

Abstract
The design and synthesis of alanine-rich α-helical peptides, constrained in a partially unfolded state by incorporation of the S,S-tetrazine phototrigger, has been achieved to permit, upon photochemical release, observation by 2D-IR spectroscopy of the sub-nanosecond conformational dynamics that govern the early steps associated with α-helix formation. Solid-phase peptide synthesis was employed to elaborate the requisite fragments, with full peptide construction via solution–phase fragment condensation. The fragment union tactic was also employed to construct 13C=18O isotopically edited amides to permit direct observation of conformational motion at or near specific peptide bonds.
INTRODUCTION
Peptides and proteins comprise dynamic entities that, in many cases, exploit conformational changes and/or transient molecular interactions to achieve their biological function.1–5 Understanding the early kinetic events that govern conformational dynamics in peptides, and in turn proteins, is critical for the development and refinement of models that relate dynamic structure to the observed biological function.6–8 Structural snapshots of such biomolecules can be achieved at atomic resolution by X-ray crystallography and nuclear magnetic resonance measurements. These static structures however do not provide information on transiently sampled non-equilibrium structures. Structural change on the millisecond timescale can however be defined by nuclear magnetic resonance spectroscopy,9 while advances in time-resolved X-ray crystallography10–14 permit temporal resolution on sub-nanosecond time scales within a crystal, although this approach remains technically challenging. Two-Dimensional Infrared Spectroscopy (2D-IR) developed and pioneered by Hochstrasser and colleagues,15–17 in conjunction with ultra-fast photochemical release (i.e., triggering),5,18,19 provides an attractive alternative method to initiate and subsequently observe the earliest kinetic processes associated with conformational change (i.e., protein folding and misfolding). For example, incorporation of a phototrigger to confine a peptide/protein within a narrow distribution of conformers, followed by ultrafast photochemical release of the geometric constraint, would permit precise temporal control. In particular, the peptide conformation would evolve, with some initial spatial coherence, toward a different distribution, ultimately reaching the native equilibrium distribution of peptide/protein conformations.
Previously developed phototriggers include non-native disulfide bonds, pioneered by Hochstrasser and DeGrado,20,21 and the methoxybenzoin-based systems developed by Chan and co-workers.22–25 These triggers however have limitations, including rapid recombination of the initial photoproducts,20,26 the inherent chemical reactivity of the derived photoproducts and, in the case of methoxybenzoin-based systems, the introduction of obtrusive non-native chemical appendages as a result of the phototrigger structure. Criteria for the ideal phototrigger, as previously defined,18 include: (1) photochemical fragmentation on a timescale shorter than the conformational changes under observation; (2) a reasonably high photochemical yield; (3) inert products with negligible side reactions along the photochemical pathway; (4) functionality permitting ready incorporation within peptides and proteins; (5) sufficient stability to permit synthetic manipulations and purification both in solution, as well as on solid-supports; and (6) photochemical cleavage with light frequencies compatible with the amide backbone and amino acid side chains.
To meet these criteria we introduced the S,S-tetrazine chromophore (Figure 1A).27–29 Importantly, the photoproducts comprise inert and unobtrusive thiocyanates and nitrogen; moreover, the S,S-tetrazine phototrigger can be inscribed readily between two cysteine residues to constrain a peptide within a narrow distribution of conformers.28 We have also described UV-pump transient 2D-IR-probe experiments to monitor the structural relaxation of a 24 residue alanine-rich peptide containing a kink induced by the S,S-tetrazine phototrigger (Figure 1B).30 Upon release of the constraint, initiated by a sub-picosecond pulse of UV irradiation (λ = 355 nm), formation of a single α-helix turn was directly observed by 2D-IR spectroscopy without ‘unzipping’ of nearby helix turns (vide infra).
Figure 1.

A. Photolysis of the S,S-tetrazine phototrigger can be achieved with laser pulses at various wavelengths that afford two thiocyanates and molecular nitrogen in less than 50 ps. B. Schematic representation of the S,S-tetrazine phototriggering applied to a S,S-Tet-AKA. Photolysis of the S,S-tetrazine (shown in space filling representation) removes the structural constraint to furnish the bis-SCN photoproduct peptide. The 2D-IR measurements at various time delays following the photolysis pulse (Topt) directly monitor the conformational relaxation within the central CAC motif (the main chain of the peptide in the CAC region is shown in sticks).
Herein, we describe the design and requisite synthesis of the constrained 24 residue alanine-rich α-helical peptide possessing the S,S-tetrazine phototrigger, employing a solution-phase fragment coupling tactic (S,S-Tet-AKA, cf. Figure 2). The synthesis of 13C=18O edited isotopomers is also described.30 Going forward, the general fragment coupling protocol developed to construct S,S-Tet-AKA can now be applied to peptides and proteins of greater complexity to initiate conformational changes that occur on sub-nanosecond timescales.
Figure 2.
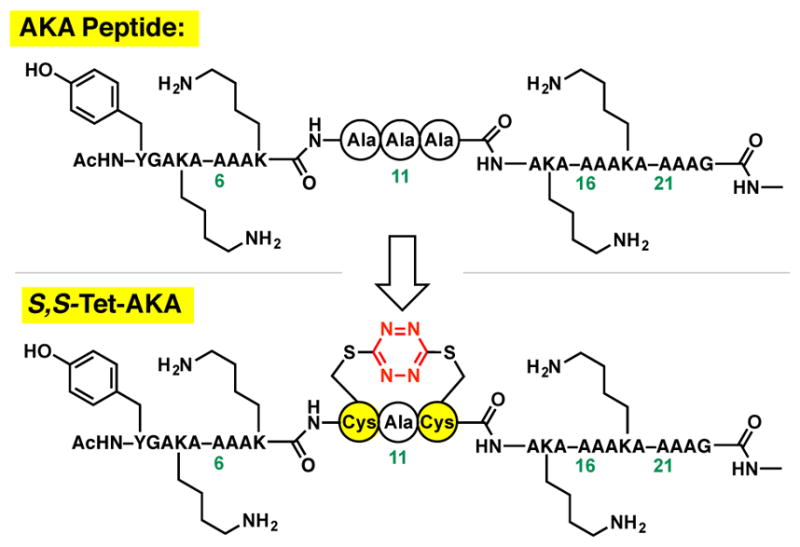
A. The AKA peptide was employed for the design of an S,S-tetrazine constrained synthetic target: S,S-Tet-AKA.
RESULTS AND DISCUSSION
Design and Synthesis
We initiated studies to construct peptides containing the S,S-tetrazine within a 24-residue alanine-rich AKA peptide (Figure 2), a system introduced by Baldwin and colleagues31 to adopt in the ground state of an α-helical structure that has subsequently often been employed in folding and relaxation studies. We reasoned that introduction of the S,S-tetrazine moiety within the middle of a 24-residue peptide would permit both the N- and C-terminal segments to maintain an α-helical conformation, while the amino acids between the cysteine residues would remain non-helical due to the geometric constraints enforced by incorporating the S,S-tetrazine phototrigger (Figure 1B). Upon photochemical release of the constraint imposed by the S,S-tetrazine phototrigger, the terminal α-helices would direct the central kinked residues to equilibrate rapidly to the preferred extended α-helical conformation. In contrast to peptide “stapling,” wherein the introduced crosslink(s) typically aim to introduce minimal change to the ground state conformation of the peptide,32 we require that the S,S-tetrazine “staple” introduces a well-defined local perturbation to the peptide conformation. Of interest, the S,S-tetrazine staple can be rapidly released upon photolysis.
A variety of (i, i+2/4) spacings between the cysteine residues inscribing the S,S-tetrazine system were initially investigated by molecular dynamics (MD) simulations, with distance constraints between the sulfur atoms employed to model the phototrigger (see Supporting Information). The resultant constrained MD simulations predicted that the S,S-Tet-AKA peptide would assume a kinked α-helical conformation, wherein the S,S-tetrazine moiety would disrupt the helical structure between Cys10 and Cys12 (cf. Figure 1B, Topt = 0 ps).30 Our synthetic targets thus became S,S-Tet-AKA, as well as isotopically edited versions thereof (Figure 2).
By constraining the center of an α-helix with an S,S-tetrazine moiety, a perturbation not dissimilar to that induced by a proline residue was envisioned.33 To estimate the potential helicity of the designed target peptides, relative to the original AKA peptide, model peptides AKAA11P and AKAA10C/A12C were constructed via solid-phase peptide synthesis (SPPS), employing the N-methyl indole aminomethyl resin (Scheme 1). The proline mutant AKAA11P was specifically constructed to mimic the S,S-tetrazine constrained peptide before photolysis (i.e., S,S-Tet-AKA), while the double cysteine mutant AKAA10C/A12C was synthesized to mimic the bis-thiocyanate photoproduct peptide.
Scheme 1.

The Synthesis of AKA variants AKAA11P and AKAA10C/A12C.
