

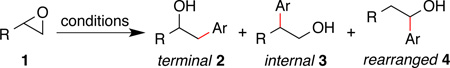
Abstract
Epoxides are versatile intermediates in organic synthesis, but have rarely been employed in cross-coupling reactions. We report that bipyridine-ligated nickel can mediate the addition of functionalized aryl halides, a vinyl halide, and a vinyl triflate to epoxides under reducing conditions. For terminal epoxides, the regioselectivity of the reaction depends upon the co-catalyst employed. Iodide co-catalysis results in opening at the less hindered position via an iodohydrin intermediate. Titanocene co-catalysis results in opening at the more hindered position, presumably via TiIII-mediated radical generation. 1,2-Disubstituted epoxides are opened under both conditions to form predominantly the trans product.
The formation of carbon-carbon bonds by the opening of epoxides with aryl nucleophiles has long played an important role in organic synthesis.1, 2 Three major addition products are commonly observed (eq 1). Although the opening of epoxides with heteroatom nucleophiles and stabilized or unstabilized carbon nucleophiles (most often cuprates) is well known,1,2, 3 transition-metal catalyzed coupling of epoxides with less reactive nucleophiles or carbon electrophiles is rare, despite the obvious synthetic utility.4
 |
(1) |
The transition-metal-catalyzed coupling of epoxides with π-systems, such as alkenes,5 alkynes,6 aldehydes,7 and CO,8 have been developed recently, but few such reactions that couple simple aryl groups to epoxides are known.9 Doyle reported a nickel-catalyzed coupling with arylboronic acids that formed rearranged products 4,10 similar to related reactions with allylmetal reagents.11 While Flowers and Gansäuer recently extended Ti(III)-epoxide chemistry12 to the intra-molecular, internal arylation of epoxides,13 the intermolecular arylation of epoxides to form products 2 and 3 remains limited to traditional methods.
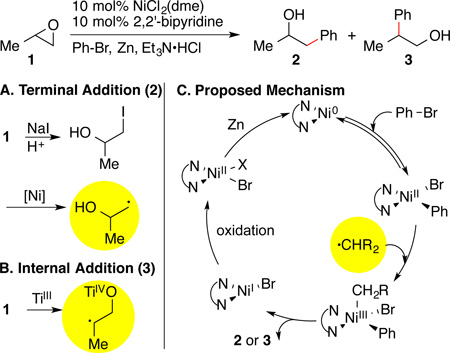
In order to bypass the difficulties associated with the reaction of epoxides with nucleophiles, we sought out an alternative cross-electrophile approach14 – the coupling of organic halides with epoxides. Initial attempts provided low conversion and primarily biaryl and arene were formed (Table 1, entry 1). In analogy to our proposed mechanism for the cross-electrophile coupling of aryl halides with alkyl halides,15, 16 it was evident that conversion of the epoxide into a radical was inefficient (Table 1C). Decomposition of the arylnickel intermediate forms biphenyl and benzene. Co-catalysis by iodide (Table 1A and entry 2) or titanium (Table 1B and entry 3) could enable the regioselective opening of epoxides by forming 2 via an iodohydrin17, 18 or 3 via a secondary radical.12, 19
Table 1.
Regiodivergent opening of epoxides.a
 | |||
|---|---|---|---|
| entry | additives | yield (%) | 2:3 |
| 1 | none | 9 | ND |
| 2 | 50 mol % NaI, 20 mol % py | 50 | 91:9 |
| 3 | 10 mol % Cp2TiCl2 | 40 | 1:4 |
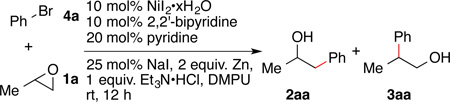
The iodide co-catalyzed reactions produced the highest yields when NiI2 was combined with a small amount of additional NaI (Table 2, entry 1). A suitable protic acid is also essential for high turnover number and frequency (entry 2), presumably because it assists in halohydrin formation.18 A number of acids were examined and only acids with a pKa around 9 in DMSO were effective (TEA•HX, DABCO•HCl, Me3N•HCl, i-Pr2NEt•HCl). Stronger acids (DABCO•2HCl, 2,4,6-collidine•HCl) and weaker acids (DBU•HCl) provided no cross product.20 Even though nickel is known to isomerize epoxides to aldehydes,10, 21 no rearranged products (4) were observed.
