

Abstract
Recent human genetic studies have provided evidences that sporadic or inherited missense mutations in four-and-a-half LIM domain protein 1 (FHL1), resulting in alterations in FHL1 protein expression, are associated with rare congenital myopathies, including reducing body myopathy and Emery–Dreifuss muscular dystrophy. However, it remains to be clarified whether mutations in FHL1 cause skeletal muscle remodeling owing to gain- or loss of FHL1 function. In this study, we used FHL1-null mice lacking global FHL1 expression to evaluate loss-of-function effects on skeletal muscle homeostasis. Histological and functional analyses of soleus, tibialis anterior and sternohyoideus muscles demonstrated that FHL1-null mice develop an age-dependent myopathy associated with myofibrillar and intermyofibrillar (mitochondrial and sarcoplasmic reticulum) disorganization, impaired muscle oxidative capacity and increased autophagic activity. A longitudinal study established decreased survival rates in FHL1-null mice, associated with age-dependent impairment of muscle contractile function and a significantly lower exercise capacity. Analysis of primary myoblasts isolated from FHL1-null muscles demonstrated early muscle fiber differentiation and maturation defects, which could be rescued by re-expression of the FHL1A isoform, highlighting that FHL1A is necessary for proper muscle fiber differentiation and maturation in vitro. Overall, our data show that loss of FHL1 function leads to myopathy in vivo and suggest that loss of function of FHL1 may be one of the mechanisms underlying muscle dystrophy in patients with FHL1 mutations.
INTRODUCTION
Four-and-a-half LIM domain protein 1 (FHL1) is a member of the gene family encoding LIM domain containing proteins (1,2). A LIM domain is characterized by two conserved cysteine-rich zinc-finger motifs, which cooperatively bind zinc atoms to mediate protein–protein interactions. Distinct splice variants of FHL1 have been reported, namely FHL1A (also known as Slim1, KyoT1 or transcript variant 3 in the mouse), FHL1B (Slimmer, KyoT3 or transcript variant 1) and FHL1C (KyoT2 or transcript variant 4), which are composed of a half LIM followed by four, three and two full LIM domains, respectively (3–5). Differential splicing leads to the presence of a nuclear-addressing sequence and/or RPB-J-binding domain in the C terminus of FHL1B and FHL1C isoforms. The FHL1A isoform can also be transcribed from an alternative upstream start codon, producing a transcript that bears 48 additional nucleotides or 16 extra amino acids at the N terminus of the protein (transcript variant 2 in the mouse) (Fig. 1A). Previous studies demonstrated that FHL1 is ubiquitously expressed and interacts with a broad range of proteins involved in oncogenicity, cell structure, migration, differentiation and cell signaling (reviewed in 6).
Figure 1.
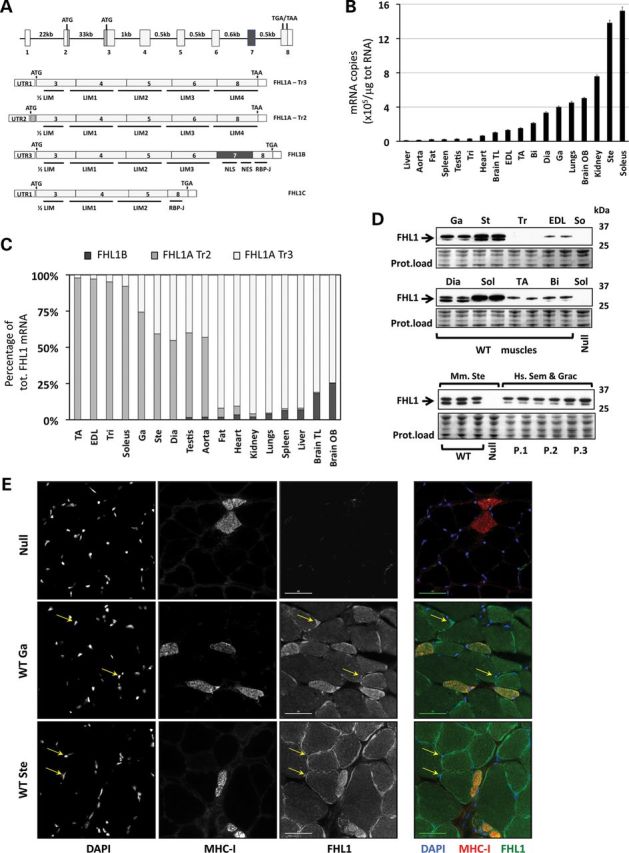
FHL1 expression in WT mice. (A) FHL1 gene structure and splicing isoforms in the mouse. The FHL1 gene is situated on the X chromosome in both humans and rodents. Gene expression leads to the production of at least four protein-coding splicing isoforms in the mouse, namely FHL1A transcript variant 3 (FHL1A-Tr3), FHL1A transcript variant 2 (FHL1A-Tr2), FHL1B and FHL1C. FHL1A-Tr3 and FHL1C share the same 5′ untranslated region (UTR1), while FHL1A-Tr2 and FHL1B have distinct 5′ untranslated regions (UTR2 and UTR3). FHL1B contains nuclear localization and exporting sequences (NLS and NES) and an RPB-J binding domain, while FHL1C contains only the RPB-J binding domain. (B) Absolute quantization of total FHL1 expression by RT-Quantitative polymerase chain reaction (qPCR) in adult mouse tissues. Bars (mean ± SEM) represent the number of mRNA copies of total FHL1 mRNA in each adult tissue. For each single experiment, amplification values were compared with a standard curve made of known concentrations of cloned FHL1 cDNA sequences (N = 4–6 mice; mean ± SEM). (C) Relative percentage of FHL1 mRNA isoform expression through various skeletal muscles and other tissues, as measured by primers recognizing different 5′ UTR regions. Of note, FHL1-Tr3 abundance could also include FHL1C levels, as both isoforms would be amplified by the same set of primers recognizing a shared UTR region and exon 3 (see Supplementary Material, Fig. S1). (D) Qualitative FHL1 protein expression analysis for various skeletal muscles (two replicates). The antibody (Aviva, against the C-terminal region of FHL1) detected two separate bands at 31–33 kDa (arrow). FHL1 expression levels for WT triceps muscles (Tri) are below detection levels after 10 s exposure time, but are revealed at longer exposures. In the lowest panel, the same antibody is used to compare FHL1 expression levels between mouse sternohyoideus (three replicates) muscle and human gracilis and semitendinosus hamstrings (three patients, in duplicate). Protein load for the 25–37 kDa ranges is shown for each western blot (Ponceau S staining). (E) Single channel and composite color pictures illustrating nuclear (DAPI), FHL1 (Aviva antibody) and MHC-I immunostaining in FHL1-null (negative control) and WT adult skeletal muscles (scale bar = 40 μm). The yellow arrows indicate FHL1 nuclear staining. Sol, soleus; Tri, triceps; Brain TL, brain temporal lobe; EDL, extensor digitorum longorum; TA, tibialis anterior; Bi, biceps; Dia, diaphragm; Ga, gastrocnemius; Brain OB, brain olfactory bulb; Ste, sternohyoideus; Mm, mus musculus; Hs, Homo sapiens.
The reported expression patterns and subcellular locations of FHL1 (sarcomeric I-band and focal adhesions for FHL1A, nucleus and cytoplasm for FHL1B and FHL1C) have suggested an important role for FHL1 in muscle fiber development, function and homeostasis. Previous studies have demonstrated a correlation between muscle growth and levels of FHL1, and have suggested that different muscle types, bearing different proportions of oxidative and glycolytic fibers, could express various levels of FHL1 protein expression (5,7–9). Most importantly, recent human genetic studies have linked FHL1 missense mutations to rare congenital myopathies, providing additional evidence of an important role for FHL1 in striated muscle development and disease: in the cases of reducing body myopathy (10–12), X-linked myopathy with postural muscle atrophy (13,14), rigid spine syndrome (15), scapuloperoneal muscular dystrophy (16) and Emery–Dreifuss muscular dystrophy (17), sporadic and inherited FHL1 missense mutations or deletions were thought to lead to loss of FHL1 protein levels and function, resulting in muscular dystrophy.
Clinical analyses of skeletal muscles from patients with FHL1 mutations also provided evidence that premature onset and severity of disease correlated with global downregulation of FHL1 protein levels (13,17), expression of truncated or mutant forms of FHL1 isoforms (17,18) and accumulation of menadione-nitro blue tetrazolium (NBT) positive ‘reducing bodies’ (10,11). Reducing bodies are thought to be aggresome-like cell inclusions that incorporate mutant and wild-type (WT) FHL1, as well as other proteins involved in cell structure, signaling and ubiquitination (19,20). It was hypothesized that these aggregates could either ‘sequester’ WT proteins away from their regular function or play a direct cytotoxic effect by impairing protein turnover and accelerating muscle wasting. Altogether, these clinical observations suggested that FHL1 mutations could confer a complex phenotype through non-exclusive mechanisms of ‘loss of function’ (reduction of FHL1 protein levels or impairment to protein partner binding) and mechanisms of ‘gain of function’ (accumulation of cytotoxic aggregates).
In the present study, we exploited an FHL1-null mouse model to evaluate the effects of FHL1 loss of function on skeletal muscle development and homeostasis. FHL1-null mice were previously engineered to disrupt FHL1 gene through a LacZ/Neo knock-in strategy, resulting in global loss of FHL1 expression in all tissues (21). Our data show that loss of FHL1 function is sufficient to cause an age-dependent myopathy, associated with myofibrillar (sarcomere) and intermyofibrillar (mitochondria and sarcoplasmic reticulum, SR) compartmental disorganization. Analyses of primary myoblasts isolated from FHL1-null muscles also demonstrates early defects in myofiber differentiation and growth in vitro that could be rescued with restoration of the FHL1A isoform. These studies along with the unique endogenous fiber-type-specific expression pattern of FHL1A in highly oxidative fibers suggest an important correlation between FHL1A and skeletal muscle disease pathogenesis in muscles bearing increased oxidative capacity and suggest that mechanisms of ‘loss of function’ could be causally related to the development of muscle dystrophy in patients with FHL1 mutations.
