

Abstract
Dengue has become a major global health threat, especially in tropical and subtropical regions. The development of antiviral agent targeting viral replication is really needed at this time. NS5 methyltransferase presents as a novel antiviral target. This enzyme plays an important role in the methylation of 5'-cap mRNA. Inhibition of the NS5 methyltransferase could inhibit dengue virus replication. In this research, two sites of NS5 methyltransferase (S-Adenosyl methionine/SAM binding site and RNA-cap site) were used as targets for inhibition. As much as 300 commercial cyclic peptides were screened to these target sites by means of molecular docking. Analysis of ligand-enzyme binding free energy and pharmacological prediction revealed two best ligands, namely [Tyr123] Prepro Endothelin (110-130), amide, human and Urotensin II, human. According to molecular dynamic simulation, both ligands maintain a stable complex conformation between enzyme and ligand at temperature 310 K and 312 K. Hence, Urotensin II, human is more reactive at 312 K than at 310 K. However, both ligands can be used as potential inhibitor candidates against NS5 methyltransferase of dengue virus with Urotensin II, human exposes more promising activity at 312 K.
Keywords: Dengue virus, NS5 methyltransferase, commercial cyclic peptides, molecular dynamics
Background
Dengue virus infection has become a very important global health problem, especially in tropical and subtropical countries. This disease is endemic in more than 100 countries and nearly half of the world's populations are at risk of infection. As much as 50-100 million cases of dengue fever occur every year, including 500,000 cases of dengue haemorrhagic fever that lead to hospitalization even death, mainly among children [1].
The agents that cause these infections are four serotypes of dengue virus i.e., DENV-1, DENV-2, DENV-3, and DENV-4. These serotypes differ in their antigenic complexes. People whom infected with one dengue serotype do not automatically acquire cross-protective immunity to other serotypes of dengue. The presence of these four serotypes of DENV has made the development of effective DENV vaccine become challenging [2]. Current methods of treatment are limited to support medical care and symptomatic treatment. Hence, new treatments to control the severity of dengue infection, such as antiviral agents, are obligatory.
The structural and non-structural proteins of DENV have become the major target of antiviral design. The structural proteins (capsid (C), premembrane (prM) and envelope (E)) play vital roles in viral formation and life cycle. While DENV non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) involve in genome replication, virion assembly and avoiding innate immune responses. Among DENV nonstructural proteins, NS5 is the largest (900 amino acid residues) and the most conserved protein in DENV (67% amino acids sequence identity among dengue serotypes) [3]. NS5 has also been an attractive target for antiviral development, as it is required for RNA capping and DENV genome replication. The NS5 protein consists of two domains: a methyltransferase (MTase) domain at residues 1-296 of its N-terminal region and an RNA-dependent RNA polymerase (RdRp) domain at residues 320-900 of its C-terminal region. NS5 MTase exhibits two methylation activities: guanine N-7 and nucleoside 2'-Omethylation, with guanine N-7 occurs before nucleoside 2'-Omethylation. Both of these activities depend on S-adenosyl-Lmethionine (SAM) as methyl donor for methylation process. After each of methylation process, S-adenosyl-L-homocysteine (SAH) is generated as a by-product [4]. A study suggested that N-7 methylation is critical for viral life cycle, while failure to perform both methylation processes is lethal to flavivirus [5]. Therefore, NS5 MTase might represent as an ideal target for dengue virus therapy. NS5 MTase has two binding sites: the SAM binding site, which lies in the same site for SAH, and RNA cap site, which is also a GTP- and GTP analogues-binding site. In this research, both of these sites became targets of antiviral design.
Currently, the use of peptide as drug becomes a very promising strategy to develop antiviral drug. Peptide-based drugs have several advantages including higher bioactivity, high specificity to their target, low interaction with other drugs and low toxicity [6]. Peptide molecule showed antiviral activity against Avian influenza virus subtype H9N2 [7] and subtype H5N1 [8]. Estimated market for peptide-based drugs is over $40 billion annually [6]. In this research, we screened 300 commercial cyclic peptides that are currently available on the market against two binding sites of NS5 methyltransferase in order to develop new hit inhibitors against dengue virus.
