

Abstract
Background
Diabetes and hypertension independently contribute to renal injury, and the major mechanisms involved are increased reactive oxygen species (ROS) bioavailability and renin-angiotensin system (RAS) activation. We investigated the role of adenosine in controlling ROS production and RAS activation associated with renal dysfunction in hypertension and diabetes.
Methods
Fourteen days after induction of diabetes with streptozotocin in 12-week-old male Wistar and spontaneously hypertensive (SHR) rats, animals were treated during 7 days with 2-chloroadenosine (CADO group, 5 mg/kg/d), a stable analogue of adenosine, or underwent a sham operation procedure. At the end of the study (day 21), intra-arterial systolic blood pressure (SBP) was measured, and 24-h urine and plasma samples and renal tissue were collected.
Results
CADO treatment decreased the plasma glucose concentration and glucose and protein excretion by more than 30% in both strains. CADO treatment decreased SBP in diabetic SHR rats (143 ± 8 versus 114 ± 4 mmHg, p < 0.05), but not in diabetic Wistar rats. The hypotensive effect of CADO was associated to a ∼70% increase in plasma angiotensinogen (AGT) concentration and a ∼50% decrease in urinary AGT excretion. CADO also caused a decrease in medullary and cortical hydrogen peroxide production of about 40%, which was associated with a proportional increase in glutathione peroxidase (GPx) activity in diabetic Wistar but not in diabetic SHR animals.
Conclusions
These results suggest that activation of adenosine receptors improves renal antioxidant capacity in diabetic Wistar but not SHR rats, although it improves glucose metabolism in both strains. Furthermore, activation of adenosine receptors does not seem to be directly influencing AGT production.
Keywords: Adenosine, angiotensinogen, hydrogen peroxide, hyperglycemia, kidney, systolic blood pressure
Introduction
Diabetes and hypertension often coexist and increase the risk of chronic kidney disease and cardiovascular complications (1). Increased oxidative stress and renin-angiotensin system (RAS) activation in the kidney are two important contributors to the initiation and progression of diabetic and hypertensive renal damage (2,3). Therefore, the control of these pathophysiological mechanisms should be required for renoprotection in both diseases.
Adenosine, an important regulatory nucleoside known for its cytoprotective role in tissues under stress, has been shown to improve glucose homeostasis and insulin signalling/secretion (4-7). Besides influencing glucose metabolism, adenosine also protects the kidney under hyperglycemic conditions. Adenosine exerts its effects through activation of four G-protein coupled receptors: the high-affinity A1 subtype, the moderate-affinity A2A and A3 subtypes, and the low-affinity A2B subtype (8). Both A1 and A2A receptors appear to be renoprotective in diabetic rats, as evidenced by the fact that diabetic nephropathy is more pronounced in adenosine A1 (9-11) and A2A (12) receptor knockout mice. Also, in streptozotocin (STZ)-diabetic rats, systemic administration of selective adenosine A2A receptor agonists (12) or daily intraperitoneal (i.p.) treatment with a non-selective agonist of adenosine receptors (13) improves renal function by reducing inflammation (12,13).
The renoprotection afforded by adenosine may also involve the control of oxidative stress and RAS activation in the kidney. Adenosine confers increased resistance to oxidative damage. Oxidative stress enhances adenosine concentration in the kidney (14), and activation of adenosine receptors has been reported to improve the antioxidant capacity, by increasing the activity of hydrogen peroxide (H2O2)-metabolizing enzymes in the heart (15) and in endothelial cells (16). In STZ-induced diabetes there is increased renal oxidative stress (17), the concentration of adenosine is increased in the renal glomeruli (18), and the distribution of adenosine receptors is considerably altered (19), probably reflecting an adaptive defence mechanism. Noteworthy, increasing adenosine extracellular concentrations prevent diabetes-induced alterations of kidney function in rats (20).
The ability of adenosine to modulate the RAS has also been shown in several studies. Adenosine A1 receptor activation restrains RAS activity (21,22). Accordingly, we have previously demonstrated that the non-selective antagonism of adenosine receptors causes hypertension (23) accompanied by increased systemic RAS activity (24,25) and augmented reactive oxygen species (ROS) production (26). In renal microcirculation, adenosine and the RAS also co-operate in the regulation of renal hemodynamics by controlling the renal vascular tone through a unifying pathway that requires both adenosine A1 and angiotensin II AT1 receptors (27). Renal RAS is increased in diabetic and hypertensive nephropathy (3,28-32), contributing to impaired renal hemodynamics, proteinuria, and fibrogenesis (3). Angiotensin II also exacerbates renal redox dysfunction since it stimulates the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-derived production of ROS within the kidney (33,34). Thus, the putative attenuation of renal RAS activation by adenosine would expectedly improve renal function.
Despite the evidence for adenosine to modulate oxidative stress and the RAS, the associated mechanisms have not been explored in hypertensive diabetic nephropathy. We tested the hypothesis that adenosine ameliorates renal dysfunction associated to hyperglycemia and hypertension by attenuating the production of ROS, namely H2O2, in the renal cortex and/or medulla, and by decreasing renal RAS activation. To assess renal RAS activation, urinary angiotensinogen (AGT) was quantified, since it is described as a marker of renal RAS activation in animal models (35-37) and in humans (38-40).
Materials and methods
All chemicals were from Sigma Aldrich (St. Louis, MO, USA) and of the highest grade available if not otherwise stated.