The far-UV CD spectra of the S,S-Tet-AKA (prepared below), AKAA11P, and AKAA10C/A12C peptides were recorded; an absorption band at 222 nm, diagnostic of helical structure, was observed for the native AKA peptide (Figure 3).31 The fractional helicity [θ222] of the native AKA was determined to be 0.72 in aqueous phosphate buffer (data not shown). The band at 222 nm nearly disappears upon incorporation of the proline residue within AKAA11P, suggesting that the fractional helicity has been significantly reduced (cf. 0.05).33 To ensure that a desirable degree of fractional helicity can be achieved for a kinked AKA-type peptide system, trifluoroethanol (TFE), known to maximize the peptide helicity and increase solubility,34 was employed to enhance the helicity of AKAA11P. Pleasingly, a value of 0.34 was observed for the AKAA11P peptide in TFE solution. A slight increase in fractional helicity to 0.49 was observed for the AKAA10C/A12C peptide in TFE. These results were highly encouraging, suggesting that following photolysis of the constrained S,S-tetrazine AKA peptide (i.e., S,S-Tet-AKA), a more helical conformation should evolve, as suggested by the helicity of the analogous AKAA10C/A12C peptide as indicated by the CD spectra.
Figure 3.

The far-UV CD spectra of the AKAA11P in phosphate buffer (dark blue) and TFE (magenta); the AKAA10C/A12C peptide in TFE (red); and final synthetic target S,S-Tet-AKA in TFE (green).
Turning to the synthesis of the 24-residue target peptide S,S-Tet-AKA, we recently introduced a SPPS synthetic protocol to incorporate the S,S-tetrazine phototrigger within short tri-, tetra-, and peptapeptide linchpins.29 The compatibility of a lysine in the i+1 position, relative to the S,S-tetrazine, however, had not been explored. To validate the compatibly of a protected Lys side chain under the conditions required to insert the S,S-tetrazine, tetrapeptide 3 was constructed via SPPS, both with the lysine side chain baring the standard tert-butyl carbamate protecting group and with the cysteine side chain thiols masked with the 4-monomethoxy trityl (Mmt) protecting groups (Scheme 2). The synthesis proceeded without incident. Equally important, we demonstrated that tetrapeptide 3 underwent steady-state photolysis to yield solely bis-thiocyanate peptide 4, thus confirming that the proximal basic Lys side chain does not perturb the steady-state photolysis of the electron-deficient S,S-tetrazine. Disappearance of the 13C-NMR signals for the tetrazine ring (δc ~ 170 ppm), with concomitant appearance of the SCN resonances at δc ~ 113 ppm27 proved to be particularly diagnostic for monitoring the S,S-tetrazine photolysis (see Supporting Information).
Scheme 2.
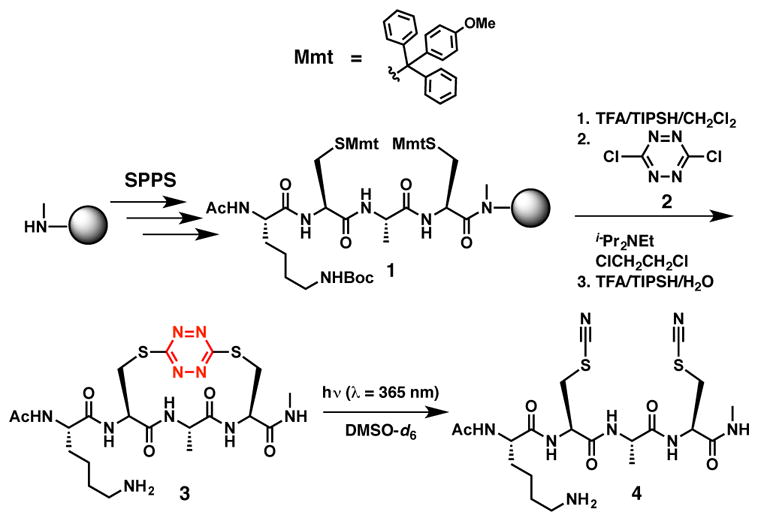
The SPPS of tetrapeptide 3 and steady state photolysis to afford bis-thiocyante tetrapeptide 4.
Having achieved the successful construction of a peptide model system and validated the photochemical fragmentation, we turned to the synthesis of S,S-Tet-AKA. The solid-phase conditions employed earlier to incorporate the S,S-tetrazine component in the model peptide27,29 unfortunately proved ineffective when the fully constructed peptide backbone remained immobilized on the solid support. A number of reaction modifications, such as including denaturants, elevating the reaction temperature, or employing more polar solvent systems failed to improve the situation. An alternative synthetic strategy was thus explored, wherein orthogonally protected Cys residues35 [i.e., thio-tertbutyl (St-Bu) at Cys10 and Mmt at Cys12] were incorporated during SPPS, in an attempt to introduce first the S,Cl-tetrazine (cf. 6, Scheme 3), followed by removal of the second thiol protecting group, with concomitant formation of the S,S-tetrazine moiety. This tactic also proved ineffective (Scheme 3). Equally unsuccessful, our tactic employed to replace the disulfide bond in the peptide hormone oxytocin with the S,S-tetrazine,27 which called for treatment of the AKAA10C/A12C peptide in an aqueous 0.1 M ammonium bicarbonate solution with dichlorotetrazine (2), failed to furnish the desired S,S-Tet-AKA peptide. Presumably the immobilized peptide has a preference to adopt the α-helical structure on the solid support, a conformational bias that cannot be sufficiently perturbed to incorporate the S,S-tetrazine phototrigger.
Scheme 3.
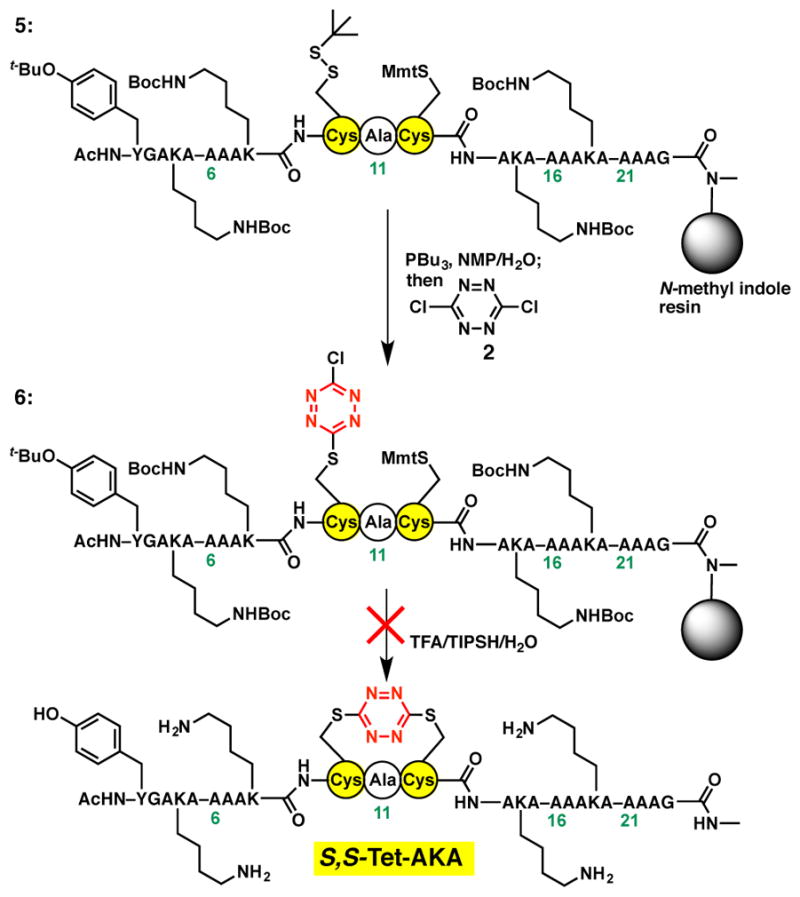
Attempted synthesis of the S,S-Tet-AKA peptide on the N-methyl indole solid support.
Undeterred, we turned to a fragment condensation strategy carried out in solution to construct the S,STet-AKA peptide. Early studies in our laboratory had demonstrated that the S,S-tetrazine moiety was stable to amide bond coupling protocols.29 Initially a model system comprising a two fragment union tactic was explored. To this end, S,S-tetrazine peptide 8, readily constructed on the Wang resin with the Lys side chain re-protected as the tert-butyl carbamate, furnished coupling partner 9 (Scheme 4A). The requite dodecapeptide coupling partner 10, corresponding to the C-terminal peptide fragment, was next prepared via a standard SPPS protocol. With fragments 9 and 10 in hand, our previously developed fragment union tactic utilizing HBTU and oxyma [ethyl 2-cyano-2-(hydroxyimino)acetate]29,36 was employed to achieve union at the Cys12–Ala13 amide bond; the yield was 53 % (Scheme 4B). Subsequent treatment with TFA furnished peptide 11 in near quantitative yield. Importantly, peptide 11 corresponds to residues 7–24 of the full synthetic target, S,S-Tet-AKA.
Scheme 4.

A. Preparation of Lys-protected S,S-tetrazine hexapeptide 9. B. Union of S,S-tetrazine hexapeptide 9 to the C-terminal peptide fragment 10 of the S,S-Tet-AKA target peptide, with the corresponding residue numbers shown, to furnish 18-mer 11.