Table 2.
Nickel/iodide co-catalyzed epoxide ring opening with aryl halidesa
 | |||
|---|---|---|---|
| entry | deviation from above | yield (%)b | 2:3 |
| 1 | none | 81 | > 95:5 |
| 2 | No TEA•HCl | 32 | 89:11 |
| 3 | 15 mol % NaI | 67 | > 95:5 |
| 4 | 25 mol % Bu4NI in place of NaI | 79 | 95:5 |
| 5 | 12.5 mol % ZnI2 in place of NaI | 46 | 88:12 |
| 6 | 12.5 mol % MnI2 in place of NaI | 61 | 95:5 |
| 7 | No NaI, 12 hours | 68 | > 95:5 |
| 8 | No NaI, 24 hours | 75 | > 95:5 |
| 9 | 25 mol % NaBr in place of NaI | 52 | > 95:5 |
| 12 | NiI2 in place of NiI2·xH2O | 86 | > 95:5 |
| 13 | 2 equiv. of Mn in place of Zn | 59 | 95:5 |
| 14c | 2 equiv. of TDAE in place of Zn | 48d | >95:5 |
| 15 | Heated to 60 °C for 12 h | 50 | 93:7 |
| 16 | I-Ph in place of Br-Ph | 51 | 91:9 |
Reactions were run with 1 equiv of Et3N•HCl and 2 equiv of zinc dust; 0.1 equiv nickel catalyst, ligand; 0.2 equiv pyridine, 0.25 equiv sodium iodide, and 1.34 equiv of 1a in 3 mL DMPU.
Uncorrected GC yield of 2aa.
TDAE = tetrakis(dimethylamino)ethylene.
Bromobenzene (52%) remained, but no biaryl or benzene was formed.
The iodide source was optimally sodium iodide or Bu4NI (entry 1 and entry 4). Reactions run with zinc or manganese iodide were notably slower. Reactions conducted with a smaller amount (entry 3) or no sodium iodide (entry 7) were slower, but would produce the product in nearly the same yield after an extended reaction time (entry 8). The source of iodide was presumably the nickel source in the latter reaction. The addition of sodium bromide in place of sodium iodide decreased the yield and selectivity for cross product (entry 9 and Figure S1). Dimethylpropylene urea (DMPU) was found to be the optimal solvent and reactions in impure DMPU provided lower yields. Finally, zinc could be replaced by manganese or the organic reductant tetrakis(dimethylamino)ethylene (entries 13 and 14).22 The latter result argues against organozinc intermediates.
Examination of the scope for aryl and vinyl halides (Table 3) and for epoxides (Scheme 1) demonstrates the generality of the method. Not only electron-rich and electron-poor bromoarenes (Table 3, entries 2 and 5–6), but also bromoarenes with a variety of functional groups provided good yields of product. This synthesis of 2ea is four steps shorter than the only literature report.23 Bromoarenes bearing acidic functional groups (TsNHAr, entry 3; -OH, entry 9) were well tolerated. Reactions with phenols under basic conditions could result in ring-opening of the epoxide by the phenolate nucleophile, but none of this product was observed. Unprotected ketone, nitrile, and aldehyde groups were well tolerated (entries 6–8). Finally, a vinyl bromide and a vinyl triflate both coupled in high yield under our standard conditions (entries 12–13).
Table 3.
