

Abstract
Phage display is a well-established procedure to isolate binders against a wide variety of antigens that can be performed on purified antigens, but also on intact cells. As selection steps are performed in vitro, it is possible to focus the outcome of the selection on relevant epitopes by performing some additional steps, such as depletion or competitive elutions. However in practice, the efficiency of these steps is often limited and can lead to inconsistent results. We have designed a new selection method named masked selection, based on the blockade of unwanted epitopes to favor the targeting of relevant ones. We demonstrate the efficiency and flexibility of this method by selecting single-domain antibodies against a specific portion of a fusion protein, by selecting binders against several members of the seven transmembrane receptor family using transfected HEK cells, or by selecting binders against unknown breast cancer markers not expressed on normal samples. The relevance of this approach for antibody-based therapies was further validated by the identification of four of these markers, Epithelial cell adhesion molecule, Transferrin receptor 1, Metastasis cell adhesion molecule, and Sushi containing domain 2, using immunoprecipitation and mass spectrometry. This new phage display strategy can be applied to any type of antibody fragments or alternative scaffolds, and is especially suited for the rapid discovery and identification of cell surface markers.
Hybridoma (1) and phage-display recombinant antibody systems (2) are currently the predominant methods for isolating monoclonal antibodies (Abs).1 Display of recombinant Abs on the surface of bacteriophage M13 has numerous advantages compared with conventional hybridoma technology. When combined with the use of large non-immune libraries, phage Ab selection represents a rich source of binders that can be isolated in a fraction of the time needed for hybridoma-based approaches. Moreover, this in vitro selection method permits the selection of binders against toxic, non-immunogenic or highly conserved antigens, which is not easily performed using the conventional hybridoma techniques. Importantly, it can be used to isolate fully human antibody fragments (3). Consequently, phage display rapidly became an established procedure for the isolation of binders against a wide variety of antigens.
Phage display-based antibody isolation typically relies on the use of recombinant proteins for several steps, including immunizations (if needed), library enrichment by selection on immobilized antigen, screening, and characterization of antibodies in terms of specificity and affinity (4). This procedure is efficient but depends on the availability of purified recombinant proteins. Unfortunately, some surface molecules, such as G-protein coupled receptors, cannot be easily expressed and purified in their native conformation. Some molecules with large extracellular domains may adopt a specific conformation upon interaction with other cell surface proteins, thereby forming complexes that are cumbersome to produce by recombinant expression. Moreover, many standard screening practices, such as the adsorption of recombinant proteins on plastic, may significantly alter protein conformations (5). For these reasons, Abs selected on the basis of binding to a recombinant protein may not bind the native conformation of this protein. It is thus of high interest to develop procedures entirely based on the use of intact cells expressing the receptor of choice. However, in this case, an extra step is necessary to enrich for phage-Abs binding to the receptor of interest rather than to other cell surface proteins. Because selection steps are performed in vitro, it is possible to influence the outcome of a selection by performing some additional steps such as deletion steps (also named negative selection) prior to positive selections to remove unwanted specificities or cross-reactions (6), by alternating the source of the antigen (7), or by using a competitive elution with a ligand or an existing monoclonal antibody to favor the selection of binders against a precise epitope (8).
Along this line, it would be of very high interest to establish a procedure able to reliably guide the selection toward an unknown but relevant antigen within a complex mixture, such as a tumor maker overexpressed at the surface of intact cells, or in a cell lysate. Indeed, during the past two decades, there has been a growing interest in approaches aiming at discovering new diagnosis biomarkers and identifying new potential surface markers for targeted therapy. Several studies have described the use of phage display and libraries of recombinant antibodies for the isolation of tumor specific binders (9–15), leading in some cases to the identification of new tumor markers (16, 17). Most of these strategies are relying on the use of depletion steps on normal samples followed by a selection step on tumor samples. Unfortunately, this procedure often leads to inconsistent results and its efficiency can be a limiting factor in complex situations such as the selection of antibodies against unknown overexpressed tumor antigens.
We have designed a new selection method, named masked selection, which is relying on the blockade of unwanted epitopes to favor the targeting of relevant ones. We demonstrate the efficiency of this method by selecting binders against a specific portion of a fusion protein, by selecting binders against two members of the seven transmembrane receptor family and a tyrosine kinase receptor using intact transfected HEK cells, or by selecting binders against unknown breast cancer markers not expressed on normal samples, as shown by flow cytometry and immunohistochemistry. The universality and efficiency of this approach should ultimately lead to the rapid selection of specific binders and the development of diagnostic and targeted therapies in various settings.
MATERIALS AND METHODS
Cells Lines, Biopsies
MCF7, SK-BR-3, and T47D are a kind gift of Daniel Olive (CRCM, Marseille, France). MDA-MB-231 and HCC1937 are a kind gift of Marie Alix Poul (IRCM, Montpellier). BrCa-Mz-01, HCC1806, HCC1954, and BT474 are a kind gift of Jean Imbert (INSERM, U928, TAGC, Marseille, France). C4.3 is a kind gift of Ralph Willemsen (Erasmus MC, Rotterdam). Jurkat, HEK293T, HeLa, HT29, PC3, and BxPc3 cells were obtained from ATCC. Cells lines MDA-MB-231, MCF7, T47D, HCC1937, HEK293T, HeLa, HT29, and PC3 were cultured in DMEM complemented with 10% (v/v) fetal calf serum. Cells lines SK-BR-3, HCC1954, BrCaMz01, BT474, BxPc3, C4.3, Jurkat, and HCC1806 were cultured in RPMI complemented with 10% (v/v) fetal calf serum. HME1 cell line was purchased from ATCC and grown as recommended by the manufacturer. All cell lines were grown at 37 °C in a humidified atmosphere and with 5% CO2. PBMC were isolated from the blood of healthy donors (collected in 500 ml blood bags containing citrate phosphate dextrose as anticoagulant and no additive) by ficoll gradient centrifugation method as described (18). For transfection assay, HEK/293T were transfected with Lipofectamine (Invitrogen), following the recommendation of the manufacturer.
Breast cancer biopsies (5801, 5772e, 5766, 5586, 5572i, 5592, 5011, 5712, 5713, 5033, and 5627) are a kind gift of S. Garcia, CRCM, Marseille. Samples of human origin and associated data were obtained from the IPC/CRCM Tumor Bank, that operates under authorization #AC-2007–33 granted by the French Ministry of Research (Ministère de la Recherche et de l'Enseignement Supérieur). Prior to scientific use of samples and data, patients were appropriately informed and asked to consent in writing, in compliance with French and European regulations. The project was approved by the IPC Institutional Review Board (Comité d'Orientation Stratégique, COS). Biopsies or cells were lysed with a potter in lysis buffer : 150 mm NaCl, 1% Triton X-100, 50 mm Tris-HCl pH8 with protease inhibitor mixture (Complete, Roche). The lysate was centrifuged for 10 min at 13,000 × g at 4 °C. Supernatant was the final cell lysate. Total protein concentration (average between 2–5 mg/ml) was determined spectrophotometrically using a protein assay kit (Bio-Rad Laboratories, Hercules, CA).
Production and Purification of sdAbs
For polyclonal production of soluble sdAbs, 10 μl of output from selection round 1 and 2 were used to inoculate 200 ml of 2YT/ampicillin (100 μg/ml). Cells were grown at 37 °C (250 rpm) until the OD600 reached 0.5. SdAb expression was induced by the addition of 0.1 mm IPTG (isopropyl-h-d-thiogalactopyranoside) at 30 °C (250 rpm) for 20 h.
SdAbs were purified by metal affinity chromatography as described (19).
In Vitro Biotinylation
The in vitro biotinylation of protein was performed using Ez-link micro NMHS-PEO4- biotinylation kit (Perbio science) following the recommendation of the manufacturer.
Llama Immunization and Library Construction
Three young adult llama (Lama glama) were immunized subcutaneously at days 1, 10, 20, and 30 with breast cancer biopsy lysate (two llamas) or with healthy breast biopsy (one llama). One llama was immunized with HER2-Fc protein and HEK-mGluR4 cells.
VHH library constructions were performed as previously described (14, 20).
Selection of Phage-sdAbs
To produce phage-sdAbs, 10 μl of the library was grown in 50 ml of 2YT/ampicillin (100 μg/ml)/glucose (2%) at 37 °C to an OD600 of 0.5. Then, the culture was infected with KM13 or hyperphage (Progen biotechnik) helper phage at a ratio of 20 phages/cell for 30 min at 37 °C without shaking. The culture was centrifuged for 10 min at 3000 × g. The bacterial pellet was resuspended in 250 ml of 2YT/ampicillin (100 μg/ml)/kanamycine (25 μg/ml), and incubated overnight at 30 °C with shaking (250 rpm). Twenty five milliliters were then centrifuged for 20 min at 3000 × g. Five ml of 20% PEG 6000, 2.5 m NaCl were added to the supernatant and incubated for 1 h on ice to precipitate phage particles. The solution was centrifuged for 15 min at 3000 × g at 4 °C and the phage-containing pellet was resuspended with 1 ml of PBS.
