

Abstract
Internal tandem duplications in the fms-like tyrosine kinase receptor (FLT3-ITDs) confer a poor prognosis in acute myeloid leukemia. We hypothesized that increased recruitment of the protein tyrosine phosphatase, Shp2, to FLT3-ITDs contributes to FLT3 ligand (FL)-independent hyperproliferation and STAT5 activation. Co-immunoprecipitation demonstrated constitutive association of Shp2 with the FLT3-ITD, N51-FLT3, as well as with STAT5. Knock-down of Shp2 in Baf3/N51-FLT3 cells significantly reduced proliferation while having little effect on WT-FLT3-expressing cells. Consistently, mutation of N51-FLT3 tyrosine 599 to phenylalanine or genetic disruption of Shp2 in N51-FLT3-expressing bone marrow low density mononuclear cells reduced proliferation and STAT5 activation. In transplants, genetic disruption of Shp2 in vivo yielded increased latency to and reduced severity of FLT3-ITD-induced malignancy. Mechanistically, Shp2 co-localizes with nuclear phospho-STAT5, is present at functional interferon-γ activation sites (GAS) within the BCL2L1 promoter, and positively activates the human BCL2L1 promoter, suggesting that Shp2 works with STAT5 to promote pro-leukemogenic gene expression. Further, using a small molecule Shp2 inhibitor, the proliferation of N51-FLT3-expressing bone marrow progenitors and primary AML samples was reduced in a dose-dependent manner. These findings demonstrate that Shp2 positively contributes to FLT3-ITD-induced leukemia and suggest that Shp2 inhibition may provide a novel therapeutic approach to acute myeloid leukemia.
Keywords: Acute Myeloid Leukemia, FLT3-ITD, Shp2, STAT5
INTRODUCTION
Acute myeloid leukemia (AML) is a lethal disease with only a 20% cure rate at 5 years post-diagnosis. Internal tandem duplications (ITDs), in-frame insertions or duplication of several amino acids near the juxtamembrane domain, in fms-like tyrosine kinase receptor (FLT3), are seen in approximately 25% of AML patients and confer a poor prognosis (1–3). FLT3-ITD receptors are constitutively phosphorylated (4–8) and function in a ligand-independent manner. Despite substantial effort devoted to the development of FLT3 kinase inhibitors, as single agents, the activity of these inhibitors in AML is limited (9). Therefore, clarification of the effector molecules promoting transformation or FLT3-ITD-expressing leukemic cells is needed to identify novel targets for therapy.
Shp2 is a protein tyrosine phosphatase containing two tandemly arranged SH2 domains at its amino terminal end (N-SH2 and C-SH2), a central phosphatase domain (PTP), and a carboxy terminal tail with tyrosine phosphorylation sites (Y580 and Y542) that potentially regulate its catalytic activity (10–12). Although Shp2 is widely expressed, homozygous mutant (Shp2−/−) embryonic stem (ES) cells are unable to contribute to hematopoietic tissues of chimeric mice, suggesting a rigorous requirement for Shp2 in hematopoietic cell growth (13). Several Shp2 gain-of-function mutations have been described in myeloid malignancies (14–18), and the Shp2 protein is overexpressed in human AML samples (19). Given that Shp2 is critical for normal hematopoiesis (13, 20), that dysregulated Shp2 function contributes to myeloid malignancies, and that Shp2 has been shown to interact with WT FLT3 tyrosine 599 (21), which is commonly duplicated in FLT3-ITDs (1, 4, 7, 22, 23), a positive role for Shp2 in FLT3-ITD-induced signaling and leukemogenesis is implied; however, a functional role for Shp2 in oncogenic FLT3-ITD-induced leukemogenesis has not previously been demonstrated.
The best-described neomorphic signal induced by FLT3-ITDs is constitutive activation of the transcription factor signal transducer and activator of transcription, STAT5. While WT FLT3 modestly activates STAT5, cells bearing FLT3-ITDs demonstrate robust constitutive STAT5 phosphorylation (4–7, 24–26). However, the collaborating molecules promoting enhanced STAT5 activation are unknown. In vitro studies indicate that Shp2 negatively regulates both STAT3 and STAT5 (27–32); however, paradoxically, Shp2 plays a positive role in activating STAT5 phosphorylation and regulating STAT5-responsive gene expression in mammary epithelial cells (33–36). Furthermore, although Shp2 is conventionally presumed to be a cytoplasmic signaling protein, robust Shp2 expression has been detected in the nuclei of human AML samples (19); however, the role of nuclear Shp2 in the setting of AML has never been investigated.
Given the well-documented role of Shp2 in WT FLT3-induced signaling (21, 37–39), in STAT5-mediated cell growth and gene expression (33–36), and in hematopoietic progenitor and stem cell function (13, 20, 40–42), we hypothesized that Shp2 contributes to FLT3-ITD-induced leukemogenesis mechanistically by working with STAT5 to promote STAT5-responsive gene expression and functionally by contributing to hematopoietic progenitor proliferation. We demonstrate that Shp2 is constitutively associated with FLT3-ITD and STAT5 in FLT3-ITD-expressing cells, that mutation of the previously-defined Shp2-binding site on FLT3, Y599 (21), reduces FLT3-ITD-induced hyperproliferation and STAT5 phosphorylation, and that genetic disruption of Shp2 increases the latency to FLT3-ITD-induced malignancy in vivo. We further demonstrate that Shp2 co-localizes with nuclear phospho-STAT5, that Shp2 is detected at levels similar to that of STAT5 at two functional STAT5-responsive elements within the human BCL2L1 promoter, and that reduced Shp2 expression results in reduced human BCL2L1 promoter activity. Lastly, we demonstrate that a novel Shp2 inhibitor, II-B08, inhibits the proliferation and STAT5 activation of FLT3-ITD-bearing cells as well as primary human AML samples. Collectively, these findings demonstrate that the protein tyrosine phosphatase, Shp2, positively contributes to FLT3-ITD-induced leukemogenesis, and provide mechanistic and functional rationale for targeting Shp2 as a novel therapeutic modality in AML.
EXPERIMENTAL PROCEDURES
Cell Lines and Patient Samples
HL60, MV411, or Baf3 cells transduced with WT-FLT3 or N51-FLT3 (43) were utilized for functional and biochemical studies. For Shp2 knock-down studies, WT FLT3- and N51-FLT3-transduced Baf3 cell lines or MV411 cells were transfected with a vector encoding U6 polymerase III–directed Shp2-specific short-hairpin RNA (shRNA) or scrambled shRNA (Origene #TR501795) and selected in 1 µg/mL puromycin (Baf3 cells) or 0.25 µg/mL puromycin (MV411 cells). Blast cells from the bone marrow of individuals with AML were obtained at the time of diagnostic testing after informed consent. Approval was obtained from the institutional review boards of Indiana University School of Medicine. Low density cells were isolated over Ficoll-Hypaque and processed as described previously (44). II-B08 was synthesized as previously described (45).
Animal Husbandry
Mice bearing floxed Ptpn11 alleles and the Mx1Cre transgene (Shp2flox/flox;MxCre+) (46) and negative control (Shp2flox/flox;MxCre−) animals received three intraperitoneal injections with 300 µg polyI:polyC (Sigma, St Louis, MO) to induce Ptpn11 recombination. Recipient mice for all transplant assays were F1 (first generation cross between C57Bl/6 and BoyJ, CD45.2+;CD45.1+) and were bred in the Indiana University In Vivo Therapeutics Core. All mice were maintained under specific pathogen-free conditions at the Indiana University Laboratory Animal Research Center (Indianapolis, IN) and this study was approved by the Institutional Animal Care and Use Committee of the Indiana University School of Medicine.
