

Abstract
Failure to reactivate stalled or collapsed DNA replication forks is a potential source of genomic instability. Homologous recombination (HR) is a major mechanism for repairing the DNA damage resulting from replication arrest. The single-strand DNA (ssDNA)-binding protein, replication protein A (RPA), plays a major role in multiple processes of DNA metabolism. However, the role of RPA2 hyperphosphorylation, which occurs in response to DNA damage, had been unclear. Here, we show that hyperphosphorylated RPA2 associates with ssDNA and recombinase protein Rad51 in response to replication arrest by hydroxyurea (HU) treatment. In addition, RPA2 hyperphosphorylation is critical for Rad51 recruitment and HR-mediated repair following HU. However, RPA2 hyperphosphorylation is not essential for both ionizing radiation (IR)-induced Rad51 foci formation and I-Sce-I endonuclease-stimulated HR. Moreover, we show that expression of a phosphorylation-deficient mutant of RPA2 leads to increased chromosomal aberrations following HU treatment but not after exposure to IR. Finally, we demonstrate that loss of RPA2 hyperphosphorylation results in a loss of viability when cells are confronted with replication stress whereas cells expressing hyperphosphorylation-defective RPA2 or wild-type RPA2 have a similar sensitivity to IR. Thus, our data suggest that RPA2 hyperphosphorylation plays a critical role in maintenance of genomic stability and cell survival after a DNA replication block via promotion of HR.
Introduction
DNA double-strand breaks (DSBs) may arise spontaneously, e.g. during DNA replication or following exposure to DNA-damaging agents (1), such as ionizing radiation (IR) or chemotherapeutic drugs. Homologous recombination (HR) is a major pathway in the repair of DSBs, especially those arising from stalled/collapsed replication forks (2–4). Defective HR may lead to genetic exchanges that result in genomic instability. In addition, cells deficient in HR are sensitive to IR and some chemotherapeutic drugs, especially S phase cells because HR preferentially repairs DSBs in this phase of the cell cycle.
The molecular mechanism of HR can be subdivided further, depending on the nature of the DNA structure. Two-ended DSBs can be caused directly by IR; however, only one-ended DSBs or no DSBs are produced as a result of damage or discontinuities during interrupted replication (5,6). The HR mechanism required for repairing two-ended DSBs has been extensively studied in the past. In general, it is believed that HR is triggered when a two-ended DSB is processed to a 3′ single-strand DNA (ssDNA) tail via resection. Once the ssDNA is generated, it is rapidly bound by the ssDNA-binding protein replication protein A (RPA) that in turn is displaced by Rad51. The resultant Rad51 filament facilitates DNA strand invasion and exchanges steps. Although the mechanisms required for HR after DNA replication stalling have not yet been defined in mammalian cells, it appears that eukaryotic cells have evolved a mechanism similar to that described in bacteria for re-establishment of replication forks after their progression has been impeded by lesions in the template. It has been suggested that similar to RuvABC complex in Escherchia coli, the endonuclease Mus81 in mammalian cells contributes to the generation of one-ended DSBs by cleavage of stalled DNA replication forks, which subsequently promotes HR (7–9). In some instances, replication may be restarted downstream of the unrepaired parental strand lesion, leaving a ‘daughter strand gap’ (DSG)—a ssDNA lesion in which no DSB-end is involved. It has been suggested that DSG repair in E.coli requires the RecFOR complex, which initiates with RecA, an equivalent of human Rad51, loading at ssDNA region (10). Studies in mammalian cells have shown the existence of DSGs during stalled DNA replication (11–14), but it is not clear how this process is regulated. Therefore, the detailed molecular mechanisms of the HR that occur in response to replication arrest in mammalian cells is much less understood compared with the HR process stimulated by two-ended DSBs.
RPA is a heterotrimer composed of three tightly associated subunits, RPA1, RPA2 and RPA3. The major ssDNA-binding subunit is RPA1. However, the other two subunits also participate in the interactions with ssDNA (15,16). RPA is required for almost all aspects of cellular DNA metabolism, including DNA replication, DNA damage checkpoint activation, DNA repair and recombination. During DNA replication, RPA binds to ssDNA and is required for the initiation as well as elongation processes (17,18). In addition to its role in DNA replication, RPA is also critical for cell cycle checkpoint activation: RPA-coated ssDNA recruits the ataxia-telangiectasia and Rad3-related (ATR)–ATR interacting protein complex, which facilitates S phase checkpoint signaling under the condition of replication arrest (19,20). RPA is also essential for multiple DNA repair pathways, including HR-mediated repair. RPA is thought to facilitate two-ended DSBs stimulated HR by removing secondary structures in ssDNA that may prevent the formation of the Rad51 filament (21,22) or by actively regulating protein–protein interactions (23). However, the role of RPA in HR after replication block remains unknown.
RPA becomes phosphorylated in cells when bound to ssDNA. Although no precise phosphorylation site has been identified in RPA1, nine potential phosphorylation sites have been suggested within the unstructured N-terminal domain of RPA2 (24–29). A novel DNA damage-induced phosphorylation site at Thr-98 in RPA2 was also recently identified (26). RPA2 is phosphorylated in both cell cycle and DNA damage-dependent manner. S phase-dependent phosphorylation of RPA2 is mediated by the cyclin-dependent kinase2 kinase. Cyclin-dependent kinase modifies RPA2 at serine residues 23 and 29 in mitotic cells. DNA damage-induced RPA2 phosphorylation, so-called RPA2 hyperphosphorylation includes modification of threonine 21, serines 4, 8 and 33 as well as at least one phosphoserine in residues 11–13. These events are carried out by the phosphatidylinositol 3-OH-kinase-related kinase family, which includes ataxia-telangiectasia mutated, ATR and DNA-dependent protein kinase (17). A recent study suggests that cyclin-dependent kinase is also required for DNA damage-induced RPA2 hyperphosphorylation (27). However, the question remains as to whether and how RPA2 hyperphosphorylation regulates the function of RPA in DNA replication, cell cycle checkpoint and DNA repair.
Earlier studies suggested that hyperphosphorylation of RPA2 may not be essential for RPA function in vitro or in vivo (18) since deletion of the N-terminus of RPA32 does not affect ssDNA-binding activity, heterotrimer complex formation or the ability to support DNA replication in vitro. However, recent evidence suggests a role for RPA2 hyperphosphorylation in DNA metabolism. It has been suggested that RPA2 phosphorylation facilitates mitotic exit in response to mitotic DNA damage (30). Several studies indicated that RPA2 hyperphosphorylation negatively regulates the role of RPA in DNA replication both in vitro and in vivo (17) although the mechanism underlying the inhibition of replication by RPA2 hyperphosphorylation is controversial. Despite the negative regulation of DNA replication, RPA2 hyperphosphorylation is likely to be involved in DNA repair, particularly HR-mediated repair due to the following reasons. First, RPA2 phosphorylation stimulates repair of chromosomal DNA damage caused either by camptothecin or bleomycin treatment (27). Second, cells expressing a RPA2 mimetic that has persistent phosphorylation exhibit normal checkpoint activation and re-enter the cell cycle normally after recovery but display a pronounced defect in the repair of DNA breaks (31). Since HR-mediated repair is a major mechanism to repair DNA damage caused by stalled/collapsed DNA replication forks, these results suggest a role for RPA2 phosphorylation in HR. Third, hyperphosphorylated RPA2 was found to directly interact with Rad51 in vitro (32) and phosphorylation of RPA2 increased the affinity between RPA and Rad51. Fourth, the observation that hyperphosphorylated RPA2 occurs predominantly in late S and G2 phases (27), where HR is a major repair mechanism, suggests a role for RPA hyperphosphorylation in HR. Therefore, we tested the biological function of RPA2 phosphorylation in HR that occurs in response to DNA damage, particularly after replication arrest.