Different strategies of panning were performed. Some phages were selected using magnetic epoxy beads (Dynabeads, invitrogen) coated with antigen or lysates immobilized on epoxy beads during 48 h at 4 °C following recommendations of the manufacturer. Other phages were selected directly on cells (30 × 106 cells). Beads or cells were washed three times in PBS (using a magnetic particle concentrator for magnetic beads and centrifugation step for cells) and phage-sdAb library (1 ml) and beads or cells were saturated in 2% milk PBS. For selection including a depletion step, phage-sdAb library were incubated with the irrelevant immobilized antigen at room temperature or with 80 × 106 irrelevant cells at 4 °C during 2 h, with rotation. Phage-sdAb libraries (depleted or not) were recovered and incubated with beads with rotation during 2 h at room temperature or at 4 °C for cells. For masked selection in the presence of soluble sdAbs, 10 μm of purified sdAbs were added during this step. Beads, cells, or plates were washed 10 times with 1 ml 0,1% Tween PBS (without Tween for cells) and two times with PBS. Bound phages were eluted with tryspin solution (Sigma) at 1 mg/ml during 30 min at room temperature with rotation. Eluted phages were incubated without shaking with log-phase TG1 cells and plated on 2YT/ampicillin (100 μg/ml)/glucose (2%) in 15 cm Petri dishes. Some isolated colonies were grown overnight in microtiter plate containing 200 μl 2YT/ampicillin (100 μg/ml)/glucose (2%) and stored at −80 °C after the addition of 15% glycerol (masterplates). The remaining colonies were harvested from the plates, suspended in 2 ml 2YT/ampicillin (100 μg/ml)/glucose (2%) and used for phage production for the next round of selection. Two rounds of selection were systematically performed.
Monoclonal Phage-sdAb Production in Microtiter Plate
A 96-well plate replicator was used to replicate the masterplates in 150 μl of 2YT/ampicillin (100 μg/ml)/glucose (2%). Colonies were grown for 2 h at 37 °C under shaking (400 rpm) and 50 μl 2YT/ampicillin (100 μg/ml)/glucose (2%) containing 2 × 109 M13KO7 helper phage were added to each well and incubated for 30 min at 37 °C without shaking. The plate was centrifuged for 10 min at 1200 × g and bacterial pellets were suspended in 150 μl 2YT/ampicillin (100 μg/ml)/kanamycine (25 μg/ml)1) and grown for 16 h at 30 °C under shaking (400 rpm). Phage-containing supernatants were tested for binding by ELISA or flow cytometry.
Phage-sdAb ELISA on Epoxy Beads
Antigens HER2-Fc (R & D Systems) or Fc were immobilized on magnetic epoxy beads (Dynabeads, Invitrogen) during 48 h at 4 °C following recommendation of the manufacturer. For ELISA, 2 μl of beads/well was used. After three washes, beads were blocked with 5% milk-PBS (MPBS) for two hours at RT. Plates were incubated for 1 h at RT with 50 μl/well of phage-containing supernatants diluted at ½ in 4% MPBS. After three washes with 0.1% Tween PBS and three washes in PBS, plates were incubated with HRP-conjugated anti-M13 mAb (Pharmacia) diluted 1/5000 during 1 h at RT. After three washes with 0.1% Tween PBS and three washes in PBS, bound secondary antibodies were detected using ABTS. Coloration was followed at 405 nm.
Phage-sdAb ELISA with Lysate Coated on Plate
Fifty microliters/well of biopsy mixture, or breast cancer cell lines (BT474, SK-BR-3, HCC1954, MCF7, MDA-MB-231, T47D, HCC1806, BRCA-Mz-01, and HCC1937) or control cells HME1 and human PBMC lysates (200 μg/ml of total proteins) were coated overnight at 4 °C on maxisorp 96-well plate (Nunc). After three washes with PBS, plates were blocked with 5% MPBS for 2 hours at RT. For competitive assay, plates were incubated with 50 μl/well of sdAbs at 10 μg/ml during 1 h at RT. Plates were incubated for 1 h at RT with 50 μl/well of phage-containing supernatants diluted at (1/2) in 4% MPBS. After three washes with 0.1% Tween PBS and three washes in PBS, plates were incubated with HRP-conjugated anti-M13 mAb (Pharmacia) at 1/5000 during 1 h at RT. After three washes with 0.1% Tween PBS and three washes in PBS, bound secondary antibodies were detected using ABTS. Coloration was followed at 405 nm.
Flow Cytometry Assay
Experiments were performed on ice with rocking in 1% BSA PBS. Typically, 2 × 105 cells in 50 μl buffer were distributed in 96-well microtiter plates. For competitive assay, plates were incubated with 50 μl/well of sdAbs at 10 μg/ml for 1 h at 4 °C. Fifty μl of phage-containing supernatants diluted at ½ in 2% BSA PBS (for phage-sdAbs assay) or 50 μl of in vitro biotinylated purified sdAbs at 10 μg/ml (for soluble sdAbs assay) were added to each well. Plates were incubated for 1 h at 4 °C. After three washes in PBS, plates were incubated for 1h at 4 °C with PE-conjugated anti-M13 mAb at 1/200 (Santa Cruz) for phage-sdab detection or with PE-conjugated streptavidine at 1/10 (Beckman Coulter) for biotinylated sdAb detection,. After three washes in PBS, fluorescence was measured using a MACSQuant cytometer (Miltenyi) and results were analyzed with the MACSQuant software. Negative (secondary antibody only) controls were carried out.
Homogenous Time-Resolved Fluorescence (HTRF) Assay
A 96-well plate replicator was used to replicate the masterplates in 150 μl of fresh broth. Colonies were grown for 2 h at 37 °C under shaking (900 rpm) and 15 μl 2YT/ampicillin (100 μg/ml)/containing 0.1 mm IPTG were added to each well. The plate was incubated for 16 h at 30 °C with shaking (400 rpm). sdAb-containing supernatants were tested for binding by HTRF. To measure HTR-FRET signals, HEK cells were transfected 24 h prior to the assay with plasmids coding for HER2, mGluR4 or CXCR4 receptors N-terminally fused to a SNAP tag (plasmids were a kind gift of Gérard Matthis and Jean-Philippe Pin, CisBio Bioassays, Marcoule, France and IGF, Montpellier, France, respectively). Cells were labeled with Tag-lite Snap-Lumi4-Tb, according to the manufacturer's kit protocol (Cisbio Bioassays). sdAb and D2 labeled anti-6his mAb (Cisbio Bioassays) were added simultaneously. After incubation for 1 h at RT, HTR-FRET signal (665 nm) and Lumi4-Tb donor signal (620 nm) were measured using a Tecan infinite M1000. HTRF ratio (665 nm/620 nm × 104) was calculated to eliminate quenching and dispensing errors.
Immunohistochemistry Assay
In vitro biotinylated sdAbs were assayed in immunohistochemistry on 5 μm sections of paraffin-embedded cancer tissues. In addition, adjacent normal breast epithelium served as specificity control. A breast cancer tissue micro array containing 80 samples in duplicate (containing lobular and ductal breast cancer biopsies from grade I to III tumors with local lymph node invasion or not) and 14 samples of healthy breast tissues in duplicate was also used. After deparaffinization of paraffin-embedded tissues, antigen retrieval of paraffin-embedded tissues was performed in 95 °C prewarmed citrate buffer during 20 min. Endogenous peroxidase activity was blocked by incubation with 3% H2O2. Slides were incubated for 1h at room temperature with in vitro biotinylated sdAbs at 10 μg/ml and washed. Detection was performed by incubations at room temperature 30 min with streptavidin peroxidase. Finally, visualization was performed by DAB revelation peroxidase reaction (Dako) using hematoxylin as counterstain.
Antigen Identification and Mass Spectrometry Analysis
Purified nanobodies were cross-linked on NHS-activated Sepharose 4 Fast Flow beads (1 μg/μl beads) according to manufacturer instructions (GE Healthcare Life sciences). For antigen pulldown, 20 μl nanobodies coupled to beads were incubated with 1 mg/ml cells lysate prepared in HNTG buffer (50 mm Hepes pH 7.3, 150 mm Nacl, 0.1 mm EDTA, 1.5 mm MgCl2, 10% Glycerol, 1% Triton). After washing, 10% of the sample was loaded on Novex® 4–12% Bis-Tris Gel (Invitrogen) and specific antigens were visualized on the gel using silver staining protocol according to (21).
For antigens identification, duplicate gels were run and proteins stained with Imperial blue (Pierce, Thermoscientific). Bands containing antigens were cut from the gel, prepared and digested with high sequencing grade trypsin (Promega) before mass spectrometry analysis according to (21). In some case, when specific antigen were not clearly visualized after silver stained gel, running of the sample in the gel was stopped as soon as proteins were stacked as a single band in the upper part of the gel. After mass spectrometry identification of the whole proteins content, immunoprecipitated antigens were specifically identified for respective used nanobodies.