Retroviral Transduction of Murine Bone Marrow Cells
Murine bone marrow low density mononuclear cells (LDMNCs) were retrovirally transduced (MSCV-WT-FLT3, MSCV-N51-FLT3, MSCV-N51-FLT3Y599F1, MSCV-N51-FLT3Y599F1/2) as previously described (47). Cells were sorted for enhanced green fluorescent protein positive (EGFP) using fluorescence activated cell sorting (FACS) and subjected to 3H-thymidine incorporation assays (48), apoptosis assays, or differentiated to macrophages for biochemical assays (47).
Immunoprecipitation and Immunoblots
To examine the FLT3-Shp2 interaction, FLT3 was immunoprecipitated with anti-FLT3 (C-20, Santa Cruz Biotechnology) and protein A sepharose beads (CL-4B, GE Healthcare). Blots were probed with anti-Shp2 (C-18, Santa Cruz Biotechnology) and re-probed with anti-FLT3. To examine Shp2-STAT5 interaction, proteins were immunoprecipitated anti-Shp2 (C-18, Santa Cruz Biotechnology) or with anti-STAT5 (C17, Santa Cruz Biotechnology), and blots were probed reciprocally with anti-STAT5 (C17) or anti-Shp2 (C-18), respectively. To examine Shp2 and STAT5 phosphorylation, signals were detected with anti-phospho-Shp2 and anti-phospho-STAT5, respectively (Cell Signaling, Beverly, MA). Nuclear and cytoplasmic proteins were isolated using NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific). Enrichment of cytoplasmic and nuclear extracts was verified by probing with anti-GAPDH (Biodesign International, Saco, ME) or with anti-PARP1 (A-20, Santa Cruz Biotechnology).
Thymidine Incorporation and Apoptosis Assays
Transduced cells were washed and starved in 0.2% BSA for 4 to 8 hours, depending on cell type, followed by culture in IMDM plus 10% fetal bovine serum (FBS) in the absence or presence of indicated growth factors. Cells were subsequently pulsed with 1.0 μCi of [3H] thymidine for 6 to 8 hours and harvested using an automated 96-well cell harvester (Brandel, Gaithersburg, MD). For apoptosis assays, transduced EGFP+ bone marrow LDMNCs were cultured in IMDM plus 2% FBS with DMSO (control) or with 5 µM, 15 µM, or 30 µM II-B08 and incubated overnight at 37°C. Cells were stained with allophycocyanin (APC)-conjugated annexin V and propidium iodide (PI) and analyzed by flow cytometry.
Immunofluorescence Confocal Microscopy
MV411 or HL60 cells were loaded into 35 mm glass bottom microwell dishes (MatTek Cultureware, Ashland, MA), fixed with methanol/acetone (1/1) at −20° C, and air dried at room temperature. Cells were blocked with 2% BSA, 10% goat serum in PBS, incubated with anti-Shp2 (BD Biosciences, #610621) or anti-phospho-STAT5 (Cell Signaling Technology, #9314), secondarily stained with Alexa-647 goat anti-mouse and Alexa-488 goat anti-rabbit, respectively, and counterstained with DAPI 5 µg/mL. Cells were imaged on Olympus FV1000-MPE confocal/multiphoton microscope (Olympus America Inc, Center Valley, PA) using UPLAPO 60X W/NA:1.20 objective lens. Samples were scanned at 405nm, 488nm, 635nm excitation wavelength, and images were collected at 461nm, 520nm, and 668nm emission wavelength, respectively. To quantify the co-localization of Shp2 and phospho-STAT5 in MV411 and HL60 cells, co-localization analysis was performed using Metamorph software (Measure Colocalization plugin, Universal Imaging, Downington, PA).
Chromatin Immunoprecipitation Assay
Chromatin immunoprecipitaton assays were performed using a ChIP assay kit (Upstate Biotechnology, Lake Placid, NY). Extracts were sonicated to shear chromatin DNA followed by immunoprecipitation using anti-STAT5 (N20, Santa Cruz Biotechnology), anti-Shp2 (C18, Santa Cruz Biotechnology), or normal rabbit IgG (Millipore). Immuoprecipitates were washed and eluted with low and high stringency buffers. Histone-DNA crosslinks were reversed, proteins were digested with proteinase K, and DNA was recovered by phenol/chloroform extraction. Quantitative PCRs using SYBR green were conducted using forward primer 5’-CCC AGG GAG TGA CTT TCC GAG GAA-3’ and reverse primer 5’-TCG AAA GCA CCA GTG GAC TCT GA-3’, which generates a 90-bp band spanning the human BCL2L1 promoter including two interferon-γ activation sites (GAS, consensus sequence TTCN2-N4GAA) (49). The immunoprecipitated fragments were expressed as a percentage of the total chromatin used in the sample. Amplified fragments were additionally electrophoresed on agarose gels to validate fragment size and sequenced for verification of the predicted BCL2L1 promoter fragment.
Reporter Assays
Puromycin-selected MV411 cells were transfected with pBCL2L1-L (human BCL2L1 promoter, -356 to -1, cloned into pGL3-basic, Promega, driving luciferase expression) and pTK-RL (Progmega, internal control), and assays for luciferase and Renilla luciferase were conducted as previously described (50).
Transplant Studies
Bone marrow LDMNCs were isolated from Shp2flox/flox;MxCre+ animals (C57Bl/6 background, CD45.2+) and lineage depleted (Lin−) (EasySep, Stem Cell Technologies, Vancouver, CA). Lin− cells were retrovirally transduced with MSCV-N51-FLT3 and sorted to homogeneity as described previously. 1 × 106 transduced cells were injected intravenously into lethally irradiated (1100 cGy split dose) F1 recipients (CD45.2+; CD45.1+) with 1 – 1.5 × 105 F1 spleen LDMNCs for radioprotection. Transplanted animals recovered for 4 – 6 weeks, and then animals were separated into two groups for treatment with either PBS (positive control animals) or 300 µg polyI:polyC.
Statistical Analyses
For Shp2 knockdown Baf3, MV411, and HL60 cell line studies, unpaired, two-tailed, Student’s t test was used. For combined data from murine primary cell studies, comparisons were conducted using random effects ANOVA to deal with repeated measurements within each independent assay. The P-value for pairwise comparisons was adjusted using Tukey’s method (51). For transplant studies, malignancy specific survival was analyzed using Kaplan Meier estimation where malignancy-related death was the pertinent event. Mice alive at the last day of follow-up were incorporated into the analysis with time to event censored as the last day known alive. Mice whose deaths were not related to malignancy were also incorporated into the analysis with time to event censored as the death time. P values were generated using the log-rank tests. For effect of II-B08 on primary human AML cell proliferation, linear mixed effects models with random intercepts were used to evalute the effects of different II-B08 concentrations. For all analyses, statistical significance is set at 0.05.
RESULTS
Shp2 is constitutively associated with FLT3 and STAT5 in FLT3-ITD-expressing cells
To examine if Shp2 interaction with FLT3 and STAT5 is enhanced in FLT3-ITD-expressing cells, we utilized Baf3 cell lines expressing WT FLT3 or a FLT3-ITD termed “N51-FLT3” which contains an insertion of 7 amino acids (Figure 1A) (22, 26, 52). Shp2 was recruited to WT FLT3 in a FL-responsive manner (Figure 1B, lanes 1–2), while Shp2 was strongly and constitutively associated with N51-FLT3 (Figure 1B, lanes 3–4). Furthermore, the Shp2-STAT5 interaction was significantly higher in N51-FLT3-expressing cells compared to WT FLT3-expressing cells (Figures 1C and 1D), providing biochemical evidence that the contribution of Shp2 to FLT3-ITD-induced leukemogenesis may be mediated through interaction with and activation of STAT5.
Figure 1. The protein tyrosine phosphatase, Shp2, interacts constitutively with FLT3 and STAT5 in FLT3-N51-expressing Baf3 cells and functionally contributes to FLT3-ITD-induced hyperproliferation.