Here, we demonstrate that hyperphosphorylated RPA2 colocalizes with ssDNA and Rad51 in response to hydroxyurea (HU)-induced DNA replication fork stalling. Cells expressing phosphorylation-defective RPA2 exhibit a defect in Rad51 foci formation and a decreased frequency of HR in response to replication arrest but have no obvious defect on IR-induced Rad51 recruitment and I-Sce-I-induced HR. In addition, an increased frequency of chromosomal abnormalities after treatment with HU, but not upon exposure to IR, was observed in cells expressing phosphorylation-defective RPA2. Therefore, we propose that RPA2 hyperphosphorylation is required for HR, which plays a critical role in the maintenance of genomic stability and cell survival in response to replication arrest.
Materials and methods
Plasmids, cell lines, infections and transfections
MCF-7 cells were obtained from the American Type Culture Collection (Manassas, VA). Retroviral vectors carrying a HA-tagged wild-type RPA2 or phosphorylation mutant RPA2, RPA2-(7xA)-(S4A/S8A/S11A/S12A/S13A/T21A/S33A), have been described previously (33). RPA2 small hairpin RNA (shRNA) was generated based on lentiviral pLKO.1-puro vector (Sigma, St Louis, MO). Lentivirus transfer vector in which RNA polymerase III U6 promoter drives the expression of small hairpin RNAs complementary to RPA2 3′-untranslated region of messenger RNA target sequence was produced based on previously published sequence (34). In brief, the oligonucleotides encoding RPA2 was cloned into the AgeI and EcoRI sites of the pLKO.1-puro vector. All the DNA plasmid transfections were performed using Lipofectamine according to the manufacturer's recommendations (Invitrogen, Carlsbad, CA).
Generation of cell lines expressing wild-type RPA2 or hyperphosphorylation mutant RPA2
HA-tagged wild-type RPA2 (RPA2-WT) or RPA2-(7xA)-mutant form (RPA2-A) were introduced into MCF7 cells by three rounds of retroviral infection. Silencing of endogenous RPA2 in these cells was accomplished by two rounds of lentiviral infection using pLKO.1 vector expressing the sequence targeting RPA2 (RPA2 shRNA). The cells were irradiated with X-rays or treated with HU for Rad51 foci analysis or chromosome aberration and survival assay.
Immunofluorescence analysis
The cells were treated with 0.5% Triton 100 cytoskeletal buffer and fixed with 4% formaldehyde. The fixed cells were permeablized using 0.1% Triton X-100 phosphate-buffered saline for 15 min, followed by blocking with 10% fetal bovine serum and then incubated with primary antibodies. The bound antibodies were revealed by goat anti-mouse IgG Alexa fluor 594 and chicken anti-rabbit IgG Alexa Fluor 488. The slides were viewed at ×1000 magnification on an Olympus fluorescence microscope (BX40 with Magna-Fire CCD camera). The protocol for ssDNA detection was conducted according to a protocol described previously (35).
Western blotting analysis
Proteins from whole cell lysates were resolved by sodium dodecyl sulphate–polyacrylamide gel electrophoresis. The primary antibodies used for this analysis are anti-HA tag antibody (Covance, Princeton, NJ; 1:1000 dilution), rabbit polyclonal antibody phosphor RPA32 Ser4/Ser8 (Bethyl, Montgomery, TX; BL647, 1: 1000 dilution), monoclonal antibody RPA2 (EMD Bioscience, Brookfield, WI; Ab-2, 1:100 dilution) and polyclonal antibodies RPA2 (Abcam, Cambridge, MA; 1:1000 dilution). Secondary antibodies used were goat anti-mouse IgG–horseradish peroxidase conjugated and goat anti-rabbit IgG horseradish peroxidase conjugated (Pierce, Rockford, IL), both at 1:5000 dilutions.
Comet assay
The Neutral Comet Assay was performed using the Comet Assay kit from Trevigen (Gaithersburg, MD) following manufacturer's instructions. The lyses occurred at 4°C for 30 min. Comets were analyzed using CometScore software (TriTek, Sumerduck, VA).
Homologous recombination assays
The HR analysis induced by I-Sce-I expression has been described previously (36). In brief, the HR was measured in MCF7-pDR-green fluorescent protein (GFP) cells with RPA2-WT or RPA2-A expression where the endogenous RPA2 was depleted by shRPA2. The HR analysis upon replication arrest was conducted according to the protocol described previously (36). Briefly, the cells were treated for 24 h with 2 mM thymidine (Thy). The cells were subsequently washed in phosphate-buffered saline and incubated in fresh medium for 3 h and then the cells were treated with 2 mM HU for 24 h. All cells were left for 24 h to recover before being subjected to flow cytometric analysis for GFP-positive cells. For each analysis, at least 1 000 000 cells were processed.
Florescence in situ hybridization for chromosome aberration analysis
Florescence in situ hybridization was performed using pan-telomeric and pan-centromeric peptide nucleic acid probes. Telomere (C3TA2)3-specific and centromere (16-mer repeat DNA)-specific probes directly labeled with Cy3 and fluorescein isothiocyanate fluorescent dyes, respectively, were obtained from Applied Biosystems (Foster City, CA).The cells were radiated (2 Gy) or treated with HU (1 mM, 6 h) and processed with florescence in situ hybridization analysis 24 h later. The cells were incubated with colcemid (0.1 μg/ml, Sigma) for 2.5 h before fixation onto slides. The slides were treated with 1 mg/ml pepsin/0.01 N HCl for 10 min at 37°C, after fixed in 4% formaldehyde, dehydrated in ethanol series (70, 90 and 100%), stained with a telomeric and centromeric probe mixture, denatured 3 min at 80°C and hybridized in a moist chamber for 2 h at reverse transcription, then dehydrated again and sealed in anti-fading 4′,6-diamidino-2-phenylindole mounting (Vector laboratories Burlingame, CA).
Clonogenic survival assays
Cells expressing RPA2-WT or RPA2-A where endogenous RPA2 has been deleted were used in a colony formation assay to measure cell survival after HU or IR treatment. The cells were harvested, filtered with cell strainers (BD Falcon™) to achieve single-cell suspension and plated in triplicates into P60 dishes at various cell densities, with a target number of surviving colonies at 50–100 per dish. Treatment with HU or IR was carried out 18 h after cell plating. After a 24 h exposure to HU at 0.1, 0.5, 1, 2 and 10 mM of concentrations or IR at 2, 4 and 6 Gy of doses, cells were rinsed twice with phosphate-buffered saline and allowed to grow in fresh medium for 14–21 days. Visible colonies were identified with methanol fixation and 0.35% methylene blue staining. The number of colonies (>50 cells) per dish was counted, and the surviving fractions were calculated as the ratio of the plating efficiencies of treated cells to untreated cells.