Mass spectrometry analysis was carried out by LC-MSMS using a LTQ-Velos-Orbitrap (Thermo Fisher Scientific) online with a nanoLC Ultimate 3000 chromatography system (Dionex) as described (22). For direct antigen identification, 5 μl of peptides corresponding to 1/3th of digested sample were injected, preconcentrated and washed on a Dionex Acclaim PepMap 100 C18 column (2 cm × 100 μm i.d. 100 A, 5 μm particle size). Peptides were further separated on a Dionex Acclaim PepMap RSLC C18 column (15 cm × 75 μm i.d., 100 A, 2 μm particle size) at a flow rate of 300 nL/min. with a 4–45% linear gradient acetonitrile/H20; 0.1% formic acid for 30 min. For identification of more complex sample (full IP), a two steps linear gradient (4–20% acetonitrile/H20; 0.1% formic acid for 90 min and 20–45% acetonitrile/H20; 0.1% formic acid for 30 min) was used.. The separation of the peptides was monitored by a UV detector (absorption at 214 nm). For peptides ionization in the nanospray source, spray voltage was set at 1.4 kV and the capillary temperature at 275 °C. All samples were measured in a data dependent acquisition mode. Each run was preceded by a blank MS run to monitor system background. The peptide masses were measured in a survey full scan (scan range 300–1700 m/z, with 30 K FWHM resolution at m/z = 400, target AGC value of 1.00 × 10E6 and maximum injection time of 500 ms). In parallel to the high-resolution full scan in the Orbitrap, the data-dependent CID scans of the 10 most intense precursor ions were fragmented and measured in the linear ion trap (normalized collision energy of 35%, activation time of 10 ms, target AGC value of 1.00 × 10E4, maximum injection time 100 ms, isolation window 2 Da). Parent masses obtained in orbitrap analyzer were automatically calibrated on 445.1200 locked mass. The fragment ion masses were measured in the linear ion trap to have a maximum sensitivity and the maximum amount of MS/MS data. Dynamic exclusion was implemented with a repeat count of 1 and exclusion duration of 7s or 30s respectively for short or long LC gradient duration.
Raw files generated from mass spectrometry analysis were processed with Proteome Discoverer 1.3 (Thermo fisher Scientific). This software was used to search data via in-house Mascot server (version 2.4.1; Matrix Science Inc., London, UK) against the Human database subset of the SwissProt database (version 2013.05) containing 20256 entries. Database search were done using the following settings: Trysin cleavage with a maximum of one miscleavage allowed, methionine oxidation as variable modifications, cysteine carbamido-methylation as fixed modification. Mass tolerance of 6 ppm and 0.8 Da were used respectively for precursors and fragments ions during search analysis. For unambiguous protein identification, only peptides with higher Mascot threshold score (identity) were selected. FDR < 1% was used.
To confirm MS results, immunoprecipitates were analyzed by Western blot using mouse anti-human transferrin receptor antibody clone H68.4 (Zymed Laboratories Inc., anti-EpCAM mouse antibody clone C-10 (Santa Cruz), polyclonal rabbit anti-MCAM antibody (Sigma) and mouse polyclonal anti-SUSD2 (Sigma).
RESULTS
The principle of masked selection consists in performing two rounds of phage selection on the antigen-negative sample to select binders against immunodominant but nonrelevant epitopes. The corresponding non-relevant binders are subsequently produced as a pool of soluble fragments that are added in large excess during a phage display selection performed with the original library on a sample of interest containing the relevant epitope (supplemental Fig. S1). During this selection, all non-relevant epitopes are thus blocked by the corresponding non-relevant soluble binders (the mask), which greatly favors the selection of binders against the relevant epitopes. This strategy was compared with a mere depletion strategy on four types of samples: recombinant proteins, transfected cell lines, cancer cell lines, and cancer biopsies.
Selection of Phage Antibodies Against a Specific Part of Recombinant HER2-Fc Protein
The proof of principle of this new approach was first established on a simple selection procedure using a purified recombinant protein. A single-domain antibody (sdAb, see (23, 24) for review) library was built using peripheral blood mononuclear cells (PBMCs) of llamas immunized with various recombinant proteins consisting of fusion between ectodomains of relevant tumor markers and human IgG1 Fc portion, including HER2-Fc. The aim of this first part of the study was to favor the selection of HER2 binders as compared with Fc binders.
A basic strategy (i.e. direct selection of phage antibody produced using KM13 helper phage) was compared with conventional depletion strategies on irrelevant Fc bearing molecules prior to positive selection on HER2-Fc fusion. A third approach was developed using hyperphage to produce the phage-antibody particles since it was demonstrated that this helper phage can significantly increase both the percentage of phage actually displaying an antibody fragment and the number of antibody fragment displayed per particles (valency). Theoretically, these two factors should increase the efficiency of the depletion strategy. Finally a masked selection strategy was performed using a human IgG1 Fc fragment protein as nonrelevant sample.
Fig. 1A shows that the highest proportion of HER2 binders was obtained using the masked selection strategy and, to a lesser extent, after a depletion step. Using Hyperphage did not significantly improve the yield of HER2 binders. Not surprisingly, the vast majority of binders obtained by the direct selection strategy were Fc binders. DNA sequencing was performed on randomly picked clones to compare the diversity of the different outputs. The direct selection revealed 8 different clones out of 24 binders, six different clones for each of the depletion strategy (out of 41 or 37 binders) and 9 different clones out of 69 binders for the masked selection (Table I), suggesting that the masked selection on purified antigen could greatly improve the frequency of relevant binders without markedly impacting the output diversity compared with the depletion strategies.
Fig. 1.
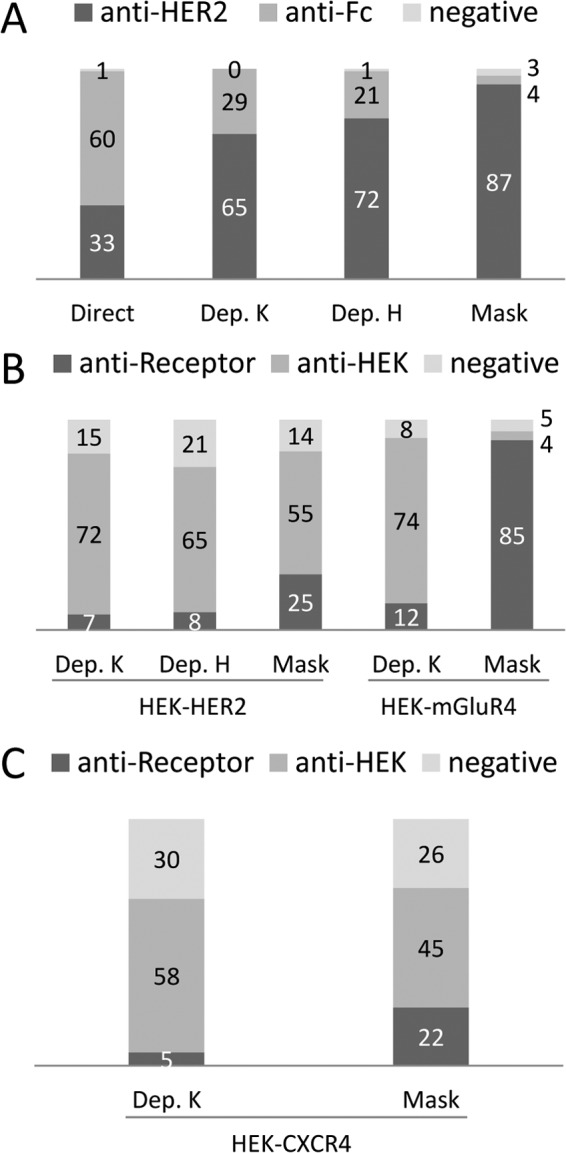
Phage ELISA analysis of clones selected on known antigens A, Screening of clones selected on recombinant HER2-Fc. Ninety-four clones randomly picked from each type of selection were assayed by monoclonal phage ELISA for binding to HER2-Fc, or Fc. Clones yielding a positive signal on HER2-Fc but not on Fc were considered anti-HER2. Those positive on HER2-Fc and Fc were considered anti-Fc and those negative on both were considered non binders (negative). Direct: simple selection. Dep. K: Depletion step, KM13 helper phage. Dep. H: Depletion step, hyperphage helper phage. Mask: Masked selection. B, Phage ELISA analysis of clones selected on transfected cells. Ninety-four clones randomly picked from each type of selection were assayed by monoclonal phage ELISA for binding to HEK cells, HEK cells transfected with HER2 (HEK-HER2) or cells transfected with mGluR4 (HEK-mGluR4). Binders yielding signals on transfected cells but not on HEK cells were considered receptor-specific (anti-Receptor), those yielding signal on all type of HEK cells were considered HEK-specific (anti-HEK) and those negative on all cells were considered negative. C, Phage ELISA screening on HEK-CXCR4 transfected cells. Ninety-three randomly picked clones from each strategy were assayed. Clones yielding a positive signal on HEK-CXCR4 but not on HEK were considered anti-CXCR4.