With construction of the truncated peptide 11 validated, we turned to a three-fragment condensation tactic to generate the target peptide possessing the full N-terminal sequence. Given that the N-terminus of our previously reported S,S-tetrazine tripeptide linchpin, employed via solution-phase fragment coupling, was most conveniently protected as the Boc carbamate,29 we sought a semi-orthogonal protecting group to mask the lysine side chains. The benzyloxycarbonyl (Cbz) protecting group was selected and proved compatible with the S,S-tetrazine insertion conditions employed to construction tetrapeptide 13 (Scheme 5A). Importantly, the Cbz-protecting groups were retained during removal of 13 from the resin by treatment with TFA/TIPSH/H2O (95:2.5:2.5) for 2.5–5 hours. Removal of the Cbz protecting group from the Lys side chain to furnish peptide 14 however required more strenuous conditions, namely treatment with TFA for 24 h.
Scheme 5.
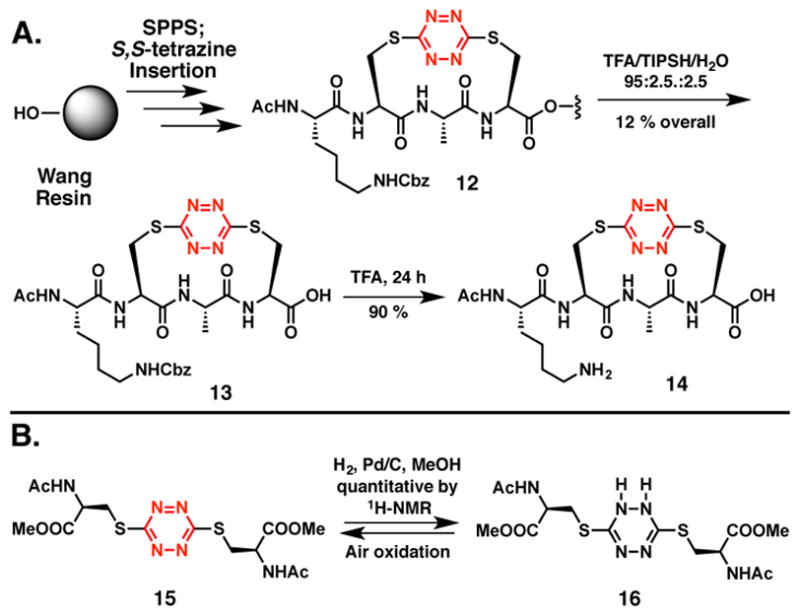
A. Evaluation of the benzyloxycarbonyl (Cbz) as an alternative protecting group for the lysine side chain and validation of deprotection conditions. B. The S,S-tetrazine ring of 15 is partially reduced to the dihydro-S,S-tetrazine system 16 upon exposure to hydrogenolysis conditions.
Turning next to the orthogonal Cbz-removal tactic employing hydrogenation, we were surprised to observe rapid loss of the orange color characteristic of the S,S-tetrazine chromophore. However, when the colorless solution was permitted to stand exposed to air, the solution slowly regained the original orange color. This observation suggested that hydrogenation had led to partial reduction of the S,S-tetrazine ring. To explore this scenario, we turned to our previously described amino acid based S,S-tetrazine 15 (Scheme 5B).27 Similar exposure of 15 to the hydrogenation conditions led to loss of the characteristic orange color, with formation of dihydro-S,S-tetrazine 16 in near quantitative yield (1H and 13C-NMR). In an effort to distinguish the tautomer obtained upon the semi-hydrogenation, density functional theory (DFT) calculations were performed using Gaussian0937 [RB3PW91, 6–31+G(d,p)] to determine the frequencies and intensities of the IR transitions corresponding to the 1,2- and 1,4-dihydro-S,S-terazine tautomers. A correction factor was applied to the frequencies based on the ratio of known experimental carbonyl frequencies versus those derived from the DFT calculations. Best agreement between the calculated and experimental infrared transitions suggested formation of 1,2-dihydro-S,S-tetrazine 16. Importantly, exposure of 16 to air over a 24 hour period demonstrated that the dihydro-S,S-tetrazine 16 does indeed oxidize to S,S-tetrazine 15.
With the use of the Cbz protecting group validated, the synthesis of the target S,S-Tet-AKA peptide employing a three fragment union tactic was undertaken (Scheme 6). The appropriate linchpin 17, was available from our model studies,29 while the requisite C- and N-terminal peptide fragments 18 and 20 were constructed via SPPS employing the N-methyl indole and 2-chlorotrityl resins, respectively. The free amine of peptide 18 was first coupled to S,S-tetrazine linchpin 27, followed by the removal of the Boc protecting group to furnish peptide 19 in 43% yield for the two steps. Peptide 19 in turn was coupled to the N-terminal peptide fragment 20, followed by treatment with TFA, to complete construction of the peptide S,S-Tet-AKA.30 Final union and deprotection proceeded in 25 % yield after HPLC purification.
Scheme 6.

The successful three-fragment union approach to synthesize S,S-Tet-AKA.
Synthetic S,S-Tet-AKA was characterized by LC-MS and MALDI-TOF. Pleasingly, the far-UV CD spectra of S,S-Tet-AKA in TFE (Figure 3) indicated the fractional helicity to be 0.43, similar to the proline-kinked mutant AKAA11P model (vide supra). For the proposed transient 2D-IR experiments, this fractional helicity was required to observe conformational reorganization to the final α-helical structure upon photolysis of the S,S-tetrazine constraint.
Photochemical Studies: Kinetics and Conformational Response to Photorelease
The synthetic S,S-Tet-AKA peptide was first subjected to steady-state photolysis (λ = 420 nm) to afford the (SCN)2-AKA peptide (see Figure 1B) in TFE. We were delighted to find that the fractional helicity of (SCN)2-AKA was 0.50, representing roughly a 0.07 increase in the helical content upon release of the S,S-tetrazine phototrigger. Given that only a single α-helix turn is formed following the S,S-tetrazine photolysis, the observed modest increase in helicity was expected. The transient 2D-IR measurements, reported elsewhere,30 also confirm the formation of the single α-helix turn following photolysis with a time constant of ~100 ps. To ensure that photolysis of the S,S-tetrazine linker was complete, formation of the SCN functionality was confirmed by the FT-IR spectrum, through observation of the SCN stretching band.30
The tripeptide model system 2129 (Figure 4, inset) was also employed to provide insights into the photochemistry of the kinked region. Upon photolysis, the yield of 21, measured by nanosecond transient absorption, was 28%, slightly larger than the 20% previously observed for the non-cyclic S,S-tetrazine peptide 15.28 The increase in photochemical yield is likely related to the conformational strain exerted on the S,S-tetrazine system when the Cys residues are in an i, i+2 relationship within cyclic peptide 21. The timescale of SCN formation upon flash photolysis of 21 was determined by femtosecond transient spectroscopy27,28 by measuring the time dependence of the increase of the SCN absorption band intensity of photoproduct peptide 22, centered at 2163 cm−1 (Figure 4). The rate of SCN formation was determined to be ~56 ps, again slightly faster than previously reported for the acyclic system 15. Based on these experiments, formation of the final photoproduct 22 is expected to be on the order of tens of picoseconds, while the initial timescale of S,S-tetrazine ring cleavage was determined by prior experiments with acyclic 15 to be ~10 ps.28
Figure 4.
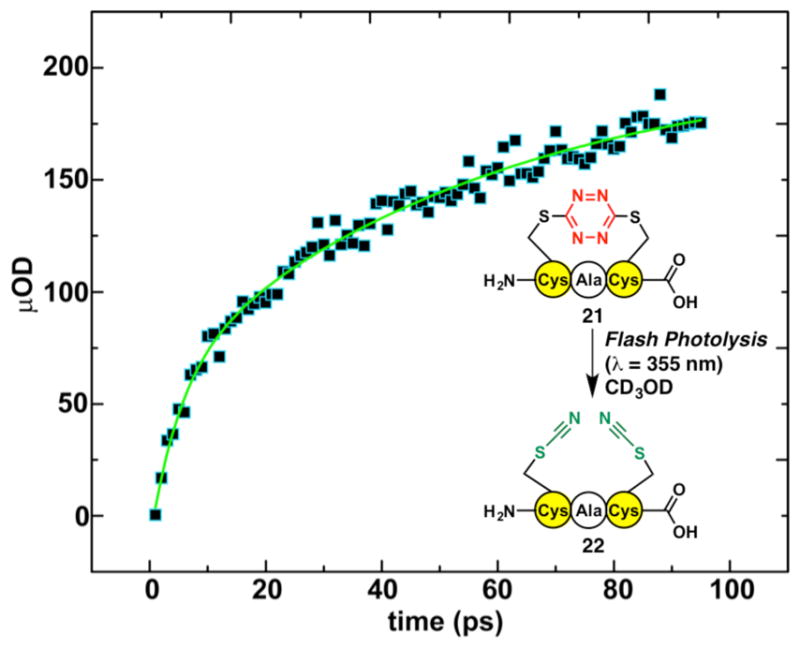
The time-dependent spectrum of the SCN transient absorption observed upon photolysis (355 nm excitation) of S,S-tetrazine tripeptide 21 to furnish bis-SCN tripeptide 22, measured at 2163 cm−1 from 0–100 ps. The raw data points are pictured in black with blue outline, while the exponential fit to data, with a time constant τ=56 ps, is shown by the green curve.