RESULTS
FHL1 expression and localization in WT muscles
To determine FHL1 abundance in adult mouse skeletal muscles, we first investigated FHL1 mRNA expression levels in various striated muscles isolated from 4- to 6-week-old Black Swiss mice and compared them with FHL1 expression in other organs. Quantitative polymerase chain reaction (qPCR) analysis using a set of SYBR Green-based primers recognizing all FHL1 isoforms (Supplementary Material, Fig. S1) showed ubiquitous expression of FHL1, with mRNA enrichment in muscles with high oxidative capacity (soleus, sternohyoideus) when compared with glycolytic muscles (e.g. tibialis anterior, triceps) (Fig. 1B). High levels of FHL1 were also detected in kidneys, the olfactory bulb and lungs. Western blot analysis confirmed protein expression levels, with oxidative and mixed muscles (soleus, diaphragm, gastrocnemius, sternohyoideus) expressing higher levels of FHL1 compared with highly glycolytic muscles (tibialis anterior, triceps, biceps, extensor digitorum longus) (Fig. 1D). qPCR analysis with SYBR Green-based primers detecting 5′ UTR sequences specific for FHL1 isoforms (Supplementary Material, Fig. S1) further revealed that FHL1A transcript variant 3 (i.e. Slim1, KyoT1) was the predominant transcript expressed in adult heart, lungs, kidney and liver, while adult skeletal muscles predominantly expressed the FHL1A transcript variant 2 (Fig. 1C). Western blot analysis using an antibody recognizing the C-terminal end of FHL1 also identified two distinct FHL1 protein bands at 31 and 33 kDa (arrow), correlating with the presence of FHL1A transcript variants 2 and 3 in respective muscle types. These data further suggest that the two distinct FHL1 proteins likely corresponded to FHL1A transcript variant 2 (∼33 kDa) and transcript variant 3 (∼31 kDa) isoforms. FHL1 transcriptional profiling by RNA-sequencing confirmed predominant expression of FHL1A transcript variant 2 (NM_001077362) in WT mouse skeletal muscles (soleus, tibialis anterior (TA) and diaphragm) and expression of FHL1A transcript variant 3 (NM_010211) in cardiac tissue (Supplementary Material, Fig. S3).
A comparative analysis of FHL1 protein expression between juvenile human gracilis and semitendinosus hamstrings, composed of ∼60% type-I and ∼20% type-IIa fibers (22), to mouse sternohyoideus revealed similar FHL1 protein expression patterns, with two identifiable bands at 31 and 33 kDa, suggesting relevance and conservation of both FHL1A isoforms in mouse and human skeletal muscles. In the mouse, FHL1B was expressed at low levels in all tissues, except in the brain where it constituted up to 15–20% of the total pool of FHL1 mRNA. qPCR analysis using TaqMan® Probe-based primers specific for FHL1A, FHL1B and FHL1C validated the relatively low abundance of FHL1B and FHL1C mRNA and the enrichment of FHL1A mRNA in adult mouse soleus muscle (Supplementary Material, Fig. S1).
Immunohistological analysis performed on cross sections of mouse sternohyoideus and medial gastrocnemius muscle, which is a mixed muscle largely composed of fast-twitch fibers, revealed weaker staining in larger fibers, with significant subsarcolemmal and moderately punctuated sarcoplasmic staining in medium-to-smaller fibers. Strong sarcoplasmic staining was observed in smaller oxidative type-I fibers, expressing myosin heavy chain I (MHC-I) (Fig. 1E). Cross-sectional analysis also suggested nuclear localization (yellow arrows). This fiber-specific pattern of expression supports mRNA and protein expression analyses showing increased FHL1 expression in oxidative fibers and establishes a correlation between muscle oxidative capacity and FHL1 expression levels in the mouse.
Survival rates in FHL1-null mice
To evaluate the effects of global FHL1 deficiency on animal lifespan, we performed a 21-month survival study on FHL1-null mice and their WT littermates (Fig. 2A). Our results showed that both FHL1-null hemizygote (y/−) males and homozygote (−/−) females (on 100% Black Swiss background) exhibited a decline in survival rates ∼6 and ∼12 months of age, respectively, when compared with WT (y/+, +/+) and heterozygote (+/−) littermate controls, suggesting that FHL1 is essential for survival.
Figure 2.
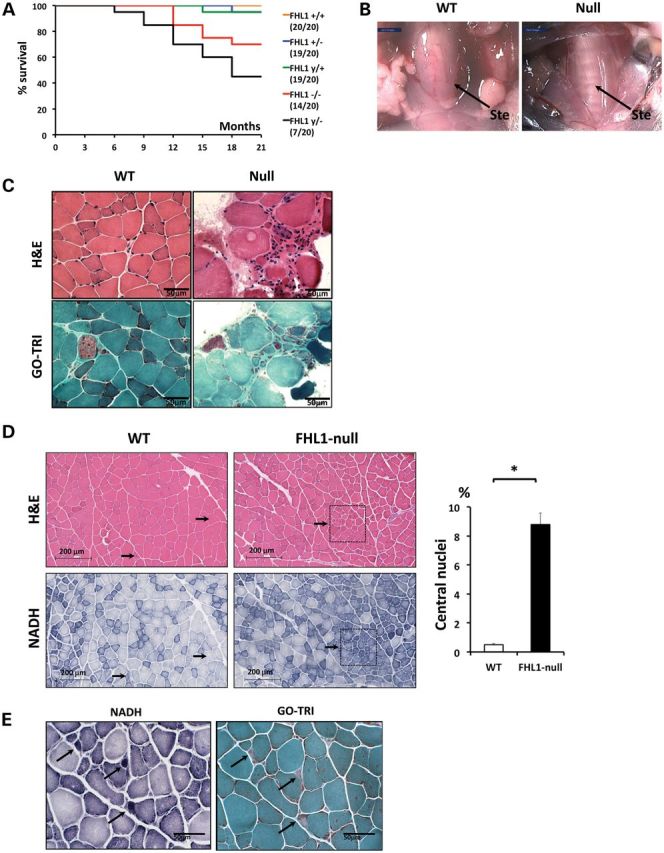
FHL1-null survival rate and muscle remodeling. (A) Survival plot of WT and FHL1-null mice. Survival expressed as percentage of the total number of animals per group surviving at observational time (in months). Plots show survival rates for WT (y/+) and hemizygote (y/−) FHL1-null males, as well as WT (+/+), heterozygote (+/−) and homozygote (−/−) FHL1-null females. In brackets are expressed, the ratios between the numbers of surviving animals at the end of the study over their initial number. (B) Surgical dissection of the supra- and infrahyoid musculature of the mouse and the detailed localization of the sternohyoideus muscle (Ste) in a WT and FHL1-null mouse. This thin muscle sheet (arrow) extends invariably from the hyoid bone and enters the tongue laterally, between the masseter and stylohyoideus muscles. In FHL1-null mice, the thinning of the sternohyoideus muscle visually exposes the trachea underneath. (C) Fresh frozen muscle cross-sections (8 µm) from the sternohyoideus muscle of 1-month-old WT and FHL1-null mice stained with hematoxylin and eosin (H&E) and modified Gomori trichrome (GO-TRI) stains. Compared with WT muscle, a marked variability in myofiber size, presence of atrophic and hypertrophic fibers, and endomysial fibrosis in areas of atrophic fibers was observed in FHL1-null mice. (D) Fresh frozen muscle sections (8 µm) from the gastrocnemius muscle of 5-month-old WT and FHL1-null mouse were stained for H&E and NADH-TR. Arrows indicate single fibers (WT) and topological regions (FHL1-null) containing centralized nuclei. Quantitation of central nucleation in WT and FHL1-null gastrocnemius: the number of muscle fibers with central nuclei is represented as % of the total number of muscle fibers analyzed (*P < 0.01; one-way ANOVA; N = 16 fields/4 animals per group). (E) Magnifications stained for NADH-TR and Gomori trichrome stain (GO-TRI) indicating punctuated areas of densely accumulated and uneven distributed mitochondria in FHL1-null muscle (black arrows).
Overt signs of muscle remodeling in FHL1-null mice
To assess whether the increased mortality in FHL1-null mice was associated with muscle structural damage, we performed histological analysis on muscle sections from 1- to 5–6-month-old FHL1-null hemizygote (y/−) versus age-matched WT littermates (y/+). Striking visual changes were observed at the level of the sternohyoideus muscle, which belongs to a group of upper airway dilator muscles (23). This muscle exhibited overt signs of muscle thinning (Fig. 2B) and dystrophy (Fig. 2C) in FHL1-null mice. This was observed with 100% penetrance at the age of 1 month. Electrophoretic separation of MHC isoforms indicated significant loss of MHC type-I (−50%), type-IIa (−30%) and type-IIb (−15%) fiber content and an increase in type-IIx content (+100%) (Supplementary Material, Fig. S4). Significant histopathological changes were also observed in 5–6-month-old distal pelvic limb muscles in FHL1-null mice. These changes included an increased percentage of oxidative fibers (staining blue/dark blue for nicotinamide adenine dinucleotide, NADH) with centralized nuclei, suggesting muscle fiber regeneration in both muscles (black arrows) (Fig. 2D). In addition, small round and dense structures staining dark blue with the nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR) reaction (black arrows) were detected in both FHL1-null TA and soleus muscles (Fig. 2E). An age-dependent fiber-type switch was observed in the glycolytic TA, but not in the oxidative soleus, as evidenced by an ∼2 fold increase in oxidative type-I fiber content, likely reflecting regeneration of oxidative fiber content in the TA, followed by an ∼8% decrease in glycolytic type-IIb fibers at the age of 5–6 months (Supplementary Material, Fig. S4). Histological analysis of the FHL1-null diaphragm also revealed muscle defects similar to FHL1-null soleus muscle and distinct from controls (data not shown).
Myofibrillar and intermyofibrillar disorganization in FHL1-null muscles
Ultrastructural analysis by transmission electron microscopy (TEM) highlighted the extensive myofibrillar and SR disarray in the sternohyoideus muscle of FHL1-null mice as early as 1 month of age (Fig. 3A). Age-dependent structural changes were also observed in pelvic limbs muscles of FHL1-null mice. They included sarcomeric Z-line streaming and regions of progressive myofibrillar degeneration in the soleus muscle (Fig. 3A), as well as SR enlargement and branching in both soleus and TA muscles, with accumulation of electron-dense SR glycogen particles in the TA (Fig. 3B). Mitochondria presenting uneven morphology was a prominent feature observed in the sternohyoideus, soleus and TA muscles (Fig. 3A and B). Altogether, these data suggest that loss of FHL1 function is sufficient to cause muscle fiber remodeling associated with myofibrillar and intermyofibrillar disorganization. These data also suggest that oxidative and glycolytic muscle types, which express different endogenous levels of FHL1, respond differently in terms of disease in the absence of FHL1.