Animals, diabetes induction, and treatment
Animals had free access to water and food (SAFE—Scientific Animal Food and Engineering, Épinay-sur-Orge, France; Na+: 2.5 g/kg; K+: 6.7 g/kg; protein: 120 g/kg) and were housed under controlled conditions of temperature (22°C), humidity (60%), and 12 h/12 h light-dark cycle. All experiments were performed in accordance with the European Union guidelines for the protection of animals used for scientific purposes (Directive 86/609/EEC and Decision 1999/575/EC) and approved by the local Institutional Committee. On day 0, male Wistar rats (11–12 weeks) and male spontaneously hypertensive rats (SHR, 12 weeks) (Charles River, Barcelona, Spain) were intraperitoneally injected with STZ (65 mg/kg). After 48 h, blood glucose concentrations were determined using an autoanalyser (Abbott Diabetes Care Ltd, Santa Clara, CA, USA), and animals with blood glucose concentrations above 300 mg/dL were considered diabetic. Two weeks after induction of diabetes animals were randomly assigned into four groups of 12 animals each (two groups of each strain). Animals were anesthetized with pentobarbital sodium (50 mg/kg; i.p.), and osmotic minipumps (Alzet model 2ML1; Alza, Palo Alto, CA, USA) were intraperitoneally implanted to allow continuous (10 µL/h) infusion of 2-chloroadenosine (CADO, 5 mg/kg/d) in one of the groups of each strain (from now on referred to as diabetic-Wistar+CADO and diabetic-SHR+CADO animals). The remaining diabetic animals underwent a sham operation procedure (and are from now on referred to as diabetic-Wistar and diabetic-SHR animals). A schematic representation of the experimental protocol is presented in Figure 1. CADO is a stable analogue of adenosine that is not a substrate for adenosine deaminase nor for cell uptake, acting on the extracellular domains of adenosine receptors (41) with high affinity for adenosine A1 and A2A receptors and, much less, for A3 and A2B receptors (8).
Figure 1.
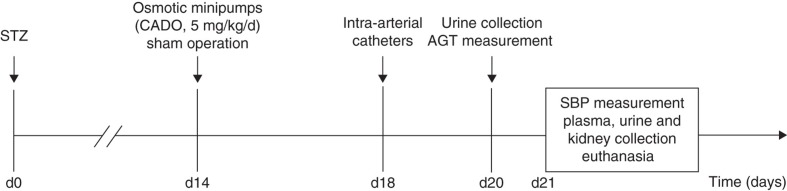
Schematic representation of the experimental protocol.
AGT = angiotensinogen; CADO = 2-chloroadenosine; d = day; SBP = systolic blood pressure; STZ = streptozotocin.
Blood pressure measurements
On day 18, half of the animals in each group were anaesthetized as described above, and an intra-arterial polyethylene catheter (PE10 connected to PE50; Bilaney, Dusseldorf, Germany) was placed on the aorta via the left femoral artery, passed subcutaneously using a trocar, and externalized at the back of the neck, where it was secured to the skin of the animal. Catheters were daily flushed with saline heparinized solution (0.1%) to prevent clotting. At the end of the study (day 21), the catheter was connected to a pressure transducer (B. Braun, Bethlehem, PA, USA) coupled with a polygraph (Letica Polygraph 6006; Letica, Barcelona, Spain) to measure intra-arterial systolic blood pressure (SBP) in conscious, unrestrained animals.
Sample collection
At the end of the study (day 21), 24-hour urine samples were collected in 0.6 mL of distilled water containing 50 µg of pepstatin A, 10 mg of sodium azide, 300 nmol of enalaprilat, and 125 mmol of ethylenediamine tetra-acetic acid (EDTA), for AGT determination (37). Additional ordinary 24-hour urine samples were collected for further measurements. Animals were then anaesthetized, as previously described, and blood samples were harvested from the left ventricle to heparinized or EDTA ice-cold tubes. After centrifugation of blood samples (3000 rpm, 15 min, 4°C), plasma was collected. Also, the left kidney was perfused with saline, via aorta, excised, and total cortex and medulla isolated and snap-frozen in liquid nitrogen. All samples were stored at -80°C until assayed.
Metabolic parameters and renal function
Glucose and creatinine concentrations were determined in urine and plasma samples by a glucose oxidase method and the colorimetric Jaffé method, respectively. Total urinary protein concentration was determined using pyrogallol red. Na+ concentration was measured using ion-selective electrodes. These assays were performed using a Cobas Mira Plus analyser (ABX Diagnostics, Geneva, Switzerland). Because high glucose concentrations interfere with the creatinine Jaffé method, creatinine concentration values were corrected using the method previously reported by Moreira-Rodrigues and co-workers (42). Glomerular filtration rate (GFR) was calculated using the formula GFR = [Ucreatinine] × V/ [Pcreatinine], where [Ucreatinine] and [Pcreatinine] denote the corrected creatinine concentration in the urine and plasma samples, respectively, and V denotes the urine flow rate (in mL/min). Although creatinine clearance gives a poor estimation of GFR, it is a non-invasive method that can provide information about the variations following treatments. Fractional Na+ excretion (FENa) was calculated using the formula [UNa] × [Pcreatinine]/ [PNa] × [Ucreatinine], where [PNa] and [UNa] represent plasma and urinary Na+ concentrations.
AGT quantification
Quantification of AGT was performed using a commercial ELISA kit, according to the protocol provided by the manufacturer (Rat Total Angiotensinogen Assay Kit; Immuno-Biological Laboratories Co. Hamburg, Germany).