Results
Hyperphosphorylated RPA2 predominately occur in response to replication arrest
To test whether hyperphosphorylation of RPA2 is critical for the DNA damage response, we first analyzed RPA2 hyperphosphorylation profiles in response to various DNA-damaging agents, including the replication inhibitors HU and Thy and the DSB inducer IR. HU inhibits ribonucleotide reductase and leads to DNA synthesis arrest. Thy disrupts DNA synthesis by altering the balance between thymidine triphosphate and deoxycytidine triphosphate through the allosteric inhibition of ribonucleotide reductase. Both, HU and Thy, inhibit the DNA elongation step of replication. While Thy does not generate detectable DSBs (37), HU causes DSBs that are obvious after ∼12 h of continuous treatment as assessed by pulsed-field gel electrophoresis (36). In contrast to the replication inhibitors HU and Thy, IR can directly cause DNA DSBs. In agreement with previous studies (33), exposure to HU and Thy significantly increased RPA2 hyperphosphorylation (Figure 1A, first panel), as shown by using an antibody specific for phosphorylated RPA2. However, this hyperphosphorylation can only be detected after exposure to a high dose of IR (Figure 1B, first panel) (39,40). We also detected the amount of DSBs in the cells treated with replication inhibitors as well as IR using the comet assay under neutral conditions, which allows detection of DSBs but not ssDNA breaks. No obvious DSBs were detected after 6 h or 24 h of continuous treatment with HU or Thy. In contrast, IR causes enormous DSBs at the doses utilized (Figure 1C). Therefore, our observation is consistent with the previous report that RPA2 hyperphosphorylation is more prevalent when replication forks are stalled rather than when DSBs are generated (33). Since it has been shown that at high doses of IR DNA replication chain elongation is blocked (41), the observation that only higher doses of IR cause RPA2 hyperphosphorylation may be caused by the blockage of DNA replication forks. Alternatively, since it has been suggested that ssDNA resection is restricted to S and G2/M phase cells and RPA2 hyperphosphorylation induced by stalled/collapsed DNA replication is ATR dependent (33), a second explanation is that higher doses of IR can cause more ssDNA, which can activate ATR and subsequently RPA2 hyperphosphorylation. Together, the results shown in Figure 1 and previous reports (33) suggest that RPA2 hyperphosphorylation is more prevalent when DNA replication forks are stalled.
Fig. 1.

Hyperphosphorylation of RPA2 predominately occurs in response to replication arrest. (A) Whole cell lysates were isolated from MCF7 cells that were mock treated or exposed to replication inhibitor, HU (2 mM, 4 h) or Thy (2 mM, 48 h) and immunoblotted with an antibody specific to phosphorylated RPA2 (RPA2-p4/8, first panel) or RPA2 (Abcam, second panel). The same blot membrane from Figure 1A was subsequently reprobed with β-actin for loading control. (B) Whole cell lysates were isolated from MCF7 cells with or without IR treatment (8, 20 or 40 Gy, 6 h after) and then immunoblotted with antibodies against phosphorylated RPA2, RPA2 or β-actin. (C) The neutral comet assay of genomic DNA of MCF7 cells treated with replication inhibitor or IR. The doses used for HU and Thy are same as Figure 1A. The number of cells in each treatment group was 150. Results were expressed as Olive moment. The amount of breaks is correlated with the tail moment values (38).
Here, we need to note that our observation and others (33) are not inconsistent with a recent publication showing that rapid hyperphosphorylation of RPA2 are observed in response to IR which is ataxia-telangiectasia mutated and DNA-dependent protein kinase dependent. This form of IR-induced RPA2 hyperphosphorylation was only observed in mitotic cells but not in interphase cells (42). Thus, hyperphosphorylation of RPA2 can take place in specific mitotic cells when DSBs are generated, a process that is dependent on ataxia-telangiectasia mutated and DNA-dependent protein kinase (42) but primarily upon stalling of replication forks, an ATR-dependent process (33).
Hyperphosphorylated RPA2 colocalizes at the ssDNA region that occurs in response to stalled/collapsed DNA replication forks
Arrest of replication fork progression should lead to the accumulation of ssDNA. We tested if hyperphosphorylated RPA2 is localized at ssDNA regions during replication block. We first examined whether HU induces ssDNA by immunofluorescence staining. ssDNA was detected by using an assay that permits identification of sites of ssDNA >1 kb in situ. This assay is based on the observation that the nucleotide base analogue bromodeoxyuridine (BrdU) is recognized by an anti-BrdU antibody when incorporated into ssDNA but not dsDNA (35). The approach has been successfully utilized in different laboratories (35,43,44). We found that HU treatment significantly increased the percentage of cells with ssDNA in vivo, indicating that ssDNA accumulated in response to replication arrest (Figure 2A). To determine if hyperphosphorylated RPA colocalized with ssDNA, cells treated with HU were costained with BrdU and phospho-specific RPA2 antibody. An extensive colocalization of hyperphosphorylated RPA2 with ssDNA foci was observed (Figure 2B) indicating that hyperphosphorylated RPA2 associates with ssDNA regions after a replication block. Given the requirement of DNA binding for RPA2 hyperphosphorylation, we reasoned that phosphorylated RPA2 foci occur later than RPA2 recruitment. Therefore, we examined the recruitment of RPA2 and hyperphosphorylated RPA2 at various time points after HU treatment by immunofluorescence staining using anti-RPA2 and specific anti-phosphorylated RPA2 antibodies. We found that RPA2 recruitment starts to increase as early as 1 h following HU treatment. In contrast, HU-induced phosphorylated RPA2 foci were not observed until 2 h after HU treatment (Figure 2C). Representative RPA2 and hyperphosphorylated RPA2 foci in response to HU are shown in Figure 2C. Moreover, RPA2 foci and phosphorylated RPA2 foci show nearly complete colocalization (Figure 2C). This result indicates that RPA2 is recruited to ssDNA and then is further hyperphosphorylated, which is consistent with previous observations that RPA2 can only be hyperphosphorylated after binding to DNA or chromatin (33,45,46).
Fig. 2.