(A) Schematic diagram showing amino acid duplication for the FLT3-N51 internal tandem duplication (ITD). (B) Total cellular protein extracts from baseline and FL-stimulated Baf3/WT-FLT3 or Baf3/N51-FLT3 cells were isolated, immunoprecipitated (IP) with anti-FLT3 and immunoblotted (IB) with anti-Shp2 and anti-FLT3. Total cellular protein extracts were immunoprecipitated (IP) with (C) anti-STAT5 or (D) anti-Shp2 and immunoblotted (IB) with anti-Shp2 or anti-STAT5, respectively. (E) 3H-thymidine incorporation and immunoblot analyses of Shp2 expression in Baf3/WT-FLT3 and Baf3/N51-FLT3 cells transfected with scrambled shRNA (SC) or shRNA specifically targeting Shp2 (KD). Representative of two independent experiments with similar results, n=4, *p<0.05 for N51 KD v. N51 SC, statistical analysis using unpaired, two-tailed, student’s t test. (F) Immunoblot analysis of Shp2 expression, STAT5 phosphorylation, and total STAT5 expression in Baf3/N51-FLT3 cells transfected with scrambled shRNA (SC) or shRNA specifically targeting Shp2 (KD). (G) Total cellular proteins from Baf3/N51-FLT3 cells transfected with scrambled shRNA (SC) or Shp2-specific shRNA (KD) were isolated, immunoprecipitated (IP) with anti-FLT3 and immunoblotted (IB) with anti-STAT5, anti-phospho-STAT5, anti-Shp2, and anti-FLT3.
Knockdown of Shp2 significantly reduces hyperproliferation of FLT3-ITD-expressing cells
Using Shp2-targeted shRNA, Shp2 expression was reduced by at least 50% in Baf3/WT-FLT3 and Baf3/N51-FLT3 cells (Figure 1E, lower panel). Baf3/WT-FLT3 cells expressing Shp2-specific shRNA demonstrated no reduction in proliferation compared to cells expressing scrambled shRNA. However, the Baf3/N51-FLT3 cells expressing Shp2-specific shRNA demonstrated significantly reduced proliferation, implying that FLT3-ITD-bearing leukemic cells may be more sensitive to Shp2 inhibition than WT FLT3-expressing cells. By immunoblot, Baf3/N51-FLT3 cells with reduced Shp2 also demonstrated diminished phospho-STAT5 (Figure 1F). Co-immunoprecipitation assays demonstrated that STAT5 and phospho-STAT5 persisted in the FLT3-ITD-containing complex even in the absence of Shp2 (Figure 1G), indicating that while Shp2 positively contributes to STAT5 constitutive activation and hyperproliferation in FLT3-ITD-expressing cells, loss of Shp2 does not ablate STAT5 interaction with FLT3-ITD.
Genetic disruption of Ptpn11 reduces FLT3-ITD-induced hyperproliferation and STAT5 hyperphosphorylation
To corroborate the shRNA studies, we utilized mice bearing a conditional deletion of Ptpn11, the gene encoding Shp2. At 4 to 6 weeks of age, the Shp2flox/flox;Mx1Cre+ and Shp2flox/flox;Mx1Cre− mice were treated with polyI:polyC to induce Cre expression in the hematopoietic compartment. Figure 2A demonstrates the predicted Ptpn11 allele recombination and reduced Shp2 expression in bone marrow LDMNCs following polyI:polyC treatment. Bone marrow LDMNCs were transduced with WT-FLT3 or N51-FLT3, sorted for EGFP (Supplemental Figure 1), and subjected to 3H-thymidine incorporation. Similar to that found with shRNA-mediated reduced Shp2 expression, genetic disruption of Shp2 caused a significant reduction in the proliferation of N51-FLT3-expressing cells (Figure 2B) and resulted in reduced STAT5 phosphorylation (Figure 2C).
Figure 2. Genetic disruption of Ptpn11 or mutation of N51-FLT3 tyrosine (Y) 599 and duplicated Y599 to phenylalanine (F) diminishes FLT3-ITD-induced hyperproliferation and STAT5 phosphorylation.

(A) Genomic DNA isolated from polyI:polyC–treated Shp2flox/flox;Mx1Cre− (negative control) and Shp2flox/flox;Mx1Cre+ tail and bone marrow LDMNCs was subjected to PCR for the original flox allele (~1300 bp) and the polyI:polyC-induced recombined null allele (~400 bp) and protein from bone marrow LDMNCs from polyI:polyC-treated Shp2flox/flox;Mx1Cre− and Shp2flox/flox;Mx1Cre+ mice was analyzed by immunoblot to examine Shp2 expression. (B) 3H-thymidine incorporation assay of transduced, sorted bone marrow progenitors in the absence and presence of FL 50 ng/mL; two independent experiments combined with n=4 replicates per experiment, ^p<0.0001 for N51-FLT3 v. WT-FLT at baseline in Cre− cells, ^^p<0.0001 for N51-FLT3 v. WT-FLT in response to FL in Cre− cells, *p<0.0001 for N51-FLT3 in Cre+ cells v. Cre− cells at baseline, and **p<0.0001 for N51-FLT3 in Cre+ cells v. Cre− cells in response to FL, statistical analysis performed using random effects ANOVA. (C) Immunoblot analysis of STAT5 phosphorylation and total STAT5 expression in exponentially growing transduced, sorted bone marrow LDMNCs from polyI:polyC-treated Shp2flox/flox;Mx1Cre− and Shp2flox/flox;Mx1Cre+ mice. (D) Schematic diagram indicating mutation of N51-FLT3 to N51-FLT3Y599F1 or N51-FLT3Y599F1/2. (E) Murine bone marrow LDMNCs were transduced with WT-FLT3, N51-FLT3, N51-FLT3Y599F1, or N51-FLT3Y599F1/2, sorted by FACS, and subjected to 3H-thymidine incorporation assay in the absence and presence of FL 50 ng/mL; representative of two independent experiments with similar results, n=5 – 8, *p<0.05 for N51-FLT3Y599F1/2 v. N51-FLT3 at baseline and in the presence of FL 50 ng/mL, statistical analysis using unpaired, two-tailed, student’s t test. (F) Immunoblot analysis of STAT5 phosphorylation, total STAT5, and FLT3 expression in retrovirally transduced and sorted WT-FLT3-, N51-FLT3-, N51-FLT3Y599F1-, or N51-FLT3Y599F1/2-expressing bone marrow-derived macrophages in the absence and presence of FL.
Mutation of N51-FLT3 tyrosine (Y) 599 and duplicated Y599 to phenylalanine (F) reduces FLT3-ITD-induced hyperproliferation and STAT5 hyperphosphorylation
As reduced Shp2 expression resulted in reduced FLT3-ITD-induced proliferation, we next examined the functional role of Y599, shown previously to bind Shp2 and promote WT FLT3 signaling (21). We generated point mutants of N51-FLT3 including N51-FLT3Y599F1 bearing tyrosine (Y) to phenylalanine (F) mutation at the original Y599, and N51-FLT3Y599F1/2 bearing Y to F mutation at both the original and duplicated Y599 (Figure 2D). While mutation of the original Y599 alone failed to reduce proliferation or STAT5 phosphorylation, mutation of both the original and duplicated Y599 significantly reduced cellular proliferation and phospho-STAT5 levels (Figure 2E and 2F). Given that Shp2 has been shown to interact with Y599 of WT FLT3 (21), these findings imply that Shp2 specifically contributes to FLT3-ITD-induced hyperproliferation, and support a model whereby increased Shp2 recruitment to FLT3-ITD original and duplicated Y599 residues promotes STAT5 hyperactivation.