To monitor the picosecond structural transitions within the amide backbone by 2D-IR upon photochemical release of S,S-Tet-AKA, the amide I stretching modes were differentiated by employing carbonyl isotopic editing. Specifically, we incorporated the 13C=18O double label at defined positions along the peptide backbone.38–42 Our modular fragment coupling strategy was well suited to prepare these peptides, having previously reported a 13C=18O isotopically edited S,S-tetrazine linchpin,29 which we utilized to prepare labeled peptide S,S-Tet-AKAC10*/A11* (Figure 5). In addition, to monitor the backbone dynamics distal to the S,S-tetrazine phototrigger, two adjacent units of 13C=18O-Ala43 were incorporated within two S,S-tetrazine peptides, the first pair within the N-terminal peptide fragment, leading to the synthesis of S,S-Tet-AKAA5*/A6*, and the second pair within the C-terminal peptide fragment to furnish S,S-Tet-AKAA17*/A18*. Standard SPPS utilizing 13C=18O-Ala at the requisite positions, followed by the fragment union tactic, was employed.
Figure 5.
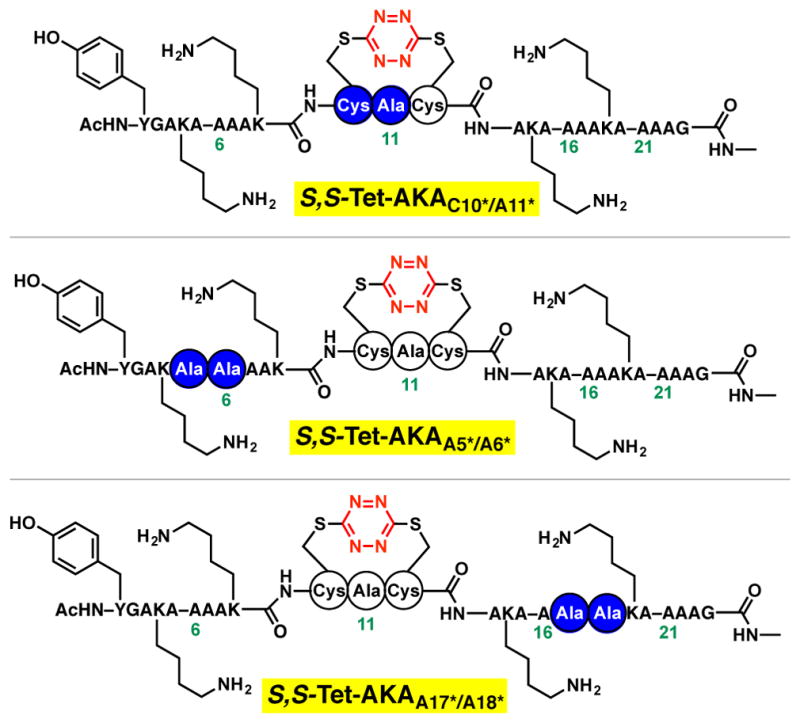
Isotopically edited peptides S,S-Tet-AKAC10*/A11*, S,S-Tet-AKAA5*/A6* and S,S-Tet-AKAA17*/A18* were synthesized to resolve the amide-I stretching mode from the remaining backbone stretching modes. The position of the 13C=18O labeled amides in the synthetic peptides is shown in blue circles.
Upon evaluation of S,S-Tet-AKAA5*/A6* and S,S-Tet-AKAA17*/A18* by standard 2D-IR methods, the coupling between these dipoles indicated that the peptides have an α-helical structure in both the N- and C-terminal domains, respectively.44,45 Subsequent evaluation by transient 2D-IR probe spectroscopy of the N- or C- terminal labeled residues revealed no changes in the angle between the dipoles following S,S-tetrazine photolysis, suggesting that these regions of the peptide do not undergo conformational changes. As previously reported, only in the case of S,S-Tet-AKAC10*/A11*, where the residues are in the domain of the peptide that is inscribed by the S,S-tetrazine constraint, did we observe significant changes in the conformation of the peptide.30 Equally important and as described in detail elsewhere,30 the phi and psi angles could be extracted from the transient 2D IR spectra of the 13C=18O amides of Cys10 and Ala11 to reveal directly the relaxation rate to the full helical structure. As the distal residues (cf. S,S-Tet-AKAA5*/A6* and S,S-Tet-AKAA17*/A18*) do not support conformational changes, we have proposed a model for this conformational transition wherein the dominant motion of the peptide is predominantly through rotation about the psi dihedral angle.30
CONCLUSION
We report here the design and validation of a general synthetic protocol that permits incorporation of the S,S-tetrazine photochemical trigger into complex peptides. Photochemical release and monitoring by 2D-IR spectroscopy permits definition, at the atomic level, of the microscopic motions that specific peptide/protein structural components explore along conformational reorganizing paths to reach a final equilibrium structure. The specific photochemical trigger, S,S-tetrazine, was introduced into the alanine-rich α-helical AKA peptide to disrupt the extended helical conformation, similar to a helix- “proline turn”-helix. The bias to assume an extended α-helix conformation however initially hindered efforts to incorporate directly the S,S-tetrazine phototrigger employing our previously developed protocols. To overcome this challenge, the separate peptide fragments were constructed through SPPS, and a fragment union tactic executed in solution to furnish the relatively large, 24 residue S,S-Tet-AKA peptide system, which pleasingly adopts a well-defined initial conformational distribution. The initial helix-turn-helix conformation, with the minor S,S-tetrazine perturbation within the middle of the sequence, permitted 2D IR observation of the microscopic structural transitions following ultra-fast photochemical release. A detailed account of the conformational dynamics observed for the S,S-Tet-AKA peptide is now available.30 Pleasingly, the synthetic methods developed herein hold the promise of wide applicability for the construction of diverse peptides and proteins possessing the ultra-fast photochemical S,S-tetrazine to define the early events in peptide/protein conformation reorganization.
EXPERIMENTAL SECTION
General Experimental Methods
Organic solvents used for reactions and washes were of reagent grade and degassed by purging with nitrogen prior to use. Di-tert-butyldicarbonate (Boc2O), diisopropylethylamine (DIPEA), acetic anhydride and N-methylimidazole, O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium (HBTU), 1-(mesitylene-2-sulfonyl)-3-nitro-1H-1,2,4-triazole (MSNT) Ethyl cyanoglyoxylate-2-oxime (oxyma), Fmoc-Ala-OH, Fmoc-Cys(Mmt), Fmoc-Asp(Ot-Bu)-OH, Fmoc-Gly-OH, Fmoc-Pro-OH, Fmoc-Ser-OH, Fmoc-Trp(N-Boc)-OH, Fmoc-Val-OH, Fmoc-Ala-OH (1-13C), L-Cysteine (1-13C) and 18O water were purchased from commercial vendors and used as received. Wang resin(LL) and N-methyl indole aminomethyl resins for peptide synthesis were obtained from Novabiochem.
Solid-phase syntheses were carried out in peptide synthesis reaction vessels (25 or 50 mL) with coarse porosity fritted glass support and Teflon stopcocks. The reaction vessels had 14/20 ground glass joints that allowed the resin to be kept under a nitrogen atmosphere while removing the monomethoxytrityl groups from cysteine. Photolysis experiments were performed in an Srinivasan-Griffin Photoreactor. Microwave irradiation was employed using 2–5 mL sealed microwave reaction vessels and an IR temperature sensor controlled the temperature.
A chloranil test was used to examine the presence of free amines on resin. A test tube containing a small amount of resin (10–12 beads) known to have a free primary amine was used as a reference along with another test tube containing a small amount of the same resin after acylation. Each resin was first washed with DMF (3×1 mL) and then to each in DMF (1 mL) was added 1 drop of 2% w/w actealdehyde/DMF, immediately followed by 1 drop of 2% w/w p-chloranil (2,3,5,6-tetrachloro-1,4-benzoquinone)/DMF and allowed to stand at room temperature for 5 min. The reaction solution was then removed by syringe and washed with DMF. The free amine resin (control) turned a deep blue color, while the acylated resin remained unchanged, suggesting that the amine had been completely acylated. The alizarin-cyanuric chloride test was used to test for alcohols on resin. Two resins in separate test tubes, one Wang resin and the other acylated Wang resin were used and each washed with DMF (3×1 mL), then a cocktail of N-methylmorpholine (1 mL), cyanuric chloride (5 mg) and DMF (3mL) was added to each tube and heated at 70 °C for 20 min. The resin beads were washed with DMF (3×1 mL). Next, a solution of N-methylmorpholine (1 mL), alizarin (5 mg) and DMF (3 mL) were added, and after 5 min the resin beads washed with DMF. The Wang resin turned dark red, while the acylated resin remained colorless.
Resin washing was conducted with the indicated solvent and was allowed to contact the resin for 30 seconds during each wash. The solvent was pushed through the frit using an “air push” apparatus made from a 15 mL disposable syringe and a 14/20 septum, or nitrogen gas was used in cases of an inert atmosphere requirement.
Preparative-scale reverse-phase chromatography was conducted using binary HPLC pumps and a UV/vis dual wavelength detector. The separations were achieved with a Waters XBridge Prep BEH 130 C18 5μm OBD 19 × 100mm column. The eluent was acetonitrile (HPLC grade) and Millipore water with 0.05% formic acid buffer unless otherwise noted. Linear gradient ellutions with a flow rate of 15 mL/min were employed, and specific gradients for each compound are noted.
