

Abstract
Proteolysis is an irreversible post-translational modification that affects intra- and intercellular communication by modulating the activity of bioactive mediators. Key to understanding protease function is the system-wide identification of cleavage events and their dynamics in physiological contexts. Despite recent advances in mass spectrometry-based proteomics for high-throughput substrate screening, current approaches suffer from high false positive rates and only capture single states of protease activity. Here, we present a workflow based on multiplexed terminal amine isotopic labeling of substrates for time-resolved substrate degradomics in complex proteomes. This approach significantly enhances confidence in substrate identification and categorizes cleavage events by specificity and structural accessibility of the cleavage site. We demonstrate concomitant quantification of cleavage site spanning peptides and neo-N and/or neo-C termini to estimate relative ratios of noncleaved and cleaved forms of substrate proteins. By applying this strategy to dissect the matrix metalloproteinase 10 (MMP10) substrate degradome in fibroblast secretomes, we identified the extracellular matrix protein ADAMTS-like protein 1 (ADAMTSL1) as a direct MMP10 substrate and revealed MMP10-dependent ectodomain shedding of platelet-derived growth factor receptor alpha (PDGFRα) as well as sequential processing of type I collagen. The data have been deposited to the ProteomeXchange Consortium with identifier PXD000503.
Historically regarded as a mechanism for unspecific degradation of proteins, proteolysis is now recognized as a specific irreversible post-translational modification that affects major intra- and intercellular signaling processes (1, 2). Proteases specifically process bioactive proteins, their receptors, and associated proteins in an interconnected interaction network termed the protease web (3). Dysregulation of the protease web might cause or result from pathologies, such as impaired tissue repair, cancer and neurodegenerative diseases. Therefore, a better understanding of the functions of individual proteases and their interconnections within proteolytic networks is a prerequisite for exploiting proteases as targets for therapeutic intervention (4).
To address this issue, several powerful technologies have been developed for the system-wide discovery of protease substrates, i.e. substrate degradomes, in complex and active proteomes (5, 6). A common principle of these mass spectrometry-based methods is the enrichment and monitoring of N-terminal peptides (protein neo-N termini) that are newly generated by a test protease (7). Protein N termini are enriched from complex proteomes either by chemical tagging and affinity resins (positive selection) or by depletion of internal peptides (negative selection) (7). Both principles have been successfully applied in various studies to characterize N-terminomes and to identify protease substrates using in vitro or cell-based systems and more recently also in vivo (8, 9). Negative enrichment approaches were further extended to the analysis of protein C termini (10, 11) and have the general advantage of recording data on naturally blocked (e.g. acetylated) N termini and internal peptides in the same experiment (8).
Even if successful in identifying novel proteolytic cleavage events, which could also be validated by orthogonal methods, high-throughput substrate discovery approaches potentially suffer from high numbers of false positive identifications, particularly when employing in vitro systems (12). These have been reduced by monitoring abundances of N-terminal peptides at multiple time points after incubation of a proteome with a test protease (12). In this SILAC-based approach the authors efficiently distinguished critical from bystander cleavages, but it was limited to three time points. Therefore, it did not allow recording kinetic profiles of the relative abundance of N-terminal peptides that are required for determination of apparent kinetic parameters for processing events. Agard et al. elegantly overcame this limitation by use of selected reaction monitoring (SRM)1 in combination with a positive N-terminal enrichment platform and determined apparent catalytic efficiencies for hundreds of caspase cleavage events in parallel (13). In a similar approach the same group characterized cellular responses to pro-apoptotic cancer drugs by recording time-courses for caspase-generated neo-N termini (14). Although very powerful and highly accurate in quantification, this method strongly exploited the canonical cleavage specificity of caspases after aspartate residues and required a two-stage process involving two types of mass spectrometers. Hence, it would be desirable to monitor the time-resolved generation of neo-N termini in complex proteomes in a single experiment by a simple and robust workflow in an unbiased manner.
The development of such an analysis platform would require a reliable method for the system-wide characterization of protein N termini that is easy to perform, fast and highly multiplexible. All these criteria are met by iTRAQ-terminal amine isotopic labeling of substrates (TAILS), a multiplex N-terminome analysis technique that has been applied in 2plex and 4plex experiments to map the matrix metalloproteinase (MMP) 2 and MMP9 substrate degradomes in vitro (15) and most recently to quantitatively analyze the proteome and N-terminome of inflamed mouse skin in the presence or absence of the immune-modulatory protease MMP2 in vivo (8).
Here, we exploited the multiplex capabilities of iTRAQ-TAILS by use of 8plex-iTRAQ reagents to monitor the generation of neo-N-terminal peptides by a test protease in complex samples over time. First, using GluC as a test protease with canonical cleavage specificity, we established a workflow for time-resolved substrate degradomics. Recording kinetic profiles significantly increased the confidence in identified cleavage events compared with binary systems and categorized primary cleavage specificities as well as secondary structure elements based on clusters of processing events with different efficiencies. By including data from before N-terminal enrichment, we extended our analysis to neo-C-terminal peptides and concomitantly monitored the generation of neo-N termini and neo-C termini as well as the decrease in abundance of the tryptic peptides spanning the cleavage sites in the same experiment. Next, we applied this approach to the time-resolved analysis of the hardly elucidated substrate degradome of matrix metalloproteinase 10 (MMP10). This important wound- and tumor-related protease is secreted by proliferating and migrating keratinocytes at the wound edge in close proximity to dermal fibroblasts and is also highly expressed in aggressive tumor cells (16–18). Our analysis revealed MMP10-dependent shedding of the platelet-derived growth factor receptor alpha (PDGFRα), processing of ADAMTS-like protein 1 (ADAMTSL1) and multiple cleavages of type I collagen, which could be validated and classified by time-resolved abundance profiles of their corresponding neo-N termini.
EXPERIMENTAL PROCEDURES
Cell Growth and Preparation of Secreted Proteins
Mmp10−/− mice were obtained from the Mutant Mouse Regional Resource Center (MMRRC) at UC Davis (strain# 011737-UCD), and immortalized murine embryonic fibroblasts (MEF) isolated from 13.5 day old embryos were established by continuous passaging following the 3T3 protocol (19). Balb/c 3T3 fibroblasts were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 1% penicillin/streptomycin and 10% fetal bovine serum (FBS), Mmp10−/− MEFs were cultured in the same medium plus 0.1 mm nonessential amino acids and 55 μm β-mercaptoethanol, and secreted proteins were collected as described previously (20). Briefly, cells were washed with phosphate buffered saline (PBS), incubated in serum-free DMEM lacking phenol red for 24 h, and supernatants supplemented with protease inhibitors (0.5 mm PMSF and 1 mm EDTA) were concentrated by ultrafiltration with 3 kDa cutoff membranes (Amicon (Millipore, Billerica, MA)). After exchange of medium to 50 mm HEPES buffer (pH 7.8), the Bradford assay (BioRad, Hercules, CA) was used to determine protein concentration, which was adjusted to 2 mg/ml protein and 250 mm HEPES (pH 7.8) with 1 m HEPES, pH 7.8. Aliquots of 0.25 mg of concentrated culture supernatants were prepared and stored at −80 °C until further use.
Digestion of Secretomes with Proteases
Concentrated (∼500×) Balb/c 3T3 secretomes were incubated with GluC (Roche, Mannheim, Germany) at an enzyme:protein ratio of 1:100 (w/w) for up to 16 h at 37 °C. Auto-activated (16 h at 37 °C) MMP10 (R&D Systems, Minneapolis, MN) was added to concentrated (∼600×) supernatants from Mmp10−/− MEFs (220 nm final concentration; 1:170 enzyme:protein ratio (w/w)) and incubated for up to 16 h at 37 °C in the presence of 10 mm CaCl2 and 100 mm NaCl. Control samples (no protease) were incubated with an equivalent volume of buffer only, and protein preparations from the same batch were used for each condition of individual experiments.
8plex-iTRAQ-TAILS Protocol
iTRAQ-TAILS analysis followed the previously published protocol (21) but used 8plex- (22) instead of 4plex-iTRAQ reagents. Aliquots of 0.25 mg of protease-treated or control secretomes were labeled using 8plex-iTRAQ reagents in a 1:4 protein:iTRAQ (w/w) ratio. After tryptic digest (Trypsin Gold (Promega, Madison, WI); 1:100 enzyme:protein (w/w)) 10% of the peptide mixture were removed for analysis prior to enrichment for N-terminal peptides. The remaining 90% were depleted for internal tryptic peptides using a 435 kDa HPG-ALD polymer (available without commercial or company restriction from Flintbox Innovation Network, The Global Intellectual Exchange and Innovation Network (http://www.flintbox.com/public/project/1948/)) in threefold excess (w/w) and under previously described conditions (21). Samples were frozen and stored at −20 °C until further use.