Scope of organic halidesa
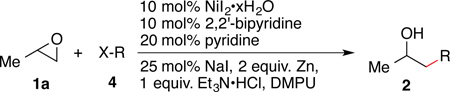 | ||||
|---|---|---|---|---|
| entry | R-X | productc | yield (%)b | |
| 1 | PhBr 4a | 2aa | 87 | |
| 2d | p-MeO-C6H4Br 4b | 2ba | 83 | |
| 3d | p-TsNH-C6H4Br 4c | 2ca | 84 | |
| 4d | p-tert-butyl-C6H4Br 4d | 2da | 72 | |
| 5 | p-CF3-C6H4Br 4e | 2ea | 79 | |
| 6 | p-CH3C(O)-C6H4Br 4f | 2fa | 84 | |
| 7 | p-NC-C6H4Br 4g | 2ga | 55 | |
| 8 | p-CHO-C6H4Br 4h | 2ha | 62e | |
| 9d | p-HO-C6H4Br 4i | 2ia | 58f | |
| 10 | o-CH3-C6H4Br 4j | 2ja | 99 | |
| 11d | 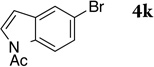 |
4k | 2ka | 99 |
| 12 | 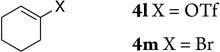 |
4l X = OTf | 2la | 93 |
| 13 | 4m X = Br | 2ma | 86 | |
As in Table 2, footnote a.
Isolated yield of purified product.
Numbering scheme: 2xy is the product of 4x and 1y.
Longer reaction time (24 h) was required.
Mixture of 2ha (55%) and 3ha (7%).
Mixture of 2ia (53%) and 3ia (5%).
Scheme 1.

Scope of epoxidesa
a As in Table 2, footnote a, but with 1 equiv of epoxide. Yield is the isolated yield of the product shown. b Mixture of 2ab (66%) and 3ab (10%). c Product of internal attack. 2ae was also isolated in 14% yield. d Longer reaction time (24 h) was required. e From 0.56 mmol of isobutene oxide.
Besides simple terminal epoxides like hexene oxide and octene oxide, terminal epoxides with protected alcohol or protected nitrogen functional groups were also good substrates (2ad, 2ag, 2ah, 2ak). Styrene oxide provided a mixture of products 2ae and 3ae, favoring the product of internal attack (3ae), consistent with the reported regioselectivity for halohydrin formation.18a More substituted 1,2 and 1,1-disubstituted epoxides form ring-opened products in good yield (2af, 2ai, 2al), but a 1,1,2-trisubstituted epoxide did not form product (data not shown). As expected, the opening of enantioenriched (R)-propylene oxide with bromobenzene afforded product 2aj with high enantiospecificity (99% es). Finally, a pre-formed iodohydrin substrate reacted to form product with regioselectivity (>95:5) and yield (64%) similar catalytic reactions (Scheme S1).
The refinement of titanium-mediated conditions required several small changes. Higher yields were obtained with manganese as the reductant. In many cases, significant amounts of epoxide remained at the end of the reaction due to inactivation of the titanium catalyst. In these reactions, we also observed that small amounts of the epoxide were converted to olefin. As reported by Gansäuer and Neese, the olefin likely arises from capture of the alkyl radical by another equivalent of titanium(III) and subsequent β-alkoxy elimination of an oxo-bridged titanium(IV) dimer, {Cp2Ti(Cl)-O-(Cl)TiCp2}.12e This titanium dimer is not easily reduced back to Ti(III), preventing further conversion of epoxide into product.
This side reaction was not eliminated by the use of additional titanium at the beginning of the reaction (2:1 Ti:Ni provided similar yield to 1:1 Ti:Ni, see Table S1), but the addition of another 4 mol% of titanium after two hours resulted in a 9% improvement in yield for cyclohexene oxide (eq 2). Note that cyclohexene oxide reacts with titanium co-catalysis to provide arylated product in higher selectivity (99:1 vs. 86:14) and yield (75% vs. 61%) than with iodide co-catalysis. The improved selectivity is likely the result of the steric bulk of –O(Cl)TiCp2shielding one face of the intermediate radical better than a simple –OH group.
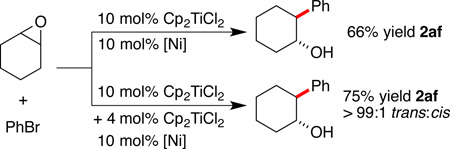 |
(2) |
These conditions were applied to several terminal epoxides (Table 4). In line with Gansäuer’s report,12d regioselectivity favored the internal product.24 Selectivity and yield with 1b was consistent with previous reports and calculations for irreversible radical formation followed by fast trapping by (L)NiII(Ar)Br (Table 1 and Table 4, entry 2), but the different regioselectivity in reactions with 1a and 1c suggests that in some cases rapid reversible radical formation followed by slower trapping is operative (entries 1 and 3).12d Functional-group compatibility is promising and electronics of the aryl halide do not dramatically alter the selectivity or yield (entries 4 and 5). For comparison with existing methods, a previous synthesis of 3na was 6 steps.25 Consistent with a titanium(III)-mediated radical process, 4-bromobenzaldehyde did not couple in high yield due to competing pinacol coupling26 and reaction of (R)-propylene oxide produced (±)-3aj.