Urinary and renal oxidative status parameters
Quantification of urinary thiobarbituric acid reactive substances (TBARS) was performed as previously reported by Sousa et al. (26). Urinary 8-isoprostane quantification was performed using a commercial kit according to the protocol provided by the manufacturer (Urinary Isoprostane ELISA Kit; Oxford Biomedical Research Inc., Oxford, MI, USA). Renal H2O2 production was quantified in cortex and medulla samples that were incubated for 60 min at 37°C in 1 mL of oxygenated KREBS-HEPES media. The media were then used for determination of H2O2 production using a commercial kit according to the protocol provided by the manufacturer (Amplex Red Hydrogen Peroxide Assay kit; Molecular Probes, Eugene, OR, USA). The activities of catalase and glutathione peroxidase (GPx) were determined in the supernatant of centrifuged (10 min, 15,700 g, 4°C) homogenized kidney samples (cold phosphate buffer, 50 mM, 7.4, containing Triton 0.1% (v/v)). Catalase activity was quantified by monitoring H2O2 decay at 240 nm, at 25°C during 40 s (26). One unit of catalase was defined as the amount of enzyme that decomposes 1 μmol of H2O2 per min. Results are expressed as units per mg of protein (extinction coefficient of 0.0394 mM/cm). GPx activity was assayed spectrophotometrically by following NADPH oxidation at 340 nm when glutathione is regenerated by glutathione reductase (26). Results are expressed as nmol of oxidized NADPH per min per mg of protein (extinction coefficient of 6.22 mM/cm).
Statistical analysis
Statistical analysis was performed using unpaired Student's t test. Values are presented as means ± SEM. P values of less than 0.05 were considered statistically significant.
Results
Metabolic parameters and renal function
CADO treatment lowered the plasma glucose concentration (Wistar 590.5 ± 42.9 versus 389.3 ± 23.0 mg/dL, and SHR 382.4 ± 44.4 versus 232.2 ± 51.0 mg/dL, p < 0.05), glucosuria (Wistar 30.0 ± 2.4 versus 18.5 ± 1.1 g/kg/24h, and SHR 23.4 ± 2.4 versus 13.2 ± 0.9 g/kg/24h, p < 0.05), and proteinuria (Wistar 197.0 ± 41.0 versus 115.7 ± 9.4 mg/kg/24h, and SHR 197.5 ± 24.2 versus 83.7 ± 12.6 mg/kg/24h, p < 0.05) in both strains (Table I). CADO also reduced urine excretion but only in diabetic-Wistar animals (Wistar 197.7 ± 12.5 versus 144.8 ± 10.2 mL/24h, p < 0.05; SHR 105.5 ± 9.7 versus 95.9 ± 5.3 mL/24h, NS) and caused no alteration in body weight, food intake, water intake, GFR, and FENa in either strain (Table I).
Table I.
Metabolic and renal function parameters of diabetic-Wistar and SHR rats treated or not with CADO.
| Plasma glucose (mg/dL) | BW (g) | Food intake (g/24h) | Water intake (mL/24h) | U-excretion (mL/24h) | U-glucose (g/kg/24h) | U-proteins (mg/kg/24h) | GFR (mL/min) | FENa (%) | |
|---|---|---|---|---|---|---|---|---|---|
| Diabetic-Wistar | 590.5 ± 42.9 | 223.9 ± 7.5 | 37.8 ± 1.7 | 180.1 ± 15.4 | 197.7 ± 12.5 | 30.0 ± 2.4 | 197.0 ± 41.0 | 2.8 ± 0.6 | 0.57±0.06 |
| Diabetic-Wistar+CADO | 389.3 ± 23.0 | 237.4 ± 7.6 | 33.1 ± 1.8 | 162.9 ± 11.1 | 144.8 ± 10.2 | 18.5 ± 1.1 | 115.7 ± 9.4 | 2.8 ± 0.3 | 0.55±0.55 |
| P value | <0.05 | NS | NS | NS | <0.05 | <0.05 | <0.05 | NS | NS |
| Diabetic-SHR | 382.4 ± 44.4 | 176.8 ± 9.1 | 25.3 ± 1.3 | 125.7 ± 9.1 | 105.5 ± 9.7 | 23.4 ± 2.4 | 197.5 ± 24.2 | 2.0 ± 0.3 | 0.80±0.12 |
| Diabetic-SHR+CADO | 232.2 ± 51.0 | 195.5 ± 6.8 | 24.7 ± 1.0 | 114.9 ± 6.4 | 95.9 ± 5.3 | 13.2 ± 0.9 | 83.7 ± 12.6 | 1.6 ± 0.1 | 0.80±0.03 |
| P value | <0.05 | NS | NS | NS | NS | <0.05 | <0.05 | NS | NS |
Results expressed as mean ± SEM, n = 9–12 for all parameters (except GFR, n = 6–8).
BW = body weight; FENa = fractional excretion of Na+; GFR = glomerular filtration rate; NS = not significant; U = urine.
Blood pressure measurements
CADO treatment did not alter SBP in diabetic-Wistar animals (122 ± 6 versus 115 ± 8 mmHg, NS) but caused a reduction of SBP (≈ 20 mmHg) in diabetic-SHR (143 ± 8 versus 114 ± 4 mmHg, p < 0.05) (Figure 2).
Figure 2.

Systolic blood pressure (SBP; mmHg; n = 4–6) of diabetic-Wistar and SHR rats treated (▪) or not (□) with 2-chloroadenosine (CADO). Results expressed as Mean±SEM*p <0.05 versus corresponding diabetic group.