Peptide Fractionation and LC-MS/MS Analysis
Peptide mixtures from both prior and after enrichment for protein N termini were fractionated by strong cation exchange chromatography (SCX) using a PolySULFOETHYL ATM 100 mm × 2.1 mm, 5 μm, 200 Å column (PolyLC Inc., Columbia, MD) and an Agilent Technologies 1100 series HPLC system (Agilent Technologies, Santa Clara, CA). Peptides were bound to the column, washed with 100% buffer A (10 mm potassium phosphate, 25% acetonitrile, pH 2.7) for 60 min and eluted by gradually increasing buffer B (buffer A + 0.5 m potassium chloride) to 5% within 5 min, then to 35% within 35 min, and finally to 100% within 10 min. Twenty-seven fractions were collected and pooled to eight fractions during sample clean up using C18 OMIX tips (Agilent Technologies, Santa Clara, CA) and according to the 214/280 nm absorption chromatogram. Peptide fractions from GluC and MMP10 experiments were analyzed on an LTQ-Orbitrap XL (XL) or an LTQ-Orbitrap Velos (Velos) mass spectrometer (Thermo Fischer Scientific, Bremen, Germany), respectively, coupled to an Eksigent-Nano-HPLC system (Eksigent Technologies, Dublin, CA). One microgram of peptides were loaded onto a self-made tip column (XL: 75 μm × 70 mm; Velos: 75 μm × 150 mm) packed with C18 material (AQ, 3 μm 200 Å, Bischoff GmbH, Leonberg, Germany) and eluted with a flow rate of 200 nl/min using a gradient from 0 to 37% of acetonitrile in 55 min (Velos: 250 nl/min, 35% acetonitrile in 62 min). Full scan MS spectra (XL: 300–2000 m/z; Velos: 300–1700 m/z) were acquired with a resolution of 60000 (XL) or 30000 (Velos) at 400 m/z after accumulation to a target value of 5E5 (XL) or 1E6 (Velos). Collision induced dissociation (CID) MS/MS spectra were recorded in a data dependent manner in the ion trap from the three (XL) or eight (Velos) most intense signals above a threshold of 500 (XL) or 1000 (Velos) using a normalized collision energy of 35% and an activation time of 30 ms (XL) or 10 ms (Velos). To obtain spectral information in the region of iTRAQ report ions, the same precursors were further fragmented using higher energy collisional dissociation (HCD) with a normalized collision energy of 42% (XL) or 45% (Velos), and the spectra were recorded at a resolution of 7500 at 400 m/z. Charge state screening was enabled, and single charge states were rejected. Precursor masses already selected for MS/MS were excluded for further selection for 90 s (XL) or 45 s (Velos), and the exclusion window was set to 20 ppm.
MS Data Analysis
Mascot Distiller v2.4.3.3 (Matrix Science, Boston, MA) was used to extract peak lists from raw files and for merging of corresponding CID/HCD spectra pairs. Peak lists (mgf) were searched by Mascot v2.3 search engine (Matrix Science, Boston, MA) against a mouse UniProtKB database (release 2012_03; 54232 entries), to which reversed decoy sequences as well as sequences for common contaminants and either for GluC or for human MMP10, respectively, had been added and with following parameters: semi-Arg-C for enzyme specificity allowing up to two missed cleavages; carbamidomethyl(C), iTRAQ(K) as fixed modifications; acetyl(N-term), iTRAQ(N-term), oxidation(M), and deamidation(NQ) as variable modifications; parent mass error at 10 ppm, fragment mass error at 0.8 Da. For GluC experiments an additional search was performed using the same parameters but with semi-GluC as enzyme and allowing up to five missed cleavages. The Trans-Proteomic Pipeline (TPP v4.6, rev 1, Build 201212051643) (23) was used to secondary validate Mascot search results and to compile a single peptide list from all peptide fractions obtained from both the pre-pullout and the pullout samples. First, data were processed by PeptideProphet setting the “minimum peptide length” to 1, using “accurate mass binning” and omitting the “NTT model.” Next, iProphet was employed for additional validation and for combining PeptideProphet results from multiple searches, and only peptides with an iProphet probability of ≥0.9 (corresponding to false discovery rate (decoy) of <1%) were included in subsequent analyses. For relative quantification iTRAQ reporter ion intensities were extracted from mgf files using a modified version of i-Tracker (24) with a mass tolerance of 0.1 Da and purity corrections supplied by the iTRAQ manufacturer and assigned to filtered peptides.
Peptide Annotation, Clustering, and Generation of Sequence Logos
Multiple CIDs were merged, and peptides were assigned to proteins and annotated for their position in the processed mature protein using the CLIPPER analysis pipeline (25). iTRAQ reporter ion intensities were normalized to the sum of all channels and a maximum of 1.0 for the highest value, and abundance clustering was performed using the Mfuzz package for R (26) and the mestimate() function to determine optimal fuzzication parameters (27). The R statistical environment (version 2.15.3) in combination with RStudio (http://www.r-project.org/) and Prism 5.0 (GraphPad Software) were used for curve fitting, and ROC curve analysis was performed using the ROCR package (28). Cleavage site specificities were calculated by WebPICS (29), and logos were generated using IceLogo (30). Secondary structures were predicted using a local installation of the PROTEUS2 algorithm (31) and visualized using WebLogo (32). Systems Biology Graphical Notation (SBGN) map was generated using CellDesigner (33, 34).
Immunoblot Analysis and Zymography
Concentrated cell culture supernatants from Mmp10−/− MEFs were incubated with recombinant MMP10 as described for iTRAQ-TAILS experiments. For immunoblot analyses equal amounts of total protein were resolved by SDS-PAGE, transferred to nitrocellulose membranes (Whatman, Clifton, NJ) and probed with antibodies directed against collagen I (Chemicon, Temecula, CA), MMP2 (Novus Biologicals, Littleton, CO) or PDGFRα (R&D Systems, Minneapolis, MN). Recombinant ADAMTSL1 was analyzed using an antibody raised against a peptide in the thrombospondin domain 2 (35). Bands were visualized using horseradish peroxidase conjugated secondary antibodies and the enhanced chemiluminescence (ECL) reaction by exposing the membranes to an x-ray film (Fuji Medical, Tokyo, Japan).
Gelatinolytic activity in sectretomes incubated with MMP10 was visualized by gelatin zymography using SDS-PAGE with 1 mg/ml gelatin, and casein zymography was performed with gels containing 1 mg/ml casein.
Substrate Cleavage Assays
Recombinant human PDGFRα (residues 24–524) was purchased from R&D Systems, Minneapolis, MN, and recombinant human ADAMTSL1 (isoform 1) was expressed and purified as described previously (35). Auto-activated MMP10 (16 h at 37 °C) was incubated with the candidate substrates in 50 mm Tris-HCl, 200 mm NaCl and 5 mm CaCl2 for 16 h at 37 °C. Reaction products were analyzed by SDS-PAGE and visualized by silver stain or immunoblot.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (36) with the data set identifier PXD000503.
RESULTS
Experimental Setup and Peptide Classification
To establish a workflow for the time-resolved analysis of proteolysis in complex samples we subjected cell culture supernatants from Balb/c 3T3 mouse fibroblasts that had been incubated with the endoproteinase GluC (Staphylococcus aureus protease V8) as test protease for 1, 2, 4, 8, and 16 h or buffer alone (0, 12, and 16 h controls) to 8plex-iTRAQ-TAILS analysis (Fig. 1A). By combining samples prior to and after enrichment of N-terminal peptides, we identified a total of 3017 semi-tryptic N-terminal or fully tryptic internal peptides (supplemental Table S1), of which 1645 could be quantified either via an N-terminal or a lysine iTRAQ-label. GluC specifically cleaves substrate proteins C-terminal to glutamate (E) and with a much lower efficiency after aspartate (D) residues (37, 38). Trypsin as the working protease generated defined quantifiable peptides of two major classes (Fig. 1B): (1) cleavage events: semi-GluC, semi-tryptic neo-N termini (E/D.iTRAQX-R) and semi-tryptic, semi-GluC neo-C termini (R.X-KiTRAQ-X-E/D); (2) non-cleavage events: non-GluC, semi-tryptic natural N termini (iTRAQ(Ac)M(X).X-(KiTRAQ)-R) and fully tryptic internal peptides (R.X-KiTRAQ-X-R). Because incubation of a nondenatured proteome with GluC yields only incomplete digestion even after 16 h (20) resulting in numerous missed cleavages, peptides of all classes could be subdivided into those harboring a glutamate or aspartate within their sequence (missed cleavage site) or not. However, because the interpretation of time-resolved data from multiple overlapping cleavage events would not allow meaningful conclusions, we only included peptides with a single missed cleavage site. This allowed also monitoring either degradation of non-GluC, semi-tryptic natural N termini and of fully tryptic internal peptides by internal cleavage as well as secondary cleavages of neo-N-terminal peptides.
