

Abstract
A PCN theranostic platform comprises a doxorubicin (DXR)-loaded liposomal core and an acid-sensitive polymer shell that is functionalized with Herceptin and GdIII-based MRI contrast agents. In vitro testing reveals a 14-fold increase in DXR-based cytotoxicity versus a non-targeted analogue and an 120-fold improvement in cellular GdIII–uptake in comparison with clinically approved DOTA-GdIII, leading to significant T1 MRI contrast enhancement.
Keywords: cancer targeting, drug delivery, magnetic resonance imaging, pH-responsive drug release, theranostics
Recent advances in nanotechnology, materials science, and biotechnology have enabled the design of theranostic nanoparticles that co-deliver therapeutic compounds and imaging probes in a single platform to heterogeneous disease sites such as tumors, simultaneously treating and enabling non-invasive monitoring of the response to treatment.[1] This can allow the clinician to detect previously unseen tumor sites and immediately adjust the treatment regimen to improve the margin for success. Magnetic resonance imaging (MRI) has emerged as an ideal imaging modality for this application due to its high-resolution, real-time 3D tomography capability, excellent penetration depth, and independence from ionizing radiation (CT) or radionuclides (PET-CT/SPECT).[2] In contrast, optical imaging techniques utilizing two-photon fluorescence or intravital microscopy/endoscopy as well as bioluminescence are generally low resolution, only provide limited (or no) tomographic information, have insufficient penetration depth, and, requires multiple labels to assess the surrounding cells or tissues, thus limiting the amount of salient information that can be obtained on the tumor and its neighboring environment.[3]
The aforementioned attributes make MRI the ideal imaging modality for evaluating the treatment of solid tumors.[4] However, to maximize the full potential of MRI for the evaluation of solid tumors, theranostic platforms have to overcome a number of limitations before they can be clinically successful. For in vivo imaging, the intrinsically low probe sensitivity, especially of T1 contrast agents, require efficient delivery and maximum cellular accumulation of contrast agents like gadolinium (Gd) chelates.[5] However, these contrast agents have poor cellular uptake, leading to insignificant contrast enhancements.[2] For therapy, the lack of specificity of chemotherapeutics to only the tumor tissue necessitates a high administered dose and reduces the efficacy of the drug.[6] This high-dosage requirement increases the prevalence of adverse side effects for patients and shrinks the therapeutic window.[7]
The conjugation of GdIII chelates to a variety of nanostructures, including inorganic nanoparticles,[8] dendrimers,[9] viral capsids,[10] lipid nanoparticles,[11] liposomes,[12] and hydrogels,[13] has been shown to increase T1 MRI contrast compared to unconjugated GdIII chelates. Many of these platforms, however, have limited therapeutic efficacy due to insufficient drug-loading capabilities and/or lack of triggered drug-release ability under the specific conditions encountered at the tumor site. Additionally, when nanoparticles accumulate passively at the tumor site through the enhanced permeability and retention (EPR) effect[1a], their poor cellular internalization[14] and inability[15] to release the payload further limit their therapeutic efficacy. These major obstacles must be overcome for nanoparticle-based theranostic platforms to reach their full potential and become clinically viable.[1a, 16]
Herein, we report the first multifunctional polymer-caged nanobin (PCN) theranostic platform that is capable of selectively targeting Her2-expressing tumor cells with high concentrations of T1 MRI contrast agents followed by the triggered release of a high payload of chemotherapeutics. We extended the PCN core-shell nanocarrier concept,[17] which comprises a doxorubicin (DXR)-loaded biocompatible liposomal core and an acid-sensitive, biodegradable polymer shell,[18] to include surface functionalization with Herceptin targeting groups and GdIII-based contrast agents (Figure 1a). In vitro testing of the Her2-targeted theranostic platform (Her-GdIII-PCNDXR) against Her2-overexpressing SK-BR-3 cells reveals an impressive 120-fold enhancement in cellular GdIII–uptake in comparison to free DOTA-GdIII, which significantly decreases T1 relaxation time and provides enhanced T1 MRI contrast for the targeted PCNs over the non-targeted nanocarriers. In addition, the 14-fold increase in cytotoxicity of the Her-GdIII-PCNDXR over the non-targeted analogue makes it a highly promising theranostic agent.