Results expressed as mean ± SEM; *p < 0.05 versus corresponding diabetic group.
Plasma and urinary AGT
CADO treatment did not alter plasma or urinary AGT in diabetic-Wistar animals (plasma 1.6 ± 0.1 versus 1.4 ± 0.1 µg/mL; urine 98.1 ± 15.5 versus 106.7 ± 18.3 ngAGT/mg Creat; NS), but in diabetic-SHR animals the same treatment evoked an increase in plasma AGT and a marked reduction in urinary AGT (plasma 1.1 ± 0.2 versus 1.9 ± 0.1 µg/mL; urine 81.2 ± 14.3 versus 41.2 ± 10.6 ngAGT/mg Creat; p < 0.05) (Figure 3).
Figure 3.

Angiotensinogen (AGT) in diabetic-Wistar and SHR rats treated (▪) or not (□) with 2-chloroadenosine (CADO). A: plasma AGT concentration (µg/mL; n = 6–7); B: urinary AGT excretion (ngAGT/mg Creat; n = 9–11). Results expressed as Mean±SEM. *p <0.05 versus corresponding diabetic group.
Results expressed as mean ± SEM; *p < 0.05 versus corresponding diabetic group.
Urinary and renal oxidative status
In diabetic-Wistar animals, CADO treatment lowered renal medullary and cortical H2O2 production (medulla 1.11 ± 0.10 versus 0.57 ± 0.09 nmol/mgprot; cortex 0.30 ± 0.03 versus 0.17 ± 0.02 nmol/mgprot; p < 0.05) and increased GPx activity (medulla 119.6 ± 5.2 versus 163.8 ± 11.6 nmolNADPH/min/mgprot; cortex 648.9 ± 71.4 versus 960.6 ± 92.2 nmolNADPH/min/mgprot; p < 0.05), without changing catalase activity, while in diabetic-SHR animals it did not affect H2O2 production or its metabolism by GPx and catalase (Figure 4). CADO administration lowered the urinary excretion of 8-isoprostane (Wistar 409.9 ± 52.7 versus 204.9 ± 22.3 ng/kg/24h, p < 0.05; and SHR 454.7 ± 38.8 versus 453.8 ± 77.7 ng/kg/24h, NS) and TBARS (Wistar 8.0 ± 0.5 versus 6.0 ± 0.6 µmol/kg/24h, p < 0.05; SHR 4.9 ± 0.8 versus 4.3 ± 0.3 µmol/kg/24h, NS) only in diabetic-Wistar animals (Figure 5).
Figure 4.

Production of hydrogen peroxide (H2O2) and activity of H2O2-neutralizing enzymes in the kidney of diabetic-Wistar and SHR rats treated (▪) or not (□) with 2-chloroadenosine (CADO). A: renal medullary H2O2 production (n = 6–8; nmol/mgprot); B: renal medullary glutathione peroxidase (GPx) activity (n = 5–6; nmolNADPH/min/mgprot); C: renal medullary catalase activity (n = 5–8; Ucat/mgprot); D: renal cortical H2O2 production (n = 5–7; nmol/mgprot); E: renal cortical GPx activity (n = 5–6; nmolNADPH/min/mgprot); F: renal cortical catalase activity (n = 5–8 Ucat/mgprot). Results expressed as Mean±SEM. *p <0.05 versus corresponding diabetic group.
Results expressed as mean ± SEM; *p < 0.05 versus corresponding diabetic group.
Figure 5.

Urinary markers of oxidative stress in diabetic-Wistar and SHR rats treated (▪) or not (□) with 2-chloroadenosine (CADO). A: 8-isoprostane (ng/kg/24h; n = 8–12); B: thiobarbituric acid reactive substances (TBARS) (µmol/kg/24h; n = 10–12). Results expressed as Mean±SEM. *p <0.05 versus corresponding diabetic group.
Results expressed as mean ± SEM; *p < 0.05 versus corresponding diabetic group.
Discussion
Our results show that the mechanisms whereby CADO may protect the kidney in diabetic nephropathy are considerably different between Wistar and SHR animals, despite the similar improvement of glucose metabolism observed in both strains. In normotensive diabetics, CADO ameliorated renal dysfunction by attenuating renal ROS production, without changing renal RAS activity or SBP. In hypertensive diabetic animals, CADO did not improve the renal redox status but significantly reduced SBP, which appeared to cause compensatory changes in systemic and renal RAS activity.
The mitigation of renal redox dysfunction elicited by the activation of adenosine receptors with CADO appears to be due to an enhancement of the antioxidant defence. Diabetic-Wistar rats treated with CADO had lower renal H2O2 production associated with enhanced renal GPx activity than those not treated with CADO. Likewise, urinary 8-isoprostanes and TBARS were lower in diabetic-Wistar animals treated with CADO. Several authors had previously described that adenosine confers increased protection against tissue injury. Adenosine has been shown to decrease total renal concentrations of TBARS in diabetic animals (13) and to protect cells from H2O2-induced oxidative injury (16,43). This cytoprotective effect has been associated with an increase in the antioxidant capacity, namely by stimulating the activities of GPx (15,16) and catalase (15). GPx are selenium-containing enzymes that play an important role in antioxidant defence by degrading H2O2 and lipid hydroperoxides. Therefore, the rise of renal GPx activity explains the reduction in both renal H2O2 levels and urinary biomarkers of lipid peroxidation observed in diabetic-Wistar animals treated with CADO. Importantly, GPx activity is dependent on glutathione availability. The glutathione antioxidant system has been reported to be impaired in vascular smooth cells (44) and kidneys (45) of SHR rats, therefore contributing to the increased oxidative stress associated to the pathogenesis of renal injury and hypertension in this strain. The importance of the glutathione antioxidant system in the regulation of blood pressure has also been underlined in some studies. For example, the administration of a glutathione synthesis inhibitor, buthionine sulfoximine, significantly increases oxidative stress and blood pressure in rats (46). Furthermore, in elderly people, antihypertensive therapy improves the glutathione antioxidant system (47). Our study also points to a deficient glutathione antioxidant system in diabetic-SHR animals that probably limits the protective antioxidant response to CADO treatment.