Fig. 1.

High confidence identification of cleavage events by time-resolved abundance profiles. A, Experimental setup. Secretomes from Balb/c 3T3 fibroblasts were incubated with GluC for increasing periods of time and analyzed by 8plex-iTRAQ-TAILS. Controls (c) were incubated with buffer alone for 0, 12 and 16 h, respectively. B, Peptide classification. GluC cleaves substrate proteins C-terminal to E and D residues generating neo-N and C termini that are monitored as semi-tryptic peptides (cleavage events) on digest with the working protease trypsin (Trp). Concomitantly, trypsin releases internal tryptic peptides and natural N termini resembling non-cleavage events. Ac: acetylated. C, Fuzzy c means clustering of abundance profiles of neo-N termini, quantifiable internal tryptic peptides and natural N termini. Peptides with a time-dependent increase in abundance after GluC incubation (cluster N.1) are separated from peptides with a relatively constant abundance over time (cluster N.2). Colorkey indicates membership value α. ctrl: 12 h and 16 h controls. D, IceLogo analysis of cleavage sites corresponding to peptides in cluster N.1 and cluster N.2, respectively. High prevalence of E in P1 position indicates correct assignment of GluC-generated neo-N termini (cleavage events) to cluster N.1 with time-dependent increase in abundance. Peptides assigned to cluster N.2 (time-independent) are either internal tryptic peptides (R in P1) or natural N termini (M in P1 (initiator methionine removed)). E, Receiver Operating Characteristic (ROC) analysis to test performance of a classifier for cleavage events. The classifier is based on degree of membership (α) to the cluster of peptides with time-dependent increase in abundance. A high true positive rate at a low false positive rate indicates high performance. F, Same analysis as in C but for neo-C termini. Colorkey indicates membership value α. ctrl: 12 h and 16 h controls.
High Confidence Classifier for Cleavage Events Based on Time-dependent Abundances
A major challenge in the interpretation of high-throughput degradomics datasets is the discrimination between cleavage events and non-cleavage events. In a previous approach the canonical cleavage site specificity of GluC was exploited to establish a classifier for cleavage events based on abundance ratios of N-terminal peptides in GluC-treated and control samples (20). Although this strongly increased the confidence in substrate identification, it was nevertheless associated with relatively high false discovery rates of around 15%. To establish a classifier with higher sensitivity and specificity, we extracted a sub-dataset of neo-N-terminal peptides derived from known single cleavage events (neo-N termini) and known non-cleavage events (natural N termini and internal tryptic peptides) (Fig. 1B; supplemental Table S2). Next, we used fuzzy c means clustering that is particularly suited for the analysis of data from time-course analyses (39) to assign peptides to two clusters based on their relative abundances in all eight samples (Fig. 1C). As expected, the abundance of peptides in cluster N.1 increased in a time-dependent manner and was low in all three control samples, whereas the abundance of peptides in cluster N.2 was relatively constant in all samples. Cleavage specificity logos generated by WebPICS (29) and IceLogo (30) revealed that cluster N.1 indeed comprised peptides derived from GluC-dependent cleavage events (E/D in P1 position; neo-N termini), whereas peptides generated by trypsin and after GluC treatment of the proteome (time-independent) were assigned to cluster N.2 (M (initiator methionine) in P1: natural N termini or R in P1: internal tryptic peptides) (Fig. 1D). As observed in previous studies, natural N termini commencing after an initiator methionine are derived from cytoplasmic proteins released into the secretome under these culture conditions (20). In fuzzy clustering peptides can belong to multiple clusters, and their degree of assignment to a specific cluster is determined by the so-called membership value α. We exploited this relation to establish a classifier for cleavage versus non-cleavage events based on cluster membership values. This classifier showed an excellent performance in receiver operating characteristic (ROC) analysis with an optimal true positive rate of 98% at a false discovery rate of <5% and a membership value threshold of 0.61 (Fig. 1E).
Monitoring Neo-C-terminal Peptides
Although TAILS focuses on N-terminal peptides and thereby on monitoring neo-N termini generated by proteolytic cleavage, we tested if including data from analysis prior to N-terminal enrichment also allows analyzing neo-C-terminal peptides. For this purpose we combined peptides with a trypsin-generated N and a GluC-derived C terminus that harbored an iTRAQ-labeled lysine within their sequence and thus were quantifiable (Fig. 1B; neo-C termini; cleavage events) with our data set of known non-cleavage events (natural N termini and internal tryptic peptides) (supplemental Table S3). Again, we applied fuzzy c means clustering to this sub-dataset and could separate neo-C termini derived from GluC activity from non-cleavage events (Fig. 1F). Furthermore, ROC analysis defined a similar performance of a classifier based on cluster membership values as for neo-N termini and an optimal threshold of 0.62 (Fig. 1E). This demonstrated the efficient identification of neo-C termini by time-resolved 8plex-iTRAQ-TAILS analysis.
Kinetic Subclassification of Cleavage Site Specificity and Structural Accessibility
In a next step, we further subclassified neo-N-terminal peptides assigned with a membership value of at least 0.61 and thus with high confidence to cluster N.1 (cleavage events). Thereby, three subclusters of peptides with distinct kinetics of time-dependent abundance after incubation with GluC could be formed (Fig. 2A) (supplemental Table S4), which was in agreement with a previous kinetic analysis of proteolysis in a complex proteome (13). For strict assignment of peptides to distinct clusters, we only included peptides with membership values of ≥0.8. Using the well-established equation for pseudo-first order kinetics we determined a range of apparent kcat/Km values of 400–800 M−1s−1 for cleavage events in subcluster N.1.2 that comprised abundance profiles in a measurable range (13) (supplemental Fig. S1). To further characterize cleavage events in each subcluster, we again generated IceLogos for cleavage site specificities (Fig. 2B). These indicated assignment of GluC cleavages after aspartate to subcluster N.1.1 of peptides derived from the least efficient cleavage events. Moreover, WebLogo representations of secondary structures showed a predominance of loops for cleavages after glutamate for highly efficient processing events when compared with cleavages with lower efficiency (Fig. 2C). Notably, for six proteins, cleavages C-terminal to glutamate within the same protein were assigned to two or three clusters, indicating multiple cleavages at different sites and with different efficiencies. Related to protein structures, low efficiency cleavage sites were located in helices or sheets, whereby sites in loops were processed with higher efficiency (Fig. 2D). Therefore, 8plex-iTRAQ-TAILS kinetic analysis of proteolytic processing in complex proteomes could effectively categorize primary cleavage site specificities and structural accessibility.
Fig. 2.

Kinetic subclassification of cleavage site specificity and structural accessibility. A, Subclustering of GluC-generated neo-N termini. Peptides assigned to cluster N.1 with a membership value of ≥0.8 were assigned to three subclusters with slow, medium and fast increase in abundance on GluC incubation. Colorkey indicates membership value α. ctrl: 12 h and 16 h controls. B, IceLogo analysis of GluC cleavage sites corresponding to peptides assigned to each subcluster. Less efficient cleavages C-terminal to aspartate were assigned to subcluster N.1.1 comprising slow cleavage events, whereas medium and fast clusters comprised neo-N-terminal peptides derived from efficient cleavages after glutamate. C, Secondary structure preferences for GluC cleavages in each subcluster. Secondary structure elements around the scissile bond were predicted by PROTEUS2 and aligned for cleavage sites in each subcluster. For all clusters only cleavages C-terminal to glutamate were included. High cleavage efficiency in subcluster N.1.3 is correlated with prevalence of extended loops. L: loop; A: helix; B: sheet. D, Correlation of structural elements and cleavage efficiency within the same protein. Models from the SWISS-MODEL Repository for P63260 (left) and P60335 (right) based on templates 3ub5A (P63260), 2jzx (P60335; residues 11–169) and 1wvn (P60335; residues 278–348), respectively. GluC cleavage sites identified by 8plex-iTRAQ-TAILS are indicated (P1 (E): red; P1′: yellow). Cleavages in loops (PIYE167.168GYAL and VMLE168.169TLSQ) were more efficient than in a helix (EYDE364.365SGPS) or a beta sheet (TTHE283.284LTIP), indicating discrimination between secondary structure elements by time-resolved iTRAQ-TAILS analysis.