Table 4.
Formation of branched products with Ni/Ti catalysisa
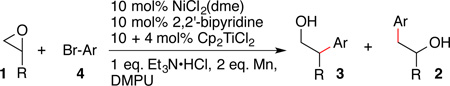 | ||||
|---|---|---|---|---|
| entry | R | Ar-Br | 3:2 | yield (%) |
| 1 | Me 1a | PhBr 4a | 3.3:1 | 70 (3aa) |
| 2 | C4H9 1b | 4a | 6:1 | 54 (3ab) |
| 3 | C6H13 1c | 4a | 99:1 | 41 (3ac) |
| 4 | 1a | p-MeO-C6H4Br 4b | 4:1 | 63 (3ba) |
| 5 | 1a | p-MeO2C-C6H4Br 4n | 3.5:1 | 62 (3na) |
Conditions: 2 equiv of Et3N•HCl and Mn dust were combined with 4 (1 equiv), 1 (1.34 equiv), and catalysts (10 mol%) in DMPU (0.17 M) and stirred for 12 h at rt. An additional portion of Cp2TiCl2was added after 2 h. Ratios of 3:2 were determined by GC analysis. Yield is an isolated mixture of 2 and 3.
In conclusion, mechanism-based reaction design has extended cross-electrophile coupling to epoxides for the first time. We have demonstrated that nickel/iodide and nickel/titanium catalyst systems can open epoxides with aryl bromides, vinyl bromides, and vinyl triflates and that the regioselectivity for reactions with terminal epoxides is governed by the co-catalyst. The results demonstrate the potential of new cross-coupling reactions based upon the interplay of radicals and transition-metal catalysis.27 Further improvement of the selectivities, including the development of enantio-selective variants12f–g and longer-lived titanium catalysts, is currently underway and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH (R01 GM097243). DJW is an Alfred P. Sloan Research Fellow.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Supplementary figures, detailed experimental procedures, characterization data, and copies of 1H, 19F, and 13C NMR spectra for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.For a recent large-scale example, see: Alam M, Wise C, Baxter CA, Cleator E, Walkinshaw A. Org. Process Res. Dev. 2012;16:435–441.
- 2.Reviews: Parker RE, Isaacs NS. Chem. Rev. 1959;59:737–799. Rosowsky A. Ethylene Oxides. In: Weissberger A, editor. Chemistry of Heterocyclic Compounds. Vol. 19. Hoboken, NJ: John Wiley & Sons, Inc.; 1964. pp. 1–523. Rao AS, Paknikar SK, Kirtane JG. Tetrahedron. 1983;39:2323–2367. Smith JG. Synthesis. 1984:629–656. Bartók M, Láng KL. Oxiranes. In: Hassner A, editor. Chemistry of Heterocyclic Compounds. Part 3. Vol. 42. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 1985. pp. 1–196. Pineschi M. Eur. J. Org. Chem. 2006:4979–4988. Hanson RM. Chem. Rev. 1991;91:437–475. Taylor SK. Tetrahedron. 2000;56:1149–1163. Hodgson DM, Gibbs AR, Lee GP. Tetrahedron. 1996;52:14361–14384.
- 3.Alkylaluminum reagents can favor internal addition, see the following and references cited therein: Schneider C, Brauner J. Eur. J. Org. Chem. 2001:4445–4450.