Genetic disruption of Ptpn11 reduces FLT3-ITD+ malignancy-induced death in vivo
Murine bone marrow cells transduced with FLT3-ITDs have been reported to induce myeloproliferative disorder (MPD) following transplantation in vivo (26). We hypothesized that genetic disruption of Ptpn11 in N51-FLT3 expressing cells in vivo would decrease the severity of and increase the latency to malignancy-induced death. Lin− bone marrow cells from Shp2flox/flox;Mx1Cre+ animals were retrovirally transduced with N51-FLT3, sorted to homogeneity, and transplanted into lethally irradiated recipients (Figure 3A). Given that previous reports indicated that polyI:polyC-induced loss of Shp2 severely inhibited the engraftment of Shp2flox/flox;Mx1Cre+ cells (42), we transplanted animals with N51-FLT3-transduced cells first, and then treated with polyI:polyC to delete Shp2 (Figure 3A). Four – six weeks following transplantation, peripheral blood was collected, and animals were divided into two groups with equal CD45.2 chimerism, EGFP expression, and peripheral WBC counts for treatment with either phosphate buffered saline (PBS, control animals) or polyI:polyC (to induce disruption of Ptpn11, experimental animals) (Figure 3A and Supplemental Figure 2A – 2C).
Figure 3.

(A) Schematic diagram showing transplant design. Lin− bone marrow cells from Shp2flox/flox;Mx1Cre+ animals (C57Bl/6 background, CD45.2+) were retrovirally transduced with N51-FLT3, sorted to homogeneity, and transplanted into lethally irradiated F1 recipients (first generation cross between C57Bl/6 and BoyJ, CD45.1+, CD45.2+) with 100,000 to 150,000 F1 splenocytes for radioprotection. (B) Spleen weight from mice at death with histopathologic diagnosis of malignancy comparing PBS-treated mice (control) to polyI:polyC-treated mice (genetic deletion of Shp2), n=16 in the PBS group and n=10 in the polyI;polyC group, p<0.05 by unpaired, two-tailed student’s t test. (C) Kaplan-Meier analysis of malignancy specific survival of mice transplanted with N51-FLT3-transduced Shp2flox/flox;Mx1Cre+ cells comparing PBS-treated mice (control) to polyI:polyC-treated mice (genetic deletion of Shp2), n=16 in the PBS group and n=10 in the polyI;polyC group, p=0.024 by log-rank test.
Animals were followed over 12 months, and spleen and bone marrow were collected for histopathologic diagnosis at the time of death. PBS-treated animals with histopathologic evaluation (n=18) all died by 41 weeks post-transplantation (Figure 3C). The majority of PBS-treated animals died of hematologic malignancy (16/18), either myeloproliferative disorder (n=8), a mixed myelo-/lymphoproliferative disorder (n=6), or lymphoproliferative disorder (n=2) (representative histopathology of myeloproliferative disorder [Supplemental Figure 2D i and 2D ii] and lymphoproliferative disorder [Supplemental Figure 2D iii]). In contrast, significantly fewer animals with Shp2 deletion (polyI:polyC-treated) succumbed to malignant disease (10/16) including myeloproliferative disorder (n=8), mixed myelo-/lymphoproliferative disorder (n=1), and lymphoproliferative disorder (n=1), and displayed significantly longer survival (Figure 3C). Furthermore, animals with deletion of Shp2 with a histopathologic diagnosis of malignancy exhibited significantly smaller spleen sizes at the time of death compared to the PBS-treated animals with malignancy (Figure 3B). Overall, these findings demonstrate that animals bearing deletion of Shp2 displayed a reduced severity of and increased latency to N51-FLT3-induced malignancy, as well as a significantly prolonged survival (Figure 3C).
Shp2 and STAT5 co-localize in the nucleus of FLT3-ITD-expressing cells
To investigate a mechanism of how Shp2 functions to promote STAT5 activation and FLT3-ITD-induced leukemogenesis, we utilized the human FLT3-ITD positive AML-derived cell line, MV411, and the human FLT3-ITD negative cell line, HL60. MV411 cells serve as a good model as they are derived from a human sample, express FLT3-ITD under endogeneous promoter regulatory elements, and demonstrate the canonical constitutive hyperactivation of STAT5 found in FLT3-ITD+ leukemias (Figure 4A). Furthermore, similar to that observed in the Baf3 cell lines stably expressing N51-FLT3 (Figure 1B), MV411 cells demonstrated constitutive interaction of Shp2 with FLT3, in contrast to the FLT3-ITD negative HL60 cells (Figure 4B). Although Shp2 is conventionally described as a cytoplasmic protein, previous studies demonstrated nuclear localization of Shp2 in AML samples (19). We used a biochemical approach to compare Shp2 and STAT5 cellular distribution between FLT3-ITD-expressing MV411 and HL60 cells. As expected, the MV411 cells had increased nuclear-localized STAT5 and activated phospho-STAT5 (Figure 4C). Total Shp2 levels were similar in the cytoplasmic and nuclear fractions in both the MV411 and HL60 cells, consistent with its previously defined obligatory role in hematopoietic cell growth. However, notably, tyrosine phosphorylated Shp2 (C-terminal tyrosine 580) was substantially higher in the FLT3-ITD-expressing MV411 cells in both the cytoplasmic- and nuclear-enriched fractions compared to the HL60 cells (Figure 4C).
Figure 4. Increased levels of phospho-Shp2 are found in the cytoplasmic and nuclear compartments of MV411 cells compared to HL60 cells.

(A) Intracellular staining of phospho-STAT5 in MV411 and HL60 cells assessed by flow cytometry. (B) Total cellular extracts from exponentially growing HL60 or MV411 cells were immunoprecipitated (IP) with anti-FLT3 and immunoblotted (IB) with anti-Shp2 and anti-FLT3. (C) Nuclear and cytoplasmic proteins were isolated from MV411 and HL-60 cells and assessed for phospho-STAT5 and phospho-Shp2 by immunoblot.
We next utilized in situ immunofluorescence to examine nuclear distribution of Shp2 and potential co-localization with phospho-STAT5. Consistent with the cell fractionation studies, nuclear expression of Shp2 was observed in both MV411 and HL60 cells (observe co-localization of DAPI and Shp2 in both cell types, Figure 5A); however, nuclear Shp2 distribution in the MV411 cells was strongly concentrated in punctated microdomains, whereas nuclear Shp2 expression in the HL60 cells was smoothly distributed. Furthermore, upon merging of images, nuclear Shp2 co-localized strongly with nuclear phospho-STAT5 at a significantly higher level in MV411 cells compared to HL60 cells (Figures 5A and 5B).
Figure 5. Shp2 and phospho-STAT5 co-localize in the nuclei of MV411 cells.