Figure 1.

(a) Schematic presentation of the preparation of DXR-loaded, Herceptin- and GdIII-conjugated PCN (Her-GdIII-PCNDXR). (b) Plots of the hydrodynamic diameters (DH) of BLDXR (black rectangle), PCNDXR (red circle), and Her-GdIII-PCNDXR (blue triangle) and (c) transmission electron microscopy (TEM) image of Her-GdIII-PCNDXR. (d) Time-dependent DXR-releasing profiles of Her-GdIII-PCNDXR at pH 5 (filled square), pH 6 (open circle), and 7.4 (open triangle).
Herceptin (Trastuzumab) is a monoclonal antibody that specifically binds to human epidermal growth factor receptor 2 (Her2), known to be overexpressed in ~25% of human breast cancers.[19] Recently, a promising clinical study[20] has reported that the Herceptin-conjugated chemotherapy agent Trastuzumab emtansine (T-DM1) significantly extends survival rate while reducing side effects in patients with aggressive Her2-overexpressing breast cancers, which suggests that targeting toxic chemotherapeutic agents to specific cell-surface receptors can indeed improve their therapeutic efficacies. We hypothesize that this strategy can be extended to our PCN-based theranostic nanoparticle platform[21] where the Her2-targeting functionality is expected to result in more efficient internalization of both drugs and imaging agents by Her2-overexpressing cancer cells through a receptor-mediated endocytosis pathway.[7, 22] Together with the pH-triggered drug-releasing capability[23] and high stability in physiological conditions,[17] the resulting enhanced cellular uptake would lead to major improvements in both cytotoxicity and MRI contrast enhancement at the target sites and would significantly advance the theranostic concept.
While high specificity to Her2-overexpressing tumor cells has spurred usage of Herceptin as a targeting ligand for delivery nanocarriers of either drugs or imaging agents,[14, 24] only a few studies[25] have reported a Her2-targeted theranostic platform that includes both chemotherapeutics and imaging probes in a single delivery nanocarrier. Most of these platforms, however, have limited theranostic efficacy due to a lack of triggered drug release and limited ability for in vivo imaging. While a Her2-targeting, iron-oxide nanoparticle-based theranostic platform demonstrated acid-triggered drug release and MR imaing abilities[25c], this system allows T2 MR imaging and exhibits premature drug release under physiological conditions (pH 7.4 and 37 °C). To the best of our knowledge, there have been no reports of Her2-targeting theranostic platforms that combine a chemotherapeutic with a T1 MRI contrast agent and show significant enhancements in both therapy and diagnosis through triggered-release and Her2-targeting. In contrast to the iron oxide nanoparticle for T2 MR imaging, T1 MRI contrast agensts such as GdIII chelates must be further conjugated to a nanocarrier to enhance their intrinsic poor cellular internalization. The work described herein demonstrates the first smart multifunctional theranostic platform that presents facile acid-triggered drug release, high stability under physiological conditions, specific targeting of Her2-overexpresssing cancers, and enhanced T1-weighted MR imaging. Together, these four characteristics lead to outstanding in vitro theranostic performance against Her2-overexpressing breast cancer cells.
Synthesis of Her-GdIII-PCN and acid-triggered drug release
The anticancer drug DXR was first loaded into the core of a liposome using an ion-gradient-mediated remote drug-loading strategy,[18a] which allows DXR to be stably encapsulated with a high density. The resulting DXR-loaded bare liposomes (BLDXR; DH = 98 ± 17 nm (Figure 1b), DXR/lipid = 0.30–0.35 mol/mol, and zeta potential = −8 ± 2 mV) were then coated with cholesterol-terminated poly(acrylic acid) (Chol-PAA; Mn = 10.7 kDa and PDI = 1.12) by a “drop-in” method[17] and then ~50% of the carboxyl groups of the Chol-PAAs were cross-linked with alkyne-functionalized diamine linkers.[18a] The resulting alkyne-functionalized, DXR-loaded PCN (PCNDXR; DH = 118 ± 22 nm (Figure 1b) and zeta potential = −9 ± 2 mV) was then conjugated to azido-functionalized, Alexa Fluor 647-labeled Herceptin, and 1-(3-azido-2-hydroxypropyl)-4,7,10-tris(carboxymethyl)-1,4,7,10-tetraazacyclododecyl-gadolinium(III) (Gd(III)HPN3-DO3A)[26] using copper(I)-catalyzed click ligation (Figure 1a).[21, 27]
Given that approximately 80,000 lipid molecules are present in a 100 nm liposome particle,[28] the resulting Herceptin-and GdIII-conjugated PCNDXR (Her-GdIII-PCNDXR) possesses 10–15 Herceptin antibodies and ~8,000 GdIII ions per particle. The aforementioned conjugation steps do not significantly change the drug loading (DXR/lipid = 0.28–0.32 mol/mol), the zeta potential (−7 ± 2 mV), and the hydrodynamic diameter (DH = 120 ± 20 nm) of the PCN. Together with the DLS data (Figure 1b), transmission electron microscope (TEM) image (Figure 1c) of the resulting Her-GdIII-PCNDXR indicate that these particles are homogenous in size and do not aggregate in buffered saline solutions.