Besides exerting antioxidant effects, adenosine may also be renoprotective by attenuating RAS activity. Adenosine is a known physiological modulator of renin release (23-25,48). In the present study, plasma concentrations and urinary excretion of AGT were used as markers of systemic and renal RAS, respectively, reflecting the angiotensin II concentration as formerly suggested by others (35-40). Even though CADO treatment altered plasma and urinary AGT in diabetic-SHR, the fact that it also markedly reduced SBP in this strain suggests that RAS changes were mainly dependent on blood pressure levels. Accordingly, in diabetic-Wistar animals neither SBP nor RAS activity were altered by CADO treatment. Acute activation of adenosine receptors causes hypotension in both Wistar and SHR rats, but the SHR rats are much more sensitive to the hemodynamic effects of adenosine (49). In fact, a six-fold higher dose was needed to induce similar SBP reduction in Wistar compared with SHR rats (49). Although we used CADO instead of adenosine, it is possible that the dose tested was enough to induce hypotension in diabetic-SHR but not in diabetic-Wistar rats. The decrease in SBP induced by CADO treatment in diabetic-SHR animals was associated with a marked increase in plasma AGT concentration. These results are probably not a direct consequence of activation of adenosine receptors by CADO, but the result of the stimulation of compensatory mechanisms, namely activation of the systemic RAS through increased AGT synthesis, to prevent further decrease in SBP. Indeed, we recently observed (28) a decrease in plasma AGT concentration following angiotensin II infusion but a marked rise in plasma AGT concentration after polyethylene glycol-catalase-induced decrease in SBP in angiotensin II-hypertensive rats. In the current study, the increased systemic AGT observed in diabetic-SHR rats treated with CADO was associated to a decrease in urinary excretion of AGT, which is in line with the hypothesis that systemic and renal AGT may be synchronized and influence SBP (unpublished data and (50)).
CADO treatment lowered the plasma glucose concentration and glucose excretion both in normotensive and hypertensive diabetic animals, reflecting adenosine's beneficial effects in glucose homeostasis and insulin action. In fact, in diabetic rats, stimulation of adenosine A1 receptors has been associated with increased glucose uptake (4) and insulin sensitivity (5), thus decreasing the plasma glucose concentration. Accordingly, the adenosine A1 receptor knockout mice develop glucose intolerance in parallel with insulin resistance (51), while this is not observed in mice overexpressing adenosine A1 receptors in the adipose tissue and fed a high-fat diet (5). In addition, the activation of adenosine A2A and A2B receptors increases beta-cell proliferation and function (7) and pancreatic insulin content (6), respectively. Thus, the effect of CADO treatment probably reflects non-insulin and insulin-dependent mechanisms that result from the activation of adenosine A1 and A2 receptors.
CADO treatment was also associated to reduced proteinuria, both in diabetic-Wistar and SHR animals. In diabetic-Wistar rats, this might be explained by the increased expression of A2A receptors in the renal cortex (19), whose activation improves glomerular structure by mediating anti-inflammatory effects (12,13) and protecting the filtration barrier (52). Here, we show for the first time that activation of adenosine receptors also improves protein handling in diabetic-SHR rats, probably through the same mechanisms that operate in diabetic-Wistar rats. CADO treatment did not alter GFR or FENa in diabetic-Wistar or SHR rats. We have previously shown that although endogenous adenosine adjusts renal vascular tonus and Na+ reabsorption, it does not influence GFR in diabetics (27). In euglycemic animals, blockade of adenosine A1 or A2 receptors is known to increase or decrease natriuresis, respectively (27,53,54). However, in diabetic animals, the natriuretic effect of activation of adenosine A2 receptors is absent (27). Thus, in diabetic animals treated with CADO we would expect a predominant anti-natriuretic effect mediated by A1 receptors. The unchanged FENa following adenosine receptor activation in diabetes has previously been reported (55). Although further studies need to be performed to elucidate this, one can speculate that it may be due to the saturation of the Na+ transport along the tubule and/or to the marked osmotic diuresis.
In conclusion, our study indicates that adenosine receptor activation ameliorates renal function in both normotensive and hypertensive diabetic animals. This renoprotection is mediated by an increase of renal antioxidant capacity in normotensive animals, by a reduction of SBP in hypertensive rats, and by the improvement in glucose metabolism and reduction of proteinuria in both strains. We found no evidence for a direct regulation of the RAS by adenosine receptor activation in diabetes. Nevertheless, the hypotensive effect of CADO in hypertensive diabetic animals appears to trigger a compensatory increase in systemic AGT paralleled by a marked decrease in renal AGT which highlights, once more, the differential regulation of these systems.