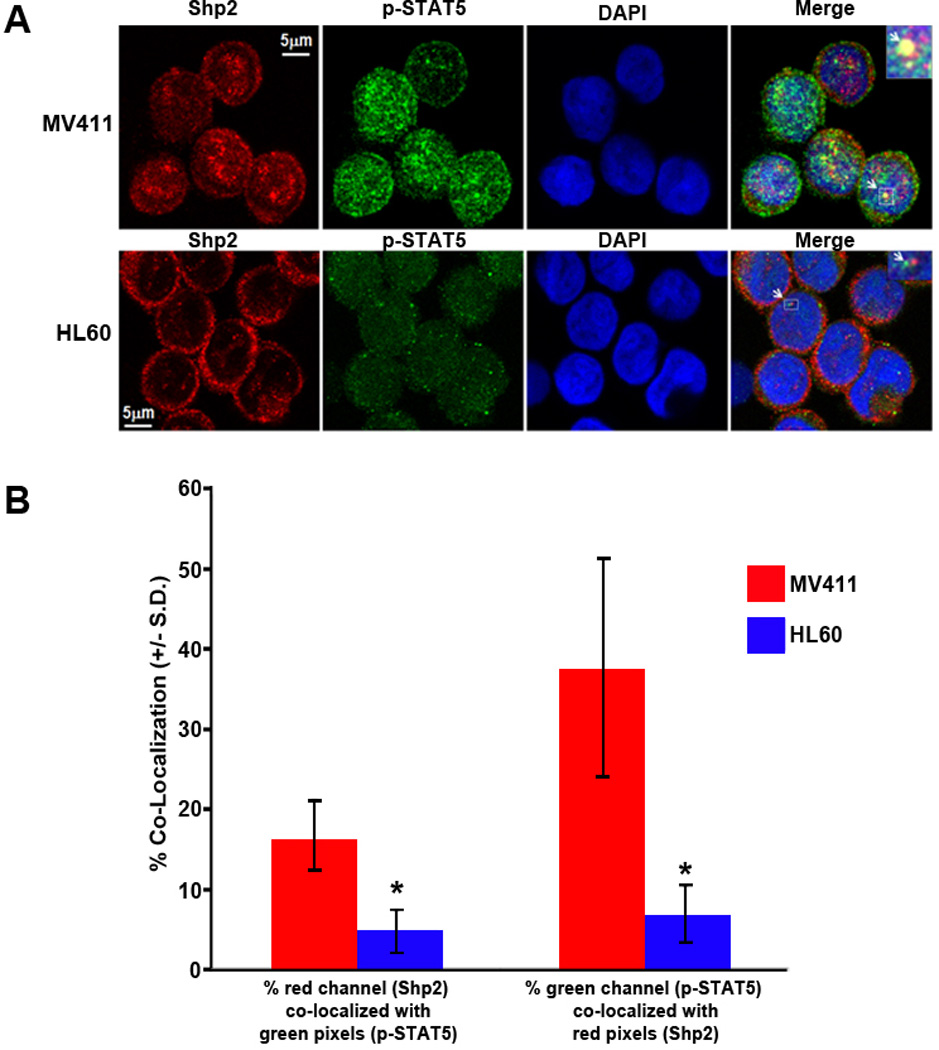
(A) Immunostaining for Shp2 and phospho-STAT5 was performed in MV411 and HL60 cells and cells were imaged using confocal microscopy. (B) Quantification of co-localization of Shp2 and phospho-STAT5. A region of interest (ROI) was selected for analysis, and the % of red channel (Shp2 staining) above threshold that co-localized with green pixels (phospho-STAT5 staining) above threshold (area A over B) was calculated. Co-localization was conversely examined calculating the % of green channel above threshold that co-localized with red pixels above threshold (area B over A). The region of overlap co-localization data were calculated, and data were logged from the entire image stack for each cell analyzed to an Excel spreadsheet, n=13 MV411 cells and 11 HL60 cells analyzed, *p<0.05 for MV411 v. HL60 cells, statistical analysis using unpaired, two-tailed, student’s t test.
FLT3-ITD-expressing hematopoietic cells demonstrate increased association of Shp2 at the BCL2L1 promoter
Given nuclear co-localization between Shp2 and STAT5 in FLT3-ITD-expressing cells, we hypothesized that Shp2 may be present in a protein complex with STAT5 at reported STAT5-regulated gene promoters known to augment leukemic cell growth. We chose to examine the BCL2L1 promoter, a STAT5-responsive promoter which regulates expression of the prosurvival protein, Bcl-XL. Bcl-XL is particularly relevant to the current studies as its expression has been shown to contribute to leukemogenesis (53, 54), to be upregulated in primitive hematopoietic progenitors expressing constitutively activated STAT5 (55), and to be downregulated in hematopoietic progenitors bearing knockdown of Shp2 expression (56). Within the human BCL2L1 promoter region, two functional interferon-γ activation sites (GAS) that bind STAT5 and regulate BCL2L1 expression have been defined (Figure 6A) (49). Serving as a positive control, immunoprecipitation with anti-STAT5 in the MV411 cells resulted in increased amplification of this region compared to the HL60 cells using ChIP analysis (Figure 6B). Immunoprecipitation with anti-Shp2 yielded levels of the amplified BCL2L1 promoter region similar to that of anti-STAT5 in the MV411 cells (Figure 6B), suggesting that Shp2 is within a protein complex binding the BCL2L1 GAS consensus sequence(s). Amplified PCR products immunoprecipitated with both anti-STAT5 and anti-Shp2 were sequenced to verify the predicted amplified BCL2L1 promoter region (data not shown). To evaluate the functional implications of Shp2 on the BCL2L1 STAT5-binding GAS response elements, the human BCL2L1 proximal promoter was assessed for its ability to direct luciferase expression in the presence and absence of Shp2 in MV411 cells. In MV411 cells lacking Shp2 expression, STAT5 activation was reduced which correlated with reduced BCL2L1 promoter activity (Figure 6C). Together, findings from the immunofluorescence, ChIP, and luciferase assays support a model that Shp2 co-localizes with STAT5 in a protein complex within the nuclei of FLT3-ITD-expressing hematopoietic cells to augment pro-leukemogenic gene expression.
Figure 6. Shp2 co-occupies the human BCL2L1 promoter with phospho-STAT5 and regulates BCL2L1 promoter activity.
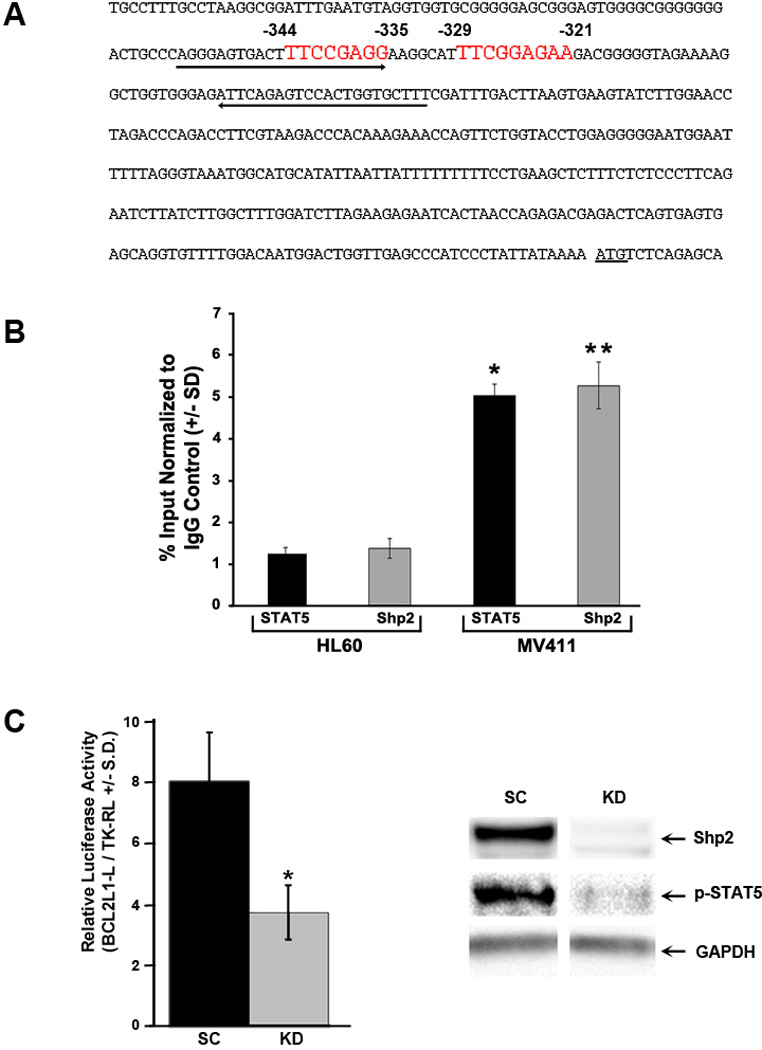
(A) Schematic diagram demonstrating the human BCL2L1 promoter with two functional interferon-γ activation sites (GAS sites, consensus sequence TTCN2-N4GAA) at -344 to -335 and -329 to – 321 upstream of the BclXL translation start codon highlighted in red with arrows indicating the primers used to amplify the region for ChIP. (B) For ChIP analysis, isolated lysates from HL60 or MV411 cells were immunoprecipitated with either anti-STAT5 or anti-Shp2 followed by quantitative PCR using of purified DNA fragments using primers specific for the human BCL2L1 promoter. The immunoprecipitated fragments were expressed as a percentage of the total chromatin used in the sample and normalized to immunoprecipitation with normal rabbit IgG. Representative of 3 independent experiments, n=3, *p<0.005 for fragments pulled down by STAT5 from MV411 v. HL60 cells and **p<0.0005 for fragments pulled down by Shp2 from MV411 v. HL60 cells, statistics performed using unpaired, two-tailed student’s t test. (C) Puromycin-selected MV411 cells (scrambled, SC or Shp2 knockdown, KD) were transfected with pBCL2L1-L and pTK-RL and protein lysates were assayed for luciferase and Renilla luciferase (left panel). Representative of 3 independent experiments with similar results, n=3, *p<0.05 for KD v. SC. Protein lysates were also analyzed for Shp2 expression and phospho-STAT5 levels (right panel, representative immunoblot from 3 independent experiments).