Table I. Frequency of anti-HER2 clones isolated by various strategies. Dep. K: Depletion step, and use of Km13 helper phage; Dep. H: Depletion step, and use of Hyperphage helper phage; Mask: Masked selection.
| Clones | C.A5 | C.G5 | A.A10 | A.F1 | A.B5 | B.F11 | A.E6 | A.E4 | C.E4 | A.D6 | C.E10 | A.H10 | C.D4 | H.F6 | H.H8 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HER2-Fc | |||||||||||||||
| Direct | 1 | 1 | 7 | 11 | 1 | 1 | 1 | 1 | |||||||
| Dep. K | 2 | 29 | 7 | 1 | 1 | 1 | |||||||||
| Dep. H | 1 | 3 | 24 | 7 | 1 | 1 | |||||||||
| Mask | 8 | 1 | 19 | 30 | 7 | 1 | 1 | 1 | 1 | ||||||
| HEK-HER2 | |||||||||||||||
| Dep. K | 1 | 5 | |||||||||||||
| Dep. H | 1 | 5 | |||||||||||||
| Mask | 1 | 9 | 12 | 1 | 1 |
Selection of Phage Antibodies Against Cell Surface Receptors on Transfected Cells
Some targets, such as members of the seven transmembrane receptors, cannot easily be recombinantly produced as soluble protein with a native conformation. In such cases, phage display can be used to select relevant binders by using a positive selection on transfected cells following a depletion step on the corresponding untransfected cell line. We decided to compare the efficiency of depletion versus masked selection for the selection of binders against various targets. The selected targets included a tyrosine kinase receptor, Human Epidermal Growth Factor Receptor 2 (HER2), and two members of the GPCR family, metabotropic glutamate receptor 4 (mGluR4, class C receptor), and the C-X-C chemokine Receptor type 4 (CXCR4, class A receptor). HER2 displays an ectodomain of about 630 amino acids (25) whereas, the two GPCRs display extracellular domains of around 610 and 62 amino acids for mGluR4 and CXCR4, respectively (26, 27). For these experiments, we used sdAb libraries that had been generated after immunization with HER2-, mGluR4-, or CXCR4- transfected HEK293T (HEK) cells. As shown in supplemental Fig. S2, the surface expression levels of these three receptors were different, mGluR4 being better expressed than HER2 and CXCR4 being very poorly expressed. Untransfected HEK293T cells were used for depletion steps and for the selection of masking sdAbs. The outputs of both strategies were analyzed by phage ELISA on transfected and untransfected cells (Fig. 1B).
In the case of HER2 receptor, the output of the depletion/selection strategy was mainly constituted of unspecific HEK binders, yielding only 7% of HER2 binders. The use of Hyperphage as helper phage for high display conditions did not improve (8%) this yield. By contrast, the masked selection approach improved more than three times the output with 26% of HER2 binders. DNA sequencing of a subset of the positive clones revealed two different clones out of six HER2 binders obtained by each of the two depletion strategies and five different clones out of 24 HER2 binders selected by masked selections (Table I). The dominant clone A.B5 was found in all different strategies. Several clones that had been isolated on purified antigens were not retrieved by any of the cell selections. Conversely, clone H.H8 was only retrieved by masked selection on cells.
All the 15 anti-HER2 binders obtained so far using recombinant antigen or transfected cells were produced as soluble fragments to be further characterized in terms of specificity and affinity. Their specificity was first confirmed by ELISA. supplemental Fig. S3 shows that all clones (including sdAb H.H8) yielded a high signal on HER2-Fc and were negative against Fc. Next, flow cytometry on HER2 positive cells was used to demonstrate the ability of these anti-HER2 sdAbs to bind their antigen in a native context. All clones were found positive (supplemental Fig. S4A), including the seven clones that were selected on recombinant antigen but not through cell selections. A competition assay was then performed to analyze the ability of each sdAb to compete with the others. The binding of phage-sdAbs on HER2 positive cells was investigated by flow cytometry in the presence of a large excess of each purified soluble sdAb (supplemental Fig. S4B). At least three groups of sdAbs binding to three independent HER2 epitopes could be established (supplemental Fig. S4C), and at least one member of each group could be isolated using masked selection.
A third independent approach based on HTRF) (28)) was finally used to clearly establish the specificity and determine the affinity of anti-HER2 sdAbs. HEK cells were transfected with HER2 fused to the SNAP tag, allowing a site-directed labeling of the receptor at the cell surface with a donor fluorophore. Bound sdAbs were detected using an anti-His tag monoclonal antibody labeled with an acceptor fluorophore. In this setting, the excitation of the donor fluorophore leads to a fluorescence emission of the acceptor fluorophore (FRET effect) if these two molecules are in close proximity, that is, if the sdAb is bound to HER2. As shown in Fig. 2A, all anti-HER2 sdAbs were found positive in this assay whereas an irrelevant (anti-Fc) sdAb did not yield any signal. SdAb H.F6 yielded a lower signal, in agreement with flow cytometry results.
Fig. 2.
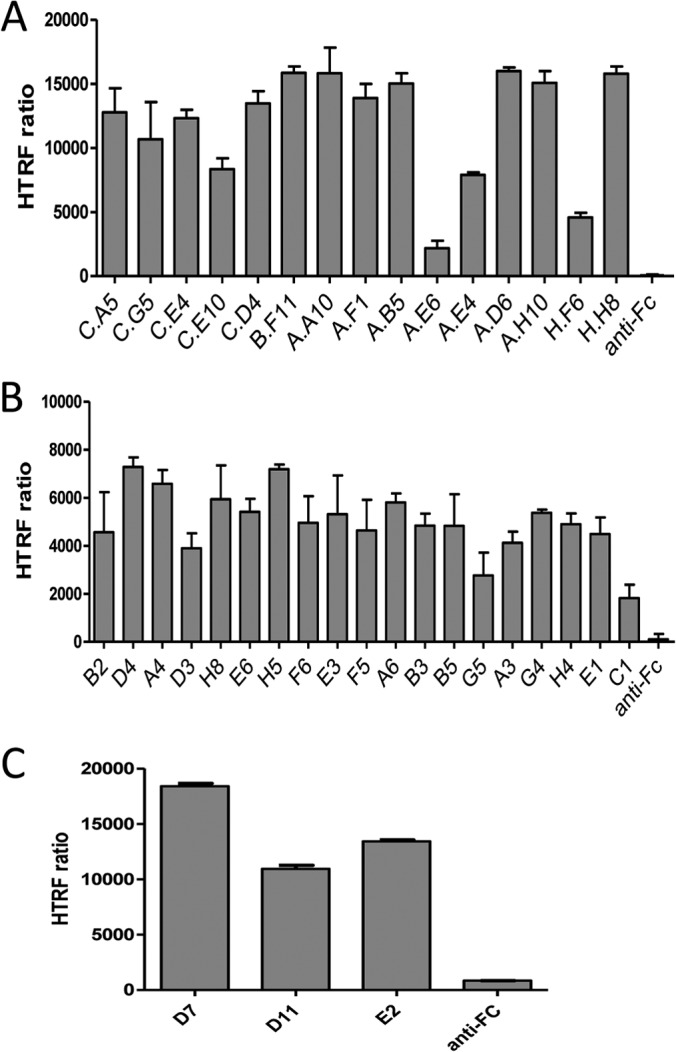
Specificity of sdAbs selected on cell surface antigens by homogeneous time resolved fluorescence (HTRF) technology. A, Analysis of anti-HER2 sdAbs. Cells transfected by SNAP-tag HER2 and labeled with donor fluorophore were incubated with sdAbs and acceptor-labeled anti-6his mAb. sdAb binding, detected by FRET, is expressed as HTRF ratio to normalize results for the HER2 receptor density (see Materials and Methods). Anti-Fc: anti human Fc sdAb used as negative control. B, Analysis of anti-mGluR4 sdAbs. (sdAb binding was followed as described in A). C, Analysis of anti-CXCR4 sdAbs (sdAb binding was followed as described in A).
Interestingly, three clones (A.E6, A.E4, and C.E10), previously shown to bind the same or overlapping epitopes yielded significantly lower FRET signals, suggesting that these epitopes are more distant to the SNAP tag than the other epitopes.
Dose response curves of the 12 best binders were used to determine the sdAb dissociation constants (supplemental Fig. S5A). The calculated KD values using a non-linear curve fitting program ranged between 0.5 and 7.3 nm (supplemental Fig. S5B).
