) Trans-activation of the DNA-damage signalling protein kinase Chk2 by T-loop exchange. EMBO J. 25, 3179–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu Y.-j., Davenport M., Kelly T. J. (2006) Two-stage mechanism for activation of the DNA replication checkpoint kinase Cds1 in fission yeast. Genes Dev. 20, 990–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu X., Tsvetkov L. M., Stern D. F. (2002) Chk2 activation and phosphorylation-dependent oligomerization. Mol. Cell. Biol. 22, 4419–4432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pike B. L., Yongkiettrakul S., Tsai M.-D., Heierhorst J. (2003) Diverse but overlapping functions of the two forkhead-associated (FHA) domains in Rad53 checkpoint kinase activation. J. Biol. Chem. 278, 30421–30424 [DOI] [PubMed] [Google Scholar]
- 28. Lee H., Yuan C., Hammet A., Mahajan A., Chen E. S., Wu M. R., Su M. I., Heierhorst J., Tsai M. D. (2008) Diphosphothreonine-specific interaction between an SQ/TQ cluster and an FHA domain in the Rad53-Dun1 kinase cascade. Mol. Cell 30, 767–778 [DOI] [PubMed] [Google Scholar]
- 29. Pike B. L., Tenis N., Heierhorst J. (2004) Rad53 kinase activation-independent replication checkpoint function of the N-terminal forkhead-associated (FHA1) domain. J. Biol. Chem. 279, 39636–39644 [DOI] [PubMed] [Google Scholar]
- 30. Old W. M., Shabb J. B., Houel S., Wang H., Couts K. L., Yen C.-Y., Litman E. S., Croy C. H., Meyer-Arendt K., Miranda J. G., Brown R. A., Witze E. S., Schweppe R. E., Resing K. A., Ahn N. G. (2009) Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol. Cell 34, 115–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guo X., Ward M. D., Tiedebohl J. B., Oden Y. M., Nyalwidhe J. O., Semmes O. J. (2010) Interdependent phosphorylation within the kinase domain T-loop regulates CHK2 activity. J. Biol. Chem. 285, 33348–33357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scales T., Derkinderen P., Leung K.-Y., Byers H., Ward M., Price C., Bird I., Perera T., Kellie S., Williamson R., Anderton B., Reynolds C. H. (2011) Tyrosine phosphorylation of tau by the SRC family kinases Lck and Fyn. Mol. Neurodegener. 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jin L. L., Tong J., Prakash A., Peterman S. M., St-Germain J. R., Taylor P., Trudel S., Moran M. F. (2010) Measurement of protein phosphorylation stoichiometry by selected reaction monitoring mass spectrometry. J. Proteome Res. 9, 2752–2761 [DOI] [PubMed] [Google Scholar]
- 34. Cordon-Preciado V., Ufano S., Bueno A. (2006) Limiting amounts of budding yeast Rad53 S-phase checkpoint activity results in increased resistance to DNA alkylation damage. Nucleic Acids Res. 34, 5852–5862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hoch N. C., Chen E. S.-W., Buckland R., Wang S.-C., Fazio A., Hammet A., Pellicioli A., Chabes A., Tsai M.-D., Heierhorst J. (2013) Molecular basis of the essential S phase function of the Rad53 checkpoint kinase. Mol. Cell. Biol. 33, 3202–3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 37. Pellicioli A., Foiani M. (2005) Signal transduction: how Rad53 kinase is activated. Curr. Biol. 15, R769–R771 [DOI] [PubMed] [Google Scholar]
- 38. Fiorani S., Mimun G., Caleca L., Piccini D., Pellicioli A. (2008) Characterization of the activation domain of the Rad53 checkpoint kinase. Cell Cycle 7, 493–499 [DOI] [PubMed] [Google Scholar]
- 39. Geiger T., Wisniewski J. R., Cox J., Zanivan S., Kruger M., Ishihama Y., Mann M. (2011) Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat. Protoc. 6, 147–157 [DOI] [PubMed] [Google Scholar]
- 40. Schleker T., Shimada K., Sack R., Pike B. L., Gasser S. M. (2010) Cell cycle-dependent phosphorylation of Rad53 kinase by Cdc5 and Cdc28 modulates checkpoint adaptation. Cell Cycle 9, 350–363 [DOI] [PubMed] [Google Scholar]
- 41. Smolka M. B., Albuquerque C. P., Chen S.-H., Schmidt K. H., Wei X. X., Kolodner R. D., Zhou H. (2005) Dynamic changes in protein-protein interaction and protein phosphorylation probed with amine-reactive isotope tag. Mol. Cell. Proteomics 4, 1358–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Albuquerque C. P., Smolka M. B., Payne S. H., Bafna V., Eng J., Zhou H. (2008) A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol. Cell. Proteomics 7, 1389–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fay D. S., Sun Z., Stern D. F. (1997) Mutations in SPK1/RAD53 that specifically abolish checkpoint but not growth-related functions. Curr. Genet. 31, 97–105 [DOI] [PubMed] [Google Scholar]
- 44. Chen S.-H., Smolka M. B., Zhou H. (2007) Mechanism of Dun1 activation by Rad53 phosphorylation in Saccharomyces cerevisiae. J. Biol. Chem. 282, 986–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vidanes G. M., Sweeney F. D., Galicia S., Cheung S., Doyle J. P., Durocher D., Toczyski D. P. (2010) CDC5 inhibits the hyperphosphorylation of the checkpoint kinase Rad53, leading to checkpoint adaptation. PLoS Biol. 8, e1000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Donnianni R. A., Ferrari M., Lazzaro F., Clerici M., Tamilselvan Nachimuthu B., Plevani P., Muzi-Falconi M., Pellicioli A. (2010) Elevated levels of the polo kinase Cdc5 override the Mec1/ATR checkpoint in budding yeast by acting at different steps of the signaling pathway. PLoS Genet. 6, e1000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sugiyama N., Masuda T., Shinoda K., Nakamura A., Tomita M., Ishihama Y. (2007) Phosphopeptide enrichment by aliphatic hydroxy acid-modified metal oxide chromatography for nano-LC-MS/MS in proteomics applications. Mol. Cell. Proteomics 6, 1103–1109 [DOI] [PubMed] [Google Scholar]