Pharmacologic inhibition of Shp2 reduces FLT3-ITD-induced cellular proliferation, survival, and STAT5 activation
Given the demonstrated positive role of Shp2 in FLT3-ITD-induced hyperproliferation, STAT5 activation, and malignant disease using genetic analyses, we next examined if pharmacologic inhibition of Shp2 would induce a similar effect. II-B08 is a Shp2-specific inhibitor that we recently identified from a library of indole salicylic acid derivatives (45). Using WT-FLT3- and N51-FLT3-transduced cells, a modest but statistically non-significant reduction in the proliferation of WT FLT3-expressing cells was observed. However, proliferation of N51-FLT3-expressing cells was substantially reduced in a dose-dependent fashion by II-B08 (Figure 7A). Furthermore, treatment with II-B08 promoted apoptosis of N51-FLT3-expressing cells in a dose-dependent manner and to a greater extent than that of WT FLT3-expressing cells (Figure 7B).
Figure 7. Pharmacologic inhibition of Shp2 diminishes FLT3-ITD-induced hyperproliferation in murine cells and inhibits primary AML cell proliferation.

(A) 3H-thymidine incorporation assay of murine bone marrow LDMNCs (C57Bl/6) transduced with either MSCV-WT-FLT3 or MSCV-N51-FLT3 in the absence or presence of increasing concentrations of II-B08; two independent experiments combined with n=4–6 replicates per experiment, *p=0.002 for N51-FLT3, 5 µM IIB-08 v. N51-FLT3, baseline, **p<0.0001 for N51-FLT3, 10 µM, 15 µM, or 20 µM v. N51-FLT3, baseline, statistical analysis performed using random effects ANOVA. (B) Apoptosis as measured by the presence of Annexin V on the surface of murine bone marrow LDMNCs transduced with either MSCV-WT-FLT3 or MSCV-N51-FLT3 in the presence of increasing concentrations of II-B08, representative of two independent experiments, p=3, statistics performed using unpaired, two-tailed student’s t test. (C) Immunoblot analysis of STAT5 and Shp2 phosphorylation in retrovirally transduced and sorted WT-FLT3- and N51-FLT3-expressing bone marrow-derived macrophages in the absence and presence of Shp2 inhibitor, II-B08. (D) 3H-thymidine incorporation of primary AML cells cultured in GM-CSF 1 ng/mL + FL 50 ng/mL, data represented as % of average proliferation in the absence of II-B08 for each independent sample, n=2 FLT3-ITD+ and n=5 FLT3-ITD− samples (n=7 total), *p<0.05 at 25 and 50 µM for II-B08-induced inhibition. (E) 3H-thymidine incorporation of primary AML cells cultured in GM-CSF 1 ng/mL + IL3 10 ng/mL, data represented as % of average proliferation in the absence of II-B08 for each independent sample, n=2 FLT3-ITD+ and n=4 FLT3-ITD− samples (n=6 total), *p<0.05 at 50 µM for II-B08-induced inhibition; statistical analysis performed using linear mixed effects models with random intercepts to evaluate the effects of different II-B08 concentrations.
We next examined if pharmacologic inhibition of Shp2 reduced STAT5 phosphorylation, similar to genetic disruption of Ptpn11 (Figure 2C). Although it was not possible to determine if II-B08 treatment reduced WT FLT3-induced STAT5 activation due to undetectable levels of phospho-STAT5 in the WT FLT3-expressing cells (Figure 7C, lanes 1 – 4), II-B08 clearly reduced the baseline and FL-stimulated phosphorylation of STAT5 in N51-FLT3-expressing cells (Figure 7C, compare lanes 7 and 8 to lanes 5 and 6). As Y580 phosphorylation has been proposed to promote Shp2 catalytic activity (10, 12), we examined the effect of II-B08 on Shp2 Y580 phosphorylation. While Shp2 phosphorylation was significantly reduced in II-B08-treated N51-FLT3-expressing cells (Figure 7C, compare lanes 7 and 8 to lanes 5 and 6), II-B08 demonstrated no effect on the FL-induced Shp2 phosphorylation in the WT FLT3-expressing cells, consistent with II-B08 exerting only a modest effect on the proliferation and survival of WT FLT3-expressing cells (Figures 7A and 7B).
We additionally found that II-B08 reduced proliferation of both FLT3-ITD+ and FLT3-ITD− primary AML samples in a dose-dependent manner in the presence of various hematopoietic growth factor cocktails (Figure 7D and Figure 7E, compiled proliferation data from both FLT3-ITD+ and FLT3-ITD− patients combined). Thus, while our studies have largely focused on the role of Shp2 in a model of FLT3-ITD+ AML, considering that Shp2 is overexpressed in a broad sampling of AML cases (19) and that II-B08 demonstrates activity against both FLT3-ITD− and FLT3-ITD+ primary AML samples, Shp2 inhibition is potentially applicable more generally for novel therapeutics in AML.
DISCUSSION
Acute myeloid leukemia (AML) continues to be an extremely challenging disease to treat, thus, new mechanisms of disease origination and progression continue to be pursued. A rational starting point for defining novel disease-inducing molecular aberrations is by extending the understanding of well-described mutations in AML, such as ITDs within FLT3. Our initial studies in Baf3 cells transduced with either WT-FLT3 or N51-FLT3 demonstrated constitutive interaction between Shp2 and FLT3 as well as with STAT5 in N51-FLT3-expressing cells. Using standard knockdown techniques, we observed a significant reduction in the proliferation of N51-FLT3 transduced Baf3 cells. Our findings are in contrast to that of Müller et al., who found that shRNA-mediated reduced Shp2 exerted little effect on FLT3-ITD-induced leukemogenesis (57). It is possible that reduced Shp2 expression was compensated by alternative pathways in the immortalized 32D cell line used in this study. To overcome these potential problems, we performed studies using primary hematopoietic progenitors bearing conditionally targeted alleles of Ptpn11. We observed a level of correction similar to the shRNA studies using the Ptpn11 knockout cells, supporting a positive role of Shp2 in FLT3-ITD-induced hyperproliferation. While we did not see complete correction of FLT3-ITD-induced hyperproliferation in the Shp2 knockdown or conditional Ptpn11 knockout studies (Figures 1E and 2B, respectively), it is possible that residual Shp2 expression after culture in vitro contributed to residual proliferation of the FLT3-ITD-expressing cells. Additionally, the persistence of STAT5 and phospho-STAT5 in the FLT3-ITD-containing complex even upon reduced Shp2 expression (Figure 1G) indicates that FLT3-ITD bears the capacity to utilize Shp2-independent means of interaction with STAT5. One potential alternative means of STAT5 interaction with FLT3-ITD is via Gab2 and/or p85α, previously shown to positively contribute to FLT3-ITD-induced signaling (58) and to interact with constitutively activated STAT5 (59).