To see if similar results could be obtained on a different type of receptors, depletion and masked strategies were applied to the selection of sdAbs against mGluR4 and CXCR4. These receptors belong to the seven-transmembrane G protein-coupled receptor family and are a good example of antigens which are difficult to produce recombinantly. In this case, all steps, that is, immunization, selection and screening, were performed using transfected HEK cells.
For mGluR4, a conventional depletion step on untransfected HEK cells followed by a selection on mGluR4-transfected cells yielded a vast majority of HEK binders and less than 13% of mGluR4 binders, as shown by phage ELISA on cells, (Fig. 1B), despite the high cell surface expression level of the transfected receptor (supplemental Fig. S2). In sharp contrast, the masked selection strategy was highly successful since 90% of the output was specific for the relevant receptors and only 4% of the outputs were HEK binders (Fig. 1B). The sequencing of 47 mGluR4 binders obtained by masked selection revealed 19 different clones, suggesting again that this approach can lead to a good diversity of binders. Soluble sdAbs corresponding to these 19 clones were produced, purified and characterized by HTRF experiments on mGluR4-transfected cells. As shown in Fig. 2B, all clones yielded high HTRF signals demonstrating their specificity for mGluR4, in contrast to an irrelevant anti-Fc sdAb that was found negative. Dose response curves and non-linear curve fitting analysis (supplemental Fig. S5C) were used to determine the dissociation constant of five binders among the best ones. KD values of these monovalent single domain antibodies ranged from 3.9 to 30 nm (supplemental Fig. S5D).
The same approach was applied to CXCR4, a receptor characterized by a low level of expression (supplemental Fig. S2) and a very short extracellular domain (∼62 amino acids). Depletion on nontransfected HEK cells followed by a selection on CXCR4-transfected cells was performed. In this case, the first round of selection results led to a significant difference between masked selection and depletion. Indeed, masked selection yielded 24% of CXCR4 specific clones, that is, fourfold higher than the conventional depletion step with 6% of receptor specific clones, as shown by phage ELISA on intact cells (Fig. 1C). Sequencing of the specific clones revealed 3 different sequences. The specificity of these binders, produced as soluble sdAbs, was demonstrated by HTRF (Fig. 2C). Dose response curves were used to determine their dissociation constants (supplemental Fig. S5E), leading to KD values ranging from 15 to 22 nm (supplemental Fig. S5F).
Selection of Phage-antibodies with Breast Cancer Specificity
The efficiency of masked selection being established on transfected HEK cells, we applied this approach to the selection of binders against unknown antigens overexpressed in cancer samples. Llamas were immunized with breast cancer biopsies and the resulting sdAb libraries were selected against either breast cancer cell lines or breast cancer biopsies, using human PBMC and the human mammary epithelium cell line HME1 (immortalized by hTERT expression) as normal sample.
Selection on Breast Cancer Biopsy Lysates
Selections were performed on a mixture of 11 breast cancer biopsy lysates (see Materials and Methods). A lysate of human peripheral blood mononuclear cells mixed with a lysate of human mammary epithelium cell line HME1 (immortalized by hTERT expression) was used as normal sample. A classical deletion/selection approach was compared with a depletion plus masked selection strategy (using sdAbs selected from the same library by panning on the normal sample as mask), and the use of KM13 or hyperphage was compared in both case.
Ninety-six clones were randomly picked after two rounds of selection for each approach. A phage ELISA screening procedure, using plastic-adsorbed biopsy or normal sample lysates, was performed to evaluate the specificity of the selected binders. The depletion strategy yielded a majority of clones binding to both biopsy and normal lysates. Only 13% (KM13) and 9% (hyperphage) of clones showed specificity for biopsy lysates (Fig. 3A). Addition of masking sdAbs during the selection step drastically decreased the number of noncancer specific clones down to 2–3%, and increased the proportion of cancer-specific clones to 40% (KM13) and 30% (hyperphage) (Fig. 3A). DNA sequencing of 53 biopsy lysate binders revealed nine different sequences (supplemental Fig. S6A). Competition experiments performed by phage ELISA in the presence of an excess of each purified sdAb indicated that four of these clones were sharing a common epitope (supplemental Fig. S6B). A representative clone of this family and the five other sdAbs targeting independent epitopes were further characterized.
Fig. 3.
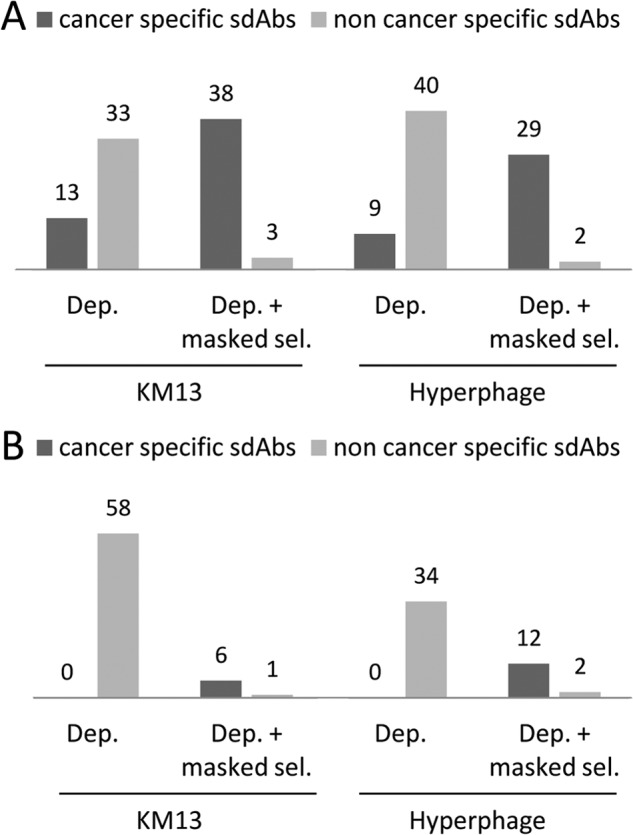
Screening of clones selected against overexpressed targets. A, Screening of clones selected against biopsy lysates. For each selection, 96 clones were randomly picked and assayed by phage ELISA for binding to biopsy or healthy cell lysates immobilized on microtiter plates. Clones positive on biopsy lysates and negative on normal lysates were considered cancer specific. Dep.: depletion. Dep. + masked sel.: depletion followed by selection performed in the presence of masking sdAbs. B, Screening of sdAbs isolated against intact breast cancer cells. For each selection, 96 clones were screened by phage-cytometry assay on a mixture of intact breast cancer cells (MDA-MB-231, MCF7, SKBr3, HCC1954). Bound phage-sdAb were detected using PE-conjugated anti-M13 mAb. Dep.: depletion. Dep. + masked sel.: depletion followed by selection performed in the presence of masking sdAbs.
To confirm the specificity of these sdAbs for breast cancer, phage ELISA was performed on a panel of immobilized lysates from 11 different breast cancer biopsies, and on a mixture of breast cancer cell line lysates (see Material and Methods). As expected, lysates of PBMCs and HME1 cell yielded no signal. Interestingly, various binding profiles were generated against the 11 breast cancer biopsy lysates (Fig 4A). To further investigate the cancer specificity of these antibody fragments, they were produced and purified as soluble fragments and tested by immunohistochemistry. Out of six tested sdAbs, only sdAb J.H6 yielded a strong signal on paraffin-embedded tissue. This sdAb was thus further characterized on larger scale using breast cancer tissue microarray including 80 breast cancer samples (lobular and ductal breast cancer biopsies) and 14 healthy breast samples. Fig. 4B shows immunohistochemistry results on representative samples (cancer biopsies and normal samples). None of the normal samples were stained by this sdAb whereas 75% of breast cancer biopsies were positive (Fig. 4C).
Fig. 4.

Fine characterization of candidate sdAbs isolated against biopsy lysates. A, Reverse phase phage-ELISA. Various biopsy lysates, a mixture of breast cancer cell lysates, (BC cell lines), a human PBMC lysate, and a healthy breast epithelial cell line HME1 lysate were coated on MaxiSorpTM 96 well plates. Phage-sdAbs were incubated and washed. Bound phages were detected using HRP-conjugated anti-M13 mAb. Data shown are representative of at least 3 independent experiments. B, Tissue micro array analysis. Paraffin embedded slide containing 80 breast cancer samples or 14 healthy breast samples were incubated with purified and in vitro biotinylated sdAb J.H6. Bound sdAbs were detected using HRP-conjugated streptavidin. Representative examples are shown. C, Staining results were classified according to their intensity. SI: staining intensity.
Selection for Cell Surface Binders on Breast Cancer Cell Lines
Although intracellular cancer specific antigens can be useful as biomarkers for diagnosis purposes, they are not compatible with some therapeutic approaches such as therapeutic antibodies. For these approaches, the identification of cancer-specific membrane antigens is required. To evaluate the potential of masked selection in this case, we compared the output of two rounds of selection performed on a mixture of four different breast cancer cell lines (MDA-MB-231, MCF7, SKBr3, and HCC1954) using the same sdAb library and either a simple depletion step on a mixture of intact human PBMCs and HME1 (PBMC+HME1), or a combination of deletion and masked selection using sdAbs from the same library selected on PBMC+HME1. Ninety-six clones from each selection strategy were screened as phage-sdAb by flow cytometry on the mixture of breast cancer cell lines, and PBMC+HME1.