Figure 3.

Myofibrillar and intermyofibrillar defects in FHL1-null muscles. (A) Transmission electron micrographs (TEMs) of longitudinally sectioned sternohyoideus (upper panels) and soleus (lower panels) muscles from WT and FHL1-null (null) mice. Sternohyoideus (Ste) muscle showed extensive sarcomere (S) and sarcoplasmic reticulum (SR) disarray, while soleus (Sol) muscle presented focal areas of Z-line streaming (horizontal arrow), as well as subsarcolemmal and intermyofibrillar accumulation of mitochondria (Mt), associated with areas of progressive sarcomere degeneration (asterisks). (B) TEM of longitudinally sectioned soleus (left panels) and TA (right panels) muscles from WT and FHL1-null (null) mice, indicating mitochondrial (Mt) and sarcoplasmic reticulum (SR) remodeling in FHL1-null tissues. (C) Citrate synthase (CS) enzyme activity in soleus (Sol), tibialis anterior (TA) and sternohyoideus (Ste) muscles of 1- and 5–6-month-old WT and FHL1-null (null) mice. Values presented as mean ± SEM, N = 5–8. Statistical analysis by two-way ANOVA (for genotype and age differences) showed an age- and genotype-dependent decrease in CS activity in FHL1-null soleus (#P < 0.01), as well as a genotype-dependent (but age-independent) decrease in CS activity in FHL1-null sternohyoideus (*P < 0.01).
Differential protein expression profiling in FHL1-null soleus and TA muscles
To evaluate global protein expression patterns associated with myofibrillar and intermyofibrillar skeletal muscle defects observed in FHL1-null oxidative soleus and glycolytic TA muscles, we performed a rigorous label free protein quantification using liquid chromatography–tandem mass spectrometry (LC–MS/MS) on protein extracts from 5- to 6-month-old FHL1-null soleus and TA muscles and compared them with age-matched WT controls (n = 4 for each muscle extract and each sample run two separate times). Statistical analysis using IdentiQuantXLTM software (24) indicated 90 and 72 unique mouse proteins being differentially regulated in FHL1-null soleus and TA muscles, respectively (Supplementary Material, Tables S1 and S2). In the soleus, 85/90 identified proteins were found to be downregulated, while 71/72 proteins identified in the TA were upregulated. The list of differentially expressed proteins was examined for overrepresentation of gene ontology (GO) annotation classes using the Database for Annotation, Visualization and Integrated Discovery (DAVID v6.7) tool (available at http://david.abcc.ncifcrf.gov/). Of note, biological processes (GOTERM_BP_FAT) like ‘generation of precursor metabolites and energy’, ‘oxidative reduction’, ‘electron transport chain’ and ‘ion transport’ achieved statistically significant EASE score (P < 0.001) among the regulated proteins in both FHL1-null soleus and TA muscles, independent of their expression levels. These data suggest a downregulation (soleus) or increased abundance (TA) of proteins associated with mitochondrial respiratory activity in FHL1-null muscles. Upregulation of mitochondrial protein content in FHL1-null TA muscle could reflect increased oxidative fiber content in this muscle as demonstrated by electrophoretic separation of MHC isoforms (Supplementary Material, Fig. S4). The cellular content categories ‘mitochondrion’, ‘mitochondrial part’ and ‘mitochondrial inner membrane’ (GOTERM_CC_FAT) confirmed overrepresentation and differential changes of mitochondrial organelle-related categories in these muscles (EASE score, P < 0.001).
Decreased citrate synthase activity in FHL1-null muscles
To assess whether observed mitochondrial defects in FHL1-null mice were associated with alterations in muscle aerobic capacity, citrate synthase (CS) activity measurements were performed on mitochondrial preparations isolated from 1- to 5–6-month-old FHL1-null soleus, sternohyoideus and TA muscles and compared with WT littermate controls. Our data demonstrate a decrease in CS activity in FHL1-null sternohyoideus and soleus muscles, but not in the TA, suggesting a reduction in skeletal muscle oxidative enzyme activity and mitochondrial content in muscles with higher aerobic capacity (Fig. 3C).
FHL1 localizes to mitochondria in muscle fibers
Given the prominent mitochondrial defects in FHL1-null muscles, we sought to assess whether FHL1 could play a direct role in mitochondrial remodeling. Immunohistological analysis was performed on longitudinal and transversal sections of the soleus muscle from 4- to 6-week-old WT Black Swiss mice. Our data showed that FHL1 could partially co-localize with mitochondrial protein markers within sarcomeric (transverse) and intersarcomeric (longitudinal) regions (white arrows) of soleus muscle (Fig. 4A). Cross-sectional analysis revealed significant subsarcolemmal and punctuated sarcoplasmic co-localization (white arrows). Diffused and pronounced sarcoplasmic and nuclear co-staining was also observed in smaller cells reminiscent of satellite cells surrounding mature muscle fibers (yellow arrow). We further showed through cellular sub-fractionation studies that FHL1 co-segregated in a mitochondrial-enriched fraction obtained from skeletal muscle tissue, suggesting a potential role for FHL1 as regulator of mitochondrial homeostasis and oxidative capacity (Supplementary Material, Fig. S5).
Figure 4.

FHL1 co-localizes with mitochondrial markers in vivo and in vitro. (A) Immunohistochemistry of longitudinal and transversal cryosections (8 μm) from 1-month-old WT mouse soleus muscle stained with DAPI (nucleus), FHL1 (Aviva antibody) and mitochondrial inner-membrane marker NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 9 (NDUFA9). On longitudinal sections, the white arrows indicate FHL1 and NDUFA9 co-localization at transversal and longitudinal sarcomeric regions occupied by mitochondria. On transversal sections, the same arrows indicate FHL1 and mitochondria co-localization at subsarcolemmal and punctuated regions within the sarcoplasm. (B) FHL1 and mitochondria partially co-localize (white arrows) within the sarcoplasmic regions of myoblasts (MB) and differentiating myotubes (MT) 3 days after serum withdrawal in vitro.
No reducing bodies but increased autophagic activity in FHL1-null muscles
Histological analysis of soleus, TA and sternohyoideus muscles in 5- to 6-month-old FHL1-null mice also showed negative staining for menadione-NBT reducing bodies and for congophilic inclusions (Supplementary Material, Fig. S6). Ultra structural TEM analysis further confirmed absence of electron-dense sarcoplasmic structures previously associated with FHL1-dependent reducing bodies (10,11). Instead, accumulation of intersarcomeric and juxtanuclear inclusion bodies reminiscent of autophagosomes or autolysosomes with accumulation of lipofuscin-like material was evident in soleus and TA muscles isolated from 5- to 6-month-old FHL1-null mice (Fig. 5A). To assess whether these membrane-delimited structures were associated with increased autophagy, we monitored LC3 conjugation by western blot analysis. Although lipid-conjugated LC3-II levels were comparable at baseline, a 6-h fasting protocol was sufficient to stimulate a marked increase in lipid-conjugated LC3-II levels in 5- to 6-month-old FHL1-null soleus, TA and sternohyoideus muscles when compared with age-matched WT (Fig. 5B). These data suggest that FHL1-null skeletal muscles have increased susceptibility to autophagic activity after a mild fasting protocol.
Figure 5.

Enhanced autophagic activity in FHL1-null muscles. (A) TEMs of longitudinally sectioned soleus (left panels) and TA (right panels) muscles from 5–6-month-old FHL1-null (null) mice, indicating accumulation of autophagosome- or autolysosome-like structures (arrows) around the nucleus (Nu) and at intersarcomeric regions in FHL1-null mice. These structures were not observed in WT muscles (data not shown). Mt, mitochondria; SR, sarcoplasmic reticulum; S, sarcomere. (B) Western blot analysis of LC3 in 5–6-month-old WT and FHL1-null sternohyoideus (Ste), soleus (Sol) and tibialis anterior (TA) muscles, at baseline (i.e. fed conditions) and after a 6-h fasting protocol. Statistical analysis by two-way ANOVA (for genotype and treatment) indicated that LC3-II/LC3-I ratio was significantly increased in FHL1-null muscles after 6 h fasting (#P < 0.01). Values presented as mean ± SEM, N = 3 mice per group.
Impaired force production and muscle fatigue in FHL1-null mice
To evaluate whether the morphological and molecular defects observed in FHL1-null muscles were associated with muscle dysfunction, an isometric stress analysis of the fast-twitch 5th toe extensor digitorum longus (EDL) muscle was performed in 1- and 5–6-month-old FHL1-null males and age-matched WT littermates. Our data showed an age-dependent decrease in average isometric stress (i.e. force normalized by physiological cross-sectional area, PCSA) and EDL fiber length, associated with an age-independent increase in muscle PCSA in FHL1-null mice (Fig. 6). These data demonstrate impaired performance during skeletal muscle contraction in 5–6-month-old FHL1 null, causing susceptibility to contraction-induced injury, as previously suggested (25). Similar to the glycolytic TA muscle, the EDL showed age-dependent fiber-type switch, as evidenced by an ∼2-fold increase in oxidative type-I and type-IIa fiber content, followed by an ∼10% decrease in glycolytic type-IIb fibers at the age of 5–6 months (Supplementary Material, Fig. S4). A fatigue exercise protocol performed on treadmills by 1-year-old mice showed that FHL1-null mice were prone to global muscular fatigue, by dropping out of the exercise protocol earlier (18 ± 2.1 min versus 27 ± 2.5 min, n = 4, P = 0.019) and at lower speed (0.17 ± 0.015 m/s versus 0.26 ± 0.020 m/s, n = 4, P = 0.008) when compared with WT littermates.
Figure 6.