While several studies have demonstrated that Shp2 is critical for normal hematopoietic primitive progenitor and stem cell function (13, 20, 29, 40–42, 56), it was unclear if genetic deletion of Shp2 would similarly reduce the function of these cells in vivo when transduced with the N51-FLT3 oncogene. However, even when delaying polyI:polyC treatment to 4 – 6 weeks post-transplant, genetic deletion of Shp2 resulted in a reduced severity of and increased latency to N51-FLT3-induced malignancy. Among the animals bearing polyI:polyC-induced genetic disruption of Shp2, the lympho- or myleoproliferative disease appeared qualitatively more indolent by histopathological evaluation (data not shown) and the spleen sizes were significantly reduced (Figure 3B). Thus, while reduced Shp2 expression likely slows the growth of hematopoietic cells via its participation in several cytokine receptor and receptor tyrosine kinase signaling pathways, our findings clearly demonstrate that loss of Shp2 expression curtails N51-FLT3-induced transformation and disease development in vivo.
Shp2 functions positively in several ways to promote cellular signaling (10, 60, 61); however, a specific mechanism of how Shp2 may be promoting leukemogenesis in the context of FLT3-ITD and hyperactivated STAT5 has never been examined. Given that Shp2 has been described to work in the nucleus with STAT5 to promote gene expression in mammary epithelial cells (35), we hypothesized that Shp2 may similarly work with STAT5 in the context of AML. We found that Shp2 co-localizes with phospho-STAT5 in the nuclei of FLT3-ITD-bearing MV411 cells and, consistently, upon immunoprecipitation with anti-Shp2, a segment of the BCL2L1 promoter containing functional STAT5 binding sites was amplified at a level similar to immunoprecipitation with anti-STAT5. To our knowledge, this is the first report that Shp2 functions at a STAT5-responsive promoter element in the context of hematopoietic or leukemic cells.
We extended our genetic findings to pharmacologic studies utilizing the Shp2 phosphatase-specific inhibitor, II-B08, and found that II-B08 treatment effectively reduced Shp2 and STAT5 phosphorylation, consistent with reduced proliferation of N51-FLT3-transduced cells. Interestingly, these studies additionally indicated that Shp2 phosphorylation is induced only upon FL stimulation in WT FLT3-expressing cells, while it is constitutively phosphorylated in the N51-FLT3-expressing cells (Figure 7C, compare lanes 5 and 6 to lanes 1 and 2). The FL-dependent phosphorylation of Shp2 in WT FLT3-expressing cells along with the constitutive, FL-independent phosphorylation of Shp2 in the N51-FLT3-expressing cells implies that Shp2 may be a direct target of FLT3-ITD kinase activity, consistent with the findings of Heiss et al. (21). The correlation of II-B08-induced reduction of both Shp2 and STAT5 phosphorylation suggests that Shp2 phosphatase function promotes STAT5 activation via the dephosphorylation, and thus activation, of a STAT5-activating kinase, such as the Src family kinases (SFK). This notion merits further investigation as it is corroborated by the findings that the SFK are also recruited to Y599 of WT FLT3 (21), that Shp2 dephosphorylation of SFK promotes Src kinase activity (10), and that SFK have been found to promote FLT3-ITD-induced hyperproliferation and STAT5 activation (62–64).
Altogether, our biochemical, genetic, and pharmacologic findings lead to a proposed model of how Shp2 may positively contribute to FLT3-ITD-induced leukemogenesis (Supplemental Figure 3). We propose that Shp2 has increased recruitment to FLT3-ITD, in part due to duplicated Y599. Furthermore, given our observation of substantial co-localization between Shp2 and phospho-STAT5 in the nuclei of FLT3-ITD-bearing MV411 cells, we propose the novel idea that in addition to promoting FLT3-ITD-induced STAT5 hyperactivation, Shp2 may also function with STAT5 to promote pro-leukemogenic gene expression. Collectively, this model supports the premise that Shp2 contributes positively to enhanced proliferation and transformation in AML, and implies that pharmacologic inhibition of Shp2 may provide a novel therapeutic approach to acute myeloid leukemias.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the Riley Children’s Foundation, the Clarian Values Fund for Research (VFR-245, RJC), and the U.S. National Institutes of Health (F31AG031648, SCN; RO1HL082981 & RO1CA134777, RJC; RO1HL075816, RO1HL077177, and RO1CA134777, RK; RO1CA069202 and RO1CA126937, ZYZ). The authors acknowledge the contribution of Baindu Bayon for generation of the pBCL2L1-L plasmid. The authors appreciate the helpful discussions and technical assistance from Dr. Karen Pollok and Tony Sinn in the Indiana University In Vivo Therapeutics Core, from Susan Rice in the Flow Cytometry Core, and from the Indiana Center for Biological Microscopy. We thank Drs. Yan Liu, Nadia Carlesso, and Merv Yoder for critical reading of the manuscript and gratefully acknowledge the administrative assistance of Linda S. Henson.
Footnotes
Author Contribution: Sarah C. Nabinger: designed and performed research, wrote manuscript
Xingjun Li: designed and performed research, wrote manuscript
Baskar Ramdas: designed and performed research
Yantao He: designed and performed research
Xian Zhang: designed and performed research
Lifan Zeng: designed and performed research
Briana Richine: designed and performed research
Joshua D. Bowling: designed and performed research
Seiji Fukuda: provided reagent
Shreevrat Goenka: designed research
Ziyue Liu: performed statistical analyses
Gen-Sheng Feng: provided reagent
Menggang Yu: performed statistical analyses
George E. Sandusky: performed pathological analyses
H. Scott Boswell: provided primary AML samples
Zhong-Yin Zhang: provided reagent
Reuben Kapur: designed research
Rebecca J. Chan: designed research and wrote manuscript
The authors have no conflicting financial interests.
Supplementary information is available at the journal's website.
REFERENCES
- 1.Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996 Dec;10(12):1911–1918. [PubMed] [Google Scholar]
- 2.Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002 Jun 15;99(12):4326–4335. doi: 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- 3.Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001 Sep 15;98(6):1752–1759. doi: 10.1182/blood.v98.6.1752. [DOI] [PubMed] [Google Scholar]
- 4.Mizuki M, Fenski R, Halfter H, Matsumura I, Schmidt R, Muller C, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. 2000 Dec 1;96(12):3907–3914. [PubMed] [Google Scholar]
- 5.Spiekermann K, Bagrintseva K, Schwab R, Schmieja K, Hiddemann W. Overexpression and constitutive activation of FLT3 induces STAT5 activation in primary acute myeloid leukemia blast cells. Clin Cancer Res. 2003 Jun;9(6):2140–2150. [PubMed] [Google Scholar]
- 6.Murata K, Kumagai H, Kawashima T, Tamitsu K, Irie M, Nakajima H, et al. Selective cytotoxic mechanism of GTP-14564, a novel tyrosine kinase inhibitor in leukemia cells expressing a constitutively active Fms-like tyrosine kinase 3 (FLT3) J Biol Chem. 2003 Aug 29;278(35):32892–32898. doi: 10.1074/jbc.M210405200. [DOI] [PubMed] [Google Scholar]
- 7.Hayakawa F, Towatari M, Kiyoi H, Tanimoto M, Kitamura T, Saito H, et al. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene. 2000 Feb 3;19(5):624–631. doi: 10.1038/sj.onc.1203354. [DOI] [PubMed] [Google Scholar]