Acknowledgements
This work was supported by national funds of the Ministério da Educação e Ciência and by Fundo Europeu de Desenvolvimento Regional via Quadro de Referência Estratégico Nacional—Programa Operacional Factores de Competitividade and Strategic Programs, through Fundação para a Ciência e a Tecnologia [project grants: PTDC/SAU-FCF/67764/2006; PEst-C/SAU/LA0002/2011; PEst-C/EQB/LA0006/2011]. D. Patinha was supported by the Fundo Social Europeu and by national founds of the Ministério da Educação e Ciência via Quadro de Referência Estratégico Nacional—Programa Operacional de Potencial Humano through the Fundação para a Ciência e a Tecnologia [grant: SFRH/BD/43187/2008].
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.UKPDS Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. UK Prospective Diabetes Study Group . BMJ. 1998;317:703–13. [PMC free article] [PubMed] [Google Scholar]
- 2.Mori T, Ogawa S, Cowely AW, Jr, Ito S. Role of renal medullary oxidative and/or carbonyl stress in salt-sensitive hypertension and diabetes . Clin Exp Pharmacol Physiol. 2012;39:125–31. doi: 10.1111/j.1440-1681.2011.05653.x. [DOI] [PubMed] [Google Scholar]
- 3.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease . Pharmacol Rev. 2007;59:251–87. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 4.Cheng JT, Chi TC, Liu IM. Activation of adenosine A1 receptors by drugs to lower plasma glucose in streptozotocin-induced diabetic rats . Auton Neurosci. 2000;83:127–33. doi: 10.1016/S0165-1838(00)00106-5. [DOI] [PubMed] [Google Scholar]
- 5.Dong Q, Ginsberg HN, Erlanger BF. Overexpression of the A1 adenosine receptor in adipose tissue protects mice from obesity-related insulin resistance . Diabetes Obes Metab. 2001;3:360–6. doi: 10.1046/j.1463-1326.2001.00158.x. [DOI] [PubMed] [Google Scholar]
- 6.Nemeth ZH, Bleich D, Csoka B, Pacher P, Mabley JG, Himer L, et al. Adenosine receptor activation ameliorates type 1 diabetes . FASEB J. 2007;21:2379–88. doi: 10.1096/fj.07-8213com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersson O, Adams BA, Yoo D, Ellis GC, Gut P, Anderson RM, et al. Adenosine signaling promotes regeneration of pancreatic beta cells in vivo . Cell Metab. 2012;15:885–94. doi: 10.1016/j.cmet.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Muller CE. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors–an update . Pharmacol Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vallon V, Schroth J, Satriano J, Blantz RC, Thomson SC, Rieg T. Adenosine A(1) receptors determine glomerular hyperfiltration and the salt paradox in early streptozotocin diabetes mellitus . Nephron Physiol. 2009;111:p30–8. doi: 10.1159/000208211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sallstrom J, Carlsson PO, Fredholm BB, Larsson E, Persson AE, Palm F. Diabetes-induced hyperfiltration in adenosine A(1)-receptor deficient mice lacking the tubuloglomerular feedback mechanism . Acta Physiol (Oxf) 2007;190:253–9. doi: 10.1111/j.1748-1716.2007.01705.x. [DOI] [PubMed] [Google Scholar]
- 11.Faulhaber-Walter R, Chen L, Oppermann M, Kim SM, Huang Y, Hiramatsu N, et al. Lack of A1 adenosine receptors augments diabetic hyperfiltration and glomerular injury . J Am Soc Nephrol. 2008;19:722–30. doi: 10.1681/ASN.2007060721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Awad AS, Huang L, Ye H, Duong ET, Bolton WK, Linden J, et al. Adenosine A2A receptor activation attenuates inflammation and injury in diabetic nephropathy . Am J Physiol Renal Physiol. 2006;290:F828–37. doi: 10.1152/ajprenal.00310.2005. [DOI] [PubMed] [Google Scholar]
- 13.Elsherbiny NM, Abd El Galil KH, Gabr MM, Al-Gayyar MM, Eissa LA, El-Shishtawy MM. Reno-protective effect of NECA in diabetic nephropathy: implication of IL-18 and ICAM-1 . Eur Cytokine Netw. 2012;23:78–86. doi: 10.1684/ecn.2012.0309. [DOI] [PubMed] [Google Scholar]
- 14.Chen YF, Li PL, Zou AP. Oxidative stress enhances the production and actions of adenosine in the kidney . Am J Physiol Regul Integr Comp Physiol. 2001;281:R1808–16. doi: 10.1152/ajpregu.2001.281.6.R1808. [DOI] [PubMed] [Google Scholar]
- 15.Husain K, Somani SM. Interaction of exercise and adenosine receptor agonist and antagonist on rat heart antioxidant defense system . Mol Cell Biochem. 2005;270:209–14. doi: 10.1007/s11010-005-5285-0. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Handy DE, Loscalzo J. Adenosine-dependent induction of glutathione peroxidase 1 in human primary endothelial cells and protection against oxidative stress . Circ Res. 2005;96:831–7. doi: 10.1161/01.RES.0000164401.21929.CF. [DOI] [PubMed] [Google Scholar]
- 17.Palm F, Cederberg J, Hansell P, Liss P, Carlsson PO. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension . Diabetologia. 2003;46:1153–60. doi: 10.1007/s00125-003-1155-z. [DOI] [PubMed] [Google Scholar]