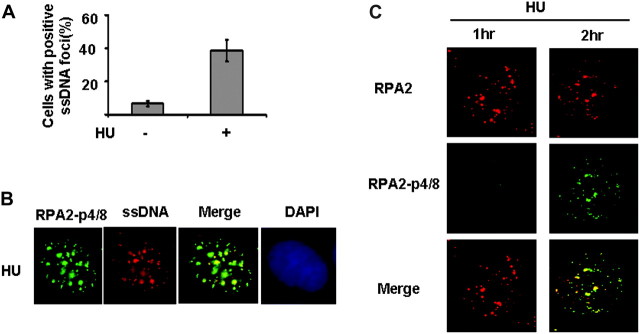
Hyperphosphorylated RPA2 colocalizes at the ssDNA region in response to stalled/collapsed DNA replication forks. (A) HU treatment results in ssDNA accumulation. The protocol for ssDNA detection has been described in a previous publication (35). In brief, the cells were grown in the presence of bromodeoxyuridine (BrdU; 10 μg/ml; Invitrogen) for 24 h. Cells with BrdU labeling were treated with 2 mM of HU or left untreated and 6 h later, the cells were fixed. After fixation, the cells will be blocked and stained with anti-BrdU mouse monoclonal antibody clone B44 (BD Biosciences, San Jose, CA) antibody. Then, the samples are incubated with an Alexa Fluor 488-conjugated goat anti-mouse antibody. Cells were scored positive when 10 nuclear foci were visible. Quantitative analysis for ssDNA foci in response to HU was shown in Figure 2A. (B) Colocalization of phosphorylated RPA2 with ssDNA. MCF7 cells are cultured 24 h in the presence of BrdU. Cells with BrdU labeling were treated with 2 mM of HU and 6 h later, the cells were fixed. The fixed cells were costained by RPA2-p-4/8 antibody and anti-BrdU antibody. Representative phosphorylated RPA2 foci and ssDNA foci in response to HU were shown in Figure 2B. (C) The appearance of phosphorylated RPA2 foci occurs later than RPA2 foci after HU treatment. Cells in exponential growth phase were treated with HU (2 mM), fixed at indicated time points and costained with anti-RPA2 (Ab-2) and anti-phosphorylated RPA2 antibody (RPA2-p4/8). Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue).
Hyperphosphorylated RPA2 is required for Rad51 recruitment following HU treatment
The accumulated ssDNA during stalled/collapsed DNA replication forks could lead to DSBs. The increased DSBs induced by HU treatment were detected by pulsed-field gel electrophoresis (36). Consistent with this report, an increase in cells with γ-H2AX foci after HU treatment has also been observed (47). DSBs and other lesions associated with DNA replication are repaired by HR in mammalian cells (37,48). A key protein in HR-mediated repair is RAD51, and cells deficient in RAD51 accumulate DSBs after replication or at stalled replication forks (49). Since it has been demonstrated that hyperphosphorylated RPA2 directly interacts with Rad51 (32), we next determined if hyperphosphorylated RPA2 colocalized with Rad51 in response to replication arrest. Hyperphosphorylated RPA2 was observed to extensively colocalize with Rad51 in response to HU treatment (Figure 3A), suggesting the association of hyperphosphorylated RPA2 with Rad51 in vivo in stalled/collapsed DNA replication response pathways. To further study the role of phosphorylated RPA2 in Rad51 recruitment, we used an RPA2 mutant construct in which seven DNA damage-associated phosphorylation sites (33) were substituted by alanine (RPA2-A). Retroviral vectors carrying a HA-tagged wild-type RPA2 or phosphorylation mutant RPA2, RPA2-(7xA)-(S4A/S8A/S11A/S12A/S13A/T21A/S33A) have been described previously (33) (Figure 3B). Like wild-type RPA2 (RPA2-WT), the RPA2-(7xA)-mutant (RPA2-A) co-immunoprecipitated with RPA1 (33) (data not shown), indicating that substitution of the serine residues in the N-terminus of the protein did not affect the interaction of RPA2 with RPA1. Also, disruption of the phosphorylation sites of RPA2 does not affect RPA heterotrimer formation, the normal S phase functions of RPA2 or cell growth (33,34). We tested Rad51 recruitment in cells expressing RPA2-WT or RPA2-A where endogenous RPA2 was depleted by shRNA targeting the 3′-untranslated region of the RPA2 messenger RNA (34). As shown in Figure 3C, endogenous RPA2 expression was reduced to undetectable levels via transduction with RPA2 shRNA. Exogenous RPA2-WT and RPA2-A proteins were resistant to shRNA RPA2 and expressed the proteins at similar levels. Strikingly, exogenous RPA2-A expression showed a major reduction in percentage of cells with positive Rad51 foci after HU treatment compared with cells expressing RPA2-WT (Figure 3D) (T-test, P < 0.01, P = 0.002), indicating that RPA2 hyperphosphorylation is required for Rad51 assembly. The specificity of Rad51 antibody was verified by the observation that depletion of Rad51 using Rad51 small interfering RNA abolished HU-induced Rad51 foci formation in cells expressing RPA2-WT (data not shown). Representative Rad51 foci are shown in Figure 3E (upper panel). The effect of RPA2-A on Rad51 recruitment was not caused by a deficiency in RPA recruitment to chromatin since normal RPA foci formation was observed in cells expressing RPA2-WT or RPA2-A in response to HU (See supplementary Figure S1, available at Carcinogenesis Online). This is consistent with previous reports showing that defective RPA2 phosphorylation doesnot affect recruitment of RPA to chromatin or the sites of damaged DNA (33,34). In order to test if RPA2 hyperphosphorylation plays a role in Rad51 recruitment in response to two-ended DSBs, the formation of Rad51 foci was compared in cells expressing RPA2-WT or RPA2-A after exposure to IR. We found that Rad51 foci formation was not affected by RPA2 hyperphosphorylation since a similar proportion of cells with IR-induced Rad51 foci was observed in cells expressing RPA2-WT or RPA2-A (Figure 3D), indicating the RPA2 hyperphosphorylation is not essential for the Rad51 recruitment in response to the classical two-ended DSBs. Representative Rad51 foci are shown in Figure 3E (bottom panel).
Fig. 3.

RPA2 hyperphosphorylation is required for Rad51 recruitment after HU treatment. (A) Phosphorylated RPA2 is colocalized with Rad51 after HU treatment. Exponentially growing MCF7 cells were treated with HU and fixed at 4 h after treatment. Cells were costained by RPA2-p4/8 and anti-Rad51 antibodies. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). (B) Schematic representation of RPA2 depicting phosphorylation sites mutant in the N-terminus of the protein. (C) Generation of cell lines expressing RPA2-WT or RPA2-A with endogenous RPA2 depletion is described in Materials and Methods. Whole cell extracts were immunoblotted with an anti-RPA2 antibody. (D) Hyperphosphorylation of RPA2 is required for Rad51 foci formation after HU treatment but not essential for IR induced Rad51 foci formation. After 18 h treatment with 2 mM HU or 8 h after 8 Gy IR, cells depleted of endogenous RPA2 and expressing RPA2-WT or RPA2-A were fixed and processed for immunocytochemistry with an anti-Rad51 antibody. Cells were scored positive when at least 10 nuclear foci were visible. Data shown are averages from three independent experiments (T-test, P < 0.01 for HU treatment; P > 0.05 for IR treatment). (E) Representative Rad51 foci in cells expressing RPA2-WT or RPA2-A. Representative Rad51 foci with HU treatment are shown in Figure 3E (upper panel). Representative Rad51 foci after IR treatment are shown in Figure 3E (bottom panel).
Notably, RPA2 hyperphosphorylation has no obvious effect on the fraction of cells with Rad51-positive foci in untreated cells (Figure 3D, T-test, P > 0.05). This finding is consistent with our observation that no obvious RPA2 hyperphosphorylation was observed in cells without any treatment (Figure 1A and B). Our finding further supports the previous study suggesting that the repair mechanism of spontaneous HR is different to that induced by replication inhibitors (50). Collectively, the data in Figure 3 demonstrate that hyperphosphorylated RPA2 accumulates in ssDNA regions and is important for the formation of Rad51 foci after replication block.