As shown in Fig. 3B, a standard depletion strategy did not succeed in isolating any cancer specific clones, whatever the helper phage used. In sharp contrast, 60 and 35% of the 96 tested clones were positive on PBMC+HME1, for KM13 and hyperphage, respectively. The addition of the masking sdAbs during the selection step not only very efficiently blocked the selection of unwanted PBMC+HME1 binders, but allowed the selection of 6 to 12 clones that specifically bound to the mixture of breast cancer cell lines, depending on the helper phage. DNA sequencing of these 18 binders revealed 11 different sequences (supplemental Fig. S6C). Flow cytometry competition experiments performed using purified sdAbs indicated that 6 different epitopes were targeted by these 11 different sdAbs (supplemental Fig. S6D). A representative binder of each epitope was chosen for further studies. These 6 purified sdAbs were tested by flow cytometry against six different breast cancer cell lines including the four used for selection, seven cancer cell lines of various origins, normal breast epithelial cell line HME1 and human PBMCs. As shown in Fig. 5A, none of the sdAbs were positive on normal cells. Four sdAbs (M.C9, M.B9, M.H3, and M.H12) were strongly positive against most of the tested cancer cell lines. SdAb M.D5 specifically recognized two of the four breast cancer cell lines used for selection whereas sdAb M.A4 was positive only on one breast cancer cell line (MDA-MB-231), but recognized two other types of cancer cell lines such as PC3 (prostate cancer) and HeLa (cervical cancer).
Fig. 5.

Fine characterization of candidate sdAbs against intact breast cancer cells. A, A cytometry assay was performed on six breast cancer cell lines, seven cancer cell lines of various origins (cervical, pancreas, ovarian, colon, prostate, and lymphocyte), on human PBMC and normal breast epithelium cell line HME1. In vitro biotinylated sdAb were added on cells. After washing, bound sdAbs were detected with PE-conjugated streptavidin. MFI: mean fluorescence intensity. Data shown are representative of at least three independent experiments. B, Tissue micro array analysis. Paraffin embedded tissue array containing 80 breast cancer samples and 14 healthy breast samples were incubated with in vitro biotinylated sdAbs. Bound sdAbs were detected by HRP-conjugated streptavidin. Shown are representative examples. C, Staining results were classified according to their intensity. SI: staining intensity.
Immunohistochemistry characterization demonstrated that two of these sdAbs (M.H3, M.H12) were functional on paraffin-embedded tissues. These sdAbs were thus assayed on breast cancer tissue micro array. Fig. 5B displays representative results obtained with these two sdAbs. None of the 14 healthy samples were stained while 98% or 79% of breast cancer biopsies were strongly stained by sdAb M.H3 and M.H12, respectively (Fig. 5C).
Identification of the Targeted Markers
Two of sdAbs leading to strong positive signals on many cell lines, M.C9 and M.H12, and two sdAbs with more restricted patterns, M.A4 and M.D5, were chosen to demonstrate the feasibility of identifying their corresponding antigens by immunoprecipitation (IP) and mass spectrometry analysis. Purified sdAbs were covalently immobilized on NHS-activated Sepharose beads and were incubated with cell line lysates. After binding and washing, the retained proteins were loaded on SDS-PAGE. M.H12 immunoprecipitation of T47D lysates yielded several strong bands below 100 kDa that were not present on the control IP performed in the absence of sdAb (Fig. 6A). Under the same conditions, M.C9 yielded a faint but specific band at around 30 kDa. Mass spectrometry (MS) analysis was performed on both IPs and led to the identification of Epithelial cell adhesion molecule (EpCAM, CD326) and Transferrin receptor 1 (TfR1, CD71) for M.C9 and M.H12 respectively (Fig 6C). IPs using M.A4 and M.D5 with SKBr3 lysate did not lead to a clearly defined band as detected by silver staining. However, the differential MS analysis of the IP outcome could identify metastasis cell adhesion molecule (MetCAM/MUC18/MCAM/CD146) and Sushi domain containing 2 (SUSD2) being selectively present in the IP eluates of sdAbs M.A4 and M.D5, respectively (Fig. 6C). To confirm these results by an independent approach, a Western blot analysis of the IP outputs was performed using corresponding commercially available antibodies. In each case, a specific signal was obtained with the IP of the corresponding sdAb, confirming the MS results (Fig. 6B).
Fig. 6.

Target identification by immunoprecipitation and mass spectrometry. A, Sepharose beads were covalently coupled to purified, incubated with cell lysates (+) or not (-), and washed. Captured proteins were separated on SDS-Page, and detected by silver staining. Cont: negative control using uncoupled beads. B, Immunoprecipitated proteins corresponding to each sdAb and matching controls were analyzed by Western blots using commercial anti-CD71, anti-EpCAM, anti-MUC18, or anti-SUSD2 antibodies. C, Mascot Results of the LC-MSMS analysis. Only peptides with high confidence Mascot score (identities) were used. Detailed MS and MSMS results are available as supplementary data.
DISCUSSION
Beside depletion (negative selection), several techniques have been proposed to focus a phage selection on a particular antigen or epitope, including guided selection (29, 30), competitive elutions (8), or alternating of the source of antigen (7). These techniques have been successfully used in several studies but they are limited in several aspects. Guided selection depends on the availability of a cloned antibody and is labor-intensive. Competitive elution uses a known ligand or monoclonal antibody to specifically elute phage-antibodies binding to the same epitope. Unfortunately this strategy has a tendency to favor the selection of low affinity binders. The availability of various sources of an antigen of interest (expressed on cell and as purified protein, for example) can be a limiting factor. But most importantly, none of these approaches can be used for target discovery.
We have designed a new procedure to restrict the output of phage display selections toward specific epitopes of a chimeric protein, toward transfected membrane receptors and toward specific or overexpressed cancer markers using crude lysates, or intact cells when surface markers are preferred. The masked selection is based on a competition between masking antibody fragments and phage particles. The general concept underlying this approach is merely to block unwanted epitopes by addition of an excess of binders previously selected by panning the very same library against the non relevant sample. This procedure can be very efficient since the input of a selection procedure usually contains 1012 phage particle in 1 ml, leading to a phage concentration of around 2 nm. Single domain antibodies used in this study are easily produced in E. coli and we routinely use 10 μm of polyclonal sdAbs for the masking procedures, ensuring a huge excess of sdAbs over phage-sdAbs to guarantee an efficient blockade of non relevant epitopes.
In all cases, from a very simple selection on purified antigen to the selection of binders against overexpressed tumor markers, the principle of masking nonrelevant epitopes was shown to be more efficient than conventional depletion. This advantage was most obvious when using highly complex sources of antigens such as intact tumor cells. This simple procedure is very powerful because it takes advantage of the potential biases of a library. Indeed, libraries often contain a large number of binders directed against non-relevant but highly abundant or very immunoreactive epitopes. These binders often out-compete binders against scarce but potentially relevant epitopes when selections are performed on complex samples. In such case, masked selection should be very efficient because these dominant binders targeting abundant proteins would be highly represented in the polyclonal population of soluble binders selected on the nonrelevant sample (the mask). In other words, the mask will naturally be biased with binders that have to block the most problematic (abundant and/or immunoreactive) epitopes.
In this study, masked selection was compared with a depletion strategy. Our results demonstrate some efficiency of depletion but also highlight its shortcomings in difficult selections on complex samples. The principle of depletion relies on the capture of non relevant binders from the phage population before positive selection. This approach can improve the specificity of an output but cannot be totally efficient since, as for any noncovalent interaction, a fraction of the total phage-antibody to be depleted will stay in solution at a ratio dependent on the affinity of the antibody. Increasing the number of antibody fragments displayed per phage particle by using hyperphage as helper phage should increase the apparent affinity of binders and thus increase the proportion of bound phage during the depletion step. Such an effect could be seen in this study on depletion performed on Fc portion but was not visible on selections performed on complex samples.
This work demonstrates that masked selection can greatly increase the number of target-specific binders, without significantly impacting on the diversity of selected clones, or on their affinity. Indeed, several binders with nanomolar affinities were selected against several epitopes of the various antigens targeted in this study, including difficult antigens such as GPCRs. Remarkably, masked selection also significantly improved the isolation of binders against CXCR4, a class A GPCR receptor displaying only 62 amino acids above the cell membrane. Noteworthy, in the case of GPCRs, the entire procedure, from immunization to sdAb characterization, was performed using only transfected cells, that is, in the absence of purified proteins.