In drug delivery, the high stability of the nanocarriers in physiological environments (pH 7.4 and 37 °C) often means that their payloads are not easily released, which leads to low therapeutic potency in spite of improved circulation time and cellular uptake.[29] Hence, the acidic environments of tumor interstitium[30] as well as cellular endosomes[31] have been used as a way to trigger drug release and improve the therapeutic efficacy of the drug.[32] For the PCN platform, the mixed carboxyl/amide environment of the polymer cage was engineered to trigger drug release at low pH’s.[17–18, 23] As expected, the Her-GdIII-PCNDXR nanocarrier reported herein also exhibits an acid-sensitive drug-release profile (Figure 1d). At pH 5 and pH 6, about half (~60% and ~40%, respectively) of the total amount of encapsulated DXR was released from Her-GdIII-PCNDXR within the initial 24 h and near-complete (~90% and ~70%, respectively) release was achieved within 3 days. At pH 7.4 and 37 °C, however, only 10% of the drug was released after 3 days. This combination of good stability at physiological pH (7.4) and facile acid-triggered drug-release capability are key characteristics of an ideal delivery platform.[33]
Enhanced DXR cytotoxicity by Her-GdIII-PCN
In addition to the acid-triggered drug-release property, the therapeutic efficacy of Her-GdIII-PCNDXR is expected to be improved by the presence of surface Herceptin groups, which accelerates the internalization of the nanocarriers into cells with high Her2 surface expression.[14] To assess this enhancement, we exposed four different human breast cancer cell lines (SK-BR-3, MDA-MB-231-Her2, MDA-MB-231, and MDA-MB-231-Vector) to Her-GdIII-PCNDXR, GdIII-PCNDXR (GdIII-coupled PCN without Herceptins; GdIII/PCN = ~8,000 and DXR/lipid = ~0.3), or free DXRs for 24 h and assessed cell viability using a Guava ViaCount assay. (Note: MDA-MB-231-Her2 and MDA-MB-231-Vector are MDA-MB-231 cells that were transfected with a lentiviral vector, with and without a Her2-encoding gene, respectively.[19] SK-BR-3 and MDA-MB-231-Her2 cells stably overexpress Her2 receptors (> 10 × 105 and > 6 × 105 receptors per cell, respectively), while MDA-MB-231 and MDA-MB-231-Vector cells do not overexpress Her2 (< 0.7 × 105 receptors per cell for both).[19, 34]
The plots of cell viability as a function of DXR dose (Figures 2a and 2b) clearly show that the targeted Her-GdIII-PCNDXR significantly enhanced the DXR cytotoxicity against Her2-overexpressing cells (SK-BR-3 and MDA-MB-231-Her2) in comparison with the non-targeted GdIII-PCNDXR. The half-maximum inhibitory concentrations (IC50) values (Table S1 in the Supporting Information (SI)) and degrees of potentiation[18a] (DOP = [IC50 (free drug)/IC50 (drug in nanocarrier)] × 100; Figure S1 in the SI) for the targeted PCN show that it is over 14 times more cytotoxic than the non-targeted PCN for SK-BR-3. We attribute this to the enhanced cellular uptake of the encapsulated drugs (Figure S2 in the SI) in the targeted PCNs, presumably through Her2 receptor-mediated endocytosis.[14] Further supporting this hypothesis is the observation that both targeted and non-targeted PCNs show similar cytoxicities against the cells that do not overexpress Her2 (MDA-MB-231 and MDA-MB-231-Vector; Figures 2c and 2d, Table S1, and Figure S1 in the SI). The enhanced cytotoxicity of Her-GdIII-PCNDXR over GdIII-PCNDXR is more modest against MDA-MB-231-Her2 cells (~2 times), consistent with the lower number of Her2 receptors present in these cells.