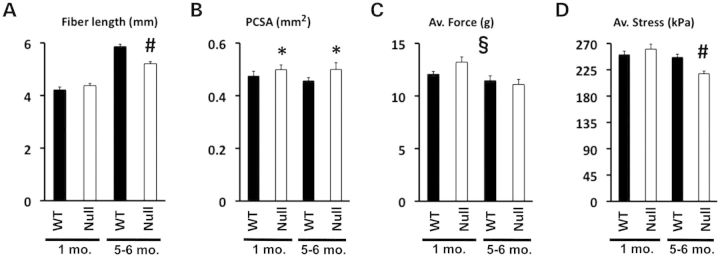
Isometric force generation in isolated 5th toes of the extensor digitorum longus (EDL). Bar graphs (mean ± SEM) represent the length (A), the PCSA (B), the average force (C) and the average isometric stress (D) of the fifth toe of isolated EDL muscles from 1- to 5–6-month-old WT and FHL1-null mice. Statistical analysis by two-way ANOVA (for differences in genotype and age) indicated an age- and genotype-dependent decrease in fiber length (#P < 0.01). The same analysis showed a genotype-dependent (but age-independent) increase in PCSA (*P < 0.01) and an age-dependent (but genotype-independent) decrease in average force generated (§P < 0.05). Finally, an age- and genotype-dependent decrease in average isometric stress was measured in FHL1-null EDL (#P < 0.01). N = 4 animals/8 muscles per group.
Loss of FHL1 function impairs myoblasts homeostasis and muscle fiber maturation in vitro
To evaluate the cellular role of FHL1 in skeletal muscle homeostasis, we isolated primary myoblasts from 4- to 5-week-old WT soleus muscles and characterized FHL1 expression during differentiation into myotubes. We showed that total FHL1 mRNA was upregulated during myotube differentiation and maturation, suggestive of a role for FHL1 in muscle development and maturation. Interestingly, upregulation of FHL1 mRNA was associated with an isoform switch from FHL1A transcript variant 3 to FHL1A transcript variant 2 as well as downregulation of FHL1B (i.e. transcript variant 1) (Fig. 7A). Similar observations were found at the protein level (Fig. 7B). Furthermore, assessment of FHL1 localization in proliferating WT myoblasts and myotubes (2–5 days old), revealed partial co-localization with mitochondrial markers, suggesting close proximity between FHL1 and mitochondrial organelles during muscle fiber maturation (Fig. 4B).
Figure 7.

FHL1 expression and role in muscle fiber differentiation in vitro. (A) Absolute quantification of FHL1 isoform expression by RT-qPCR in proliferating WT myoblasts (MB) and differentiating myotubes (MT), 5 days (5D) and 10 days (10D) after serum withdrawal (N = 4 myoblasts preparations, from four different mice per group). Bars (mean ± SEM) represent the number of mRNA copies of each FHL1 isoform, as indicated in legend of Figure 1. (B) Qualitative (left panel) and quantitative (right panel) FHL1 protein expression analyses in proliferating myoblasts (MB) and differentiating myotubes (MT), 3 days (3D) and 5 days (5D) post-serum withdrawal. The antibody (Aviva) detected two separate bands at 31–33 kDa (arrow), as previously shown in adult muscle tissue (Fig. 1). Quantification and statistical analysis by two-way ANOVA (for time-course and protein band expression) indicated a switch in FHL1 bands expression during differentiation (#P < 0.01). The 5–15 kDa range was used as a protein loading control for each western blot (Ponceau S staining). (C) Light microscopic images depicting the morphology of differentiating myoblasts 3 days post-serum withdrawal. From left to right, WT myoblasts naturally elongated and fused to form myotubes in vitro. This process was severely compromised in FHL1-null (null) myoblasts. Permanent transfection of a pCDNA-His vector containing a FHL1A transcript variant 2 (Tr2) cDNA was sufficient to rescue myotube formation in FHL1-null myoblasts. Transfection of FHL1B cDNA or the empty vector did not have the same effect in FHL1-null myoblasts. (D) Myoblast fusion assay revealed significantly lower fusion indexes in FHL1-null cells and in FHL1-null cells permanently transfected with a pCDNA-His vector containing a FHL1B cDNA or empty. Transfection of FHL1-null myoblasts with a pCDNA-His vector containing FHL1A-Tr2 was sufficient to restore fusion indexes 3 days (3D) and 5 days (5D) after serum withdrawal. Statistical analysis performed as pair-wise comparisons (one-way ANOVA) to WT condition at the same time point (*P < 0.01, N = 3–5 myoblasts preparations per group, from 3 WT and 3 FHL1-null mice). (E) Western blot detection of mitochondrially encoded NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 9 (NDUFA9) and cytochrome c oxidase I (MT-CO1) in WT and FHL1-null proliferating myoblasts (MB) and myotubes (MT), 5 days (5D) post-serum withdrawal, as well as FHL1-null myotubes expressing pCDNA-His FHL1A-Tr2 or FHL1B. For quantification, see Supplementary Material, Figure S7.
To evaluate the early consequences of loss of FHL1 on skeletal muscle development and maturation, we compared growth characteristics of FHL1-null versus WT control myoblasts from soleus muscles, following in vitro differentiation to myotubes. Loss of FHL1 function resulted in delayed muscle fiber formation and maturation, as evidenced by decreased number of forming myotubes (Fig. 7C) and decreased myoblast fusion indexes 2–5 days following serum withdrawal in FHL1-null cells (Fig. 7D). These early skeletal muscle growth defects were associated with decreased levels of selective mitochondrial electron-transfer protein markers, in both FHL1-null proliferating myoblasts and differentiating myotubes. These data suggest that loss of FHL1 impairs myoblasts homeostasis and muscle fiber formation in vitro. We further showed that restoration of the muscle-predominant isoform FHL1A transcript variant 2, but not FHL1B overexpression, was sufficient to rescue the muscle fiber maturation defects, as evidenced by the increased myoblast fusion indexes 3–5 days after serum withdrawal and re-expression of mitochondrial markers equivalent to WT controls levels (Fig. 7D and E; Supplementary Material, Figs. S7 and S8). Altogether, these data suggest that early muscle defects associated with FHL1 are causatively related to loss of FHL1A transcript variant 2 expression and its role during differentiation is necessary for muscle fiber maturation in vitro.
DISCUSSION
In this study, we provide compelling evidence that loss of FHL1 function is sufficient to induce development of a progressive myopathy associated with myofibrillar and intermyofibrillar disorganization in mice. Specifically, we demonstrate that FHL1 deficiency in vivo is associated with (i) defects in SR structure and mitochondrial content, with accumulation of intersarcomeric and juxtanuclear inclusion bodies and susceptibility to autophagy in both oxidative and glycolytic muscles; (ii) development of myofibrillar disarray and decreased aerobic activity in muscles that expressed higher oxidative capacity; (iii) decreased survival rates, associated with age-dependent impairment of muscle contractile function and lower exercise capacity and (iv) expression of FHL1A transcript variant 2 is required for proper muscle fiber differentiation and maturation in vitro.
Skeletal muscle fibers are generally categorized according to contractile and metabolic properties (26). Two major groups of fibers are the slow-contracting, highly oxidative type-I fibers and the fast-contracting type-II fibers, the latter being further divided according to metabolic/contractile properties into type-IIa (oxidative and glycolytic), type-IIx (moderately glycolytic) and type-IIb (purely glycolytic). Each of these main fiber types bears distinct profiles with regard to cross-sectional dimensions, with type-I fibers having the smallest diameter, type-IIa being intermediate in size and type-IIb being the largest. Our study confirms previous observations (7) suggesting that FHL1 is not restricted to a specific fiber type and that FHL1 expression levels are not distributed equally among all fiber types. Our expression and localization analyses showed that FHL1 levels were highest in muscles with an elevated proportion of smaller oxidative type-I/IIa fibers. That is, FHL1 levels were associated with the metabolic/oxidative capacity of the muscle rather than its contractile properties, suggestive of a potential role for FHL1 as a regulator of muscle aerobic capacity. This hypothesis was supported by our findings that FHL1 proteins co-localized and/or co-fractionated with energy-producing mitochondrial organelles in adult tissue in vivo and in forming myotubes in vitro. Moreover, FHL1 deficiency in mice caused significant mitochondrial dysmorphism and mislocalization in all muscles types, with decreased aerobic capacity and reduced expression of mitochondrial proteomic markers, such as NDUFA2, NDUFS2 and COX5A, in muscles with high oxidative capacity (Fig. 3B; Supplementary Materials, Tables S1 and S2). Finally, our in vitro data showed that both FHL1-deficient myoblasts and forming myotubes downregulated mitochondrial protein markers NDUFA9 and MT-CO1 (also known as COX1), which are important building blocks of respiratory electron chain complex I and cytochrome c oxidase subunit I. Levels of these proteins were restored by re-expressing an FHL1A isoform, suggesting that progressive skeletal muscle remodeling in FHL1-null mice could be closely related to dissipating energy stores, and that disease pathology and severity could therefore be associated with intrinsic metabolic defects directly caused by FHL1A loss of function.
Our study also revealed that FHL1 deficiency was associated with development of myofibrillar disarray in vivo and decreased myoblasts fusion and differentiation in vitro. These observations are consistent with previous reports identifying FHL1 as a necessary scaffolding protein in sarcomeric assembly and signaling cascades during muscle fiber maturation and homeostasis (8,9,21). Interestingly, muscles that presented overt signs of myofibrillar myopathy in FHL1-null mice were also the ones that expressed the highest levels of FHL1 under WT conditions (i.e. sternohyoideus and soleus). This observation suggests that severity of myofibrillar remodeling could be linked to FHL1 expression levels under physiological conditions.