Decreasing Abundances of Peptides with Internal Cleavage Sites
Having established kinetic models for monitoring the time-resolved generation of neo-N and neo-C termini by kinetic 8plex-iTRAQ-TAILS, we analyzed abundance profiles for natural N termini and internal fully tryptic peptides that harbor a single glutamate or aspartate in their sequence (Fig. 3A; cleavage events). Following the same principle of distinguishing these potential known cleavages from known non-cleavage events by their time-dependent abundance on GluC incubation, a sub-dataset comprising peptides derived from these cleavage events and known non-cleavage events (Fig. 3A; internal tryptic peptides and natural N termini; supplemental Table S5) was assigned to two clusters (Fig. 3B). As expected, 100% of peptides assigned to cluster I.1 (α ≥0.8) displaying a time-dependent decrease in abundance after GluC incubation were either natural N termini or internal tryptic peptides harboring an E or D in their sequence, whereby cluster I.2 comprised 41% natural N termini and internal tryptic peptides with and 59% without internal E or D residues. Furthermore, 78% of peptides in cluster I.1 harbored a glutamate in their sequence and 22% an aspartate, whereas this ratio was 36% (E) to 64% (D) for E or D containing peptides in cluster I.2. Again, this is in agreement with the higher efficiency of GluC toward cleavage after glutamate than aspartate and the structural inaccessibility of many potential cleavage sites to the protease. For five of the 20 peptides with time-dependent decrease in abundance in cluster I.1 we also identified the corresponding neo-N termini, which concomitantly showed an increase in abundance as exemplified for an internal tryptic peptide in Fig. 3C. This cleavage event was further complemented by detection of the neo-C-terminal peptide, revealing all three events, i.e. the decreasing abundance of the internal tryptic peptide and increasing abundances of both the neo-N-terminal and the neo-C-terminal peptides that all showed the same kinetics (Fig. 3C). Consequently, quantitative analyses of the decrease of a tryptic peptide and the increase of either the corresponding neo-N terminus or neo-C terminus allows estimating ratios of noncleaved and cleaved forms of substrate proteins.
Fig. 3.

Time-dependent decrease in abundance of cleavage site spanning peptides. A, Categorization of cleavage events. Natural N termini and internal tryptic peptides harboring a single E or D in their sequence are decreased in abundance by internal cleavage (cleavage events). The same types of peptides with no internal E or D are not affected by GluC (non-cleavage events). Solid line depicts cleaved natural N terminus and dashed line cleaved internal tryptic peptide. Black line indicates cleavage site spanning peptide, blue line neo-C and red line neo-N terminus. Trp: trypsin; Ac: acetylated. B, Fuzzy c means clustering of abundance profiles of quantifiable cleavage site spanning peptides and noncleaved internal tryptic peptides and natural N termini. Peptides with a time-dependent decrease in abundance after GluC incubation (cluster I.1) are separated from peptides with a relatively constant abundance (cluster I.2). ctrl: 12 h and 16 h controls. C, Time-resolved analysis of decrease in abundance of a cleavage site spanning peptide and concomitant increase of the corresponding neo-N and neo-C termini. D, Secondary cleavage of neo-N termini harboring an internal E or D in their sequence. Cluster NE.4 represents neo-N termini with rapid increase in abundance at 2 to 4 h of GluC incubation and subsequent decrease on secondary cleavage. For all clusters see supplemental Fig. S2. Colorkey indicates membership value α. ctrl: 12 h and 16 h controls. E, Example for rapid generation of a neo-N terminus by GluC cleavage after E and slow appearance of an internal neo-N terminus by secondary processing after D.
Influence of Secondary Cleavage Events on Neo-N Termini
A particular advantage of monitoring protease cleavage events over time is the possibility to also capture neo-N-terminal peptides that on initial increase decrease in abundance because of additional cleavage by either the generating or a different protease within the time frame of the experiment. Thereby, the decrease in abundance of a neo-N terminus could result from a second limited proteolysis event or from unspecific degradation of the truncated form of the substrate. To test if kinetic 8plex-iTRAQ-TAILS detects these events, we analyzed abundance profiles of GluC-derived neo-N termini that harbor a single glutamate or aspartate within their sequence. As expected from the high number of missed cleavages identified in our analysis of internal cleavage sites (Fig. 3B), fuzzy c means clustering of neo-N-terminal peptides with a single missed cleaved E or D residue yielded three clusters associated with increasing cleavage efficiencies similar to neo-N termini, which do not harbor an E or D in their sequence (supplemental Fig. S2; supplemental Table S6; clusters NE.1 to NE.3). However, 20 neo-N termini of this class were assigned to a fourth cluster NE.4 based on their continuous decrease in abundance after 2 to 4 h following an initial rapid increase after GluC incubation (supplemental Fig. S2 and Fig. 3D). This abundance profile could be explained by a highly efficient upstream cleavage and a much slower secondary processing event. This could be governed by differences in the primary cleavage site sequence (glutamate versus aspartate in P1). Indeed, we could identify for a secondary cleavage event also the corresponding second neo-N terminus (Fig. 3E), which was generated after an aspartate in combination with an upstream cleavage after glutamate. Thus, time-resolved 8plex-iTRAQ-TAILS allows detecting cleavage events that are affected by secondary cleavages and would be missed in a 2plex experiment monitoring only a single time point.
Identification of MMP10-dependent Cleavage Events by Time-resolved Analysis
Having established 8plex-iTRAQ-TAILS for time-resolved analysis of proteolytic cleavage events using GluC as a test protease with canonical cleavage specificity, we applied the modified method to identification of new cleavages mediated by MMP10, an important wound and tumor protease, for which only very few substrates are known (16–18). For this purpose we incubated supernatants from MMP10-deficient immortalized murine embryonic fibroblasts (MEF) with auto-activated recombinant MMP10 for 1, 2, 4, 8, 12, and 16 h or buffer alone (0 and 16 h controls) and recorded time-resolved abundance profiles of resulting peptides by 8plex-iTRAQ-TAILS (Fig. 4A). We used a concentration of recombinant MMP10 that was 100- to 500-fold higher than levels of endogenous MMP10, which have been measured in cell culture supernatants (40, 41). However, secretomes were 600× concentrated prior to incubation with the protease, resulting in a similar enzyme:protein (w/w) ratio. These parameters have been chosen based on previous studies (15, 20, 42), but actual enzyme activities might considerably differ in vivo. Importantly, activated MMP10 was stable over the entire time of incubation as shown by casein zymography and TAILS analysis of the N termini of the mature and active protease (supplemental Fig. S3). We identified 385 semi-tryptic N-terminal and 1264 fully tryptic internal peptides (supplemental Table S7), from which we extracted a sub-dataset of 221 peptides resulting from potential cleavage events (semi-tryptic neo-N termini) as well as 521 peptides derived from expected non-cleavage events (51 quantifiable natural N termini and 470 quantifiable fully tryptic internal peptides) (Fig. 4B; supplemental Table S8). Fuzzy c means clustering of this sub-dataset and a stringent α-value threshold of 0.9 robustly discriminated between 67 peptides with time-dependent increase in abundance (cluster M.1) and 464 peptides with relatively constant abundance in all samples (cluster M.2) (Fig. 4C). Based on data for neo-N termini in the GluC test experiment, we extracted 38 peptides from cluster M.1 that had relative abundance values of less than 25% of the maximum abundance in both control channels (0 and 16 h). This step efficiently excluded peptides, which might have been generated by basal proteolysis. IceLogo analysis of the 38 corresponding cleavage sites revealed high occurrences of proline in P3 and leucine or isoleucine in P1′, which are indicative of MMP-mediated processing (15, 20), suggesting that indeed most of the observed neo-N termini had been generated either directly by MMP10 or another MMP that is activated by MMP10 (Fig. 4D). The same analysis of cleavage sites corresponding to peptides in cluster M.2 yielded a high prevalence of arginine in P1 and thus of fully tryptic internal peptides, demonstrating the general stability of the proteome during the time of incubation with the test protease. Notably, 211 peptides could not be assigned to any of the two clusters by our selection criteria, strongly enhancing the confidence in identification of bona fide MMP10-dependent cleavage events.
Fig. 4.

Time-resolved analysis of the MMP10 substrate degradome. A, Experimental setup. Secretomes from Mmp10−/− MEFs were incubated with recombinant MMP10 or buffer (controls (c)) and analyzed by 8plex-iTRAQ-TAILS. B, Peptides generated either by MMP10 and trypsin (Trp) (cleavage events; time-dependent) or by trypsin alone (non-cleavage events; time-independent). Ac: acetylated. C, Fuzzy c means clustering of abundance profiles of neo-N termini, quantifiable internal tryptic peptides and natural N termini. Peptides with a time-dependent increase in abundance after MMP10 incubation (cluster M.1) are separated from peptides with a relatively constant abundance (cluster M.2). Peptides in cluster M.1 with abundances in control channels higher than 25% of the maximum abundance (gray) were omitted from subsequent analyses. Colorkey indicates membership value α. ctrl: 16 h control. D, IceLogo analysis of cleavage sites corresponding to peptides in cluster M.1 and cluster M.2, respectively. High prevalence of P in P3 and/or L/I in P1′ positions for cleavages in cluster M.1 (neo-N termini; time-dependent) correlate with an MMP cleavage specificity. Peptides assigned to cluster M.2 (time-independent) are mostly internal tryptic peptides (R in P1).