Figure 2.
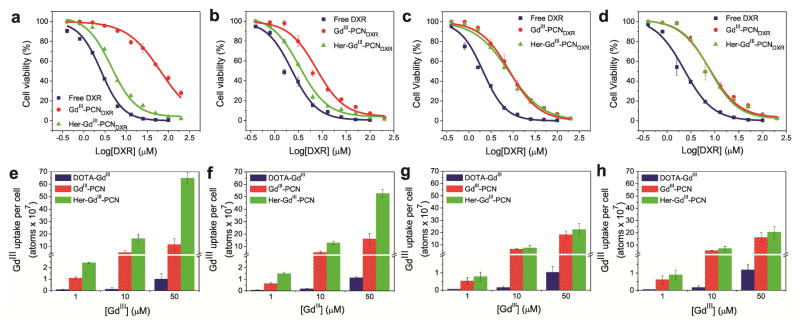
(a–d) Plots of dose-responsive cell viability of SK-BR-3 (a), MDA-MB-231-Her2 (b), MDA-MB-231 (c), and MDA-MB-231-Vector cells (d) exposed to free DXR (blue rectangle), GdIII-PCNDXR (red circle), or Her-GdIII-PCNDXR (green triangle) for 24 h at 37 °C. (e–h) Cellular GdIII-uptake profiles of SK-BR-3 (e), MDA-MB-231-Her2 (f), MDA-MB-231 (g), and MDA-MB-231-Vector cells (h) that were incubated for 24 h at 37 °C in the presence of free DOTA-GdIII (left blue bars), GdIII-PCN (middle red bars), and Her-GdIII-PCN (right green bars).
Enhanced GdIII uptake by Her-GdIII-PCN
The efficient cellular internalization of the Her2-targeted PCNs through Her2 receptor-mediated endocytosis is also expected to accelerate cellular uptake of the conjugated GdIII–based MR contrast agents, leading to an increase in T1 MRI contrast in Her2-overexpressing cells. To verify this, we incubated the same four cell types (SK-BR-3, MDA-MB-231-Her2, MDA-MB-231, and MDA-MB-231-Vector) for 4 h and 24 h in media containing DXR-free Her-GdIII-PCN (Herceptin/PCN = ~13 and GdIII/PCN = ~8,000), DXR-free GdIII-PCN (GdIII/PCN = ~8,000), or free DOTA-GdIII (DOTA = 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetate) and quantify the amount of cellular GdIII-uptake by inductively coupled plasma mass spectrometry (ICP-MS). Because DXR is cytotoxic, the use of DXR-free PCNs is critical for the exact quantification of GdIII uptake into live cells, especially for the 24 h incubation time; however, data for DXR-loaded PCNs have also been obtained (Figure S4 in the SI).
Consistent with the aforementioned cytotoxicity experiments, the GdIII-uptake data (Figures 2e and 2f; Figures S3a and S3b in the SI) clearly show that Herceptin conjugation greatly accelerates cellular internali internalization of Her-GdIII-PCN particles and results in large accumulation of GdIII ions in Her2-overexpressing cells. As a result, cellular GdIII-uptake by SK-BR-3 cells that have been exposed to Her-GdIII-PCN is 120 fold higher than that observed for cells exposed to free DOTA-GdIII and 6 fold greater than that observed for cells exposed to the non-targeted GdIII-PCN. Even when the GdIII-uptake experiments were carried out with DXR-loaded PCN, which caused cell death, significant enhancement is still observed (Figure S4 in the SI). Similar to that observed in the aforementioned cytotoxicity studies, the cells that do not overexpress Her2 (MDA-MB-231 and MDA-MB-231-Vector) show comparable GdIII-uptake when exposed to either the targeted or the non-targeted PCNs (Figures 2g and 2h; Figures S3c and S3d in the SI). These results again reinforce our hypothesis that the targeting functionality is highly effective at driving the internalization of Her-GdIII-PCNDXR into Her2-overexpressing cells.