Our study showed that the sternohyoideus was the most dystrophic muscle in FHL1-null mice. Consistent with its fiber-type distribution, this muscle is classified as a moderately fast-twitch muscle (27) but with high oxidative capacity, as confirmed by significant levels of type-IIa fibers content and high CS activity. Importantly, the sternohyoideus belongs to a group of upper airway dilator muscles that regulate airway patency and prevent collapse of upper airway following negative pressure generated during inspiration (28). Abnormal pharyngeal muscle function and morphology is frequently observed in clinical cases of Duchenne muscular dystrophy or oculopharyngeal muscular dystrophy where it causes upper airway occlusion, dysphagia and obstructive apneas (29,30). Experimentally, severe sternohyoideus dysfunction and remodeling were also reported in two other mouse models of muscular dystrophy, namely the dystrophin-deficient mdx mouse and the merosin-deficient dy/dy mouse model (31,32), suggesting that this phenotype is not specific to FHL1 deficiency but may also reveal that FHL1-linked mechanisms could be affected in other myopathies. The mechanisms, by which the sternohyoideus degenerates faster than limb muscles in the absence of FHL1 (or other structural proteins-like dystrophin), are unknown. One possible explanation is that chronic and regular rhythmical activity, combined with a specific fiber-type distribution (an elevated combination of type-IIa and type-IIb), could contribute to the more pronounced deleterious changes observed for this muscle in vivo.
Clinical studies of FHL1 mutations provided evidence that early onset and severity of muscle disease, especially in cases of FHL1-dependent reducing body myopathy, could correlate with progressive development and accumulation of menadione-NBT positive ‘reducing bodies’ which contains mutant FHL1 proteins. Reducing bodies are thought to be aggresome-like cell inclusions that incorporate proteins of various origins (10,11,19). In the present study, we reported that loss of FHL1 function did not cause emergence of menadione-NBT positive reducing bodies or electron-dense protein aggregates in FHL1-null muscles, thus suggesting that formation of reducing bodies in patients with FHL1 mutations is likely due to a ‘gain of function’ of mutant FHL1. Instead, we observed development and accumulation of electron-lucent juxtanuclear inclusion bodies strongly resembling membrane-limited autophagic vesicles in structure. We speculate that accumulation of these autophagic vesicles could be secondary to mechanisms of selective sequestration and subsequent degradation of dysfunctional mitochondria and other byproducts related to myofibrillar disassembly in FHL1-null muscle fibers.
Our results also highlighted a crucial role for FHL1 in mouse survival rates. The specific reasons associated for increased mortality in FHL1-null mice are not known. In this study, we presented evidence that skeletal muscle remodeling is associated with generalized muscle fatigue and age-dependent susceptibility to contraction-induced injury. Also, manifestation of lethargy and lack of movement was regularly observed in older FHL1-null mice (data not shown). Potential respiratory complications associated with impairment in sternohyoideus muscle function could explain the observed decrease in survival rates in FHL1-null mice. However, it is important to note that FHL1-null mice lack FHL1 expression in all organs, thus additional systemic effects cannot be discarded. In a previous study, we demonstrated that 8- to 10-week-old FHL1-null mice exhibited a blunted and beneficial response to pressure overload cardiac hypertrophy induced by transverse aortic constriction (21). Long-term echocardiographic assessment of 1-year-old FHL1-null mice further revealed the absence of heart dysfunction in FHL1-null mice when compared with WT littermate controls (data not shown). Thus, increased mortality due to heart pump dysfunction is unlikely, although cardiac involvement, including conduction defects, development of arrhythmogenic events and hypertrophic cardiomyopathy are observed in clinical cases of FHL1-dependent Emery–Dreifuss muscular dystrophy, X-linked myopathy with postural muscle atrophy, rigid spine syndrome and scapuloperoneal muscular dystrophy (13–16,33,34). Interestingly, we observed the development of a pelvic limb-clasping reflex that progressed to the thoracic limbs with ageing in FHL1-null mice (not shown). This reflex is generally observed in mice with brain lesions (35), suggesting possible neuropathic complications in FHL1-null mice.
In conclusion, the present study provides compelling evidence that absence of FHL1 expression is sufficient to induce development of an age-dependent myopathy associated with myofibrillar and intermyofibrillar disorganization in mice, suggesting that ‘loss of FHL1 function’ could lead to myopathy in patients with FHL1 nonsense or frame-shift mutations. However, it should be noted that the vast majority of mutations identified in patients with FHL1 mutations are missense mutations or in-frame deletions. In these cases, mutant phenotypes may result from loss-of-function alone (FHL1 downregulation and impairment of protein partner binding), or perhaps from simultaneous loss of native FHL1 function and gain of function owing to aberrant folding of mutant FHL1 proteins. Misfolded protein can result in protein aggregates/reducing body formation, which is associated with more severe disease manifestation in patients with FHL1 mutations. Production of mutant mouse models mimicking different FHL1 mutations, in addition to FHL1-null mutant mice, will provide further insights into molecular mechanisms relating to FHL1 function and dysfunction in the clinical setting.
METHODS
Generation of FHL1-null mice and human muscle biopsies
We previously reported the generation of FHL1-null mice, through a lacZ/neo knock-in strategy, resulting in loss of total FHL1 protein in all tissues (21). Experiments were conducted on ∼1 and 5–6-month-old males within a pure Black Swiss background. Survival studies were collected for both male and female mice over a period of 21 months. Littermates for our experiments were obtained by breeding WT (FHL1 +/y) Black Swiss males and heterozygote (FHL1 +/−) females, to produce 50% hemizygote (FHL1 −/y) and 50% WT (FHL1 +/y) males. For our survival study, homozygote (FHL1 −/−) and heterozygote (FHL1 +/−) females were generated by crossing hemizygote (FHL1 −/y) males with heterozygote (FHL1 +/−) females. Offspring from FHL1 intercrosses were genotyped by PCR analysis performed on DNA extracted from ear clips. Specific primers detecting FHL1 WT allele (forward, FW: 5′-ACCATGTCGGAGAAGTTCGACT-3′; reverse, RV: 5′-AGGCTGCAGGTATACACCTGAA-3′) and FHL1 mutant allele (FW: 5′-CCTCTGAGTCAGCAGCGTGATT-3′; RV: 5′-AGATGAAACGCCGAGTTAACGC-3′) were used in standard semi-quantitative PCR procedures. The UCSD Animal Care and Use Committee approved all animal procedures. The use of human skeletal muscle biopsy samples was approved under the UCSD Human Research Protections Program Project number #121700XX.
Analysis of FHL1 mRNA expression
Total RNA was isolated from excised FHL1-null and WT skeletal muscles or cell culture preparations using Trizol reagent (Life Technologies—Invitrogen) according to manufacturer's recommendations. First-strand cDNA synthesis from purified RNAs and qPCR amplification were performed using the GoTaq 2-Step RT-qPCR System kit (Promega) according to manufacturer's protocols. Resulting cDNAs were subjected to SYBR Green-based real-time amplification using a C1000 Thermal Cycler apparatus (Bio-Rad). Each PCR amplification reaction (20 µl volume) contained 100 ng of starting cDNA template and 0.5 µm of FW and RV primers. Cycling conditions for SYBR Green primers consists of an initial step at 50°C (2 min) and a first denaturizing step at 95°C (2 min), followed by 50 cycles of a thermal step protocol consisting of 95°C (20 s), 60°C (20 s) and 72°C (20 s). A standard melt curve profiling consisting of a 65–95°C thermal ramp was performed at the end of each protocol. Cycling conditions for FAM-based primers consisted of a first denaturizing step at 95°C (2 min), followed by 50 cycles of a thermal step protocol consisting of 95°C (15 s) and 60°C (1 min). Primers for FHL1 were designed using the Primer3 software according to cDNA sequences of reference (RefSeq) reported in GenBank (http://www.ncbi.nlm.nig.gov/) and confirmed by independent cloning. Amplification of total FHL1 was performed using a set of SYBR Green-based primers spanning exons 3 and 4 (primer set P0). Amplification of FHL1 splicing isoforms was performed using SYBR Green-based primers recognizing distinct 5′ UTR regions (primer sets P1, P2 and P3) or isoform-specific exon boundaries (primer sets P4, P5 and P6) (Supplementary Material, Fig. S1): P0 (FW: 5′-CAACACCTGCGTGGACTG-3′; RV: 5′-CGTGCCAGTAGCGATTCTTA-3′; 77 bp product), P1 (FW: 5′-GGTGCCGCCTAGACAGCTGC-3′; RV: 5′-CAGTCGAACTTCTCCGACAT-3′; 127 bp product), P2 (FW: 5′-CTGGGGTTGAGGGAAGACAG-3′; RV: same as P1; 123 bp product) and P3 (FW: 5′-CCTGGGGCTCCAAGGTCCCT-3′; RV: same as P1; 127 bp product). The following primers were designed to amplify with FAM-labeled TaqMan® internal probes: P4 (FW: 5′-CTGCGTGGATTGCTACAAGA-3′; probe: 5′-TGTGCTGGATGCAAGAACCCCA-3′; RV: 5′-CCTTCATAGGCCACCACACT-3′; 102 bp product); P5 (FW: same as P4; probe: 5′-TGTGCTGGATGCAAGAACCCCA-3′; RV: 5′-CACACTGGGGACTTCCTAGC-3′; 126 bp product); P6 (FW: 5′-TGACTTGCCATGAGACCAAG-3′; probe: 5′-TGCAACAAGGGTTTGGTAAAGGC-3′; RV: 5′-GCAACAAGGGTTTGGTAAAGGC-3′; 100 bp product); beta-2-microglobulin (Life Technologies, product Mm00437762_m1). Absolute quantification was calculated using the standard curve method where amplification values were compared with a standard curve made of known concentrations of cloned and linearized plasmid-based FHL1 cDNA sequences. Data reported in Result were calculated as FHL1 mRNA copies per microgram of total transcribed RNA.