Probably one of the most remarkable applications of masked selection is the isolation of binders against unknown but differentially expressed targets. In this study, we have isolated binders against differentially expressed antigens using both biopsy lysates and intact breast cancer cell lines, thus insuring that the targeted markers are accessible membrane proteins. Two sdAbs leading to strong flow cytometry signals on various cancer cell lines (M.C9 and M.H12) and two sdAbs with more restricted expression patterns (M.A4 and M.D5) were chosen to demonstrate the possibility of identifying the corresponding antigen by immunoprecipitation and mass spectrometry (MS). The antigen targeted by M.C9 was identified as the epithelial cell adhesion molecule (EpCAM, CD326). This result further validates our approach since EpCAM is known to be a tumor associated antigen, abundantly expressed in primary tumors and metastases of most epithelial malignancies (31), including breast cancer (32, 33). More recently, EpCAM was also shown to be expressed on cancer stem cells and circulating tumor cells, which further raised enthusiasm to use EpCAM for drug delivery (34, 35). The second sdAb, M.H12, binds the human transferrin receptor 1 (TfR1, CD71), a receptor involved in cellular iron homeostasis. Numerous studies have shown that TfR1 is highly expressed in cancer cells (36), including tumors of the nervous system, chronic lymphocytic leukemia, non-Hodgkin's lymphoma, lung adenocarcinoma but also breast cancer (37). A proposed explanation for its wide overexpression in tumor cells is that these rapidly growing cells require more iron as nutrient and cofactor of ribonucleotide reductase for their massive DNA synthesis (36). In addition, TfR1 was also shown to be a signaling molecule that enhances anti-apoptosis and potentiates breast cancer cells survival through tyrosine phosphorylation by Src (38). M.A4 is binding MetCAM. This receptor was first identified as a human melanoma antigen, rarely expressed on normal epidermal melanocytes, but abundantly expressed in advanced primary and metastatic melanoma (39). Phage display selection on mesothelioma cell lines also led to the isolation of anti-MetCAM antibody fragments (40). It that has been shown to play an important role during melanoma progression to metastasis (41). However, its role in breast cancer is much less characterized and appears to be controversial (42). Finally sdAb M.D5 binds SUSD2, a 822-amino acid type I membrane protein. Very little is known about this human receptor. It was indeed recently shown by mass spectrometry to interact with galectin-1, a protein extensively studied for its role in tumor immune-escape mechanisms and angiogenesis (43), and was even more recently isolated using a breast cancer cDNA library based approach (44). In this work, the authors show weak or no expression of SUSD2 in normal epithelial cells, but staining of lobular and ductal breast carcinomas, thereby confirming our findings on the expression of this protein. Altogether, these results clearly warrant further investigations to fully evaluate the potential of SUSD2 as innovative therapeutic target.
Thus, our results highlight the strong benefit of the masked selection strategy for isolating high affinity binders against overexpressed receptors. In this work aiming at establishing a proof of concept of masked selection efficiency, we screened around two hundreds clones selected on intact cells, which already led to the isolation of 11 different tumor specific binders. Because phage display can be easily adapted to high throughput, a straightforward scale-up of the procedure has the potential to lead the isolation of large numbers of tumor specific binders. It would also be of high interest to perform this approach on more clinically relevant samples than tumor cell lines, such as patient derived xenografts (45).
By facilitating the identification of new targetable markers for antibody-based therapy while simultaneously providing the specific antibody, masked selection has a great potential in the field of cancer diagnostic and therapy. It should also be of high interest for many other purposes, such as the identification of specific cell surface receptors of some cell types, such as normal and cancer stem cells or regulatory T cells, for example. In principle, this technique is very flexible and can be used with any kind of antibody fragments (scFv, Fab fragments), alternative scaffolds (darpins, monobodies, affibodies, anticalins) (46, 47), or even other type of display such as ribosome display (48).
Supplementary Material
Acknowledgments
We would like to thank Stéphane Garcia for biopsies and TMA, Gérard Matthis and Jean-Philippe Pin for the kind gift of SNAP tag vectors and Daniel Olive, Marie Alix Pool, Ralph Willemsen and Jean Imbert for the gift of cell lines.
Footnotes
Author contributions: P.C. designed research; K.E., D.N., M.L., and K.A. performed research; J.B., S.A., B.K., D.B., and P.C. analyzed data; K.E., D.N., S.A., and P.C. wrote the paper.
* This work was supported by the ANR (Agence Nationale de Recherche) program ‘Nanosciences and Nanotechnologies’ under the grant ANR-07-PNANO-051-01 and by the ARC (Association pour la Recherche contre le Cancer). DB and PC are members of the IBiSA nanobody platform supported by INSERM transfert, OSEO and FP7 program NAMDIATREAM. SA and JPB are members of Marseille Proteomic (MaP), an IBiSA platform supported by Institut Paoli-Calmettes, Canceropôle PACA, INCa, and Région Provence Alpes Côte d'Azur.
 This article contains supplemental Figs. S1 to S6.
This article contains supplemental Figs. S1 to S6.
Competing interests: The author(s) declare no competing interests.
1 The abbreviations used are:
- Abs
- antibodies
- HTRF
- homogenous time-resolved fluorescence
- PBMC
- peripheral blood mononuclear cells.
REFERENCES
- 1. Kohler G., Milstein C. (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 [DOI] [PubMed] [Google Scholar]
- 2. McCafferty J., Griffiths A. D., Winter G., Chiswell D. J. (1990) Phage antibodies: filamentous phage displaying antibody variable domains. Nature 348, 552–554 [DOI] [PubMed] [Google Scholar]
- 3. Weiner L. M. (2006) Fully human therapeutic monoclonal antibodies. J. Immunother. 29, 1–9 [DOI] [PubMed] [Google Scholar]
- 4. Hoogenboom H. R. (2005) Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 23, 1105–1116 [DOI] [PubMed] [Google Scholar]
- 5. Ngai P. K., Ackermann F., Wendt H., Savoca R., Bosshard H. R. (1993) Protein A antibody-capture ELISA (PACE): an ELISA format to avoid denaturation of surface-adsorbed antigens. J. Immunol. Methods 158, 267–276 [DOI] [PubMed] [Google Scholar]
- 6. Siva A. C., Kirkland R. E., Lin B., Maruyama T., McWhirter J., Yantiri-Wernimont F., Bowdish K. S., Xin H. (2008) Selection of anti-cancer antibodies from combinatorial libraries by whole-cell panning and stringent subtraction with human blood cells. J. Immunol. Methods 330, 109–119 [DOI] [PubMed] [Google Scholar]
- 7. Even-Desrumeaux K., Fourquet P., Secq V., Baty D., Chames P. (2012) Single-domain antibodies: a versatile and rich source of binders for breast cancer diagnostic approaches. Mol. Biosyst. 8, 2385–2394 [DOI] [PubMed] [Google Scholar]
- 8. Veggiani G., Ossolengo G., Aliprandi M., Cavallaro U., de Marco A. (2011) Single-domain antibodies that compete with the natural ligand fibroblast growth factor block the internalization of the fibroblast growth factor receptor 1. Biochem. Biophys. Res. Commun. 408, 692–696 [DOI] [PubMed] [Google Scholar]
- 9. Stefan N., Martin-Killias P., Wyss-Stoeckle S., Honegger A., Zangemeister-Wittke U., Pluckthun A. (2011) DARPins recognizing the tumor-associated antigen EpCAM selected by phage and ribosome display and engineered for multivalency. J. Mol. Biol. 413, 826–843 [DOI] [PubMed] [Google Scholar]
- 10. Liew P. X., Ge F., Gullo C., Teoh G. K., Hwang W. Y. (2009) Use of phage display to isolate specific human monoclonal antibody fragments against a potential target for multiple myeloma. Ann. Acad. Med. Singapore 38, 621–629 [PubMed] [Google Scholar]
- 11. Omidfar K., Rasaee M. J., Modjtahedi H., Forouzandeh M., Taghikhani M., Golmakani N. (2004) Production of a novel camel single-domain antibody specific for the type III mutant EGFR. Tumour Biol. 25, 296–305 [DOI] [PubMed] [Google Scholar]
- 12. Mazuet C., Lerouge D., Poul M. A., Blin N. (2006) Breast carcinoma specific antibody selection combining phage display and immunomagnetic cell sorting. Biochem. Biophys. Res. Commun. 348, 550–559 [DOI] [PubMed] [Google Scholar]