Subclassification of MMP10-dependent Cleavage Events by Kinetic Subclustering
Next, we assigned 23 of the 38 filtered peptides in cluster M.1 to two subclusters with either slow (subcluster M.1.1; 13 peptides) or fast (subcluster M.1.2; 10 peptides) increase in abundance over time (Fig. 5A; supplemental Table S9). Applying a stringent threshold for the α value of 0.8 further narrowed in on neo-N termini that were generated in response to MMP10-mediated processing. Analysis of cleavage site specificities by IceLogo determined different types of substrates in each subcluster (Fig. 5B). Although the majority of cleavages in subcluster M.1.1 (slow) exhibited the typical GPXG.L/I cleavage motif in collagen α chains (43), substrates in subcluster M.1.2 (fast) were cleaved at sites with P in P3 and/or L/I in P1′ but were almost all non-collagen proteins (Fig. 5C). Interestingly, the only collagen cleavage assigned to subcluster M.1.2 and thus to the group of more efficient cleavages corresponded to the canonical MMP processing site in the collagen α2(I) chain after position 871, which leads to generation of the typical ¾ and ¼ fragments (43). Since cleavages within the same protein were assigned to both subclusters, we could discriminate between a known efficient processing event at a structurally accessible site and additional less efficient cleavages at more occluded sites by time-resolved iTRAQ-TAILS analysis (Fig. 5D).
Fig. 5.

Subclassification of MMP10-dependent cleavage events. A, Subclustering of MMP10-generated neo-N termini. Peptides assigned to cluster M.1 with a membership value of ≥0.9 were assigned to two subclusters with slow and fast increase in abundance on MMP10 incubation. Colorkey indicates membership value α. ctrl: 16 h control. B, IceLogo analysis of cleavage sites corresponding to peptides assigned to subcluster M.1.1 and subcluster M.1.2, respectively. Low efficient cleavages in subcluster M.1.1 resemble the typical MMP cleavage motif GPXG.L/I in collagens. P in P3 and/or L/I in P1′ for cleavages in subcluster M.1.2 indicate MMP-dependent cleavage events. C, Examples for proteins and MMP10-dependent cleavages in each subcluster. Predominant assignments of collagens to subcluster M.1.1 demonstrate slow processing of these structurally occluded cleavage sites. Only the canonical MMP cleavage site in the type I collagen α2 chain is processed with high efficiency as indicated by assignment to subcluster M.1.2. See supplemental Table S9 for a full list of MMP10-dependent cleavages. D, Kinetic profiles of cleavages in the collagen α2(I) chain. Time-resolved subclustering distinguished the canonical MMP cleavage site (red) from additional MMP10-dependent cleavages within the same collagen chain. E, Validation of multiple processing of type I collagen and concomitant increase in abundance of active MMP2. Upper panel: Immunoblot analysis of MMP10 incubated secretomes from Mmp10−/− MEFs using an anti-collagen I antibody showed appearance of cleavage fragments (arrows) with distinct kinetic profiles. Middle panel: Gelatin zymography revealed MMP10-dependent increase in abundance of a band matching the size of activated MMP2 (arrow) in MMP10-incubated secretomes. Lower panel: Immunoblot analysis of the same samples using an antibody directed against MMP2 confirmed time-dependent activation of MMP2 (arrow) on MMP10 incubation.
Confirming these data immunoblot analysis of MMP10-treated secretomes from Mmp10−/− MEFs revealed appearance of type I collagen cleavage fragments at different time points of incubation (Fig. 5E). Although MMP10 can cleave type III, IV, and V collagen, it has never been shown to have direct activity toward type I collagen (44). This suggested processing of the collagen α2(I) chain by an endogenous collagenolytic MMP on activation by exogenous MMP10. Indeed, gelatin zymography analysis of MMP10-incubated secretomes from Mmp10−/− MEFs identified a band that increased with time of incubation and correlated to the molecular weight of activated MMP2. This result could be further validated by immunoblot analysis of the same samples using an MMP2 specific antibody (Fig. 5E).
Indirect Cleavage of PDGFRα and Direct Processing of ADAMTSL1 by MMP10
Among proteins that were efficiently processed on incubation of secretomes from Mmp10−/− MEFs with MMP10 (subcluster M.1.2), we identified the platelet-derived growth factor receptor alpha (PDGFRα) by a neo-N-terminal peptide starting at position 417 within the extracellular domain (Fig. 6A). Immunoblotting of MMP10-treated Mmp10−/− MEF secretomes with a PDGFRα specific antibody confirmed this cleavage and the MMP10-dependent generation of two fragments of expected molecular weights of ∼80 and ∼40 kDa, respectively. However, recombinant human PDGFRα protein (residues 24–524; extracellular domain) was not cleaved by direct incubation with recombinant human MMP10 in vitro, indicating a similar indirect processing event as for collagen α2(I) by an MMP10-activated endogenous protease. Interestingly, recombinant MMP2 also did not directly process the extracellular domain of PDGFRα (supplemental Fig. S4), suggesting the activity of a different protease toward this protein.
Fig. 6.
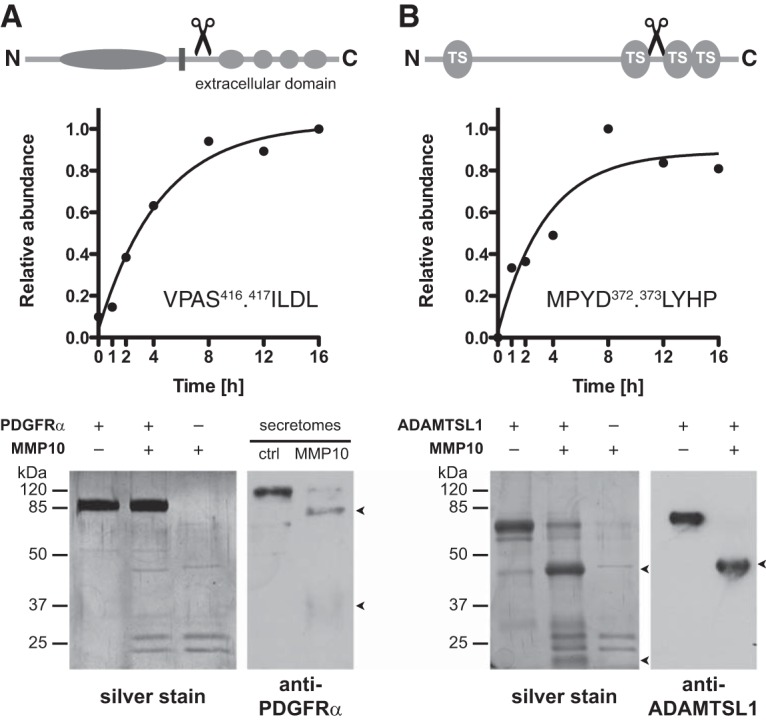
Indirect processing of PDGFRα and direct cleavage of ADAMTSL1 by MMP10. A, Domain structure of PDGFRα with annotated cleavage site in the extracellular domain, time-dependent abundance profile of the generated neo-N terminus, and characterization of processing by incubation of recombinant proteins and immunoblot analysis of MMP10-incubated secretomes. Lack of cleavage of recombinant PDGFRα by MMP10 indicates indirect cleavage in MMP10-incubated cell supernatants. B, Domain structure of ADAMTSL1 (isoform 1) with annotated cleavage site in an extended loop between thrombospondin domains (TS) 2 and 3, time-dependent abundance profile of the generated neo-N terminus, and validation of processing by incubation of recombinant proteins. Detection of MMP10-generated fragments with expected molecular weights by silver stain and immunoblot analysis identified ADAMTSL1 as a direct MMP10 substrate.
Within the same cluster of efficient MMP10-dependent cleavages the extracellular matrix protein ADAMTS-like protein 1 (ADAMTSL1), also known as Punctin-1 (35), was identified by a neo-N terminus corresponding to a cleavage site between the thrombospondin domains 2 and 3 (Fig. 6B). SDS-PAGE and silver staining revealed cleavage fragments of the expected molecular weights (∼40 and ∼20 kDa) of human recombinant ADAMTSL1 on incubation with active recombinant MMP10 and thus identified ADAMTSL1 as a direct MMP10 substrate. This cleavage could be further confirmed by immunoblot analysis using an antibody directed against a peptide within the thrombospondin domain 2 of ADAMTSL1 that detected the ∼40 kDa N-terminal cleavage fragment. Although less efficiently, MMP2 also cleaved ADAMTSL1 into fragments of the same sizes (supplemental Fig. S4), suggesting cooperative processing of ADAMTSL1 by both MMP10 and MMP2.