Methodology
Preparation of commercial cyclic peptides as ligands:
Commercial cyclic peptides were sought online through the database of peptide-seller chemical companies like Genesis Peptides, Bachem, Selleck Chem, and American Peptide. Then the structures of these peptides were drawn using ChemSketch ACDLabs. The ligands were imported into MOE database viewer and saved in .mdb format using the software MOE 2009.10. Ligands optimisation and energy minimisation were conducted in order to adjust the structure of ligands and the position of hydrogen atoms. AMBER94 forcefield was used in the process of geometry optimisation. Energy minimisation was performed by choosing the value 0.001 kkal/Ǻ mol for RMS gradient.
Preparation of NS5 Methyltransferase as target protein:
Amino acid sequences of DENV NS5 methyltranferase were obtained in FASTA format from the National Center of Biotechnology Information (NCBI) database, while 3D structure of NS5 MTase was downloaded from the Research Collaboratory for Structural Bioinformatics (RSCB) database. Protein structure with PDB code 2P41 was chosen as target protein. The protein structure was adjusted and optimised through sequential steps of protonate 3D, partial charge and energy minimization that are available in MOE. Forcefield AMBER94 was also used in the process of energy minimization of protein, but the selected value of RMS gradient for protein is 0.05 kkal/Ǻ mol.
Molecular Docking:
The docking process was performed using software MOE 2009.10. Triangle matcher was assigned as placement method. Scoring function used in this process was London dG that ranked 100 best poses of ligand-protein complex. Refinement was conducted based on forcefield parameter. Only one best pose was taken out of 100 poses. The complexes with the lowest ΔGbinding value were visualized using LigX-interaction option in MOE to expose their contact residues.
ADME and Toxicity Prediction of Cyclic Peptides:
Prediction of ADME (Absorption, Distribution, Metabolism and Excretion) character of cyclic peptides as drug candidates was performed using online software, ACD I-Labs/Percepta (http://ilab.acdlabs.com/iLab2/index.php). Toxicity analysis was performed using offline software Toxtree v-2.5.0 based on Benigni/Bossa rulebase for mutagenicity and carcinogenicity [9].
Molecular Dynamics Simulation of Ligand-Protein Complex:
Molecular dynamics simulation comprises of three stages: initialisation, equilibration and production. Before conducting those steps, the complex must be prepared by adjusting forcefield parameter into AMBER94 and solvation system into born solvation. Molecular dynamics simulation was performed towards the best ligands obtained from molecular docking and ADME-Toxicity prediction steps. This simulation was also done using MOE 2009.10.
Discussion
Screening of commercially available cyclic peptides was performed to find potential inhibitors against two binding sites of NS5 methyltransferase (SAM site and RNA-cap site). Through online searching, 300 commercial cyclic peptides were found and used as ligands to target NS5 MTase. All of these ligands, along with the standards, were docked into SAM site and RNA-cap site of NS5 MTase using MOE software. Amino acid residues at the binding site of SAM are Ser 56, Lys 61, Cys 82, Gly 86, Trp 87, Thr 104, Lys 105, Asp 131, Val 132, Phe 133, Asp 146, Ile 147, Lys 181 and Glu 217 [10]. Meanwhile, amino acid residues with significant role at RNA-cap site are Lys 14, Leu 17, Asn 18, Leu 20, Phe 25, Lys 29, Ser 150 and Ser 151 [11].
Standard molecules used at SAM-site were SAM, SAH and TWY. SAM (S-adenosyl-L-methionine) is natural substrate of this site, while SAH (S-adenosyl-L-homocysteine) is a byproduct and an analogue of SAM that was used in [10] as inhibitor of SAM site. Standard molecules used at RNA-cap site were RTP and YEF. RTP (Ribavirin Triphosphate) is also an analogue of natural substrate of this site and also used in [10] as inhibitor. TWY and YEF are cyclic peptides designed by Tambunan et al [12] to target SAM site and RNA-cap site of NS5 MTase.