1H NMR (500 MHz), 13C NMR (125 MHz) and 2D NMR spectra were recorded with either a 5 mm dual inverse probe or 5 mm DCH CryoProbe. Analytical LC-MS analyses were conducted binary gradient HPLC system connected to a diode array detector and a mass spectrometer with electro-spray ionization. The HPLC-MS samples were analyzed as solutions in water or acetonitrile, prepared at 0.15–0.20 mg/mL concentration. The analytical HPLC-MS analyses were conducted with an Atlantis–C18 column (4.6 ×50 mm; 5 μm) and linear gradients of 0.05% formic acid in acetonitrile and 0.05% formic acid water were utilized with a flow rate of 2 mL/min. High resolution mass spectra (HRMS) were obtained with a MALDI-TOF-TOF mass spectrometer using electrospray ionization in positive or negative mode, depending upon the analyte. All FTIR spectra were obtained using defalt parameters. Absorption spectra were taken with a UV-Vis spectrometer and CD spectra were obtained with a 1 mm sample holder.
Femtosecond IR Tranisent Absorption Method
The optical density at the excitation wavelength (355 nm) of the samples ranges from 0.09–0.12 in a 400 μm path length CaF2 cell for femtosecond transient IR experiments. Fourier-transform limited 70-fs pulses with center frequencies of 2150 cm−1 were used in the femtosecond IR transient absorption experiments for the samples in chloroform. The energy of the 355 nm excitation pulse, 5 μJ, was attenuated by an iris. The beam radius of the IR probe pulse was 75% of the visible pump pulse radius at the sample cell. The beam radius of the IR probe pulse was 75% of the visible pump pulse radius at the sample cell. The visible beam is passed through a hole in the center of the parabolas used to collect the infrared probe pulses and the overlap between the pump and probe pulses is achieved by first using 200 um pinhole at the sample holder, followed by maximizing the transient absorption signal of a Si wafer. The transient absorption of the carbonyl stretch of fluorenone is utilized as a standard to optimize the overlap of the pump beam. Timing between the pump and probe pulses is varied by using an automated translation stage with fs time resolution. The transmitted probe pulse is focused on to the focal plane of a monochromator equipped with a 64 element mercury-cadmium-telluride array detector. The appropriate monochromator grating was chosen, comprising 150 lines per mm groove. The kinetic trace was collected (averaged over 16 scans, 1600 shots per scan) over 100 ps time range with 1 ps steps.
General Resin Loading Procedures
Wang resin
The resin (0.22 mmol) was placed in a peptide synthesis vessel and swelled with CH2Cl2 (10 mL) for 1 h. The solvent was drained and a pre-mixed solution of Fmoc-AA-OH (0.88 mmol, 4.0 equiv), MSNT (0.88 mmol, 4.0 equiv) and methyl imidazole (0.66 mmol, 3.0 equiv) dissolved in CH2Cl2 (5 mL) was added to the resin. The contents were gently rocked for 2 h, then drained and the resin washed with CH2Cl2 (3 × 5 mL). The procedure was repeated and the resin carried on to the next step.
N-Methyl Indole AM Resin
N-Methyl indole resin (0.22 mmol) was placed in a peptide synthesis vessel and swelled with CH2Cl2 (10 mL) for 1 h. The solvent was drained and the resin was then treated with a solution of 20% piperidine/DMF (2 × 7 mL), allowing each treatment to contact the resin for 10 min. The resin was then washed with DMF (3 × 5 mL), then a pre-mixed solution of Fmoc-AA-OH (1.1 mmol, 5 equiv), HBTU (1.1 mmol, 5 equiv), oxyma (1.1 mmol, 5 equiv) and DIPEA (2.2 mmol, 10 equiv) dissolved in DMF (5 mL) was added to the resin. The contents were rocked gently for 1 h, then drained and the resin washed with DMF (3 × 5 mL). The coupling procedure was repeated and the resin carried on to the next step.
2-Chlorotrityl Chloride Resin
2-Chlorotrityl chloride resin (0.22 mmol) was placed in a peptide synthesis vessel and swelled with CH2Cl2 (10 mL) for 1 h. The solvent was drained and the resin was then a pre-mixed solution of Fmoc-AA-OH (0.26 mmol, 1.2 equiv), and DIPEA (0.88 mmol, 4 equiv) dissolved in CH2Cl2 (5 mL) was added to the resin. The contents were rocked gently for 1 h, then drained and the resin washed with DMF (3 × 5 mL). The coupling procedure was repeated and the resin carried on to the next step.
General Procedures for Solid-Phase Peptide Synthesis
The resin-bound Fmoc-Amino Acid (0.22 mmol) was washed with DMF (3 × 5 mL) and then treated with a solution of 20% piperidine/DMF (2 × 7 mL) allowing each treatment to contact the resin for 10 min. The resin was washed with DMF (3 × 5 mL), then a pre-mixed solution of Fmoc-protected amino acid (1.1 mmol, 5.0 equiv), HBTU (1.1 mmol, 5.0 equiv), oxyma (1.1 mmol, 5.0 equiv) and DIPEA (2.2 mmol, 10.0 equiv) dissolved in DMF (5 mL) was added to the resin. The contents were rocked gently for 1 h, then drained and the resin washed with DMF (3 × 5 mL). The Fmoc de-protection procedure was repeated followed by the coupling of the amino acid sequence to synthesize the desired peptide. The N-terminus was protected as the Boc- or Ac-derivative by first employing the Fmoc de-protection procedure and washing the resin with DMF (3 × 5 mL). A pre-mixed solution of Boc-anhydride (2.2 mmol, 10 equiv)/acetic anhydride (2.2 mmol, 10 equiv) and DIPEA (4.4 mmol, 20 equiv) in DMF (5 mL) was added to the resin. The contents were rocked gently for 1 h then drained and the resin was sequentially washed with DMF (3 × 5 mL) and CH2Cl2 (3 × 5 mL).
S,S-Tetrazine Tethering During SPPS
The resin-bound peptide (0.22 mmol) containing two S-Mmt protected Cys grous was swelled with CH2Cl2 (10 mL) for 1 h. The peptide synthesis vessel was then sealed with a septum, vented to a mineral oil filled bubbler and attached to a two-neck collection flask. The solvent was drained and a pre-mixed solution of CH2Cl2/TIPSH/TFA (92.5:5.0:2.5 peptides on the Wang resin; 95:4:1 for loaded N-methyl indole AM resin) was added to the resin and a stream of nitrogen was introduced through the collection flask and bubbled through the resin bed to agitate the orange reaction mixture for 3 min. The contents were drained by pushing the liquid contents into the collection flask using a stream of nitrogen and the resin was washed with CH2Cl2 (10 mL). The procedure was repeated 4 additional times until the reaction solution became clear, suggesting the monomethoxytrityl groups were completely removed. The resin was washed with 10% DIPEA/ClCH2CH2Cl (3 × 5 mL). To the resin was added a solution of dichlorotetrazine (2, 0.24 mmol, 1.1 equiv) in ClCH2CH2Cl (5 mL) followed by the addition of DIPEA (0.5 mL) neat. The peptide vessel was wrapped in foil to exclude ambient light and the contents were agitated for 18 h. The solvent was drained and the resin washed with CH2Cl2 (3 × 5 mL), MeOH (3 × 5 mL) and CH2Cl2 (3 × 5 mL).
Synthesis and Isolation of Peptides
AKA Peptide
This previously reported peptide31 was prepared from 0.050 mmol of loaded methyl indole AM resin following the general proceedures. The final product was purified by reverse-phase HPLC (gradient 5–35% organic over 10 min) to give 13.7 mg (13%) of a white amorphous powder after lyophilization: HRMS (ES) m/z 2093.1575 [(M+Na)+; calcd for C91H155N29O26Na: 2093.1596]; MALDI-TOF m/z 2071.014 [(M+H)+; calcd for C92H156N29O26: 2071.1776].
AKAA11P peptide
The AKAA11P peptide was prepared from 0.050 mmol loaded methyl indole AM resin utilizing the general proceedures, and was purified by reverse-phase chromatography (gradient 5–35% organic over 10 min) to afford 27.8 mg (27%) of AKAA11P as a white amorphous powder after lyophilization: HRMS (ES) m/z 2097.1933 [(M+H)+; calcd for C93H158N29O26: 2097.1980]; MALDI-TOF m/z 2097.123 [(M+H)+; calcd for C93H158N29O26: 2097.1980].
AKAA10C/A12C peptide
The AKAA10C/A12C was prepared from 0.036 mmol loaded methyl indole AM resin employing the general proceedures, and was purified by reverse-phase chromatography (gradient 5–35% organic over 10 min) to give 10.5 mg (15%) of AKAA10C/A12C as a white amorphous powder after lyophilization: HRMS (ES) m/z 2157.1082 [(M+Na)+; calcd for C91H155N29O26NaS2: 2157.1037]; MALDI-TOF m/z 2157.315 [(M+Na)+; calcd for C91H155N29O26NaS2: 2157.1037].