DISCUSSION
Recent advances in positional proteomics have enabled the reliable identification of protease substrates in complex biological samples (5). Evolving these powerful technologies further, we present a workflow for the time-resolved monitoring of protein termini by exploiting the multiplex capabilities of the recently introduced iTRAQ-TAILS analysis platform (15, 21). Time-resolved 8plex-iTRAQ-TAILS significantly enhanced the confidence in validity of recorded cleavage events for both protein N and C termini and efficiently classified cleavages by sequence and structural accessibility of the cleavage site. Moreover, we identified cleavage events by decrease in abundance of cleaved peptides and determined ratios for cleaved and noncleaved forms of target proteins by concomitant quantification of the neo-N terminus, the neo-C terminus and the peptide spanning the cleavage site. Finally, we applied this approach to the analysis of the MMP10 substrate degradome in fibroblast secretomes and revealed direct processing of ADAMTSL1 as well as MMP10-dependent ectodomain shedding of PDGFRα and sequential processing of type I collagen, which might be mediated by activation of proMMP2 (Fig. 7).
Fig. 7.
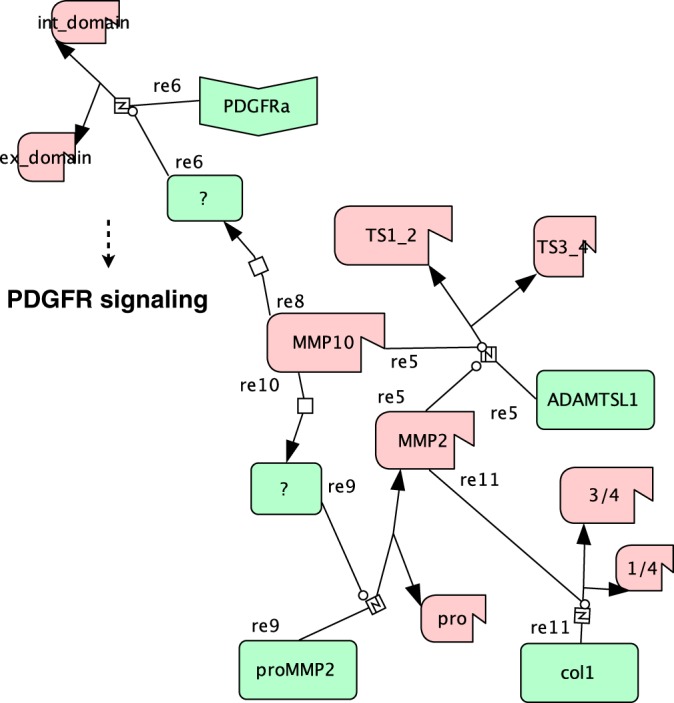
Systems Biology Graphical Notation map of MMP10-dependent processing events. Activated MMP10 (propeptide removed; truncated) activates an unknown protease (reaction (re) 8) that sheds the PDGFRα extracellular (ex_domain) from the intracellular (int_domain) domain (re6) interfacing with PDGFR signaling. Concomitantly, MMP10 activates an alternative protease (re10), which proteolytically activates MMP2 (re9) leading to type I collagen processing (re11). ADAMTSL1 is cleaved either by MMP10 or MMP2 (re5) in an N-terminal and a C-terminal part comprising thrombospondin domains 1 and 2 (TS1_2) and 3 and 4 (TS3_4), respectively.
With our time-resolved 8plex-iTRAQ-TAILS analysis we determined relative proteolytic efficiencies of multiple cleavage events in complex proteomes, a major challenge in current substrate degradomics (6). Extending a recent study that used SRM to record catalytic efficiencies of pre-determined cleavage events (13), we exploited kinetic abundance profiles of N-terminal peptides to significantly enhance sensitivity in distinguishing cleavage from non-cleavage events. This decreased the total number of identified cleavage sites compared with previous analyses (15, 20) and concomitantly removed many bystander events, which are presumably contaminating datasets obtained in experiments that compare treatment with a single protease to a control sample (12). In the original bioinformatics analysis pipeline for TAILS data (‘CLIPPER’), only N-terminal peptides were included in the datasets, whereas separate analyses of samples prior to N-terminal enrichment were primarily used to increase confidence in protein identification (25). In our modified approach, we composed an integrated dataset of all peptides before and after enrichment of protein N termini. Thereby, the dataset is vastly expanded by quantifiable internal tryptic peptides, which could be released from proteins by the working protease (trypsin) with only low variability in abundance at each time point of incubation with the test protease (GluC). This gives some indication of the relative stability of the proteome over time, because multiple unspecific cleavages would have led to decrease in abundance of a significant number of internal tryptic peptides. Moreover, the expanded dataset allowed reliable identification of neo-C termini by monitoring quantifiable N-terminally tryptic and C-terminally nontryptic peptides and identification of cleavage events by decrease in abundance of processed peptides. If these peptides spanning the cleavage site were quantified in addition to neo-N and/or neo-C termini, it was even possible to determine relative abundances of full-length and truncated forms of substrate proteins.
Most importantly, time-resolved analysis allowed subclassification of protease cleavage events by catalytic efficiency. Our results obtained by fuzzy c means clustering are comparable to data from Agard et al., yielding clusters of cleavage events mainly below, within and above a measurable range (13). Within a measurable range we determined an apparent kcat/Km value for GluC of 4–8 * 102 m−1s−1, which is two orders of magnitude lower than the published value for a chromogenic peptide substrate (45). This is expected, because in our experimental setup GluC acts on protein substrates within a complex and native proteome, resulting in much lower catalytic efficiencies and a high number of missed cleavages. Hence, time-resolved 8plex-iTRAQ-TAILS provides a powerful method to determine intrinsic catalytic efficiencies of multiple cleavage events in complex proteomes in parallel.
Because processing by the GluC endoproteinase is hardly or not at all affected by specific noncatalytic protease substrate interactions, differences in catalytic efficiencies are either mediated by sequence or structural constraints of the cleavage site. Thereby, GluC cleaves with two orders of magnitude lower efficiency after aspartate than glutamate residues (46). Time-resolved 8plex-iTRAQ-TAILS analysis resolved this specificity by constraining cleavages with aspartate in P1 to the cluster resembling the least efficient cleavage events. Using N-terminomics Timmer et al. identified extended loops and helices as preferred secondary structures for cleavage by GluC (47). Our time-resolved analysis corroborates this finding and extends it by the expected observation that GluC cleaves extended loops with higher efficiency than helices and sheets, which is also the case for multiple cleavages within the same protein. Strikingly, kinetic 8plex-iTRAQ-TAILS distinguished the canonical MMP processing site in triple-helical collagen from additional cleavages in structurally less accessible regions of the protein (48). Thus, adding time-resolved quantitative information to N-terminomics analyses provides data to probe for cleavage site specificity and structural accessibility in native protein substrates.
To validate and prove the extended capabilities of our workflow for time-resolved monitoring of proteolytic events in complex proteomes, we analyzed the MMP10 substrate degradome. MMP10 is an important wound and tumor protease, which is highly expressed in migrating keratinocytes and believed to mediate epithelial cell migration by controlling processes at the epidermal-dermal interface in the healing skin wound and to modulate tumor-stroma interactions in carcinogenesis (16, 49, 50). By applying our newly devised strategy for discriminating between cleavage and non-cleavage events based on kinetic abundance profiles of neo-N-terminal peptides, we selectively narrowed in on MMP10-dependent processing events. Subclassification not only revealed two distinct subclusters of less and more efficient cleavages, but also elucidated classes of cleaved proteins with distinct structural properties and functionalities. Interestingly, processing of collagens, the classical MMP substrates, was almost exclusively assigned to the group of less efficient cleavages, whereas non-collagen substrates were more efficiently cleaved. Notably, only the classical collagenase cleavage site in the type I collagen α2 chain was assigned to the cluster of efficient cleavages. Most identified MMP10-dependent collagen cleavages are presumably indirect rather than direct, particularly because the inability of this MMP to process type I collagen is well established (44). Our data indicate that the actual active collagenase in fibroblast secretomes on incubation with MMP10 is MMP2, which has proven activity against type I collagen (43, 51). Supporting this hypothesis, two cleavage events identified in the type I collagen α1 and α2 chains, respectively, had also been detected in MMP2 degradomics studies (15, 20). However, although described as upstream activator of several proMMPs (52, 53), MMP10 does not directly activate MMP2 (53). This suggests activation of intermediate proteases or modulation of cofactors within the native fibroblast secretome on incubation with active MMP10, leading to proteolytic conversion of proMMP2 to the active protease, which ultimately processes type I collagen.
As a particularly interesting processing event, time-resolved 8plex-iTRAQ-TAILS identified cleavage of PDGFRα within its extracellular domain close to the plasma membrane, which is characteristic for receptor ectodomain shedding. Although dependent on MMP10, this cleavage was neither directly mediated by MMP10 nor by activated MMP2, indicating activity of an additional unknown protease. Interestingly, a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) can shed the ectodomain of PDGFRβ (54), and a recent study described proteolytic activation of ADAM10 by meprin β through processing of the propeptide that remains bound to the active site after furin cleavage (55). Importantly, PDGFs and their receptors are pivotal players in controlling fibroblast proliferation and migration during cutaneous wound healing (56). Thus, ectodomain shedding of PDGFRα could be an additional mechanism to fine-tune activity of these potent growth factors by sequestering the active ligands.