- 48. Cobb J. A., Schleker T., Rojas V., Bjergbaek L., Tercero J. A., Gasser S. M. (2005) Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev. 19, 3055–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Durocher D., Taylor I. A., Sarbassova D., Haire L. F., Westcott S. L., Jackson S. P., Smerdon S. J., Yaffe M. B. (2000) The molecular basis of FHA domain:phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol. Cell 6, 1169–1182 [DOI] [PubMed] [Google Scholar]
- 50. Liao H., Yuan C., Su M.-I., Yongkiettrakul S., Qin D., Li H., Byeon I.-J. L., Pei D., Tsai M.-D. (2000) Structure of the FHA1 domain of yeast Rad53 and identification of binding sites for both FHA1 and its target protein Rad9. J. Mol. Biol. 304, 941–951 [DOI] [PubMed] [Google Scholar]
- 51. Liao H., Byeon I.-J. L., Tsai M.-D. (1999) Structure and function of a new phosphopeptide-binding domain containing the FHA2 of rad53. J. Mol. Biol. 294, 1041–1049 [DOI] [PubMed] [Google Scholar]
- 52. Smolka M. B., Chen S.-H., Maddox P. S., Enserink J. M., Albuquerque C. P., Wei X. X., Desai A., Kolodner R. D., Zhou H. (2006) An FHA domain-mediated protein interaction network of Rad53 reveals its role in polarized cell growth. J. Cell Biol. 175, 743–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Aucher W., Becker E., Ma E., Miron S., Martel A., Ochsenbein F., Marsolier-Kergoat M.-C., Guerois R. (2010) A strategy for interaction site prediction between phospho-binding modules and their partners identified from proteomic data. Mol. Cell. Proteomics 9, 2745–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guillemain G., Ma E., Mauger S., Miron S., Thai R., Guerois R., Ochsenbein F., Marsolier-Kergoat M.-C. (2007) Mechanisms of checkpoint kinase Rad53 inactivation after a double-strand break in Saccharomyces cerevisiae. Mol. Cell Biol. 27, 3378–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pike B. L., Yongkiettrakul S., Tsai M. D., Heierhorst J. (2004) Mdt1, a novel Rad53 FHA1 domain-interacting protein, modulates DNA damage tolerance and G(2)/M cell cycle progression in Saccharomyces cerevisiae. Mol. Cell Biol. 24, 2779–2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Matthews L. A., Jones D. R., Prasad A. A., Duncker B. P., Guarné A. (2012) Saccharomyces cerevisiae Dbf4 has unique fold necessary for interaction with Rad53 kinase. J. Biol. Chem. 287, 2378–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Usui T., Foster S. S., Petrini J. H. J. (2009) Maintenance of the DNA-damage checkpoint requires DNA-damage-induced mediator protein oligomerization. Mol. Cell 33, 147–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Usui T., Petrini J. H. J. (2007) The Saccharomyces cerevisiae 14-3-3 proteins Bmh1 and Bmh2 directly influence the DNA damage-dependent functions of Rad53. Proc. Natl. Acad. Sci. U.S.A. 104, 2797–2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lottersberger F., Rubert F., Baldo V., Lucchini G., Longhese M. P. (2003) Functions of Saccharomyces cerevisiae 14-3-3 proteins in response to DNA damage and to DNA replication stress. Genetics 165, 1717–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Roberts T. M., Zaidi I. W., Vaisica J. A., Peter M., Brown G. W. (2008) Regulation of Rtt107 recruitment to stalled DNA replication forks by the cullin Rtt101 and the Rtt109 acetyltransferase. Mol. Biol. Cell 19, 171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mordes D. A., Nam E. A., Cortez D. (2008) Dpb11 activates the Mec1–Ddc2 complex. Proc. Natl. Acad. Sci. U.S.A. 105, 18730–18734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rouse J. (2004) Esc4p, a new target of Mec1p (ATR), promotes resumption of DNA synthesis after DNA damage. EMBO J. 23, 1188–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Breslow D. K., Cameron D. M., Collins S. R., Schuldiner M., Stewart-Ornstein J., Newman H. W., Braun S., Madhani H. D., Krogan N. J., Weissman J. S. (2008) A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat. Methods 5, 711–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jiang Y. W., Kang C. M. (2003) Induction of S. cerevisiae filamentous differentiation by slowed DNA synthesis involves Mec1, Rad53 and Swe1 checkpoint proteins. Mol. Biol. Cell 14, 5116–5124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shi Q.-M., Wang Y.-M., Zheng X.-D., Teck Ho Lee R., Wang Y. (2007) Critical role of DNA checkpoints in mediating genotoxic-stress-induced filamentous growth in Candida albicans. Mol. Biol. Cell 18, 815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.