S,S-Tetrazine Tetrapeptide 3
The tetrapeptide 3 was synthesized from 0.22 mmol loaded methyl indole AM resin employing the general proceedures, and was purified by reverse-phase chromatography (gradient 5–45% organic over 12 min) to give 12.2 mg (10%) of 3 as an orange amorphous powder after lyophilization. All the resonances for 3 were assigned using 2D NMR (COSY, HMBC, and HSQC): 1H NMR (500 MHz, DMSO-d6) δ 8.49 (d, Ala-3-NH, J = 8.5 Hz, 1H), 8.48 (d, Cys-4-NH, J = 7.0 Hz, 1H), 8.19 (d, Lys-1-NH, J = 8.0 Hz, 1H), 8.01 (q, Me-NH, J = 4.5 Hz, 1H), 7.84 (d, Cys-2-NH, J = 6.5 Hz, 1H), 4.71 (dt, Cys-2-Hα, J = 6.5, 3.5 Hz, 1H), 4.68 (ddd, Cys-4-Hα, J = 6.5, 4.5, 2.0 Hz, 1H), 4.50 (dd, Cys-4-Hβ, J = 15.0, 4.5 Hz, 1H), 4.46 (dd, Cys-2-Hβ, J = 15.0, 2.5 Hz, 1H), 4.26 (dq, Lys-1-Hα, J = 8.0, 4.5 Hz, 1H), 4.14 (dq, Ala-3-Hα, J = 8.5, 7.0 Hz, 1H), 3.45 (dd, Cys-4-Hβ, J = 14.5, 2.0 Hz, 1H), 3.34 (dd, Cys-2-Hβ, J = 14.5, 4.0 Hz, 1H), 2.76 (t, -CH2-NH2, J = 7.5 Hz, 2H), 2.64 (d, CH3-NH, J = 4.5 Hz, 3H), 1.88 (s, CH3-CO, 3H), 1.54–1.47 (m, 4H), 1.36–1.28 (m, 2H), 1.04 (d, Ala-3-CH3, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 171.4, 171.2, 170.3, 169.6, 168.9, 166.9, 52.5, 51.3, 51.0, 47.6, 38.8, 32.4, 31.1, 30.3, 26.7, 26.0, 22.6, 22.5, 19.8; HRMS (ES) m/z 557.2072 [(M+H)+; calcd for C20H33N10O5S2: 557.2077].
Steady-state photolysis of tetrapeptide 3
A solution of 3 in CD3OD (~5 mg/mL) was photolyzed under steady state irradition for 4 h to afford bis-SCN peptide 4. 1H NMR (500 MHz, CD3OD) δ 4.72 (dd, J = 8.5, 4.5 Hz, 1H), 4.66 (dd, J = 7.5, 5.0 Hz, 1H), 4.34 (q, J = 7.0 Hz, 1H), 4.30 (dd, J = 8.0, 5.5 Hz, 1H), 3.64 (dd, J = 14.0, 4.5 Hz, 1H), 3.54 (dd, J = 14.0, 5.0 Hz, 1H), 3.37 (dd, J = 14.0, 9.0 Hz, 1H), 3.36 (dd, J = 14.0, 7.5 Hz, 1H), 2.99–2.94 (m, 2H), 2.77 (brs, 3H), 2.05–2.01 (overlapping m, 2H), 2.04 (overlapping s, 3H), 1.73–1.68 (m, 2H), 1.44 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 172.7, 172.5, 170.3, 169.1, 169.0, 113.6, 113.5, 53.3, 52.8, 52.7, 49.5, 39.14, 35.6, 31.5, 27.1, 26.3, 23.0, 22.8, 18.8; HRMS (ES) m/z 529.2007 [(M+H)+; calcd for C20H33N8O5S2: 529.2015].
S,S-Tetrazine Peptide 8
Peptide 8 was prepared from 0.13 mmol of loaded Wang resin employing the general proceedures, and was purified by reverse-phase chromatography (gradient 5–30% organic over 12 min) to give 17.8 mg (20%) of 8 as an orange amorphous powder after lyophilization: HRMS (ES) m/z 686.2522 [(M+H)+; calcd for C25H40N11O8S2: 686.2503].
Peptide 9
A 25 mL round bottom flask was charged with 8 (6.0 mg, 0.0088 mmol) and NaHCO3 (1.8 mg, 0.022 mmol, 2.5 equiv) then dissolved in water (2.0 mL). A pre-mixed solution of Boc2O (2.9 mg, 0.013 mmol, 1.5 equiv) dissolved in 1,4-dioxane (1.0 mL) was added, and the reaction solution was heated to 45 °C with stirring. After 30 min, completion of the reaction was observed by LC-MS and the reaction solution was directly purified by reverse-phase chromatography (gradient 5–50% organic over 10 min) to give 5.5 mg (80%) of 9 as a red/orange amorphous powder after lyophilization: HRMS (ES) m/z 808.2853 [(M+Na)+; calcd for C30H47N11O10NaS2: 808.2847].
Peptide building block 10
The peptide sequence comprising the N-terminal fragment of the target peptide was assembled from 0.10 mmol loaded methyl indole AM resin employing the general proceedures, was cleaved from the resin to retain the N-terminal Fmoc protecting group, and was purified by reverse-phase chromatography (gradient 5–35% organic over 10 min) to give 65.4 mg (54%) of 10 a white amorphous powder after lyophilization: HRMS (ES) m/z 1206.6654 [(M+H)+; calcd for C57H88N15O14: 1206.6635]. Next, to install the requisite N(ε)-Boc protecting groups, a 25 mL round bottom flask was charged with the above peptide (30.0 mg, 0.025 mmol) and NaHCO3 (10.5 mg, 0.125 mmol, 5 equiv) then dissolved in water (2.0 mL). A pre-mixed solution of Boc2O (16.4 mg, 0.075 mmol, 3 equiv) dissolved in 1,4-dioxane (2.0 mL) was added, and the reaction solution was heated to 45 °C with stirring. After 2 h, completion of the reaction was observed by LC-MS and the solvent was evaporated under reduced pressure. The crude reaction mixture was dissolved in DMF (2.0 mL), and then using a syringe, added dropwise with stirring to a 25 mL round bottom flask charged with 10% piperidine/DMF (2.0 mL). Once addition was completed, the reaction solution was allowed to stir at room temperature for 10 min before it was concentrated under reduced pressure. The resulting residue was dissolved in acetonitrile/water (1:1) and then purified by reverse-phase chromatography (gradient 5–60% organic over 10 min) to give 19.8 mg (67%) of 10 as an amorphous powder after lyophilization: HRMS (ES) m/z 1184.6713 [(M+H)+; calcd for C52H94N15O16: 1184.6998].
Peptide 11
To a 10 mL tear-drop flask containing Peptide 10 (3.3 mg, 0.0028 mmol) in DMF (1 mL) was added dropwise a pre-mixed solution of 9 (4.4 mg, 0.0056 mmol, 2.0 equiv), Oxyma (0.80 mg, 0.0056 mmol, 2.0 equiv), HBTU (2.1 mg, 0.0056 mmol, 2.0 equiv) and DIPEA (2.3 μL, 0.014 mmol, 5 equiv) in DMF (1.0 mL). After stirring for 3 h, completion of the reaction was observed by LC-MS and the solution was concentrated under reduced pressure. The resulting residue was taken into acetonitrile/water (1:1) and then purified by reverse-phase chromatography (gradient 5–70% organic over 10 min) to give 2.9 mg (53%) of the protected peptide intermediate as a light orange amorphous powder after lyophilization: HRMS (ES) m/z 1973.9673 [(M+Na)+; calcd for C82H138N26O25NaS2: 1973.9666]; MALDI-TOF m/z 1973.761 [(M+Na)+; calcd for C82H138N26O25NaS2: 1973.9666]. Next, a 10 mL tear-drop flask charged with the protected peptide intermediate (2.9 mg, 0.0015 mmol) and a solution of 1:3 TFA/CH3CN (2 mL) was added in one portion. After stirring for 2 h, completion of the reaction was observed by LC-MS and the solution was concentrated under reduced pressure to afford, without any purification, 2.5 mg of peptide 11 as a light orange amorphous powder: HRMS (ES) m/z 1651.8291 [(M+H)+; calcd for C67H1114N26O19S2: 1651.8273]; MALDI-TOF m/z 1651.355 [(M+H)+; calcd for C67H1114N26O19S2: 1651.8273].
S,S-Tetrazine Tetrapeptide 13
The Cbz-protected peptide 13 was synthesized from 0.11 mmol loaded Wang resin employing the general proceedures and was purified by reverse-phase chromatography (gradient 10–80% organic over 10 min) to give 11.9 mg (16%) of an orange amorphous powder after lyophilization: HRMS (ES) m/z 700.1962 [(M+Na)+; calcd for C27H35N9O8NaS2: 700.1948]. The 1H-NMR specturum (DMSO-d6) indicated the presence of the diagnostic β-Cys resonances for peptides containing the S,S-tetrazine phototrigger; however, multiple conformers were observed for this protected peptide intermediate (See Supporting Information).