Docking process generated 4 ligands with better affinity than standards for each binding sites, as indicated by their lowest ΔGbinding score of all protein-ligand complexes Table 1 (see supplementary material). Negative value of ΔG in a reaction indicates that a reaction is favourable. The most negative values of ΔGbinding were shown by the complex of NS5 MTase – [Tyr123] Prepro Endothelin (110-130), amide, human at SAM site and the complex of NS5 MTase − Urotensin II, human at RNA-cap site. Hence, these ligands have higher affinity with their target sites than standards. Their interactions with contact residues of SAM site and RNA-cap site were displayed at (Table 1). The number of hydrogen bonds between ligands and catalytic residues of RNA-cap site is less than that of SAM site, due to smaller size of RNA-cap site than the size of SAM site. Visualisation of ligand-protein interaction was shown at (Figure 1a & b).
Figure 1.
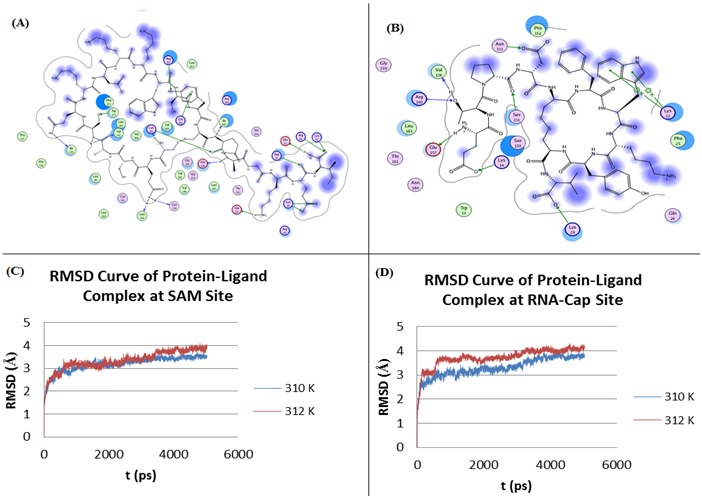
Visualisation of the interaction between two best ligands and each site of NS5 MTase. (A) SAM site - [Tyr123] Prepro Endothelin (110-130), amide, human, (B) RNA-cap site - Urotensin II, human, (C) RMSD value vs. simulation time of protein-ligand complex at SAM site, and (D) RMSD value vs. simulation time of protein-ligand complex at RNA-Cap site.
Prediction of ADME-Toxicity was conducted on 4 ligands with the lowest ΔGbinding generated during docking stage. The softwares used in this prediction were ACD I-Labs/Percepta and Toxtree-v2.5.0. The properties observed using ACD I-Labs were oral bioavailability, active transport, permeability glycoprotein (PGP inhibitor), central nervous system (CNS) active and probability of health effect on blood, cardiovascular, gastrointestinal, kidney, liver and lungs. Meanwhile, the properties observed using Toxtree-v2.5.0 was genotoxic and nongenotoxic carcinogenicity based on Benigni/Bossa rulebase. Based on these properties, analysis of ADME-Toxicity generated one best ligand for each binding site, namely [Tyr123] Prepro Endothelin (110-130), amide, human for SAM site and Urotensin II, human, for RNA-cap site (see supplementary material). These ligands possess good ADME characters, show minimum effect on health and have no carcinogenicity character.
Molecular dynamics simulation was conducted to study the effect of solvent on the stability of complexes between the best ligands and protein. This simulation was applied at temperature 310 K and 312 K on two complexes, i.e. between the complex of NS5 MTase − [Tyr123] Prepro Endothelin (110- 130), amide, human at SAM site and the complex of NS5 MTase − Urotensin II, human at RNA-cap site. These temperatures represent human body temperature at normal and fever condition. Molecular dynamics simulation differs from molecular docking in that it uses born solvation which takes into account the presence of solvent molecules. Algorithm used in this simulation is NPA (Nosé−Poincaré-Andersen), while the forcefield used is AMBER 94. The pressure of the simulation process is 101 kPa.
Molecular dynamics simulation comprises of three steps including initialisation, equilibration and production. The aim of initialisation step is to prepare the solvent before entering the next steps in molecular dynamics. Determination of the equilibration time was conducted during 100 pico second (ps) of initialisation step. Based on this process, potential energy of protein−ligand complex remained steady after 40 ps. Hence, after 40 ps the complexes between protein and ligand have adjusted their conformation with the solvent. The initialisation step was followed by heating and equilibration step. During heating stage, temperature of the system was raised towards the equilibrium stage during 20 ps. Based on the equilibration time that was determined previously, the equilibration step of this simulation was performed for 40 ps. The production steps of molecular dynamics simulation were performed at 310 K and 312 K during 5000 ps. After each of production steps, the complexes were brought into cooling stages for 20 ps to find the lowest conformational energy of the molecules. This process is called annealing. Cooling stage brings the temperature of simulation into 1 K. The calculated position, velocity and acceleration of the simulation were saved every 0.5 ps.