In contrast to PDGFRα, ADAMTSL1 is directly processed by MMP10 within a linker domain between thrombospondin domains 2 and 3 in very close proximity to a previously described putative cleavage site for an unknown serine protease (35). Concomitant processing by MMP2 presumably at the same site as MMP10 suggests a general susceptibility of this region to proteolysis, whereby overlapping activities for multiple MMPs have already been described for several substrates (15). So far, the functions of ADAMTSL1 and the closely related ADAMTSL3 remain unknown. However, the C. elegans ortholog MADD4 has been implicated in midline-oriented guidance of migrating cells (57), and a similar function of ADAMTSL1 might be modulated by MMP10-mediated processing.
In conclusion, with time-resolved 8plex-iTRAQ-TAILS we introduce a workflow for substrate degradomics that significantly increases confidence in substrate identification and classifies cleavage sites by primary sequence and structural accessibility in secondary structure elements and multimeric higher-order structures, such as collagen fibrils. Moreover, time-resolved analysis of the MMP10 substrate degradome identified novel candidate substrates whose processing might mediate important MMP10 functions in tissue repair and carcinogenesis.
Supplementary Material
Acknowledgments
We thank S. Werner (ETH Zurich) for continuous support, very helpful suggestions, discussions, and comments on the manuscript and J. Marinho for help with zymography.
Footnotes
Author contributions: U.a.d.K. designed research; P.S. and P.N. performed research; L.W.W., J.N.K., and S.S.A. contributed new reagents or analytic tools; F.E.E. and U.a.d.K. analyzed data; P.S. and U.a.d.K. wrote the paper.
* This work was supported by grants from the Swiss National Science Foundation (31003A_140726) and from the European Commission (Marie Curie International Reintegration Grant; FP7-PEOPLE-2010-RG/SkiNterminomics) to U.a.d.K., by National Institutes of Health (HL107147 and EY021151) to S.S.A., and by funds from the ETH Zurich. J.N.K. acknowledges a Michael Smith Foundation for Health Research Career Investigator Scholar Award.
 This article contains supplemental Figs. S1 to S4 and Tables S1 to S9.
This article contains supplemental Figs. S1 to S4 and Tables S1 to S9.
COMPETING FINANCIAL INTERESTS: The authors declare no competing financial interests.
1 The abbreviations used are:
- SRM
- Selected Reaction Monitoring
- TAILS
- Terminal Amine Isotopic Labeling of Substrates
- MMP
- Matrix Metalloproteinase
- PDGFR
- Platelet-Derived Growth Factor Receptor
- ADAM
- A Disintegrin And Metalloproteinase domain-containing protein
- ADAMTS
- ADAM with Thrombospondin motifs
- ADAMTSL
- ADAMTS-like protein
- iTRAQTM
- Isobaric Tag for Relative and Absolute Quantitation
- GluC
- Endoproteinase GluC
- MMRRC
- Mutant Mouse Regional Resource Center
- MEF
- Murine Embryonic Fibroblasts
- DMEM
- Dulbecco's Modified Eagle's Medium
- FBS
- Fetal Bovine Serum
- HPG-ALD
- Hyperbranched Polyglycerol Aldehydes
- HCD
- Higher Energy Collisional Dissociation
- TPP
- Trans-Proteomic Pipeline
- ROC
- Receiver Operating Characteristic
- PICS
- Proteomic Identification of Protease Cleavage Sites
- SBGN
- Systems Biology Graphical Notation.
REFERENCES
- 1. Turk B., Turk D. S. A., Turk V. (2012) Protease signalling: the cutting edge. EMBO J. 31, 1630–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Khokha R., Murthy A., Weiss A. (2013) Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 13, 649–665 [DOI] [PubMed] [Google Scholar]
- 3. auf dem Keller U., Doucet A., Overall C. M. (2007) Protease research in the era of systems biology. Biol. Chem. 388, 1159–1162 [DOI] [PubMed] [Google Scholar]
- 4. Overall C. M., Kleifeld O. (2006) Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer. 6, 227–239 [DOI] [PubMed] [Google Scholar]
- 5. Rogers L., Overall C. M. (2013) Proteolytic post translational modification ofproteins: proteomic tools and methodology. Mol. Cell. Proteomics. 10.1074/mcp.M1113.031310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Plasman K., Van Damme P., Gevaert K. (2013) Contemporary positional proteomics strategies to study protein processing. Curr. Opin. Chem. Biol. 17, 66–72 [DOI] [PubMed] [Google Scholar]
- 7. Impens F., Colaert N., Helsens K., Plasman K., Van Damme P., Vandekerckhove J., Gevaert K. (2010) Mass spectrometry-driven protease substrate degradomics. Proteomics 10, 1284–1296 [DOI] [PubMed] [Google Scholar]
- 8. auf dem Keller U., Prudova A., Eckhard U., Fingleton B., Overall C. M. (2013) Systems-level analysis of proteolytic events in increased vascular permeability and complement activation in skin inflammation. Sci. Signal. 6, rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tholen S., Biniossek M. L., Gansz M., Gomez-Auli A., Bengsch F., Noel A., Kizhakkedathu J. N., Boerries M., Busch H., Reinheckel T., Schilling O. (2013) Deletion of cysteine cathepsins B or L yields differential impacts on murine skin proteome and degradome. Mol. Cell. Proteomics. 12, 611–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van Damme P., Staes A., Bronsoms S., Helsens K., Colaert N., Timmerman E., Aviles F. X., Vandekerckhove J., Gevaert K. (2010) Complementary positional proteomics for screening substrates of endo- and exoproteases. Nat. Methods. 7, 512–515 [DOI] [PubMed] [Google Scholar]
- 11. Schilling O., Barré O., Huesgen P. F., Overall C. M. (2010) Proteome-wide analysis of protein carboxy termini: C terminomics. Nat. Methods. 7, 508–511 [DOI] [PubMed] [Google Scholar]
- 12. Plasman K., Van Damme P., Kaiserman D., Impens F., Demeyer K., Helsens K., Goethals M., Bird P. I., Vandekerckhove J., Gevaert K. (2011) Probing the efficiency of proteolytic events by positional proteomics. Mol. Cell. Proteomics. 10, 10.1074/mcp.M1110.003301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Agard N. J., Mahrus S., Trinidad J. C., Lynn A., Burlingame A. L., Wells J. A. (2012) Global kinetic analysis of proteolysis via quantitative targeted proteomics. Proc. Natl. Acad. Sci. U.S.A. 109, 1913–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shimbo K., Hsu G. W., Nguyen H., Mahrus S., Trinidad J. C., Burlingame A. L., Wells J. A. (2012) Quantitative profiling of caspase-cleaved substrates reveals different drug-induced and cell-type patterns in apoptosis. Proc. Natl. Acad. Sci. U.S.A. 109, 12432–12437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Prudova A., auf dem Keller U., Butler G. S., Overall C. M. (2010) Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol. Cell. Proteomics. 9, 894–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krampert M., Bloch W., Sasaki T., Bugnon P., Rülicke T., Wolf E., Aumailley M., Parks W. C., Werner S. (2004) Activities of the matrix metalloproteinase stromelysin-2 (MMP-10) in matrix degradation and keratinocyte organization in wounded skin. Mol. Biol. Cell. 15, 5242–5254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Madlener M., Mauch C., Conca W., Brauchle M., Parks W. C., Werner S. (1996) Regulation of the expression of stromelysin-2 by growth factors in keratinocytes: implications for normal and impaired wound healing. Biochem. J. 320 (Pt 2), 659–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Muller D., Breathnach R., Engelmann A., Millon R., Bronner G., Flesch H., Dumont P., Eber M., Abecassis J. (1991) Expression of collagenase-related metalloproteinase genes in human lung or head and neck tumours. Int. J. Cancer. 48, 550–556 [DOI] [PubMed] [Google Scholar]
- 19. Todaro G. J., Green H. (1963) Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17, 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. auf dem Keller U., Prudova A., Gioia M., Butler G. S., Overall C. M. (2010) A statistics-based platform for quantitative N-terminome analysis and identification of protease cleavage products. Mol. Cell. Proteomics. 9, 912–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kleifeld O., Doucet A., Prudova A., auf dem Keller U., Gioia M., Kizhakkedathu J. N., Overall C. M. (2011) Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates. Nat. Protoc. 6, 1578–1611 [DOI] [PubMed] [Google Scholar]