S,S-Tetrazine Peptide 14
To a 10 mL tear-drop flask charged with 13 (2.0 mg, 0.0030 mmol) was added a solution of 19:1 TFA/TIPSH (3mL). After stirring for 2 h, completion of the reaction was observed by LC-MS and the solution was concentrated under reduced pressure. The resulting residue was taken into water (1 mL) and then purified by reverse-phase chromatography (gradient 5%–45% organic over 10 min) to give 1.5 mg (90%) of 14 an orange amorphous powder after lyophilization. 1H NMR (500 MHz, DMSO-d6) δ 8.44 (d, J = 6.5 Hz, 1H), 8.48 (d, J = 8.5 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 7 Hz, 1H), 7.74 (bs, 1H), 6.51 (s, 2H), 4.79 (bm, 1H), 4.74 (dt, J = 6.5, 3.5 Hz, 1H), 4.54 (m, 2H), 4.25 (ddd, J = 8.6, 4.8, 3.5 Hz, 1H), 4.17 (ddd, J = 7.6, 6.4, 1.0 Hz, 1H), 3.52 (dd, J = 14.9, 1.8 Hz, 1H), 3.37 (dd, J = 15.2, 3.5 Hz, 1H), 2.76 (bm, 2H), 1.88 (s, 3H), 1.63 (bm, 1H), 1.52 (bm, 4H), 1.37–1.31 (bm, 3H), 1.00 (d, J = 6.5 Hz, 3H); 13C-NMR (125 MHz, DMSO-d6) δ 171.1, 170.8, 170.6, 170.4, 169.4, 166.8, 52.4, 51.6, 50.9, 47.4, 38.6, 32.3, 30.9, 30.3, 26.6, 22.5, 22.3, 19.7 (note one peak overlaps with the solvent signal); HRMS (ES) m/z 544.1777 [(M+H)+; calcd for C19H30N9O6S2: 544.1760].
Dihydro-S,S-tetrazine 16
A 10 mL tear drop flask equipped with a stir bar was charged with a solution of 1527 (15.0 mg, 0.035 mmol) in methanol (5 mL). A spatula tip of 10% palladium on carbon (50% wet with water) was added to the solution. The heterogeneous mixture was flushed with hydrogen gas using a balloon and then stirred under a hydrogen gas environment for 5 min. The mixture was then filtered through a pad of silica gel and the filterate concentrated in vacuo to afford, without any required purification, 14.9 mg (98%) of 16 as a colorless oil: IR (CH2Cl2) 3047 (s), 2985 (s), 2306 (m), 1745 (s), 1679 (s), 1623 (m), 1510 (s); 1H NMR (500 MHz, CD3OD) δ 4.69 (dd, J = 8.0, 4.5 Hz, 1H), 3.73 (s, 3H), 3.46 (dd, J = 14.0, 4.5 Hz, 1H), 3.22 (dd, J = 14.0, 8.0 Hz, 1H), 1.99 (s, 3H); 1H NMR (500 MHz, DMSO-d6) δ 8.89 (s, 1H), 4.43 (d, J = 7.5 Hz, 1H), 4.89 (ddd, J = 9.0, 7.5, 5.0 Hz, 1H), 3.63 (s, 3H), 3.33 (dd, J = 13.5, 5.0 Hz, 1H), 3.06 (dd, J = 13.5, 9.0 Hz, 1H), 1.86 (s, 3H); 13C NMR (125 MHz, CD3OD) δ 173.5, 172.3, 151.8, 54.0, 53.2, 33.2, 22.5; 13C NMR (125 MHz, DMSO-d6) δ 170.7, 169.3, 149.4, 52.1, 51.5, 31.2, 22.2; HRMS (ES) m/z 435.1129 [(M+H)+; calcd for C14H23N6O6S2: 435.1121].
To a 10 mL tear drop flask charged with 16 (5.2 mg, 0.012 mmol) was added methanol (5 mL). The solution was allowed to stand exposed to air for 8 h after which the solution, which turned from colorless to orange, was concentrated in vacuo and then chromatographed on silica gel using 5% methanol/ethyl acetate as the eluent to give 5.0 mg (97%) of 15 as an orange solid, which matched the reported characterization data previously reported for 15.27 To provide evidence as to whether 16 is the 1,2- or 1,4-dihydrotetrazine, IR analysis aided with simulations, were highly suggestive of the 1,2–dihydrotetrazine (see Supporting Information).
Peptide 18
Peptide 18, comprising the C-terminal fragment of S,S-Tet-AKA, was synthesized from from 0.25 mmol of loaded methyl indole AM resin according to the general procedures and was purified by reverse-phase chromatography (gradient 5–60% organic over 10 min) to provide 116.1 mg (37%) of 18 as a white amorphous powder after lyophilization: HRMS (ES) m/z 1252.6707 [(M+H)+; calcd for C58H90N15O16: 1252.6690]; MALDI-TOF m/z 1274.399 [(M+Na)+; calcd for C58H89N15O16Na: 1274.6510].
Peptide 19
To a 25 mL tear-drop flask containing peptide 18 (53.0 mg, 0.043 mmol) in DMF (3.5 mL) was added dropwise a pre-mixed solution of 1729 (20.0 mg, 0.043 mmol, 1.0 equiv), oxyma (6.7 mg, 0.047 mmol, 1.1 equiv), HBTU (17.9 mg, 0.047 mmol, 1.1 equiv) and DIPEA (18.7 μL, 0.11 mmol, 2.5 equiv) in DMF (1.5 mL). After stirring for 8 h, the reaction solution was concentrated under reduced pressure. To the resulting residue was added a solution of 1:3 TFA/CH3CN (5 mL). After stirring for 2 h, the reaction solution was directly purified by reverse-phase chromatography (gradient 10–70% organic over 10 min) to give 32.8 mg (43%) of 19 as an amorphous powder after lyophilization: HRMS (ES) m/z 1629.7113 [(M+Na)+; calcd for C69H102N22O19NaS2: 1629.7031]; MALDI-TOF m/z 1629.665 [(M+Na)+; calcd for C69H102N22O19NaS2: 1629.7031].
Peptide 20
Peptide 20, comprising the N-terminal fragment of S,S-Tet-AKA, was prepared from 0.25 mmol loaded 2-chlorotrityl chloride resin following the general procedures and was purified by reverse-phase chromatography (gradient 10–70% organic over 10 min) to afford 109.1 mg (38%) of 20 as a white amorphous powder after lyophilization: HRMS (ES) m/z 1160.5630 [(M+H)+; calcd for C56H78N11O16: 1160.5628]; MALDI-TOF m/z 1182.469 [(M+Na)+; calcd for C56H77N11O16Na: 1182.5448].
Peptide S,S-Tet-AKAA10C/A12C
To a 10 mL tear-drop flask containing peptide 19 (30.8 mg, 0.019 mmol) in DMF (2.5 mL) was added dropwise a pre-mixed solution of 20 (22.3 mg, 0.019 mmol, 1.0 equiv), Oxyma (3.0 mg, 0.021 mmol, 1.1 equiv), HBTU (8.0 mg, 0.021 mmol, 1.1 equiv) and DIPEA (8.4 μL, 0.048 mmol, 2.5 equiv) in DMF (1.5 mL). After stirring for 8 h, the reaction solution was concentrated under reduced pressure. To the resulting residue was added neat TFA (4 mL). After stirring for 24 h, the solution was concentrated under reduced pressure, taken into acetonitrile/water (1:1), and then purified by reverse-phase chromatography (gradient 5–35% organic over 10 min) to provide 10.5 mg (25%) of S,S-Tet-AKAA10C/A12C as an amorphous powder after lyophilization: HRMS (ES) m/z 2213.1260 [(M+H)+; calcd for C93H154N33O26S2: 2213.1184]; MALDI-TOF m/z 2213.598 [(M+H)+; calcd for C93H154N33O26S2: 2213.1184].
Isotopically edited peptides containing 13C/18O amides
The proceedures described above were employed to synthesize the S,S-Tet-(C10*/A11*)AKAA10C/A12C, S,S-Tet-(A5*/A6*)AKAA10C/A12C, and S,S-Tet-(A17*/A18*)AKAA10C/A12C, peptides, utilizing either FmocHN-Cys(SSt-Bu)-13C18O2H or FmocHN-Ala-13C18O2H, which were prepared according to previously reported methods.29,43 Mass spectra and LC-MS chromatograms of the synthetic intermediates is provided in the Supporting Information.
Peptide S,S-Tet-(C10*/A11*)AKAA10C/A12C
HRMS (ES) m/z 2219.1348 [(M+H)+; calcd for 12C9113C2H154N3316O2418O2S2: 2219.1336]; MALDI-TOF m/z 2219.522 [(M+H)+; calcd for 12C9113C2H154N3316O2418O2S2: 2219.1336].
Peptide S,S-Tet-(A5*/A6*)AKAA10C/A12C
HRMS (ES) m/z 2219.1357 [(M+H)+; calcd for 12C9113C2H154N3316O2418O2S2: 2219.1336]; MALDI-TOF m/z 2219.781 [(M+H)+; calcd for 12C9113C2H154N3316O2418O2S2: 2219.1336].
Peptide S,S-Tet-(A17*/A18*)AKAA10C/A12C
HRMS (ES) m/z 2219.1414 [(M+H)+; calcd for 12C9113C2H154N3316O2418O2S2: 2219.1336]; MALDI-TOF m/z 2219.756 [(M+H)+; calcd for 12C9113C2H154N3316O2418O2S2: 2219.1336].