RPA2 hyperphosphorylation is specifically required for HR after replication arrest but is not necessary for HR after the induction of DSB by I-Sce-I expression
The observation that RPA2 hyperphosphorylation affects Rad51 recruitment in response to replication arrest led us to speculate that RPA2 hyperphosphorylation plays a role in HR after replication arrest. One of the main systems established for the study of HR processes is the measurement of the occurrence of HR in artificial recombination substrates. This assay is broadly used to study two-ended DSBs stimulated HR by overexpressing the I-Sce-I endonuclease protein (51) or to study the HR in response to replication block by HU and Thy or topoisomerase I inhibitor camptothecin (36,50,52–54). Therefore, we next examined the biological consequence of expressing RPA2-A on a previously established HR assay system (51). The established MCF7 cell line containing an integrated copy of the pDR-GFP reporter (MCF7 DR-GFP) was described previously (55). It is based on reconstruction of the wild-type GFP from two non-functional fragments of GFP complementary DNA (51). The protocol for HR induced by stalled/collapsed DNA replication forks was conducted according to a previous publication (36). To improve the frequency of HR, the cells were first synchronizated by double thy block that results in 70–90% of cells in the S phase (data not shown). The resulting S phase cells were treated with HU. We found that cells expressing RPA2-A displayed a decreased frequency of HR triggered by HU treatment in comparison to cells expressing RPA2-WT (Figure 4A) (T-test, P < 0.05, P = 0.006), suggesting that RPA2 hyperphosphorylation is critical for HR-mediated repair after replication arrest.
Fig. 4.

RPA2 hyperphosphorylation is required for the HR stimulated by HU but not for HR induced by I-Sce-I. (A) RPA2 hyperphosphorylation is specifically critical for HR after replication arrest. The frequencies of HR were calculated in cells expressing RPA2-WT or RPA2-A after HU treatment by flow cytometry. Results are means from three independent experiments with standard deviation. (B) Cells expressing phosphorylation-defective RPA2 show similar HR frequency induced by I-Sce-I expression in comparison to cells with wild-type RPA2. The HR frequency was measured by flow cytometric analyses (see Materials and Methods) in cells expressing RPA2-WT or RPA2-A. Results are means from three independent experiments, with standard deviation. (C) Immunoblot analysis of RPA2 hyperphosphorylation expression in MCF7 cells with I-Sce-I plasmid transfection or HU and Thy. Cell lysates were harvested 24 h after I-Sce-I transfection or after HU and Thy treatment. The doses used for HU and Thy are same as Figure 1A. Proteins were resolved by sodium dodecyl sulphate–polyacrylamide gel electrophoresis and visualized by immunoblot with anti-RPA2-p4/8 antibody or anti-RPA2 antibody (Abcam). (D) Foci of phosphorylated RPA2 in cells with I-Sce-I transfection or HU treatment. The cells were fixed 24 h after I-Sce-I transfection or 4 h after HU treatment. Fixed cells were stained by a specific anti-RPA2 phosphorylation antibody and anti-RPA2-p4/8 antibody. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue).
We next examined the biological consequence of expressing RPA2-A on HR induced by rare-cutting I-Sce-I endonuclease. We found no changes in the frequency of HR in cells expressing RPA2-A compared with cells expressing RPA2-WT (Figure 4B) (T-test, P > 0.05, P = 0.18), indicating that RPA2 hyperphosphorylation has no obvious role on HR induced by I-Sce-I expression. This data suggests that RPA2 hyperphosphorylation is specifically required for HR after replication block but not in response to two-ended DSBs. Our findings could be explained by the fact that no detectable RPA2 hyperphosphorylation or RPA2 phosphorylation foci were observed by I-Sce-I overexpression, whereas they were easily detected in cells after HU treatment (Figures 1A and 4C and D). We need to note that the frequency of the HR induced by HU is lower than that stimulated by I-Sce-I expression due to the genome-wide effect of replication inhibition compared with the targeted DSB induced with I-Sce-I (50). Together, these findings strongly support a key role for RPA2 hyperphosphorylation in HR triggered by DNA replication arrest.
RPA2 hyperphosphorylation prevents HU-induced chromosomal aberrations
Failure to restart stalled/collapsed replication forks is a potential source of genomic instability. In cells without DNA damage, DNA replication duplicates each chromosome during S phase to create a sister chromatid. If the chromosomal DNA replication is blocked, the ssDNA generated due to replication arrest could be processed into DSBs (36). In normal cells, the DSBs are repaired by HR since only one-end DSBs are created, which is not an appropriate substrate for non-homologous end joining. However, if DSBs are not repaired by HR, these persistent DSBs will result in chromatid breaks or chromosome breaks. The deficient HR and the lack of HU-induced Rad51 focus formation in cells expressing RPA2-A indicates a defective response to stalled/collapsed DNA replication forks, which could lead to genomic instability. Therefore, we examined whether a deficiency in RPA2 hyperphosphorylation results in the accumulation of chromosomal abnormalities in response to HU using florescence in situ hybridization. We observed that cells expressing RPA2-A displayed higher frequencies of chromatid and chromosome breaks after exposure to HU (Figure 5A and B) compared with cells expressing RPA2-WT. The representative metaphases spread after HU treatment in cells expressing RPA2-A is shown in Figure 5B.
Fig. 5.

RPA2 hyperphosphorylation is required for maintenance of genomic stability. (A) Frequencies of HU-induced chromosome aberrations are indicated. The data shown is from one of two independent experiments. Chid and Chme represent chromatid and chromosome, respectively. Fifty metaphases for each sample were analyzed. (B) Representative metaphase spreads in cells treated with 1 mM HU for 6 h and incubated in fresh medium for 24 h. Florescence in situ hybridization using telomeric and centromeric probes reveals the red/pink and green color. Chromatid-type aberrations indicated in the white arrow. Chromosome-type aberrations indicated in the yellow arrow. (C) Frequencies of HU-induced radial structures are indicated. Fifty metaphases for each sample were analyzed. The data shown is from one of two typical independent experiments. (D) A typical radical structure induced by HU from cells expressing RPA2-A. Y-shaped tri-radial chromosomes and star-shaped quadri-radial chromosomes were indicated by arrowheads. (E) Frequencies of IR-induced chromosome aberration are indicated. Fifty metaphases for each sample were analyzed. The data shown is from one of two typical independent experiments.
Most importantly, cells expressing RPA2-A exhibited a higher rate of radial structures (Figure 5C), such as tri-radial and quadri-radial aberrations (Figure 5D), generated by the increased frequency of chromatid breaks and subsequent interchromatid fusions. The presence of radial chromosomes in cells expressing RPA2-A further reveals a link between RPA2 hyperphosphorylation and HR since cells deficient in HR often show similar chromosome aberrations, such as the cells deficient in the FA/BRCA2 pathway (56,57). These findings highlight an important role of RPA2 hyperphosphorylation in maintenance of genomic stability during DNA replication. Next, we tested the role of hyperphosphorylated RPA2 in maintenance of genomic stability after IR. We found that cells expressing RPA2-A display similar frequencies of chromatid and chromosome breaks after exposure to IR (Figure 5E). Thus, Figure 5 indicates that RPA2 hyperphosphorylation is specially required for maintenance of genomic stability in response to replication arrest, which perfectly fits its role in Rad51 foci formation and HR induced by HU (Figures 2 and 3).