- 13. Nielsen U. B., Marks J. D. (2000) Internalizing antibodies and targeted cancer therapy: direct selection from phage display libraries. Pharm. Sci. Technolo. Today 3, 282–291 [DOI] [PubMed] [Google Scholar]
- 14. Behar G., Chames P., Teulon I., Cornillon A., Alshoukr F., Roquet F., Pugniere M., Teillaud J. L., Gruaz-Guyon A., Pelegrin A., Baty D. (2009) Llama single-domain antibodies directed against nonconventional epitopes of tumor-associated carcinoembryonic antigen absent from nonspecific cross-reacting antigen. FEBS J. 276, 3881–3893 [DOI] [PubMed] [Google Scholar]
- 15. Roovers R. C., van der Linden E., de Bruine A. P., Arends J. W., Hoogenboom H. R. (2001) Identification of colon tumour-associated antigens by phage antibody selections on primary colorectal carcinoma. Eur. J. Cancer 37, 542–549 [DOI] [PubMed] [Google Scholar]
- 16. Jensen K. B., Jensen O. N., Ravn P., Clark B. F., Kristensen P. (2003) Identification of keratinocyte-specific markers using phage display and mass spectrometry. Mol. Cell. Proteomics 2, 61–69 [DOI] [PubMed] [Google Scholar]
- 17. Geuijen C. A., Bijl N., Smit R. C., Cox F., Throsby M., Visser T. J., Jongeneelen M. A., Bakker A. B., Kruisbeek A. M., Goudsmit J., de Kruif J. (2005) A proteomic approach to tumour target identification using phage display, affinity purification and mass spectrometry. Eur. J. Cancer 41, 178–187 [DOI] [PubMed] [Google Scholar]
- 18. Rozan C., Cornillon A., Petiard C., Chartier M., Behar G., Boix C., Kerfelec B., Robert B., Pelegrin A., Chames P., Teillaud J. L., Baty D. (2013) Single-Domain Antibody-Based and Linker-Free Bispecific Antibodies Targeting FcgammaRIII Induce Potent Antitumor Activity without Recruiting Regulatory T Cells. Mol. Cancer Ther. 12, 1481–1491 [DOI] [PubMed] [Google Scholar]
- 19. Even-Desrumeaux K., Baty D., Chames P. (2010) Strong and oriented immobilization of single domain antibodies from crude bacterial lysates for high-throughput compatible cost-effective antibody array generation. Mol. Biosyst. 6, 2241–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alvarez-Rueda N., Behar G., Ferre V., Pugniere M., Roquet F., Gastinel L., Jacquot C., Aubry J., Baty D., Barbet J., Birkle S. (2007) Generation of llama single-domain antibodies against methotrexate, a prototypical hapten. Mol. Immunol. 44, 1680–1690 [DOI] [PubMed] [Google Scholar]
- 21. Shevchenko A., Wilm M., Vorm O., Mann M. (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858 [DOI] [PubMed] [Google Scholar]
- 22. Belotti E., Puvirajesinghe T. M., Audebert S., Baudelet E., Camoin L., Pierres M., Lasvaux L., Ferracci G., Montcouquiol M., Borg J. P. (2012) Molecular characterisation of endogenous Vangl2/Vangl1 heteromeric protein complexes. PLoS One 7, e46213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muyldermans S. (2001) Single domain camel antibodies: current status. J. Biotechnol. 74, 277–302 [DOI] [PubMed] [Google Scholar]
- 24. Harmsen M. M., De Haard H. J. (2007) Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 77, 13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cho H. S., Mason K., Ramyar K. X., Stanley A. M., Gabelli S. B., Denney D. W., Jr., Leahy D. J. (2003) Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756–760 [DOI] [PubMed] [Google Scholar]
- 26. Bessis A. S., Bertrand H. O., Galvez T., De Colle C., Pin J. P., Acher F. (2000) Three-dimensional model of the extracellular domain of the type 4a metabotropic glutamate receptor: new insights into the activation process. Protein Sci. 9, 2200–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu B., Chien E. Y., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., Stevens R. C. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Degorce F., Card A., Soh S., Trinquet E., Knapik G. P., Xie B. (2009) HTRF: A technology tailored for drug discovery - a review of theoretical aspects and recent applications. Curr. Chem. Genomics 3, 22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Beiboer S. H., Reurs A., Roovers R. C., Arends J. W., Whitelegg N. R., Rees A. R., Hoogenboom H. R. (2000) Guided selection of a pan carcinoma specific antibody reveals similar binding characteristics yet structural divergence between the original murine antibody and its human equivalent. J. Mol. Biol. 296, 833–849 [DOI] [PubMed] [Google Scholar]
- 30. Figini M., Obici L., Mezzanzanica D., Griffiths A., Colnaghi M. I., Winter G., Canevari S. (1998) Panning phage antibody libraries on cells: isolation of human Fab fragments against ovarian carcinoma using guided selection. Cancer Res. 58, 991–996 [PubMed] [Google Scholar]
- 31. Simon M., Stefan N., Pluckthun A., Zangemeister-Wittke U. (2013) Epithelial cell adhesion molecule-targeted drug delivery for cancer therapy. Expert Opin. Drug Delivery 10, 451–468 [DOI] [PubMed] [Google Scholar]
- 32. Gastl G., Spizzo G., Obrist P., Dunser M., Mikuz G. (2000) Ep-CAM overexpression in breast cancer as a predictor of survival. Lancet 356, 1981–1982 [DOI] [PubMed] [Google Scholar]
- 33. Schmidt M., Hasenclever D., Schaeffer M., Boehm D., Cotarelo C., Steiner E., Lebrecht A., Siggelkow W., Weikel W., Schiffer-Petry I., Gebhard S., Pilch H., Gehrmann M., Lehr H. A., Koelbl H., Hengstler J. G., Schuler M. (2008) Prognostic effect of epithelial cell adhesion molecule overexpression in untreated node-negative breast cancer. Clin. Cancer Res. 14, 5849–5855 [DOI] [PubMed] [Google Scholar]
- 34. Allard W. J., Matera J., Miller M. C., Repollet M., Connelly M. C., Rao C., Tibbe A. G., Uhr J. W., Terstappen L. W. (2004) Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 10, 6897–6904 [DOI] [PubMed] [Google Scholar]
- 35. Pantel K., Brakenhoff R. H., Brandt B. (2008) Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat. Rev. Cancer 8, 329–340 [DOI] [PubMed] [Google Scholar]
- 36. Daniels T. R., Delgado T., Rodriguez J. A., Helguera G., Penichet M. L. (2006) The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin. Immunol. 121, 144–158 [DOI] [PubMed] [Google Scholar]
- 37. Yang D. C., Wang F., Elliott R. L., Head J. F. (2001) Expression of transferrin receptor and ferritin H-chain mRNA are associated with clinical and histopathological prognostic indicators in breast cancer. Anticancer Res. 21, 541–549 [PubMed] [Google Scholar]
- 38. Jian J., Yang Q., Huang X. (2011) Src regulates Tyr(20) phosphorylation of transferrin receptor-1 and potentiates breast cancer cell survival. J. Biol. Chem. 286, 35708–35715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lehmann J. M., Holzmann B., Breitbart E. W., Schmiegelow P., Riethmuller G., Johnson J. P. (1987) Discrimination between benign and malignant cells of melanocytic lineage by two novel antigens, a glycoprotein with a molecular weight of 113,000 and a protein with a molecular weight of 76,000. Cancer Res. 47, 841–845 [PubMed] [Google Scholar]
- 40. Bidlingmaier S., He J., Wang Y., An F., Feng J., Barbone D., Gao D., Franc B., Broaddus V. C., Liu B. (2009) Identification of MCAM/CD146 as the target antigen of a human monoclonal antibody that recognizes both epithelioid and sarcomatoid types of mesothelioma. Cancer Res. 69, 1570–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zigler M., Villares G. J., Dobroff A. S., Wang H., Huang L., Braeuer R. R., Kamiya T., Melnikova V. O., Song R., Friedman R., Alani R. M., Bar-Eli M. (2011) Expression of Id-1 is regulated by MCAM/MUC18: a missing link in melanoma progression. Cancer Res. 71, 3494–3504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zeng G. F., Cai S. X., Wu G. J. (2011) Up-regulation of METCAM/MUC18 promotes motility, invasion, and tumorigenesis of human breast cancer cells. BMC Cancer 11, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Camby I., Le Mercier M., Lefranc F., Kiss R. Galectin-1: a small protein with major functions. Glycobiology 16, 137R–157R, 2006 [DOI] [PubMed] [Google Scholar]
- 44. Watson A. P., Evans R. L., Egland K. A. (2013) Multiple functions of sushi domain containing 2 (SUSD2) in breast tumorigenesis. Mol. Cancer Res. 11, 74–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Siolas D., Hannon G. J. (2013) Patient-Derived Tumor Xenografts: Transforming Clinical Samples into Mouse Models. Cancer Res. 73, 5315–5319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stumpp M. T., Amstutz P. (2007) DARPins: a true alternative to antibodies. Curr. Opin. Drug Discov. Devel. 10, 153–159 [PubMed] [Google Scholar]
- 47. Gebauer M., Skerra A. (2009) Engineered protein scaffolds as next-generation antibody therapeutics. Curr. Opin. Chem. Biol. 13, 245–255 [DOI] [PubMed] [Google Scholar]
- 48. Zahnd C., Amstutz P., Pluckthun A. (2007) Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat. Methods 4, 269–279 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.