- 18.Roa H, Gajardo C, Troncoso E, Fuentealba V, Escudero C, Yanez A, et al. Adenosine mediates transforming growth factor-beta 1 release in kidney glomeruli of diabetic rats . FEBS Lett. 2009;583:3192–8. doi: 10.1016/j.febslet.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 19.Pawelczyk T, Grden M, Rzepko R, Sakowicz M, Szutowicz A. Region-specific alterations of adenosine receptors expression level in kidney of diabetic rat . Am J Pathol. 2005;167:315–25. doi: 10.1016/S0002-9440(10)62977-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vallon V, Osswald H. Dipyridamole prevents diabetes-induced alterations of kidney function in rats . Naunyn Schmiedebergs Arch Pharmacol. 1994;349:217–22. doi: 10.1007/BF00169840. [DOI] [PubMed] [Google Scholar]
- 21.Tagawa H, Vander AJ. Effects of adenosine compounds on renal function and renin secretion in dogs . Circ Res. 1970;26:327–38. doi: 10.1161/01.res.26.3.327. [DOI] [PubMed] [Google Scholar]
- 22.Weihprecht H, Lorenz JN, Schnermann J, Skott O, Briggs JP. Effect of adenosine1-receptor blockade on renin release from rabbit isolated perfused juxtaglomerular apparatus . J Clin Invest. 1990;85:1622–8. doi: 10.1172/JCI114613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Albino-Teixeira A, Matias A, Polonia J, Azevedo I. Blockade of adenosine receptors causes hypertension and cardiovascular structural changes in the rat . J Hypertens Suppl. 1991;9:S196–7. [PubMed] [Google Scholar]
- 24.Sousa T, Morato M, Albino-Teixeira A. Angiotensin converting enzyme inhibition prevents trophic and hypertensive effects of an antagonist of adenosine receptors . Eur J Pharmacol. 2002;441:99–104. doi: 10.1016/s0014-2999(02)01488-7. [DOI] [PubMed] [Google Scholar]
- 25.Morato M, Sousa T, Guimarães S, Moura D, Albino-Teixeira A. The role of angiotensin II in hypertension due to adenosine receptors blockade . Eur J Pharmacol. 2002;455:135–41. doi: 10.1016/s0014-2999(02)02587-6. [DOI] [PubMed] [Google Scholar]
- 26.Sousa T, Pinho D, Morato M, Marques-Lopes J, Fernandes E, Afonso J, et al. Role of superoxide and hydrogen peroxide in hypertension induced by an antagonist of adenosine receptors . Eur J Pharmacol. 2008;588:267–76. doi: 10.1016/j.ejphar.2008.04.044. [DOI] [PubMed] [Google Scholar]
- 27.Patinha D, Fasching A, Pinho D, Albino-Teixeira A, Morato M, Palm F. Angiotensin II contributes to glomerular hyperfiltration in diabetic rats independently of adenosine type I receptors . Am J Physiol Renal Physiol. 2013;304:F614–22. doi: 10.1152/ajprenal.00285.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sousa T, Oliveira S, Afonso J, Morato M, Patinha D, Fraga S, et al. Role of H(2) O(2) in hypertension, renin-angiotensin system activation and renal medullary disfunction caused by angiotensin II . Br J Pharmacol. 2012;166:2386–401. doi: 10.1111/j.1476-5381.2012.01957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shiono K, Sokabe H. Renin-angiotensin system in spontaneously hypertensive rats . Am J Physiol. 1976;231:1295–9. doi: 10.1152/ajplegacy.1976.231.4.1295. [DOI] [PubMed] [Google Scholar]
- 30.Price DA, Porter LE, Gordon M, Fisher ND, De'Oliveira JM, Laffel LM, et al. The paradox of the low-renin state in diabetic nephropathy . J Am Soc Nephrol. 1999;10:2382–91. doi: 10.1681/ASN.V10112382. [DOI] [PubMed] [Google Scholar]
- 31.Cassis LA. Downregulation of the renin-angiotensin system in streptozotocin-diabetic rats . Am J Physiol. 1992;262:E105–9. doi: 10.1152/ajpendo.1992.262.1.E105. [DOI] [PubMed] [Google Scholar]
- 32.Kobori H, Ozawa Y, Suzaki Y, Nishiyama A. Enhanced intrarenal angiotensinogen contributes to early renal injury in spontaneously hypertensive rats . J Am Soc Nephrol. 2005;16:2073–80. doi: 10.1681/ASN.2004080676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Onozato ML, Tojo A, Goto A, Fujita T, Wilcox CS. Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: effects of ACEI and ARB . Kidney Int. 2002;61:186–94. doi: 10.1046/j.1523-1755.2002.00123.x. [DOI] [PubMed] [Google Scholar]
- 34.Chabrashvili T, Tojo A, Onozato ML, Kitiyakara C, Quinn MT, Fujita T, et al. Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney . Hypertension. 2002;39:269–74. doi: 10.1161/hy0202.103264. [DOI] [PubMed] [Google Scholar]