RPA2 hyperphosphorylation is required for resistance to HU but not IR
The HR-mediated repair is required to restart stalled or collapsed replication forks under replication stress conditions. The role of hyperphosphorylated RPA2 in Rad51 recruitment and HR in response to HU indicates that RPA2 hyperphosphorylation plays a role in cell survival after replication arrest. To test this hypothesis, we determined the effect of RPA2 hyperphosphorylation on survival following HU treatment or IR exposure. As shown in Figure 6A, cells expressing RPA2-A produced a phenotype of greater sensitivity to HU by clonogenic cell survival (Figure 6A). In contrast, the cells expressing hyperphosphorylation mutant RPA2 showed a similar sensitivity to IR (Figure 6B). Thus, Figure 6 suggests that RPA2 hyperphosphorylation is required for cell survival under conditions of replication arrest, which is correlated with its role in Rad51 recruitment and HR stimulated by HU treatment (Figure 3). It should be noted that although hyperphosphorylation of RPA2 plays a role in inhibition of DNA replication after activation of S phase checkpoint during replication stress, the defects of HR in cells expressing phosphorylation defective RPA2 may explain the sensitivity to HU (Figure 6A) since it has been indicated that S phase checkpoint is not required for cell viability under replication stress conditions (58).
Fig. 6.

RPA2 hyperphosphorylation is critical for cell survival in response to HU but not IR. Clonogenic survival in MCF7 cells expressing RPA2-WT or RPA2-A after HU treatment (A) or exposure to IR (B). The cells expressing RPA2-WT or RPA2-A were treated with HU or IR at the indicated doses. The plotted points are on the averages of three to four experiments with standard deviations.
Discussion
The activation of oncogenes and inactivation of tumor suppressor genes frequently results from cancer-promoting mutations. These genetic alterations are often mediated by impaired DSB repair. RPA is an essential factor for DSB repair and cell cycle checkpoint activation. In the present report, we demonstrate that hyperphosphorylated RPA2 colocalizes with ssDNA and Rad51 and is essential for Rad51 recruitment and HR in response to replication arrest. In addition, we also demonstrate that RPA2 hyperphosphorylation is critical for maintenance of genomic stability and cell viability after replication stalling. These findings provide direct evidence that RPA2 hyperphosphorylation facilitates HR and is important for maintenance of genomic stability and cell survival under conditions of replication stress in addition to its role on mitotic exit in response to mitotic DNA damage (30).
Hyperphosphorylation of RPA2 is specifically required for HR in response to replication arrest
Consistent with previous reports, we found that RPA2 hyperphosphorylation profoundly occurs following replication arrest (Figure 1). This result can be explained by studies showing that it is not only the ssDNA but also the nucleic acid structure at stalled replication forks that regulates function of ATR (59–61). The single-strand/double-strand junctions, which reflect the structure at the replication forks, are able to stimulate RPA2 hyperphosphorylation by ATR more effectively than ssDNA fragment (61). In our study, we observed that hyperphosphorylation of RPA2 is required for HR in response to replication arrest but not two-ended DSBs-induced HR. Our work further supports the notion that the HR mechanisms required for repairing classical two-end DSBs and restart for stalled/collapsed DNA replication are different (36,50,53). Several models have been proposed in response to the stalled or collapsed DNA replication in E.coli, including one-ended DSBs-stimulated recombination-dependent replication and direct-restart mechanism of reactivating forks without any DSBs creation (4). A potential mechanism required for repair of ssDNA gaps, DSG, has also been proposed (4,62). Whether RPA2 hyperphosphorylation regulates any or all these subpathways of HR in mammalian cells is a very interesting topic to be addressed in the future.
A question raised by our studies is why RPA2 hyperphosphorylation is specifically required for HR after replication arrest. It has been shown that hyperphosphorylation induced a conformational change in RPA (63). Because RPA hyperphosphorylation does not significantly alter the high affinity of RPA for ssDNA (28,63), a likely scenario is that hyperphosphorylated RPA located at DNA damage sites after replication arrest facilities the recruitment of factors for DNA repair, such as Rad51, a key player for HR-mediated repair. The components that associate with RPA during replication arrest most likely differ from those associated with RPA in response to two-ended DSB. Rad51 may not be efficiently recruited to the sites of stalled/collapsed DNA replication forks. The conformational change caused by RPA2 hyperphosphorylation is necessary for Rad51 recruitment to the damaged DNA sites under the conditions of replication arrest. This speculation is supported by a recent study showing that the affinity of RPA to Rad51 is increased when RPA is hyperphosphorylated. In addition, we observed that deficient RPA2 phosphorylation failed to recruit Rad51 (Figure 3) although RPA is recruited to the damaged DNA sites normally (supplementary Figure S1 is available at Carcinogenesis Online) (33,34).
Hyperphosphorylated RPA2 is a central factor in response to replication arrest
Given the complexity of DNA replication, S phase is a vulnerable period for the genome in the cell-division cycle. During replication arrest, long regions of RPA-coated ssDNA at replication forks are necessary for ATR checkpoint signaling. Interestingly, RPA2 is also a direct substrate of ATR (33). The evidence suggests a regulated process in which ssDNA-bound RPA activates the ATR kinase, which in turn induces RPA hyperphosphorylation. In addition, it has been documented that hyperphosphorylation of RPA2 prevents RPA from participating in DNA replication in response to DNA damage. In the present study, we show that RPA2 hyperphosphorylation is essential for HR after stalled or collapsed DNA replication forks (Figure 4). Therefore, hyperphosphorylated RPA2 appears to serve as a central regulator in response to replication arrest, which is evidenced by the increased chromosome instability and cell toxicity observed in cells lacking RPA2 hyperphosphorylation (Figures 5 and 6). Therefore, a possible scenario under conditions of replication stress is that ssDNA bound by RPA actives ATR and subsequently RPA2 hyperphosphorylation induced by ATR promotes HR via facilitation of Rad51 recruitment (supplementary Figure S2 is available at Carcinogenesis Online).
Interestingly, a recent study suggests that dephosphorylation of RPA2 is also important to DNA repair process after replication arrest, and persistent RPA2 phosphorylation leads to the increased DSBs (31). The complexity of DNA DSB repair processing requires that the individual reaction steps are ordered. The HR-mediated repair process in a highly organized process. RPA2 hyperphosphorylation and dephosphorylation could be an important mechanism to regulate the assembly and/or disassembly of protein complexes at the site of stalled/collapsed DNA lesions in a sequential order. Therefore, HR-mediated repair in response to stalled/collapsed DNA replication forks is a strictly regulated process in which both phosphorylation and dephosphorylation of RPA is required. Defects in either part of the regulation process results in genomic instability.