The conformational changes of protein-ligand complex can be studied from the curve of simulation time versus RMSD (rootmean- square deviation). Protein conformation is a set of three dimensional coordinates of its atomic constituents. The magnitude of conformational change between these coordinates is described as RMSD value. The RMSD value of protein-ligand complex versus simulation time was shown on (Figure 1C & D).
According to the graph, the complex of NS5 MTase − [Tyr123] Prepro Endothelin (110-130), amide, human did not undergo much conformational changes as the temperature increased. RMSD value of the complex at 310 K and 312 K showed no significant difference, hence there are similarities in the structure of complex at the given temperatures. Conversely, the complex of NS5 MTase − Urotensin II, human maintained more conformational stability of the complex at 312 K (temperature during fever) rather than at 310 K (normal body temperature).
Conclusion
Commercial cyclic peptides obtained from online websites were screened against SAM site and RNA-cap site of NS5 MTase. Screening through molecular docking process generated 4 best ligands for each binding sites as shown by their lowest ΔGbinding compared to standards. The result of ADME-Toxicity prediction revealed that [Tyr123] Prepro Endothelin (110-130), amide, human and Urotensin II, human possess the best ADME-Toxicity characters among other ligands. During molecular dynamics simulation [Tyr123] Prepro Endothelin (110-130), amide, human maintained a stable interaction with the SAM site of NS5 MTase at the tested temperatures (310 K and 312 K), while the complex of NS5 MTase − Urotensin II, human at the RNA-cap site was more reactive at 312 K rather than at 310 K. However, from this study, it could be inferred that these two commercial cyclic peptides could serve as potential candidates to be developed into antiviral agents against DENV although Urotensin II, human is showing better reactivity at 312 K compared with [Tyr123] Prepro Endothelin (110-130), amide, human.
Supplementary material
Acknowledgments
This research was supported by grant from Hibah Penguatan Riset Berbasis Kolaborasi Nasional Universitas Indonesia Tahun 2014. USFT and AAP supervised this research, HZ prepared the manuscript and BBU work on the technical details. We would like to express our gratitude to Dr. Kiki Ariyanti Sugeng from Department of Mathematics University of Indonesia for proofreading the manuscript.
Footnotes
Citation:Tambunan et al, Bioinformation 10(1): 023-027 (2014)
References
- 1.Halstead SB, et al. Lancet. 2007;370:1644. doi: 10.1016/S0140-6736(07)61687-0. [DOI] [PubMed] [Google Scholar]
- 2.Alen MMF, Schols D. J Trop Med. 2012;2012:1. doi: 10.1155/2012/628475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perera R, Kuhn RJ. Curr Op Microbiol. 2008;11:369. doi: 10.1016/j.mib.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noble CG, et al. Antiviral Res. 2010;85:450. doi: 10.1016/j.antiviral.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 5.Zhou Y, et al. J Virol. 2007;81:3891. doi: 10.1128/JVI.02704-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Craik DJ, et al. Chem Biol Drug Des. 2013;81:136. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 7.Rajik M, et al. Virol J. 2009;6:74. doi: 10.1186/1743-422X-6-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones JC, et al. J Virol. 2006;80:11960. doi: 10.1128/JVI.01678-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benigni R, et al. Environ Mol Mutagen. 2007;48:754. doi: 10.1002/em.20355. [DOI] [PubMed] [Google Scholar]
- 10.Podvinec M, et al. J Med Chem. 2010;53:1483. doi: 10.1021/jm900776m. [DOI] [PubMed] [Google Scholar]
- 11.Benarroch D, et al. J Biol Chem. 2004;279:35638. doi: 10.1074/jbc.M400460200. [DOI] [PubMed] [Google Scholar]
- 12.Tambunan USF, et al. Bioinformation. 2012;8:348. doi: 10.6026/97320630008348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.