- 35.Kobori H, Katsurada A, Miyata K, Ohashi N, Satou R, Saito T, et al. Determination of plasma and urinary angiotensinogen levels in rodents by newly developed ELISA . Am J Physiol Renal Physiol. 2008;294:F1257–63. doi: 10.1152/ajprenal.00588.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobori H, Nishiyama A, Harrison-Bernard LM, Navar LG. Urinary angiotensinogen as an indicator of intrarenal angiotensin status in hypertension . Hypertension. 2003;41:42–9. doi: 10.1161/01.hyp.0000050102.90932.cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kobori H, Harrison-Bernard LM, Navar LG. Urinary excretion of angiotensinogen reflects intrarenal angiotensinogen production . Kidney Int. 2002;61:579–85. doi: 10.1046/j.1523-1755.2002.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kobori H, Alper AB, Jr, Shenava R, Katsurada A, Saito T, Ohashi N, et al. Urinary angiotensinogen as a novel biomarker of the intrarenal renin-angiotensin system status in hypertensive patients . Hypertension. 2009;53:344–50. doi: 10.1161/HYPERTENSIONAHA.108.123802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishiyama A, Konishi Y, Ohashi N, Morikawa T, Urushihara M, Maeda I, et al. Urinary angiotensinogen reflects the activity of intrarenal renin-angiotensin system in patients with IgA nephropathy . Nephrol Dial Transplant. 2011;26:170–7. doi: 10.1093/ndt/gfq371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Urushihara M, Kondo S, Kagami S, Kobori H. Urinary angiotensinogen accurately reflects intrarenal renin-angiotensin system activity . Am J Nephrol. 2010;31:318–25. doi: 10.1159/000286037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pawlowska D, Granger JP, Knox FG. Effects of adenosine infusion into renal interstitium on renal hemodynamics . Am J Physiol. 1987;252:F678–82. doi: 10.1152/ajprenal.1987.252.4.F678. [DOI] [PubMed] [Google Scholar]
- 42.Moreira-Rodrigues M, Quelhas-Santos J, Serrao P, Fernandes-Cerqueira C, Sampaio-Maia B, Pestana M. Glycaemic control with insulin prevents the reduced renal dopamine D1 receptor expression and function in streptozotocin-induced diabetes . Nephrol Dial Transplant. 2010;25:2945–53. doi: 10.1093/ndt/gfq150. [DOI] [PubMed] [Google Scholar]
- 43.Lee HT, Emala CW. Adenosine attenuates oxidant injury in human proximal tubular cells via A(1) and A(2a) adenosine receptors . Am J Physiol Renal Physiol. 2002;282:F844–52. doi: 10.1152/ajprenal.00195.2001. [DOI] [PubMed] [Google Scholar]
- 44.Wu L, Juurlink BH. The impaired glutathione system and its up-regulation by sulforaphane in vascular smooth muscle cells from spontaneously hypertensive rats . J Hypertens. 2001;19:1819–25. doi: 10.1097/00004872-200110000-00016. [DOI] [PubMed] [Google Scholar]
- 45.Lee SK, Arunkumar S, Sirajudeen KN, Singh HJ. Glutathione system in young spontaneously hypertensive rats . J Physiol Biochem. 2010;66:321–7. doi: 10.1007/s13105-010-0038-2. [DOI] [PubMed] [Google Scholar]
- 46.Vaziri ND, Wang XQ, Oveisi F, Rad B. Induction of oxidative stress by glutathione depletion causes severe hypertension in normal rats . Hypertension. 2000;36:142–6. doi: 10.1161/01.hyp.36.1.142. [DOI] [PubMed] [Google Scholar]
- 47.Rybka J, Kupczyk D, Kedziora-Kornatowska K, Motyl J, Czuczejko J, Szewczyk-Golec K, et al. Glutathione-related antioxidant defense system in elderly patients treated for hypertension . Cardiovasc Toxicol. 2011;11:1–9. doi: 10.1007/s12012-010-9096-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jackson EK. Adenosine: a physiological brake on renin release . Annu Rev Pharmacol Toxicol. 1991;31:1–35. doi: 10.1146/annurev.pa.31.040191.000245. [DOI] [PubMed] [Google Scholar]
- 49.Ohnishi A, Biaggioni I, Deray G, Branch RA, Jackson EK. Hemodynamic effects of adenosine in conscious hypertensive and normotensive rats . Hypertension. 1986;8:391–8. doi: 10.1161/01.hyp.8.5.391. [DOI] [PubMed] [Google Scholar]
- 50.Ramkumar N, Kohan DE. Proximal tubule angiotensinogen modulation of arterial pressure . Curr Opin Nephrol Hypertens. 2013;22:32–6. doi: 10.1097/MNH.0b013e328359dbed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Faulhaber-Walter R, Jou W, Mizel D, Li L, Zhang J, Kim SM, et al. Impaired glucose tolerance in the absence of adenosine A1 receptor signaling . Diabetes. 2011;60:2578–87. doi: 10.2337/db11-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Awad AS, Rouse M, Liu L, Vergis AL, Rosin DL, Linden J, et al. Activation of adenosine 2A receptors preserves structure and function of podocytes . J Am Soc Nephrol. 2008;19:59–68. doi: 10.1681/ASN.2007030276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zou AP, Nithipatikom K, Li PL, Cowley AW., Jr Role of renal medullary adenosine in the control of blood flow and sodium excretion . Am J Physiol. 1999;276:R790–8. doi: 10.1152/ajpregu.1999.276.3.R790. [DOI] [PubMed] [Google Scholar]
- 54.Vallon V, Muhlbauer B, Osswald H. Adenosine and kidney function . Physiol Rev. 2006;86:901–40. doi: 10.1152/physrev.00031.2005. [DOI] [PubMed] [Google Scholar]
- 55.Pflueger AC, Berndt TJ, Knox FG. Effect of renal interstitial adenosine infusion on phosphate excretion in diabetes mellitus rats . Am J Physiol. 1998;274:R1228–35. doi: 10.1152/ajpregu.1998.274.5.R1228. [DOI] [PubMed] [Google Scholar]