- 4.Nielsen LPC, Jacobsen EN. Chapter 7: Catalytic Asymmetric Epoxide Ring-opening Chemistry. In: Yudin AK, editor. Aziridines and Epoxides in Organic Synthesis. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA; 2006. pp. 229–269. [Google Scholar]
- 5.Ikeda Y, Yorimitsu H, Shinokubo H, Oshima K. Adv. Synth. Catal. 2004;346:1631–1634. [Google Scholar]
- 6.(a) Beaver MG, Jamison TF. Org. Lett. 2011;13:4140–4143. doi: 10.1021/ol201702a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. J. Am. Chem. Soc. 2004;126:4130–4131. doi: 10.1021/ja0491735. [DOI] [PubMed] [Google Scholar]; (c) Miller KM, Huang W-S, Jamison TF. J. Am. Chem. Soc. 2003;125:3442–3443. doi: 10.1021/ja034366y. [DOI] [PubMed] [Google Scholar]
- 7.Molinaro C, Jamison TF. Angew. Chem. Int. Ed. 2005;44:129. doi: 10.1002/anie.200461705. [DOI] [PubMed] [Google Scholar]
- 8.Examples with CO and isocyanates: Getzler YDYL, Mahadevan V, Lobkovsky EB, Coates GW. J. Am. Chem. Soc. 2002;124:1174–1175. doi: 10.1021/ja017434u. Mahadevan V, Getzler YDYL, Coates GW. Angew. Chem. Int. Ed. 2002;41:2781–2784. doi: 10.1002/1521-3773(20020802)41:15<2781::AID-ANIE2781>3.0.CO;2-S. Kramer JW, Lobkovsky EB, Coates GW. Org. Lett. 2006;8:3709–3712. doi: 10.1021/ol061292x. Church TL, Byrne CM, Lobkovsky EB, Coates GW. J. Am. Chem. Soc. 2007;129:8156–8162. doi: 10.1021/ja069065d.
- 9.The coupling of vinyl epoxides with a variety of nucleophiles via allylmetal intermediates is well developed. See Muzart J. Eur. J. Org. Chem. 2011:4717–4741. Trost BM, Molander GA. J. Am. Chem. Soc. 1981;103:5969–5972. Tsuji J, Kataoka H, Kobayashi Y. Tetrahedron Lett. 1981;22:2575–2578.
- 10.Nielsen DK, Doyle AG. Angew. Chem. Int. Ed. 2011;50:6056–6059. doi: 10.1002/anie.201101191. [DOI] [PubMed] [Google Scholar]
- 11.(a) Jiang N, Hu Q, Reid CS, Lu Y, Li C-J. Chem. Commun. 2003:2318–2319. doi: 10.1039/b305161g. [DOI] [PubMed] [Google Scholar]; (b) Roy UK, Roy S. Tetrahedron. 2006;62:678–683. [Google Scholar]
- 12.(a) Gansäuer A, Narayan S. Adv. Synth. Catal. 2002;344:465–475. [Google Scholar]; (b) RajanBabu TV, Nugent WA. J. Am. Chem. Soc. 1988;110:8561–8562. [Google Scholar]; (c) RajanBabu TV, Nugent WA. J. Am. Chem. Soc. 1994;116:986–997. [Google Scholar]; (d) Gansäuer A, Barchuk A, Keller F, Schmitt M, Grimme S, Gerenkamp M, Mück-Lichtenfeld C, Daasbjerg K, Svith H. J. Am. Chem. Soc. 2007;129:1359–1371. doi: 10.1021/ja067054e. [DOI] [PubMed] [Google Scholar]; (e) Gansäuer A, Fleckhaus A, Lafont MA, Okkel A, Kotsis K, Anoop A, Neese F. J. Am. Chem. Soc. 2009;131:16989–16999. doi: 10.1021/ja907817y. [DOI] [PubMed] [Google Scholar]; (f) Gansäuer A, Shi L, Otte M. J. Am. Chem. Soc. 2010;132:11858–11859. doi: 10.1021/ja105023y. [DOI] [PubMed] [Google Scholar]; (g) Gansäuer A, Fan C-A, Keller F, Karbaum P. Chem.—Eur. J. 2007;13:8084–8090. doi: 10.1002/chem.200701021. [DOI] [PubMed] [Google Scholar]
- 13.Gansäuer A, Behlendorf M, von Laufenberg D, Fleckhaus A, Kube C, Sadasivam DV, Flowers RA. Angew. Chem. Int. Ed. 2012;51:4739–4742. doi: 10.1002/anie.201200431. [DOI] [PubMed] [Google Scholar]
- 14.(a) Everson DA, Shrestha R, Weix DJ. J. Am. Chem. Soc. 2010;132:920–921. doi: 10.1021/ja9093956. [DOI] [PubMed] [Google Scholar]; (b) Everson DA, Jones BA, Weix DJ. J. Am. Chem. Soc. 2012;134:6146–6159. doi: 10.1021/ja301769r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Everson DA, George DT, Weix DJ, Buergler JF, Wood JL. Org. Synth. 2013;90:200–214. doi: 10.15227/orgsyn.090.0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biswas S, Weix DJ. J. Am. Chem. Soc. 2013;135:16192–16197. doi: 10.1021/ja407589e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.See also Hu’s reaction of [NiII](Alkyl) with an alkyl radical to form cross-product: Breitenfeld J, Ruiz J, Wodrich MD, Hu X. J. Am. Chem. Soc. 2013;135:12004–12012. doi: 10.1021/ja4051923.