Western blotting
Unless otherwise stated, proteins were extracted from tissue or cells by briefly sonicating samples suspended in custom-made RIPA buffer (1.752 g NaCl, 2 ml NP-40, 1 g deoxycholic acid, 1 ml 20% SDS, 6.67 ml 1.5 M Tris, pH 8, to 200 ml with ddH2O), followed by 4°C centrifugation (9838g, table-top centrifuge with Eppendorf FA-45-24-11 rotor) to remove membrane components. Protein samples were stored in custom-made RIPA buffer at −80°C. 20 µg of total proteins were separated using NuPAGE 4–12% Bis–Tris gels with 20× NuPAGE MES or MOPS SDS Running buffers and antioxidants, according to manufacturer's protocols (Novex—Life Technologies). After separation, proteins were transferred to nitrocellulose membrane in a Tris–Glycine buffer complemented with 20% methanol. To visualize bands and check for transfer efficiency, membranes were stained with Ponceau S. Membranes were then blocked in a buffer containing 10 mm Tris, 0.15 m NaCl, 0.1% Tween-20 and skim milk and rocked with the primary antibody (1:500 to 1:1000 dilution) overnight at 4°C. After washing procedures, membranes were incubated with peroxidase-conjugated secondary antibodies (1:1000 dilution) for 1 h at room temperature and bands identified using chemiluminescence on autoradiography film. Bands were analyzed by densitometry and values expressed as arbitrary values of protein levels, normalized by total protein transferred on nitrocellulose membrane (as visualized with Ponceau S). Primary antibodies were used as follows: rabbit FHL1 targeting the 4th LIM domain (Aviva—C-terminal region; ARP34378_T100), mouse FHL1 N-terminal region (abcam; ab58067), mouse MHC-I/slow (Sigma; M8421, clone NOQ7.5.4D), rabbit LC3B (Cell Signaling; #3868, D11), mouse NDUFA9 (abcam/MitoSciences; ab14713/MS111), mouse MT-CO1 (abcam/MitoSciences; ab14705/MS404), mouse His-tag (Invitrogen; R931-25, clone 3D5), rabbit histone H3 (Cell Signaling; #9717) and mouse sarcomeric alpha-actinin/ACTN2 (Sigma; A7811, clone EA-53).
RNA-sequencing library construction and analysis
RNA sequencing was performed as previously described with no RNA or cDNA fragmentation and without any normalization (36). RNA-sequencing libraries were constructed with random hexamer priming. To reduce biological variation, RNA was pooled from three biological replicates. Uniform amplification of the cDNA library was achieved with amplification cycles before the reaction reached saturation determined with qPCR. Following sequencing, alignment of reads was performed with Tophat (version 1.0.14) (24). Read-depth profiles were constructed with Tophat's ‘wiggles’ tool and values were normalized to total aligned reads and uploaded on the UCSC browser (http://genome.ucsc.edu/index.html).
Subcellular fractionation and CS activity
Myofibrils were isolated from mouse sternohyoideus muscle by homogenization in a glass dounce in 20 vol (w/v) of a borate–KCl buffer (pH 7.1) containing 0.039 m sodium borate, 0.025 m KCl, 1 mm phenylmethylsulfonyl fluoride. The homogenate was centrifuged at 3200 rpm for 12 min, and the supernatant was discarded. Purification of the pelleted material was continued by washing twice with 1.0% Triton X-100 and by a final suspension in 0.1 m KCl, 50 mm Tris (pH 7.4), and 1 mm dithiothreitol (DTT). Before storage, they were washed in a low-salt buffer containing 0.1 m KCl, 2 mm MgCl2, 0.01 m Tris–maleate (pH 7.0), 50% glycerol, and 1 mm DTT. All procedures were performed at 4°C. To isolate nuclei and mitochondrial fractions, muscles were homogenized in a glass dounce in buffer containing 10 mm Tris (pH 7.4), 250 mm sucrose, 1 mm EDTA, 0.15% Nonidet P-40, 10 mm sodium butyrate, 0.1 mm phenylmethylsulfonyl fluoride, protease inhibitor mixture (Roche) and phosphatase inhibitors (Roche). Subcellular fractionation was carried out by centrifugation at 600g to pellet crude nuclear fraction. The crude nuclear pellet was resuspended in homogenization buffer, layered on a 2 m sucrose pad and centrifuged at 7500g for 5 min to isolate the enriched nuclear fraction. The first supernatant from the crude nuclear pellet was centrifuged at 5000g for 30 min to yield the mitochondria, which were further purified using a 28% Percoll gradient and 14 000g spin for 40 min (mitochondria are in the pellet). All steps were carried out at 4°C. The purity of these individual fractions was evaluated by western blotting of specific fraction markers, namely histone H3 for the nuclear fraction, NDUFA9 for the mitochondrial fraction and muscle alpha-actinin for the myofibrillar fraction. Western blot analysis was also performed to test expression of FHL1 proteins in isolated subcellular fractions and in unfractionated samples. Living mitochondria preparations for CS activity measurements were obtained using the Mitochondria Isolation Kit MITOISO1 (Sigma). Activity was determined in triplicate based on the formation of 2-nitro-5-thiobenzoic acid (TNB) at a wavelength of 412 nm at 25°C on a spectrophotometer plate reader (SpectraMax 340 PC). In each well, 8 µl of sample was added to a reaction medium containing 178 µl assay buffer, 2 µl of 30 mm acetyl coenzyme A and 10 mm TNB acid. The baseline assay solution absorbance was recorded, reactions were initiated by addition of 10 µl oxaloacetic acid, and the change in absorbance measured every 15 s for 2 min.
LC–MS/MS
Snap frozen soleus and TA muscle samples were ground into a fin powder using a mortar and pestle and immersed in 8 volumes of freshly prepared lysis buffer (8 m urea, 10 mm dithiothreitol DTT solution) and vortexed until complete dissolution. Muscle homogenates were centrifuged at 15 000g for 20 min at 4°C to remove any insoluble materials and fully solubilized samples were stored at −80°C until analysis. 100 µg aliquots of protein samples were adjusted to 200 µl with 4 m urea and then reduced and alkylated by triethylphosphine and iodoethanol as previously described (37). A 150 µl aliquot of a 20 µg/ml trypsin solution was added to the sample and incubated at 35°C for 3 h, after which another 150 µl of trypsin was added, and the solution incubated at 35°C for 3 h. Exactly 20 µg of each tryptic digest sample were injected randomly as two technical replicates onto a C18 reversed-phase column (TSK gel ODS-100V, 3 µm, 1.0 × 150 mm) at a flow rate of 50 µl/min as part of the Surveyor autosampler and MS HPLC system (Thermo-Electron, Waltham, MA, USA) coupled to a Thermo-Finnigan linear ion-trap mass spectrometer. The mobile phases A, B and C were 0.1% formic acid in water, 50% ACN with 0.1% formic acid in water and 80% ACN with 0.1% formic acid in water, respectively. The gradient elution profile was as follows: 10% B (90% A) for 7 min, 10–67.1% B (90–32.9% A) for 163 min, 67.1–100% B (32.9–0% A) for 10 min and 100–50% B (0–50% C) for 10 min. Data were collected in the ‘data-dependent MS/MS’ mode with the ESI interface using normalized collision energy of 35%. Dynamic exclusion settings were set to repeat count 1, repeat duration 30 s, exclusion duration 120 s and exclusion mass width 0.6 m/z (low) and 1.6 m/z (high). Acquired data were searched against the most recent UniProt protein sequence database of mouse using SEQUEST (v.28 rev.12) algorithms in Bioworks (v.3.3). General parameters were set to: mass type set as ‘monoisotopic precursor and fragments’, enzyme set as ‘trypsin (KR)’, enzyme limits set as ‘fully enzymatic—cleaves at both ends’, missed cleavage sites set at 2, peptide tolerance 2.0 amu, fragment ion tolerance 1.0 amu, fixed modification set as +44 Da on Cysteine and no variable modifications used. The searched peptides and proteins were validated by PeptideProphet and ProteinProphet in the Trans-Proteomic Pipeline (v.3.3.0) (http://tools.proteomecenter.org/software.php). Only proteins with probability ≥0.9 and peptides with probability ≥0.8 were used for quantitation, which was performed using IdentiQuantXL™ software as previously described (38). One-way analysis of variance (ANOVA) was performed to determine the significance of differences between the two groups (WT versus FHL1 null) for each muscle type. A P-value ≤ 0.05 was considered for significantly different protein expression between groups.
Measurement of fiber-type distributions
The distribution of fiber types was determined by separating MHC isoforms using SDS–PAGE as previously described (39). Muscles were homogenized and centrifuged, and the myofibril-rich pellets were washed and resuspended in buffers supplemented with protease inhibitor cocktail (Roche). Protein was then diluted in sample buffer (2% SDS, 80 mm Tris, 10% glycerol and 1.2% (wt/vol) bromophenol blue) containing 100 mm DTT as a reducing agent to a concentration of 0.125 mg/ml proteins and boiled for 5 min. Isoforms were separated on polyacrylamide gels (16 × 22 cm, thickness = 0.75 mm) for 22 h at 275 V at 4°C. Stacking and resolving gels were 4 and 8% polyacrylamide, respectively. Gels were silver-stained according to the manufacturer's protocol (SilverQuest Staining Kit, Invitrogen). Densitometry was performed to quantify bands and compute isoforms distribution. Data in Results were expressed as percentage (%) of the total amount of MHC detected.
Histology and immunohistochemistry
Excised muscle tissues were snap frozen in isopentane precooled in liquid nitrogen and stored at −80°C until further analysis. Transversely or longitudinally oriented sections (8 µm) were cut at mid point and stained according to published protocols and procedures (40). Standard histochemical stains included hematoxylin and eosin (H&E), NADH-TR, Congo-red, Gomori trichrome (Go-Tri) and Menadione-NBT staining. Whole histological sections were imaged and recorded with a digital slide scanner Hamamatsu NanoZoomer 2.0-HT and analyzed with the ImageJ software package. H&E transversal sections were assessed for the number of fibers containing central nuclei over the total number of fibers counted per field images taken at 15× magnification. On average, ∼300 fibers were counted per field (two separated fields per section, one section per limb, four animals per group;, i.e. N = 16 fields per group). Immunodetection was performed on 8 µm cryosections fixed in −20°C acetone for 10 min and blocked in serum for at least 8 h at 4°C. Primary antibodies were applied overnight at 4°C (1:100–1:200 dilutions). Sections were then stained with secondary antibodies at room temperature for 1 h (Invitrogen; 1:100–1:300 dilution). Sections requiring fluorescent detection were visualized under an epifluorescent microscope (Olympus IX70) and deconvolved using the SoftworX analysis package.