Supplementary Material
Acknowledgments
Financial support was provided by the NIH GM 12694, and the RLBL facility grant (NIH P41 RR 001348). The MALDI-TOF-TOF instrumentation was supported by NSF MRI-0820996. The authors also thank Drs. George Furst and Rakesh Kohli at the University of Pennsylvania for assistance in obtaining NMR and high-resolution MS, respectively.
Footnotes
Supporting Information. HPLC-MS, MALDI-TOF-TOF, a description of the simulated IR spectra for dihydro-S,S-tetrazine 16, characterization data for synthetic intermediates related to the 13C/18O isotopically labeled peptides and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.McCammon JA, Gelin BR, Karplus M. Nature. 1977;267:585. doi: 10.1038/267585a0. [DOI] [PubMed] [Google Scholar]
- 2.Henzler-Wildman KA, Lei M, Thai V, Kerns SJ, Karplus M, Kern D. Nature. 2007;450:913. doi: 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- 3.Henzler-Wildman K, Kern D. Nature. 2007;450:964. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 4.Smock RG, Gierasch LM. Science. 2009;324:198. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kolano C, Helbing J, Bucher G, Sander W, Hamm P. J Phys Chem B. 2007;111:11297. doi: 10.1021/jp074184g. [DOI] [PubMed] [Google Scholar]
- 6.Frauenfelder H, Sligar SG, Wolynes PG. Science. 1991;254:1598. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 7.Brooks CL, Gruebele M, Onuchic JN, Wolynes PG. Proc Natl Acad Sci USA. 1998;95:11037. doi: 10.1073/pnas.95.19.11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolynes PG, Eaton WA, Fersht AR. Proc Natl Acad Sci USA. 2012;109:17770. doi: 10.1073/pnas.1215733109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sborgi L, Verma A, Sadqi M, Alba E, Muñoz V. Protein Supersecondary Structures. In: Kister AE, editor. Methods in Molecular Biology. Vol. 932. Springer Science+Business Media; New York: 2013. pp. 205–218. [DOI] [PubMed] [Google Scholar]
- 10.Henzler-Wildman KA, Thai V, Lei M, Ott M, Wolf-Watz M, Fenn T, Pozharski E, Wilson Ma, Petsko Ga, Karplus M, Hübner CG, Kern D. Nature. 2007;450:838. doi: 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- 11.Cammarata M, Levantino M, Schotte F, Anfinrud PA, Ewald F, Choi J, Cupane A, Wulff M, Ihee H. Nat Methods. 2008;5:881. doi: 10.1038/nmeth.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho HS, Dashdorj N, Schotte F, Graber T, Henning R, Anfinrud P. Proc Natl Acad Sci USA. 2010;107:7281. doi: 10.1073/pnas.1002951107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kern J, Alonso-Mori R, Hellmich J, Tran R, Hattne J, Laksmono H, Glöckner C, Echols N, Sierra RG, Sellberg J, Lassalle-Kaiser B, Gildea RJ, Glatzel P, Grosse-Kunstleve RW, Latimer MJ, McQueen TA, DiFiore Dr, Fry AR, Messerschmidt M, Miahnahri A, Schafer DW, Seibert MM, Sokaras D, Weng T-C, Zwart PH, White WE, Adams PD, Bogan MJ, Boutet Sb, Williams GJ, Messinger J, Sauter NK, Zouni A, Bergmann U, Yano J, Yachandra VK. Proc Natl Acad Sci USA. 2012;109:9721. doi: 10.1073/pnas.1204598109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung YO, Lee JH, Kim J, Schmidt M, Moffat K, Šrajer V, Ihee H. Nat Chem. 2013;5:212. doi: 10.1038/nchem.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamm P, Lim MH, Hochstrasser RM. J Phys Chem B. 1998;102:6123. [Google Scholar]
- 16.Hamm P, Lim M, DeGrado WF, Hochstrasser RM. Proc Natl Acad Sci USA. 1999;96:2036. doi: 10.1073/pnas.96.5.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asplund MC, Zanni MT, Hochstrasser RM. Proc Natl Acad Sci USA. 2000;97:8219. doi: 10.1073/pnas.140227997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Volk M. Eur J Org Chem. 2001:2605. [Google Scholar]
- 19.Kolano C, Helbing J, Kozinski M, Sander W, Hamm P. Nature. 2006;444:469. doi: 10.1038/nature05352. [DOI] [PubMed] [Google Scholar]
- 20.Lu HSM, Volk M, Kholodenko Y, Gooding E, Hochstrasser RM, DeGrado WF. J Am Chem Soc. 1997;119:7173. [Google Scholar]
- 21.Volk M, Kholodenko Y, Lu HSM, Gooding EA, DeGrado WF, Hochstrasser RM. J Phys Chem B. 1997;101:8607. [Google Scholar]
- 22.Rock RS, Chan SI. J Org Chem. 1996;61:1526. [Google Scholar]
- 23.Rock RS, Chan SI. J Am Chem Soc. 1998;120:10766. [Google Scholar]
- 24.Hansen KC, Rock RS, Larsen RW, Chan SI. J Am Chem Soc. 2000;122:11567. [Google Scholar]
- 25.Chen RPY, Huang JJT, Chen HL, Jan H, Velusamy M, Lee CT, Fann WS, Larsen RW, Chan SI. Proc Natl Acad Sci USA. 2004;101:7305. doi: 10.1073/pnas.0304922101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milanesi L, Waltho JP, Hunter CA, Shaw DJ, Beddard GS, Reid GD, Dev S, Volk M. Proc Natl Acad Sci USA. 2012;109:19563. doi: 10.1073/pnas.1211764109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tucker MJ, Courter JR, Chen J, Atasoylu O, Smith AB, III, Hochstrasser RM. Angew Chem Int Ed. 2010;49:3612. doi: 10.1002/anie.201000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tucker MJ, Abdo M, Courter JR, Chen J, Smith AB, III, Hochstrasser RM. J Photochem Photobiol, A. 2012;234:156. doi: 10.1016/j.jphotochem.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abdo M, Brown SP, Courter JR, Tucker MJ, Hochstrasser RM, Smith AB., III Org Lett. 2012;14:3518. doi: 10.1021/ol301490h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tucker MJ, Abdo M, Courter JR, Chen J, Brown SP, Smith AB, Hochstrasser RM. Proc Natl Acad Sci USA. 2013;110:17314. doi: 10.1073/pnas.1311876110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marqusee S, Robbins VH, Baldwin RL. Proc Natl Acad Sci USA. 1989;86:5286. doi: 10.1073/pnas.86.14.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verdine GL, Hilinski GJ. Methods in Enzymology: Protein Engineering for Therapeutics Part B. In: Wittrup KD, Verdine GL, editors. Methods in Enzymology. Vol. 503. Elsevier Academic Press Inc; San Diego: 2012. pp. 3–33. [Google Scholar]
- 33.Chakrabartty A, Kortemme T, Baldwin RL. Protein Sci. 1994;3:843. doi: 10.1002/pro.5560030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo PZ, Baldwin RL. Biochemistry. 1997;36:8413. doi: 10.1021/bi9707133. [DOI] [PubMed] [Google Scholar]
- 35.Barlos K, Gatos D, Koutsogianni S. J Pept Res. 1998;51:194. doi: 10.1111/j.1399-3011.1998.tb01216.x. [DOI] [PubMed] [Google Scholar]
- 36.Subiros-Funosas R, Prohens R, Barbas R, El-Faham A, Albericio F. Chem-Eur J. 2009;15:9394. doi: 10.1002/chem.200900614. [DOI] [PubMed] [Google Scholar]
- 37.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision D.01. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 38.Torres J, Adams PD, Arkin IT. J Mol Biol. 2000;300:677. doi: 10.1006/jmbi.2000.3885. [DOI] [PubMed] [Google Scholar]
- 39.Torres J, Kukol A, Goodman JM, Arkin IT. Biopolymers. 2001;59:396. doi: 10.1002/1097-0282(200111)59:6<396::AID-BIP1044>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 40.Fang C, Wang J, Charnley AK, Barber-Armstrong W, Smith AB, Decatur SM, Hochstrasser RM. Chem Phys Lett. 2003;382:586. [Google Scholar]
- 41.Fang C, Wang J, Kim YS, Charnley AK, Barber-Armstrong W, Smith AB, Decatur SM, Hochstrasser RM. J Phys Chem B. 2004;108:10415. [Google Scholar]
- 42.Fang C, Hochstrasser RM. J Phys Chem B. 2005;109:18652. doi: 10.1021/jp052525p. [DOI] [PubMed] [Google Scholar]
- 43.Marecek J, Song B, Brewer S, Belyea J, Dyer RB, Raleigh DP. Org Lett. 2007;9:4935. doi: 10.1021/ol701913p. [DOI] [PubMed] [Google Scholar]
- 44.Remorino A, Korendovych IV, Wu Y, DeGrado WF, Hochstrasser RM. Science. 2011;332:1206. doi: 10.1126/science.1202997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Remorino A, Hochstrasser RM. Acc Chem Res. 2012;45:1896. doi: 10.1021/ar3000025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.