Consistent with the observed GdIII uptake results, the T1-weighted MR images of SK-BR-3 cells (Figure 3; Figure S5 in the SI) that were exposed to the Herceptin-targeted PCNs show the highest contrast enhancement (corresponding to increased GdIII uptake resulting in a significant decrease in T1, as shown in Tables S2 and S3 in the SI); as opposed to the MR images of the cells exposed to non-targeted PCNs and DOTA-GdIII. A similar trend was observed for MDA-MB-231-Her2 cells that were exposed to Herceptin-targeted PCNs, non-targeted PCNs, and DOTA-GdIII (Figure S6 in the SI). As expected, the MR images of the cells that do not overexpress Her2 (MDA-MB-231) show no discernible contrast between those exposed to the Herceptin-targeted PCNs and those exposed to the non-targeted PCNs (Figure S7 in the SI). Together with the aforementioned enhanced cytotoxicity, the MR contrast enhancement shown by the targeted PCNs demonstrates their exciting promises as a theranostic platform.
Figure 3.
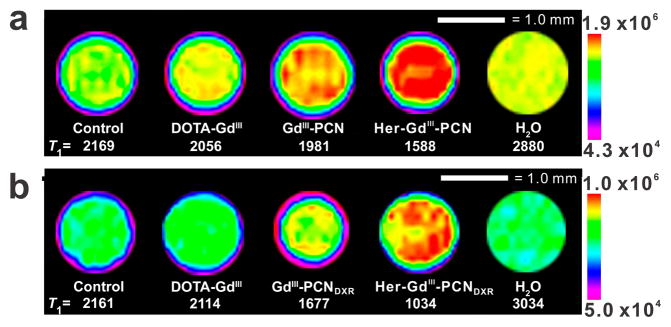
T1-weighted MR images (at 9.4 T and 25 °C) of SK-BR-3 cells incubated for 24 h with media alone (control) or with Gd-containing ([GdIII] = 50 μM) formulations (free DOTA-GdIII, GdIII-PCN, or Her-GdIII-PCN), with (a) and without (b) DXR. The maximum and minimum signal intensities are shown in the color-mapped calibration bar on the right side of each image.
In conclusion, we have demonstrated that the multifunctional PCN platform fulfills the criteria for an ideal theranostic platform that efficiently and selectively delivers both chemotherapeutic and MRI contrast agents to Her2-overexpressing cancer cells when guided by a cancer-targeting ligand (Herceptin). Together with the excellent acid-triggered drug-release property of the PCN platform, the accelerated cellular internalization of the Herceptin-conjugated PCN nanocarrier (Her-GdIII-PCNDXR) allows for site-specific and enhanced accumulation of anti-cancer drugs in Her2-overexpressing cells, resulting in 14-fold increase in cytotoxicity compared to the non-targeted nanocarrier (GdIII-PCNDXR). Herceptin-targeting also greatly improves GdIII-uptake (up to 120 times better than free DOTA-GdIII) by Her2-overexpressing cells and thus, enhances T1 MRI contrast. The high specificity of Herceptin-conjugated PCN for Her2-overexpressing cells demonstrates its excellent promise as a theranostic agent for the simultaneous detection and treatment of metastatic and primary Her2-overexpressing tumors.