Ultrastructural analysis (TEM)
Muscle segments were first fixed in 4% paraformaldehyde in 0.1 m sodium cacodylate (SC) buffer (pH 7.3) and then moved into 2% glutaraldehyde in 0.1 m SC buffer for 24 h and rinsed three times in 0.1 m SC buffer. Muscle segments were post-fixed and stained with 1% osmium tetroxide (OsO4) and 1.5% potassium ferrocyanide (K4Fe(CN)6) in 0.1 m SC buffer for 90 min at 4°C. The use of reduced osmium tetroxide-containing potassium ferrocyanide favored a high electron density of glycogen particles (41). After postfixation, samples were rinsed three times in 0.1 m SC buffer at 4°C followed by a final rinse in ddH2O and dehydrated through graded series of alcohol at 4–20°C. Samples were then immersed two times 10 min in acetone, followed by a 50:50 mixture of acetone and epoxy resin Durcupan (Sigma-Aldrich) for 2 h on shaker and finally in a 100% epoxy resin Durcupan solution changed every 24 h for up to 3 days on shaker. Samples were sectioned at 60 nm on a Leica UCT ultramicrotome, and picked up on Formvar and carbon-coated copper grids. Sections were stained with 2% uranyl acetate for 5 min and Sato's lead stain for 1 min. Grids were viewed using a JEOL 1200EX II (JEOL, Peabody, MA, USA) transmission electron microscope and photographed using a Gatan digital camera (Gatan, Pleasanton, CA, USA) at different magnifications.
Isometric stress measurements
The model used for biomechanical testing was the 5th toe belly of the multibellied EDL. This belly was selected for its fiber length homogeneity, fiber-type heterogeneity and robust origin and insertion tendons (42). After mouse sacrifice, hindlimbs were transected at the proximal femur, skinned and immersed in ice-cold mammalian Ringer solution. The 5th toe belly of the EDL was excised and transferred to a custom-made muscle-testing apparatus (42). The distal tendon was tied down with silk sutures to a rigid post interfaced with dual-mode ergometer (Aurora Scientific, Aurora, ON, Canada). The proximal tendon was secured, also with silk sutures, to a rigid frame attached to a horizontally adjustable platform. Muscle length (Lm) was adjusted such that mechanical slack was eliminated, and laser diffraction verified slack sarcomere length (∼3.0 µm). Lm was then measured through a dissecting microscope fitted with an eyepiece crosshair reticule, by using a digital micrometer to translate the chamber under the field of view from the proximal EDL origin to the distal myotendinous junction. An electrical stimulator (FHC, Bowdoinham, ME, USA) provided muscle activation via platinum plate electrodes extending across the entire muscle. Muscle twitches were imposed at successively higher stimulation voltages, beginning with 5 V, until maximum twitch force was achieved (typically at ∼8 V). Voltage was then doubled to guarantee recruitment of all fibers. Maximum isometric force was imposed by applying a 400 ms train of 0.3 ms pulses delivered at 100 Hz while maintaining constant muscle length. A computer algorithm in LabVIEW (National Instruments, Austin, TX, USA) was used to trigger the stimulator, acquire signals from the force transducer and analyze all force-time records. Three contractions were imposed, each spaced 2 min apart and successive isometric force values were averaged. After testing, the muscle was removed from the chamber and weighed. To determine the longitudinal stress within the muscle, force was normalized to PCSA, an anatomical parameter whose value is directly proportional to force-generating capacity. Fiber length (Lf in mm) was determined for each muscle by multiplying Lm by the characteristic 5th toe EDL Lf:Lm ratio of 0.69. Muscle mass (M in mg), muscle density (ρ = 1.056 g/cm3), fiber pennation angle (θ = 11.3°), and Lf were used to compute PCSA (in mm2), which is equal to (M cos θ)/(ρLf). The constants in this formula have been published previously (43).
Mouse exercise protocol
Four pairs of 1-year-old FHL1-null and WT male littermates were subjected to a single round of treadmill running protocol (Model: exer-6M Treadmill with Treadmill Shock Detection Unit). Mice were placed in their respective lanes and allowed to adjust to the surroundings for 15 min. During the adjustment period, the treadmill was set to 0.09 m/s. The treadmill incline was set to 25° at all times. Every 2 min, the speed of the treadmill was ramped up in 0.03 m/s steps from the initial 0.09 m/s. Mice were left running until exhaustion, defined as inability to continue running despite a mild electrical stimulation provided by an electro shocking device.
Satellite cell isolation, culture, transfection and differentiation
Satellite cell-derived primary myoblasts were isolated from soleus muscles by dissection and sequential digestions in Collagenase B – Dispase II solution (Roche) at 37°C, as previously published (44). Myoblasts were separated from non-myogenic cells by two rounds of differential plating of 1 h each. Enriched myoblast populations were plated at a density of 0.1 × 106 cells/cm2 on gelatin-coated wells and cultured in complete medium containing Ham's F-10 (Cellgro) supplemented with l-glutamine, 20% FBS (Life Technologies), 10 mg/ml streptomycin, 100 units/ml penicillin (GIBCO) and 2.5 ng/ml bFGF (Life Technologies). Proliferating myoblasts were grown to ∼70% confluency and further purified by one additional round of differential plating. Cells were passaged at least two to three times before being used for experimental procedures. Differentiation of myoblasts was achieved by replacing the high-serum medium with one containing DMED (Cellgro) supplemented with l-glutamine, 5% horse serum (Life Technologies), 10 mg/ml streptomycin and 100 units/ml penicillin (GIBCO). Myoblast transfections were performed with the pcDNA6/V5-His vector (Invitrogen), containing ampicillin and blasticidin selection cassettes. In this vector, FHL1 constructs were conjugated to a V5-His sequence tag at the cDNA 3′ end. TransIT-2020 transfection reagent (Mirus) was used to transfect the pCDNA constructs to proliferating myoblasts in their ∼50% confluency, according to manufacturer's recommendations. Twenty-four hours post-transfection, myoblasts underwent blasticidin-mediated selection. After 1 week of blasticidin exposure, surviving myoblasts colonies were tested for FHL1 isoform expression.
Measurements of myoblast fusion indexes
The fusion index was defined as the total number of nuclei (excluding mono- and binucleated) in alpha-actinin positive cells divided by the total number of nuclei per field. The number of nuclei per myotube was established by counting nuclei within every myotube per microscopic field divided by the number of myotubes in the field, where a myotube is defined as an alpha-actinin positive cell with at least three nuclei.
STATISTICS
Results were expressed as mean ± SEM unless otherwise indicated; one-way and two-way ANOVAs were applied to test for differences (P < 0.05) between age-matched FHL1-null and WT groups. When statistical significance was achieved, post hoc analysis using Tukey's t-test with the Bonferroni correction was applied.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
J.C. was funded by grants from the National Institute of Arthritis and Musculoskeletal and Skin (R21AR061024 and R01AR059334). R.L.L. was funded by an R24 Center Grant (R24 HD050837). A.K.P. was supported by a Cardiovascular Physiology and Pharmacology Training Grant (5T32HL007444-27), I.B. was supported by an American Heart Association postdoctoral fellowship (12POST12030256) and D.S.G. was supported by a Development Grant from the Muscular Dystrophy Association.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Hank Chambers, from the Rady Children's Hospital in San Diego for providing human skeletal muscle biopsies for this study. We are grateful to Mrs Ying Jones and Mr Timo Meerloo from the Electron Microscopy Facility at the UCSD, for invaluable assistance with preparation and analysis of TEM sections. We acknowledge Dr Steve DePalma for his significant contribution with the RNA-sequencing experiments. In addition, we recognize the Sharon Tamor Hansen Imaging and Physiology Laboratory for assistance with fluorescent microscopy and deconvolution analysis.