Hyperphosphorylated RPA2 may be a potential indicator for sensitivity to replication arrest agents
We have shown that defective RPA2 hyperphosphorylation results in the enhanced sensitivity to HU, which is an approved therapeutic agent for chronic myelogenous leukemia. HU is commonly used for other myeloproliferative disorders and also used occasionally for metastatic melanoma and other cancers. Given that hyperphosphorylated RPA2 can also be induced by topoisomerase I inhibitor camptothecin, cross-linking agents cisplatin and other chemotherapeutic drugs (50,64), our studies should have significant clinical implications. A recent study has suggested that RPA2 hyperphosphorylation confers resistance to cisplatin and other chemotherapy drugs (65). Therefore, the evaluation of RPA2 hyperphosphorylation can be potentially used as an indicator for the sensitivity/resistance to these chemotherapeutic agents. The understanding of the molecular mechanism by which RPA2 phosphorylation promotes HR should eventually lead to the development of a novel class of anticancer drugs targeting RPA2 hyperphosphorylation.
Supplementary material
Supplementary Figures S1 and S2 can be found at http://carcin.oxfordjournals.org/
Funding
A seed grant and startup fund from the Department of Radiation Oncology, Washington University School of Medicine by the Dr J.R.R.; Stephanie Pagano Fund for Mesothelioma Research by a grant from the American Cancer Society (IRG-58-010-51); Wendy Will Case Cancer Fund to J.Z.
Supplementary Material
Acknowledgments
We apologize for those whose work was not cited due to the limited space. We thank M.Jasin for their generous contribution of materials.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- ATR
ataxia-telangiectasia mutated and Rad3-related
- BrdU
bromodeoxyuridine
- DSB
DNA double-strand break
- DSG
daughter strand gap
- GFP
green fluorescent protein
- HR
homologous recombination
- HU
hydroxyurea
- IR
ionizing radiation
- ssDNA
single-strand DNA
- RPA
replication protein A
- Thy
thymidine
- shRNA
small hairpin RNA
References
- 1.O'Driscoll M, et al. The role of double-strand break repair—insights from human genetics. Nat. Rev. Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- 2.Marians KJ. Mechanisms of replication fork restart in Escherichia coli. Philos. trans. R. Soc. Lond. 2004;359:71–77. doi: 10.1098/rstb.2003.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lovett ST. Connecting replication and recombination. Mol. Cell. 2003;11:554–556. doi: 10.1016/s1097-2765(03)00110-2. [DOI] [PubMed] [Google Scholar]
- 4.Heller RC, et al. Replisome assembly and the direct restart of stalled replication forks. Nat. Rev. 2006;7:932–943. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- 5.Wyman C, et al. DNA double-strand break repair: all's well that ends well. Annu. Rev. Genet. 2006;40:363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- 6.Helleday T, et al. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst.) 2007;6:923–935. doi: 10.1016/j.dnarep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 7.Blais V, et al. RNA interference inhibition of Mus81 reduces mitotic recombination in human cells. Mol. Biol. Cell. 2004;15:552–562. doi: 10.1091/mbc.E03-08-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osman F, et al. Exploring the roles of Mus81-Eme1/Mms4 at perturbed replication forks. DNA Repair (Amst.) 2007;6:1004–1017. doi: 10.1016/j.dnarep.2007.02.019. [DOI] [PubMed] [Google Scholar]
- 9.Hanada K, et al. The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. EMBO J. 2006;25:4921–4932. doi: 10.1038/sj.emboj.7601344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grompone G, et al. Requirement for RecFOR-mediated recombination in priA mutant. Mol. Microbiol. 2004;52:551–562. doi: 10.1111/j.1365-2958.2004.03997.x. [DOI] [PubMed] [Google Scholar]
- 11.Mezzina M, et al. Replication of simian virus 40 DNA after UV irradiation: evidence of growing fork blockage and single-stranded gaps in daughter strands. J. Virol. 1988;62:4249–4258. doi: 10.1128/jvi.62.11.4249-4258.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Svoboda DL, et al. Differential replication of a single, UV-induced lesion in the leading or lagging strand by a human cell extract: fork uncoupling or gap formation. Proc. Natl Acad. Sci. USA. 1995;92:11975–11979. doi: 10.1073/pnas.92.26.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cordeiro-Stone M, et al. Replication fork bypass of a pyrimidine dimer blocking leading strand DNA synthesis. J. Biol. Chem. 1997;272:13945–13954. doi: 10.1074/jbc.272.21.13945. [DOI] [PubMed] [Google Scholar]
- 14.Cordeiro-Stone M, et al. Analysis of DNA replication forks encountering a pyrimidine dimer in the template to the leading strand. J. Mol. Biol. 1999;289:1207–1218. doi: 10.1006/jmbi.1999.2847. [DOI] [PubMed] [Google Scholar]
- 15.Sibenaller ZA, et al. The 32- and 14-kilodalton subunits of replication protein A are responsible for species-specific interactions with single-stranded DNA. Biochemistry. 1998;37:12496–12506. doi: 10.1021/bi981110+. [DOI] [PubMed] [Google Scholar]
- 16.Bochkarev A, et al. The crystal structure of the complex of replication protein A subunits RPA32 and RPA14 reveals a mechanism for single-stranded DNA binding. EMBO J. 1999;18:4498–4504. doi: 10.1093/emboj/18.16.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Binz SK, et al. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair (Amst.) 2004;3:1015–1024. doi: 10.1016/j.dnarep.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 18.Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 19.Zou L, et al. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 20.Namiki Y, et al. ATRIP associates with replication protein A-coated ssDNA through multiple interactions. Proc. Natl Acad. Sci. USA. 2006;103:580–585. doi: 10.1073/pnas.0510223103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baumann P, et al. The human Rad51 protein: polarity of strand transfer and stimulation by hRP-A. EMBO J. 1997;16:5198–5206. doi: 10.1093/emboj/16.17.5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugiyama T, et al. Rad52 protein associates with replication protein A (RPA)-single-stranded DNA to accelerate Rad51-mediated displacement of RPA and presynaptic complex formation. J. Biol. Chem. 2002;277:31663–31672. doi: 10.1074/jbc.M203494200. [DOI] [PubMed] [Google Scholar]
- 23.Fanning E, et al. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006;34:4126–4137. doi: 10.1093/nar/gkl550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niu H, et al. Mapping of amino acid residues in the p34 subunit of human single-stranded DNA-binding protein phosphorylated by DNA-dependent protein kinase and Cdc2 kinase in vitro. J. Biol. Chem. 1997;272:12634–12641. doi: 10.1074/jbc.272.19.12634. [DOI] [PubMed] [Google Scholar]
- 25.Zernik-Kobak M, et al. Sites of UV-induced phosphorylation of the p34 subunit of replication protein A from HeLa cells. J. Biol. Chem. 1997;272:23896–23904. doi: 10.1074/jbc.272.38.23896. [DOI] [PubMed] [Google Scholar]
- 26.Nuss JE, et al. DNA damage induced hyperphosphorylation of replication protein A. 1. Identification of novel sites of phosphorylation in response to DNA damage. Biochemistry. 2005;44:8428–8437. doi: 10.1021/bi0480584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anantha RW, et al. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J. Biol. Chem. 2007;282:35910–35923. doi: 10.1074/jbc.M704645200. [DOI] [PubMed] [Google Scholar]