- 17.Oshima invoked a bromohydrin in reactions with styrenes.5
- 18.(a) Chini M, Crotti P, Gardelli C, Macchia F. Tetrahedron. 1992;48:3805. [Google Scholar]; (b) Soroka M, Goldeman W. Tetrahedron. 2005;61:4233. [Google Scholar]; (c) Mathieu-Pelta I, Evans SA. J. Org. Chem. 1992;57:3409. [Google Scholar]; (d) Eisch JJ, Liu ZR, Ma X, Zheng GX. J. Org. Chem. 1992;57:5140–5144. [Google Scholar]; (e) Bonini C, Righi G. Synthesis. 1994;1994:225–238. [Google Scholar]
- 19.Coupling of these intermediates with metal hydrides is known: Gansäuer A, Fan C, Piestert F. J. Am. Chem. Soc. 2008;130:6916–6917. doi: 10.1021/ja801232t. Gansäuer A, Otte M, Shi L. J. Am. Chem. Soc. 2011;133:416–417. doi: 10.1021/ja109362m.
- 20.(a) Kolthoff IM, Chantooni MK, Bhowmik S. J. Am. Chem. Soc. 1968;90:23–28. [Google Scholar]; (b) Benoit RL, Lefebvre D, Fréchette M. Can. J. Chem. 1987;65:996–1001. [Google Scholar]; (c) Bordwell FG. Acc. Chem. Res. 1988;21:456–463. [Google Scholar]
- 21.Miyashita A, Shimada T, Sugawara A, Nohira H. Chem. Lett. 1986;15:1323–1326. [Google Scholar]
- 22.Kuroboshi M, Tanaka M, Kishimoto S, Goto K, Mochizuki M, Tanaka H. Tetrahedron Lett. 2000;41:81–84. [Google Scholar]
- 23.Schadt FL, Lancelot CJ, Schleyer PvR. J. Am. Chem. Soc. 1978;100:228–246. [Google Scholar]
- 24.Although TiIVX4 has been reported to form halohydrins from internal attack,18d–e the conditions were different (hydrocarbon solvent, low temperature, slow addition of epoxide) and the formation of Cp2TiIIICl is evident from the color change of the reaction (dark red to olive green).
- 25.Matsumoto T, Ishida T, Yoshida T, Terao H, Takeda Y, Asakawa Y. Chem. Pharm. Bull. 1992;40:1721–1726. doi: 10.1248/cpb.40.1721. [DOI] [PubMed] [Google Scholar]
- 26.(a) Hirao T. Top. Curr. Chem. 2007;279:53–75. [Google Scholar]; (b) Gansäuer A. Chem. Commun. 1997:457–458. [Google Scholar]
- 27.(a) Ford L, Jahn U. Angew. Chem. Int. Ed. 2009;48:6386–6389. doi: 10.1002/anie.200901761. [DOI] [PubMed] [Google Scholar]; (b) Jahn U. Radicals in Transition Metal Catalyzed Reactions? Transition Metal Catalyzed Radical Reactions?: A Fruitful Interplay Anyway, Parts I–III. In: Heinrich M, Gansäuer A, editors. Radicals in Synthesis III. Vol. 320. Springer Berlin Heidelberg; 2012. pp. 191–451. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.