- 22. Choe L., D'Ascenzo M., Relkin N. R., Pappin D., Ross P., Williamson B., Guertin S., Pribil P., Lee K. H. (2007) 8-plex quantitation of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer's disease. Proteomics. 7, 3651–3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deutsch E. W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B., Eng J. K., Martin D. B., Nesvizhskii A. I., Aebersold R. (2010) A guided tour of the Trans-Proteomic Pipeline. Proteomics 10, 1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shadforth I. P., Dunkley T. P., Lilley K. S., Bessant C. (2005) i-Tracker: for quantitative proteomics using iTRAQ. BMC Genomics 6, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. auf dem Keller U., Overall C. M. (2012) CLIPPER-An add-on to the Trans-Proteomic Pipeline for the automated analysis of TAILS N-terminomics data. Biol. Chem. 393, 1477–1483 [DOI] [PubMed] [Google Scholar]
- 26. Kumar L., M E. F. (2007) Mfuzz: a software package for soft clustering of microarray data. Bioinformation 2, 5–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schwammle V., Jensen O. N. (2010) A simple and fast method to determine the parameters for fuzzy c-means cluster analysis. Bioinformatics 26, 2841–2848 [DOI] [PubMed] [Google Scholar]
- 28. Sing T., Sander O., Beerenwinkel N., Lengauer T. (2005) ROCR: visualizing classifier performance in R. Bioinformatics 21, 3940–3941 [DOI] [PubMed] [Google Scholar]
- 29. Schilling O., auf dem Keller U., Overall C. M. (2011) Factor Xa subsite mapping by proteome-derived peptide libraries improved using WebPICS, a resource for proteomic identification of cleavage sites. Biol. Chem. 392, 1031–1037 [DOI] [PubMed] [Google Scholar]
- 30. Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 31. Montgomerie S., Cruz J. A., Shrivastava S., Arndt D., Berjanskii M., Wishart D. S. (2008) PROTEUS2: a web server for comprehensive protein structure prediction and structure-based annotation. Nucleic Acids Res. 36, W202–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Crooks G. E., Hon G., Chandonia J. M., Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kitano H., Funahashi A., Matsuoka Y., Oda K. (2005) Using process diagrams for the graphical representation of biological networks. Nat. Biotechnol. 23, 961–966 [DOI] [PubMed] [Google Scholar]
- 34. Funahashi A., Morohashi M., Kitano H., Tanimura N. (2003) CellDesigner: a process diagram editor for gene-regulatory and biochemical networks. BIOSILICO 1, 159–162 [Google Scholar]
- 35. Hirohata S., Wang L. W., Miyagi M., Yan L., Seldin M. F., Keene D. R., Crabb J. W., Apte S. S. (2002) Punctin, a novel ADAMTS-like molecule, ADAMTSL-1, in extracellular matrix. J. Biol. Chem. 277, 12182–12189 [DOI] [PubMed] [Google Scholar]
- 36. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schilling O., Overall C. M. (2008) Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat. Biotechnol. 26, 685–694 [DOI] [PubMed] [Google Scholar]
- 38. Houmard J., Drapeau G. R. (1972) Staphylococcal protease: a proteolytic enzyme specific for glutamoyl bonds. Proc. Natl. Acad. Sci. U.S.A. 69, 3506–3509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 40. Kim H. S., Shang T., Chen Z., Pflugfelder S. C., Li D. Q. (2004) TGF-beta1 stimulates production of gelatinase (MMP-9), collagenases (MMP-1, −13) and stromelysins (MMP-3, −10, −11) by human corneal epithelial cells. Exp. Eye Res. 79, 263–274 [DOI] [PubMed] [Google Scholar]
- 41. Li D. Q., Shang T. Y., Kim H. S., Solomon A., Lokeshwar B. L., Pflugfelder S. C. (2003) Regulated expression of collagenases MMP-1, −8, and −13 and stromelysins MMP-3, −10, and −11 by human corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 44, 2928–2936 [DOI] [PubMed] [Google Scholar]
- 42. Kleifeld O., Doucet A., auf dem Keller U., Prudova A., Schilling O., Kainthan R. K., Starr A. E., Foster L. J., Kizhakkedathu J. N., Overall C. M. (2010) Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat. Biotechnol. 28, 281–288 [DOI] [PubMed] [Google Scholar]
- 43. Aimes R. T., Quigley J. P. (1995) Matrix metalloproteinase-2 is an interstitial collagenase. Inhibitor-free enzyme catalyzes the cleavage of collagen fibrils and soluble native type I collagen generating the specific 3/4- and 1/4-length fragments. J. Biol. Chem. 270, 5872–5876 [DOI] [PubMed] [Google Scholar]
- 44. Nicholson R., Murphy G., Breathnach R. (1989) Human and rat malignant-tumor-associated mRNAs encode stromelysin-like metalloproteinases. Biochemistry 28, 5195–5203 [DOI] [PubMed] [Google Scholar]
- 45. Park J. W., Park J. E., Park J. K., Lee J. S. (2011) Purification and biochemical characterization of a novel glutamyl endopeptidase secreted by a clinical isolate of Staphylococcus aureus. Int. J. Mol. Med. 27, 637–645 [DOI] [PubMed] [Google Scholar]
- 46. Wehofsky N., Wissmann J., Alisch M., Bordusa F. (2000) Engineering of substrate mimetics as novel-type substrates for glutamic acid-specific endopeptidases: design, synthesis, and application. Biochim. Biophys. Acta 1479, 114–122 [DOI] [PubMed] [Google Scholar]
- 47. Timmer J. C., Zhu W., Pop C., Regan T., Snipas S. J., Eroshkin A. M., Riedl S. J., Salvesen G. S. (2009) Structural and kinetic determinants of protease substrates. Nat. Struct. Mol. Biol. 16, 1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Salsas-Escat R., Nerenberg P. S., Stultz C. M. (2010) Cleavage site specificity and conformational selection in type I collagen degradation. Biochemistry 49, 4147–4158 [DOI] [PubMed] [Google Scholar]
- 49. van Kempen L. C. L., Rhee J.-S., Dehne K., Lee J., Edwards D. R., Coussens L. M. (2002) Epithelial carcinogenesis: dynamic interplay between neoplastic cells and their microenvironment. Differentiation 70, 610–623 [DOI] [PubMed] [Google Scholar]
- 50. P O. C., Rhys-Evans P. H., Eccles S. A. (2001) Expression of matrix metalloproteinases and their inhibitors correlates with invasion and metastasis in squamous cell carcinoma of the head and neck. Arch. Otolaryngol. Head Neck Surg. 127, 813–820 [PubMed] [Google Scholar]
- 51. Patterson M. L., Atkinson S. J., Knauper V., Murphy G. (2001) Specific collagenolysis by gelatinase A, MMP-2, is determined by the hemopexin domain and not the fibronectin-like domain. FEBS Lett. 503, 158–162 [DOI] [PubMed] [Google Scholar]
- 52. Barksby H. E., Milner J. M., Patterson A. M., Peake N. J., Hui W., Robson T., Lakey R., Middleton J., Cawston T. E., Richards C. D., Rowan A. D. (2006) Matrix metalloproteinase 10 promotion of collagenolysis via procollagenase activation: implications for cartilage degradation in arthritis. Arthritis Rheum. 54, 3244–3253 [DOI] [PubMed] [Google Scholar]
- 53. Nakamura H., Fujii Y., Ohuchi E., Yamamoto E., Okada Y. (1998) Activation of the precursor of human stromelysin 2 and its interactions with other matrix metalloproteinases. Eur. J. Biochem. 253, 67–75 [DOI] [PubMed] [Google Scholar]
- 54. Mendelson K., Swendeman S., Saftig P., Blobel C. P. (2010) Stimulation of platelet-derived growth factor receptor beta (PDGFRbeta) activates ADAM17 and promotes metalloproteinase-dependent cross-talk between the PDGFRbeta and epidermal growth factor receptor (EGFR) signaling pathways. J. Biol. Chem. 285, 25024–25032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jefferson T., Auf dem Keller U., Bellac C., Metz V. V., Broder C., Hedrich J., Ohler A., Maier W., Magdolen V., Sterchi E., Bond J. S., Jayakumar A., Traupe H., Chalaris A., Rose-John S., Pietrzik C. U., Postina R., Overall C. M., Becker-Pauly C. (2013) The substrate degradome of meprin metalloproteases reveals an unexpected proteolytic link between meprin beta and ADAM10. Cell. Mol. Life Sci. 70, 309–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Donovan J., Shiwen X., Norman J., Abraham D. (2013) Platelet-derived growth factor alpha and beta receptors have overlapping functional activities towards fibroblasts. Fibrogenesis Tissue Repair 6, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Seetharaman A., Selman G., Puckrin R., Barbier L., Wong E., D'Souza S. A., Roy P. J. (2011) MADD-4 is a secreted cue required for midline-oriented guidance in Caenorhabditis elegans. Dev. Cell 21, 669–680 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.