Supplementary Material
Acknowledgments
This work is financially supported by the NIH (NCI CCNE Grant C54CA151880, CCNP Grant U01CA151461, and Core Grant P30CA060553 to the Lurie Cancer Center of Northwestern University). Instruments in the Northwestern University IMSERC, Keck, and NUANCE facilities were purchased with grants from NSF-NSEC, NSF-MRSEC, the Keck Foundation, the State of Illinois, and Northwestern University. Metal analysis was performed at the Northwestern University QBIC facility, which is generously supported by NASA Ames Research Center NNA06CB93G. We thank Prof. Vincent L. Cryns for the MDA MB-231 cell line used in this work.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
References
- 1.a) Koo H, Huh MS, Sun IC, Yuk SH, Choi K, Kim K, Kwon IC. Acc Chem Res. 2011;44:1018–1028. doi: 10.1021/ar2000138. [DOI] [PubMed] [Google Scholar]; b) Xie J, Lee S, Chen X. Adv Drug Delivery Rev. 2010;62:1064–1079. doi: 10.1016/j.addr.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bardhan R, Lal S, Joshi A, Halas NJ. Acc Chem Res. 2011;44:936–946. doi: 10.1021/ar200023x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song Y, Xu X, MacRenaris KW, Zhang XQ, Mirkin CA, Meade TJ. Angew Chem, Int Ed. 2009;48:9143–9147. doi: 10.1002/anie.200904666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ntziachristos V. Nat Methods. 2010;7:603–614. doi: 10.1038/nmeth.1483. [DOI] [PubMed] [Google Scholar]
- 4.Aime S, Cabella C, Colombatto S, Geninatti Crich S, Gianolio E, Maggioni F. J Magn Reson Imaging. 2002;16:394–406. doi: 10.1002/jmri.10180. [DOI] [PubMed] [Google Scholar]
- 5.Major JL, Meade TJ. Acc Chem Res. 2009;42:893–903. doi: 10.1021/ar800245h.; b) We note that both T1 and T2 MRI contrast agents are needed for accurate cancer diagnostic (see: J. Mater. Chem. 2009, 19, 6267–6273). Thus, the development of T1 MRI-based theranostic platforms is an active research area in its own rights and must not be considered as being duplicative of T2 MRI-based platforms.
- 6.Allen TM, Cullis PR. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 7.Byrne JD, Betancourt T, Brannon-Peppas L. Adv Drug Delivery Rev. 2008;60:1615–1626. doi: 10.1016/j.addr.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 8.a) Alric C, Taleb J, Le Duc G, Mandon C, Billotey C, Le Meur-Herland A, Brochard T, Vocanson F, Janier M, Perriat P, Roux S, Tillement O. J Am Chem Soc. 2008;130:5908–5915. doi: 10.1021/ja078176p. [DOI] [PubMed] [Google Scholar]; b) Taylor KML, Kim JS, Rieter WJ, An H, Lin W, Lin W. J Am Chem Soc. 2008;130:2154–2155. doi: 10.1021/ja710193c. [DOI] [PubMed] [Google Scholar]
- 9.Xu R, Wang Y, Wang X, Jeong EK, Parker DL, Lu ZR. Exp Biol Med. 2007;232:1081–1089. doi: 10.3181/0702-RM-33. [DOI] [PubMed] [Google Scholar]
- 10.Datta A, Hooker JM, Botta M, Francis MB, Aime S, Raymond KN. J Am Chem Soc. 2008;130:2546–2552. doi: 10.1021/ja0765363. [DOI] [PubMed] [Google Scholar]
- 11.Frias JC, Williams KJ, Fisher EA, Fayad ZA. J Am Chem Soc. 2004;126:16316–16317. doi: 10.1021/ja044911a. [DOI] [PubMed] [Google Scholar]
- 12.Botta M, Tei L. Eur J Inorg Chem. 2012;2012:1945–1960. [Google Scholar]
- 13.Courant T, Roullin VG, Cadiou C, Callewaert M, Andry MC, Portefaix C, Hoeffel C, de Goltstein MC, Port M, Laurent S, Elst LV, Muller R, Molinari M, Chuburu F. Angew Chem, Int Ed. 2012;51:9119–9122. doi: 10.1002/anie.201203190. [DOI] [PubMed] [Google Scholar]
- 14.Yu MK, Park J, Jon S. Theranostics. 2012;2:3–44. doi: 10.7150/thno.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith D, Clark SH, Bertin PA, Mirkin BL, Nguyen ST. J Mater Chem. 2009;19:2159–2165. doi: 10.1039/B817511J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janib SM, Moses AS, MacKay JA. Adv Drug Delivery Rev. 2010;62:1052–1063. doi: 10.1016/j.addr.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee SM, Chen H, Dettmer CM, O’Halloran TV, Nguyen ST. J Am Chem Soc. 2007;129:15096–15097. doi: 10.1021/ja070748i. [DOI] [PubMed] [Google Scholar]