REFERENCES
- 1.Morgan M.J., Madgwick A.J., Charleston B., Pell J.M., Loughna P.T. The developmental regulation of a novel muscle LIM-protein. Biochem. Biophys. Res. Commun. 1995;212:840–846. doi: 10.1006/bbrc.1995.2045. [DOI] [PubMed] [Google Scholar]
- 2.Morgan M.J., Madgwick A.J. Slim defines a novel family of LIM-proteins expressed in skeletal muscle. Biochem. Biophys. Res. Commun. 1996;225:632–638. doi: 10.1006/bbrc.1996.1222. [DOI] [PubMed] [Google Scholar]
- 3.Taniguchi Y., Furukawa T., Tun T., Han H., Honjo T. LIM protein KyoT2 negatively regulates transcription by association with the RBPJ DNA binding protein. Mol. Cell. Biol. 1998;18:644–654. doi: 10.1128/mcb.18.1.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown S., McGrath M.J., Ooms L.M., Gurung R., Maimone M.M., Mitchell C.A. Characterization of two isoforms of the skeletal muscle LIM protein 1, SLIM1. Localization of SLIM1 at focal adhesions and the isoform slimmer in the nucleus of myoblasts and cytoplasm of myotubes suggests distinct roles in the cytoskeleton and in nuclear-cytoplasmic communication. J. Biol. Chem. 1999;274:27083–27091. doi: 10.1074/jbc.274.38.27083. [DOI] [PubMed] [Google Scholar]
- 5.Chu P.H., Ruiz-Lozano P., Zhou Q., Cai C., Chen J. Expression patterns of FHL/SLIM family members suggest important functional roles in skeletal muscle and cardiovascular system. Mech. Dev. 2000;95:259–265. doi: 10.1016/s0925-4773(00)00341-5. [DOI] [PubMed] [Google Scholar]
- 6.Shathasivam T., Kislinger T., Gramolini A.O. Genes, proteins and complexes: the multifaceted nature of FHL family proteins in diverse tissues. J. Cell. Mol. Med. 2010;14:2702–2720. doi: 10.1111/j.1582-4934.2010.01176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loughna P.T., Mason P., Bayol S., Brownson C. The LIM-domain protein FHL1 (SLIM 1) exhibits functional regulation in skeletal muscle. Mol. Cell Biol. Res. Commun. 2000;3:136–140. doi: 10.1006/mcbr.2000.0206. [DOI] [PubMed] [Google Scholar]
- 8.McGrath M.J., Cottle D.L., Nguyen M.A., Dyson J.M., Coghill I.D., Robinson P.A., Holdsworth M., Cowling B.S., Hardeman E.C., Mitchell C.A., et al. Four and a half LIM protein 1 binds myosin-binding protein C and regulates myosin filament formation and sarcomere assembly. J. Biol. Chem. 2006;281:7666–7683. doi: 10.1074/jbc.M512552200. [DOI] [PubMed] [Google Scholar]
- 9.Cowling B.S., McGrath M.J., Nguyen M.A., Cottle D.L., Kee A.J., Brown S., Schessl J., Zou Y., Joya J., Bönnemann C.G., et al. Identification of FHL1 as a regulator of skeletal muscle mass: implications for human myopathy. J. Cell Biol. 2008;183:1033–1048. doi: 10.1083/jcb.200804077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schessl J., Zou Y., McGrath M.J., Cowling B.S., Maiti B., Chin S.S., Sewry C., Battini R., Hu Y., Cottle D.L., et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J. Clin. Invest. 2008;118:904–912. doi: 10.1172/JCI34450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schessl J., Taratuto A.L., Sewry C., Battini R., Chin S.S., Maiti B., Dubrovsky A.L., Erro M.G., Espada G., Robertella M., et al. Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. Brain. 2009;132:452–464. doi: 10.1093/brain/awn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shalaby S., Hayashi Y.K., Nonaka I., Noguchi S., Nishino I. Novel FHL1 mutations in fatal and benign reducing body myopathy. Neurology. 2009;72:375–376. doi: 10.1212/01.wnl.0000341311.84347.a0. [DOI] [PubMed] [Google Scholar]
- 13.Windpassinger C., Schoser B., Straub V., Hochmeister S., Noor A., Lohberger B., Farra N., Petek E., Schwarzbraun T., Ofner L., et al. An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. Am. J. Hum. Genet. 2008;82:88–99. doi: 10.1016/j.ajhg.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schoser B., Goebel H.H., Janisch I., Quasthoff S., Rother J., Bergmann M., Müller-Felber W., Windpassinger C. Consequences of mutations within the C terminus of the FHL1 gene. Neurology. 2009;73:543–551. doi: 10.1212/WNL.0b013e3181b2a4b3. [DOI] [PubMed] [Google Scholar]
- 15.Shalaby S., Hayashi Y.K., Goto K., Ogawa M., Nonaka I., Noguchi S., Nishino I. Rigid spine syndrome caused by a novel mutation in four-and-a-half LIM domain 1 gene (FHL1) Neuromuscul. Disord. 2008;18:959–961. doi: 10.1016/j.nmd.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 16.Quinzii C.M., Vu T.H., Min K.C., Tanji K., Barral S., Grewal R.P., Kattah A., Camaño P., Otaegui D., Kunimatsu T., et al. X-linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-and-a-half-LIM protein 1. Am. J. Hum. Genet. 2008;82:208–213. doi: 10.1016/j.ajhg.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gueneau L., Bertrand A.T., Jais J.P., Salih M.A., Stojkovic T., Wehnert M., Hoeltzenbein M., Spuler S., Saitoh S., Verschueren A., et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 2009;85:338–353. doi: 10.1016/j.ajhg.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tiffin H.R., Jenkins Z.A., Gray M.J., Cameron-Christie S.R., Eaton J., Aftimos S., Markie D., Robertson S.P. Dysregulation of FHL1 spliceforms due to an indel mutation produces an Emery-Dreifuss muscular dystrophy plus phenotype. Neurogenetics. 2013;14:113–121. doi: 10.1007/s10048-013-0359-8. [DOI] [PubMed] [Google Scholar]
- 19.Feldkirchner S., Walter M.C., Müller S., Kubny C., Krause S., Kress W., Hanisch F.G., Schoser B., Schessl J. Proteomic characterization of aggregate components in an intrafamilial variable FHL1-associated myopathy. Neuromuscul. Disord. 2013;23:418–426. doi: 10.1016/j.nmd.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 20.Liewluck T., Hayashi Y.K., Ohsawa M., Kurokawa R., Fujita M., Noguchi S., Nonaka I., Nishino I. Unfolded protein response and aggresome formation in hereditary reducing-body myopathy. Muscle Nerve. 2007;35:322–326. doi: 10.1002/mus.20691. [DOI] [PubMed] [Google Scholar]
- 21.Sheikh F., Raskin A., Chu P.H., Lange S., Domenighetti A.A., Zheng M., Liang X., Zhang T., Yajima T., Gu Y., et al. An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J. Clin. Invest. 2008;118:3870–3880. doi: 10.1172/JCI34472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tirrell T.F., Cook M.S., Carr J.A., Lin E., Ward S.R., Lieber R.L. Human skeletal muscle biochemical diversity. J. Exp. Biol. 2012;215:2551–2559. doi: 10.1242/jeb.069385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greene E. Anatomy of the Rat. New York: Hafner Publishing Co.; 1935. 1955. [Google Scholar]
- 24.Trapnell C., Pachter L., Salzberg S.L. Tophat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramaswamy K.S., Palmer M.L., van der Meulen J.H., Renoux A., Kostrominova T.Y., Michele D.E., Faulkner J.A. Lateral transmission of force is impaired in skeletal muscles of dystrophic mice and very old rats. J. Physiol. 2011;589:1195–1208. doi: 10.1113/jphysiol.2010.201921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scott W., Stevens J., Binder-Macleod S.A. Human skeletal muscle fiber type classifications. Phys. Ther. 2001;81:1810–1816. [PubMed] [Google Scholar]
- 27.McGuire M., Dumbleton M., MacDermott M., Bradford A. Contractile and electrical properties of sternohyoid muscle in streptozotocin diabetic rats. Clin. Exp. Pharmacol. Physiol. 2001;28:184–187. doi: 10.1046/j.1440-1681.2001.03433.x. [DOI] [PubMed] [Google Scholar]
- 28.Remmers J.E., deGroot W.J., Sauerland E.K., Anch A.M. Pathogenesis of upper airway occlusion during sleep. J. Appl. Physiol. 1978;44:931–938. doi: 10.1152/jappl.1978.44.6.931. [DOI] [PubMed] [Google Scholar]
- 29.Khan Y., Heckmatt J.Z. Obstructive apnoeas in Duchenne muscular dystrophy. Thorax. 1994;49:157–161. doi: 10.1136/thx.49.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dedrick D.L., Brown L.K. Obstructive sleep apnea syndrome complicating oculopharyngeal muscular dystrophy. Chest. 2004;125:334–336. doi: 10.1378/chest.125.1.334. [DOI] [PubMed] [Google Scholar]
- 31.Attal P., Lambert F., Marchand-Adam S., Bobin S., Pourny J.C., Chemla D., Lecarpentier Y., Coirault C. Severe mechanical dysfunction in pharyngeal muscle from adult mdx mice. Am. J. Respir. Crit. Care Med. 2000;162:278–281. doi: 10.1164/ajrccm.162.1.9905063-r2. [DOI] [PubMed] [Google Scholar]
- 32.van Lunteren E., Moyer M. Sternohyoid muscle fatigue properties of dy/dy dystrophic mice, an animal model of merosin-deficient congenital muscular dystrophy. Pediatr. Res. 2003;54:547–553. doi: 10.1203/01.PDR.0000081762.51546.16. [DOI] [PubMed] [Google Scholar]
- 33.Knoblauch H., Geier C., Adams S., Budde B., Rudolph A., Zacharias U., Schulz-Menger J., Spuler A., Yaou R.B., Nürnberg P., et al. Contractures and hypertrophic cardiomyopathy in a novel FHL1 mutation. Ann. Neurol. 2010;67:136–140. doi: 10.1002/ana.21839. [DOI] [PubMed] [Google Scholar]
- 34.Friedrich F.W., Wilding B.R., Reischmann S., Crocini C., Lang P., Charron P., Müller O.J., McGrath M.J., Vollert I., Hansen A., et al. Evidence for FHL1 as a novel disease gene for isolated hypertrophic cardiomyopathy. Hum. Mol. Genet. 2012;21:3237–3254. doi: 10.1093/hmg/dds157. [DOI] [PubMed] [Google Scholar]
- 35.Lalonde R., Strazielle C. Brain regions and genes affecting limb-clasping responses. Brain Res. Rev. 2011;67:252–259. doi: 10.1016/j.brainresrev.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 36.Christodoulou D.C., Gorham J.M., Herman D.S., Seidman J.G. Construction of normalized RNA-seq libraries for next-generation sequencing using the crab duplex-specific nuclease. Curr. Protoc. Mol. Biol. 2011 doi: 10.1002/0471142727.mb0412s94. Chapter 4, Unit 4.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lai X., Bacallao R.L., Blazer-Yost B.L., Hong D., Mason S.B., Witzmann F.A. Characterization of the renal cyst fluid proteome in autosomal dominant polycystic kidney disease (ADPKD) patients. Proteomics Clin. Appl. 2008;2:1140–1152. doi: 10.1002/prca.200780140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lai X., Wang L., Tang H., Witzmann F.A. A novel alignment method and multiple filters for exclusion of unqualified peptides to enhance label-free quantification using peptide intensity in LC-MS/MS. J. Proteome Res. 2011;10:4799–4812. doi: 10.1021/pr2005633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Talmadge R.J., Roy R.R. Electrophoretic separation of rat skeletal muscle myosin heavy-chain isoforms. J. Appl. Physiol. 1993;75:2337–2340. doi: 10.1152/jappl.1993.75.5.2337. [DOI] [PubMed] [Google Scholar]
- 40.Dubowitz V., Sewry C.A. Muscle Biopsy: A Practical Approach. 3rd edn. London: Saunders Elsevier; 2007. pp. 21–39. [Google Scholar]
- 41.Ørtenblad N., Nielsen J., Saltin B., Holmberg H.C. Role of glycogen availability in sarcoplasmic reticulum Ca2+ kinetics in human skeletal muscle. J. Physiol. 2011;589:711–725. doi: 10.1113/jphysiol.2010.195982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chleboun G.S., Patel T.J., Lieber R.L. Skeletal muscle architecture and fiber-type distribution with the multiple bellies of the mouse extensor digitorum longus muscle. Acta Anat. (Basel) 1997;159:147–155. doi: 10.1159/000147977. [DOI] [PubMed] [Google Scholar]
- 43.Sam M., Shah S., Fridén J., Milner D.J., Capetanaki Y., Lieber R.L. Desmin knockout muscles generate lower stress and are less vulnerable to injury compared with wild type muscles. Am. J. Physiol. Cell. Physiol. 2000;279:C1116–C1122. doi: 10.1152/ajpcell.2000.279.4.C1116. [DOI] [PubMed] [Google Scholar]
- 44.Rando T.A., Blau H.M. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J. Cell. Biol. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.