- 28.Oakley GG, et al. RPA phosphorylation in mitosis alters DNA binding and protein-protein interactions. Biochemistry. 2003;42:3255–3264. doi: 10.1021/bi026377u. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, et al. Modulation of replication protein A function by its hyperphosphorylation-induced conformational change involving DNA binding domain B. J. Biol. Chem. 2005;280:32775–32783. doi: 10.1074/jbc.M505705200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anantha RW, et al. RPA phosphorylation facilitates mitotic exit in response to mitotic DNA damage. Proc. Natl Acad. Sci. USA. 2008;105:12903–12908. doi: 10.1073/pnas.0803001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng J, et al. Protein phosphatase 2A-dependent dephosphorylation of replication protein A is required for the repair of DNA breaks induced by replication stress. Mol. Cell. Biol. 2009;29:5696–5709. doi: 10.1128/MCB.00191-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu X, et al. Preferential localization of hyperphosphorylated replication protein A to double-strand break repair and checkpoint complexes upon DNA damage. Biochem. J. 2005;391:473–480. doi: 10.1042/BJ20050379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olson E, et al. RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. J. Biol. Chem. 2006;281:39517–39533. doi: 10.1074/jbc.M605121200. [DOI] [PubMed] [Google Scholar]
- 34.Vassin VM, et al. Replication protein A (RPA) phosphorylation prevents RPA association with replication centers. Mol. Cell. Biol. 2004;24:1930–1943. doi: 10.1128/MCB.24.5.1930-1943.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raderschall E, et al. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc. Natl Acad. Sci. USA. 1999;96:1921–1926. doi: 10.1073/pnas.96.5.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saintigny Y, et al. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001;20:3861–3870. doi: 10.1093/emboj/20.14.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lundin C, et al. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol. Cell. Biol. 2002;22:5869–5878. doi: 10.1128/MCB.22.16.5869-5878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olive PL, et al. Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the “comet” assay. Radiat. Res. 1990;122:86–94. [PubMed] [Google Scholar]
- 39.Liu VF, et al. The ionizing radiation-induced replication protein A phosphorylation response differs between ataxia telangiectasia and normal human cells. Mol. Cell. Biol. 1993;13:7222–7231. doi: 10.1128/mcb.13.12.7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boubnov NV, et al. scid cells are deficient in Ku and replication protein A phosphorylation by the DNA-dependent protein kinase. Mol. Cell. Biol. 1995;15:5700–5706. doi: 10.1128/mcb.15.10.5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Painter RB. Inhibition of mammalian cell DNA synthesis by ionizing radiation. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1986;49:771–781. doi: 10.1080/09553008514552981. [DOI] [PubMed] [Google Scholar]
- 42.Stephan H, et al. Ionizing radiation-dependent and independent phosphorylation of the 32-kDa subunit of replication protein A during mitosis. Nucleic Acids Res. 2009;37:6028–6041. doi: 10.1093/nar/gkp605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schlegel BP, et al. BRCA1 promotes induction of ssDNA by ionizing radiation. Cancer Res. 2006;66:5181–5189. doi: 10.1158/0008-5472.CAN-05-3209. [DOI] [PubMed] [Google Scholar]
- 44.Sartori AA, et al. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henricksen LA, et al. Recombinant replication protein A: expression, complex formation, and functional characterization. J. Biol. Chem. 1994;269:11121–11132. [PubMed] [Google Scholar]
- 46.Fotedar R, et al. Cell cycle regulated phosphorylation of RPA-32 occurs within the replication initiation complex. EMBO J. 1992;11:2177–2187. doi: 10.1002/j.1460-2075.1992.tb05277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Feraudy S, et al. Pol eta is required for DNA replication during nucleotide deprivation by hydroxyurea. Oncogene. 2007;26:5713–5721. doi: 10.1038/sj.onc.1210385. [DOI] [PubMed] [Google Scholar]
- 48.Johnson RD, et al. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J. 2000;19:3398–3407. doi: 10.1093/emboj/19.13.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sonoda E, et al. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 1998;17:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saleh-Gohari N, et al. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol. Cell. Biol. 2005;25:7158–7169. doi: 10.1128/MCB.25.16.7158-7169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pierce AJ, et al. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wen Q, et al. A mutant allele of MRE11 found in mismatch repair-deficient tumor cells suppresses the cellular response to DNA replication fork stress in a dominant negative manner. Mol. Biol. Cell. 2008;19:1693–1705. doi: 10.1091/mbc.E07-09-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bolderson E, et al. ATM is required for the cellular response to thymidine induced replication fork stress. Hum. Mol. Genet. 2004;13:2937–2945. doi: 10.1093/hmg/ddh316. [DOI] [PubMed] [Google Scholar]
- 54.Sorensen CS, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 55.Zhang J, et al. MDC1 interacts with Rad51 and facilitates homologous recombination. Nat. Struct. Mol. Biol. 2005;12:902–909. doi: 10.1038/nsmb991. [DOI] [PubMed] [Google Scholar]
- 56.Niedernhofer LJ, et al. Fanconi anemia (cross)linked to DNA repair. Cell. 2005;123:1191–1198. doi: 10.1016/j.cell.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 57.Venkitaraman AR. Tracing the network connecting BRCA and Fanconi anaemia proteins. Nat. Rev. Cancer. 2004;4:266–276. doi: 10.1038/nrc1321. [DOI] [PubMed] [Google Scholar]
- 58.Tittel-Elmer M, et al. The MRX complex stabilizes the replisome independently of the S phase checkpoint during replication stress. EMBO J. 2009;28:1142–1156. doi: 10.1038/emboj.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumagai A, et al. Claspin and the activated form of ATR-ATRIP collaborate in the activation of Chk1. J. Biol. Chem. 2004;279:49599–49608. doi: 10.1074/jbc.M408353200. [DOI] [PubMed] [Google Scholar]
- 60.Cortez D. Unwind and slow down: checkpoint activation by helicase and polymerase uncoupling. Genes Dev. 2005;19:1007–1012. doi: 10.1101/gad.1316905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu JS, et al. Phosphorylation of replication protein A by S-phase checkpoint kinases. DNA Repair (Amst.) 2006;5:369–380. doi: 10.1016/j.dnarep.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 62.Lopes M, et al. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell. 2006;21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 63.Binz SK, et al. The phosphorylation domain of the 32-kDa subunit of replication protein A (RPA) modulates RPA-DNA interactions. Evidence for an intersubunit interaction. J. Biol. Chem. 2003;278:35584–35591. doi: 10.1074/jbc.M305388200. [DOI] [PubMed] [Google Scholar]
- 64.Cruet-Hennequart S, et al. Enhanced DNA-PK-mediated RPA2 hyperphosphorylation in DNA polymerase eta-deficient human cells treated with cisplatin and oxaliplatin. DNA Repair (Amst.) 2008;7:582–596. doi: 10.1016/j.dnarep.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 65.Manthey KC, et al. Hyperphosphorylation of replication protein A in cisplatin-resistant and -sensitive head and neck squamous cell carcinoma cell lines. Head Neck. 2009 doi: 10.1002/hed.21234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.