- 18.a) Lee SM, Chen H, O’Halloran TV, Nguyen ST. J Am Chem Soc. 2009;131:9311–9320. doi: 10.1021/ja9017336. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee SM, Ahn RW, Chen F, Fought AJ, O’Halloran TV, Cryns VL, Nguyen ST. ACS Nano. 2010;4:4971–4978. doi: 10.1021/nn100560p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strohecker AM, Yehiely F, Chen F, Cryns VL. J Biol Chem. 2008;283:18269–18282. doi: 10.1074/jbc.M802156200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh DY, Diéras V, Guardino E, Fang L, Lu MW, Olsen S, Blackwell K. N Engl J Med. 2012;367:1783–1791. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee SM, Song Y, Hong BJ, MacRenaris KW, Mastarone DJ, O’Halloran TV, Meade TJ, Nguyen ST. Angew Chem, Int Ed. 2010;49:9960–9964. doi: 10.1002/anie.201004867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hatakeyama H, Akita H, Ishida E, Hashimoto K, Kobayashi H, Aoki T, Yasuda J, Obata K, Kikuchi H, Ishida T, Kiwada H, Harashima H. Int J Pharm. 2007;342:194–200. doi: 10.1016/j.ijpharm.2007.04.037. [DOI] [PubMed] [Google Scholar]
- 23.Lee SM, Lee OS, O’Halloran TV, Schatz GC, Nguyen ST. ACS Nano. 2011;5:3961–3969. doi: 10.1021/nn200478m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Wang AZ, Gu F, Zhang L, Chan JM, Radovic-Moreno A, Shaikh MR, Farokhzad OC. Expert Opin Biol Ther. 2008;8:1063–1070. doi: 10.1517/14712598.8.8.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Anhorn MG, Wagner S, Kreuter J, Langer K, von Briesen H. Bioconjugate Chem. 2008;19:2321–2331. doi: 10.1021/bc8002452. [DOI] [PubMed] [Google Scholar]
- 25.a) Yang J, Lee CH, Ko HJ, Suh JS, Yoon HG, Lee K, Huh YM, Haam S. Angew Chem, Int Ed. 2007;46:8836–8839. doi: 10.1002/anie.200703554. [DOI] [PubMed] [Google Scholar]; b) Hwang JY, Park J, Kang BJ, Lubow DJ, Chu D, Farkas DL, Shung KK, Medina-Kauwe LK. PLoS One. 2012;7:e34463. doi: 10.1371/journal.pone.0034463. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lim EK, Huh YM, Yang J, Lee K, Suh JS, Haam S. Adv Mater. 2011;23:2436–2442. doi: 10.1002/adma.201100351. [DOI] [PubMed] [Google Scholar]
- 26.Mastarone DJ, Harrison VSR, Eckermann AL, Parigi G, Luchinat C, Meade TJ. J Am Chem Soc. 2011;133:5329–5337. doi: 10.1021/ja1099616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong V, Presolski SI, Ma C, Finn MG. Angew Chem, Int Ed. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Y, Drummond DC, Zou H, Hayes ME, Adams GP, Kirpotin DB, Marks JD. J Mol Biol. 2007;371:934–947. doi: 10.1016/j.jmb.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bandak S, Goren D, Horowitz A, Tzemach D, Gabizon A. Anti-Cancer Drugs. 1999;10:911–920. doi: 10.1097/00001813-199911000-00007. [DOI] [PubMed] [Google Scholar]
- 30.Tannock IF, Rotin D. Cancer Res. 1989;49:4373–4384. [PubMed] [Google Scholar]
- 31.Casey JR, Grinstein S, Orlowski J. Nat Rev Mol Cell Biol. 2010;11:50–61. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- 32.Ganta S, Devalapally H, Shahiwala A, Amiji M. J Controlled Release. 2008;126:187–204. doi: 10.1016/j.jconrel.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 33.Adair JH, Parette MP, Altinoğlu Eİ, Kester M. ACS Nano. 2010;4:4967–4970. doi: 10.1021/nn102324e. [DOI] [PubMed] [Google Scholar]
- 34.Costantini DL, Bateman K, McLarty K, Vallis KA, Reilly RM. J Nucl Med. 2008;49:1498–1505. doi: 10.2967/jnumed.108.051771. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.