- 8.Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene. 2002 Apr 11;21(16):2555–2563. doi: 10.1038/sj.onc.1205332. [DOI] [PubMed] [Google Scholar]
- 9.Pratz KW, Levis MJ. Bench to bedside targeting of FLT3 in acute leukemia. Current drug targets. Jul;11(7):781–789. doi: 10.2174/138945010791320782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neel BG, Gu H, Pao L. The 'Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003 Jun;28(6):284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 11.Lai LA, Zhao C, Zhang EE, Feng GS. Protein Phosphatases. Vol. 5. Berlin Heidelberg: Springer-Verlag; 2004. pp. 275–299. [Google Scholar]
- 12.Lu W, Gong D, Bar-Sagi D, Cole PA. Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Molecular cell. 2001 Oct;8(4):759–769. doi: 10.1016/s1097-2765(01)00369-0. [DOI] [PubMed] [Google Scholar]
- 13.Qu CK, Yu WM, Azzarelli B, Cooper S, Broxmeyer HE, Feng GS. Biased suppression of hematopoiesis and multiple developmental defects in chimeric mice containing Shp-2 mutant cells. Mol Cell Biol. 1998;18(10):6075–6082. doi: 10.1128/mcb.18.10.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003 Jun;34(2):148–150. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 15.Loh ML, Reynolds MG, Vattikuti S, Gerbing RB, Alonzo TA, Carlson E, et al. PTPN11 mutations in pediatric patients with acute myeloid leukemia: results from the Children's Cancer Group. Leukemia. 2004 Nov;18(11):1831–1834. doi: 10.1038/sj.leu.2403492. [DOI] [PubMed] [Google Scholar]
- 16.Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004 Dec 15;64(24):8816–8820. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- 17.Tartaglia M, Martinelli S, Iavarone I, Cazzaniga G, Spinelli M, Giarin E, et al. Somatic PTPN11 mutations in childhood acute myeloid leukaemia. Br J Haematol. 2005 May;129(3):333–339. doi: 10.1111/j.1365-2141.2005.05457.x. [DOI] [PubMed] [Google Scholar]
- 18.Loh ML, Martinelli S, Cordeddu V, Reynolds MG, Vattikuti S, Lee CM, et al. Acquired PTPN11 mutations occur rarely in adult patients with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk Res. 2005 Apr;29(4):459–462. doi: 10.1016/j.leukres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Xu R, Yu Y, Zheng S, Zhao X, Dong Q, He Z, et al. Overexpression of Shp2 tyrosine phosphatase is implicated in leukemogenesis in adult human leukemia. Blood. 2005 Nov 1;106(9):3142–3149. doi: 10.1182/blood-2004-10-4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qu CK, Shi ZQ, Shen R, Tsai FY, Orkin SH, Feng GS. A deletion mutation in the SH2-N domain of Shp-2 severely suppresses hematopoietic cell development. Mol Cell Biol. 1997;17(9):5499–5507. doi: 10.1128/mcb.17.9.5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heiss E, Masson K, Sundberg C, Pedersen M, Sun J, Bengtsson S, et al. Identification of Y589 and Y599 in the juxtamembrane domain of Flt3 as ligand-induced autophosphorylation sites involved in binding of Src family kinases and the protein tyrosine phosphatase SHP2. Blood. 2006 Sep 1;108(5):1542–1550. doi: 10.1182/blood-2005-07-008896. [DOI] [PubMed] [Google Scholar]
- 22.Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002 Jan 1;99(1):310–318. doi: 10.1182/blood.v99.1.310. [DOI] [PubMed] [Google Scholar]
- 23.Abu-Duhier FM, Goodeve AC, Wilson GA, Gari MA, Peake IR, Rees DC, et al. FLT3 internal tandem duplication mutations in adult acute myeloid leukaemia define a high-risk group. Br J Haematol. 2000 Oct;111(1):190–195. doi: 10.1046/j.1365-2141.2000.02317.x. [DOI] [PubMed] [Google Scholar]
- 24.Choudhary C, Muller-Tidow C, Berdel WE, Serve H. Signal transduction of oncogenic Flt3. International journal of hematology. 2005 Aug;82(2):93–99. doi: 10.1532/IJH97.05090. [DOI] [PubMed] [Google Scholar]
- 25.Choudhary C, Brandts C, Schwable J, Tickenbrock L, Sargin B, Ueker A, et al. Activation mechanisms of STAT5 by oncogenic Flt3-ITD. Blood. 2007 Jul 1;110(1):370–374. doi: 10.1182/blood-2006-05-024018. [DOI] [PubMed] [Google Scholar]
- 26.Rocnik JL, Okabe R, Yu JC, Lee BH, Giese N, Schenkein DP, et al. Roles of tyrosine 589 and 591 in STAT5 activation and transformation mediated by FLT3-ITD. Blood. 2006 Aug 15;108(4):1339–1345. doi: 10.1182/blood-2005-11-011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Wen R, Yang S, Schuman J, Zhang EE, Yi T, et al. Identification of Shp-2 as a Stat5A phosphatase. J Biol Chem. 2003 May 9;278(19):16520–16527. doi: 10.1074/jbc.M210572200. [DOI] [PubMed] [Google Scholar]
- 28.Yu CL, Jin YJ, Burakoff SJ. Cytosolic tyrosine dephosphorylation of STAT5. Potential role of SHP-2 in STAT5 regulation. J Biol Chem. 2000 Jan 7;275(1):599–604. doi: 10.1074/jbc.275.1.599. [DOI] [PubMed] [Google Scholar]
- 29.Chan RJ, Johnson SA, Li Y, Yoder MC, Feng GS. A definitive role of Shp-2 tyrosine phosphatase in mediating embryonic stem cell differentiation and hematopoiesis. Blood. 2003 Sep 15;102(6):2074–2080. doi: 10.1182/blood-2003-04-1171. [DOI] [PubMed] [Google Scholar]
- 30.Ke Y, Zhang EE, Hagihara K, Wu D, Pang Y, Klein R, et al. Deletion of shp2 in the brain leads to defective proliferation and differentiation in neural stem cells and early postnatal lethality. Mol Cell Biol. 2007 Oct;27(19):6706–6717. doi: 10.1128/MCB.01225-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bard-Chapeau EA, Li S, Ding J, Zhang SS, Zhu HH, Princen F, et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell. May 17;19(5):629–639. doi: 10.1016/j.ccr.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang W, Chan RJ, Chen H, Yang Z, He Y, Zhang X, et al. Negative regulation of Stat3 by activating PTPN11 mutants contributes to the pathogenesis of Noonan syndrome and juvenile myelomonocytic leukemia. J Biol Chem. 2009 Aug 14;284(33):22353–22363. doi: 10.1074/jbc.M109.020495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ali S, Chen Z, Lebrun JJ, Vogel W, Kharitonenkov A, Kelly PA, et al. PTP1D is a positive regulator of the prolactin signal leading to beta-casein promoter activation. Embo J. 1996 Jan 2;15(1):135–142. [PMC free article] [PubMed] [Google Scholar]
- 34.Berchtold S, Volarevic S, Moriggl R, Mercep M, Groner B. Dominant negative variants of the SHP-2 tyrosine phosphatase inhibit prolactin activation of Jak2 (janus kinase 2) and induction of Stat5 (signal transducer and activator of transcription 5)-dependent transcription. Molecular endocrinology (Baltimore, Md. 1998 Apr;12(4):556–567. doi: 10.1210/mend.12.4.0086. [DOI] [PubMed] [Google Scholar]
- 35.Chughtai N, Schimchowitsch S, Lebrun JJ, Ali S. Prolactin induces SHP-2 association with Stat5, nuclear translocation, and binding to the beta-casein gene promoter in mammary cells. J Biol Chem. 2002 Aug 23;277(34):31107–31114. doi: 10.1074/jbc.M200156200. [DOI] [PubMed] [Google Scholar]
- 36.Ke Y, Lesperance J, Zhang EE, Bard-Chapeau EA, Oshima RG, Muller WJ, et al. Conditional deletion of Shp2 in the mammary gland leads to impaired lobulo-alveolar outgrowth and attenuated Stat5 activation. J Biol Chem. 2006 Nov 10;281(45):34374–34380. doi: 10.1074/jbc.M607325200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang S, Broxmeyer HE. Flt3 ligand induces tyrosine phosphorylation of gab1 and gab2 and their association with shp-2, grb2, and PI3 kinase. Biochem Biophys Res Commun. 2000 Oct 14;277(1):195–199. doi: 10.1006/bbrc.2000.3662. [DOI] [PubMed] [Google Scholar]
- 38.Zhang S, Broxmeyer HE. p85 subunit of PI3 kinase does not bind to human Flt3 receptor, but associates with SHP2, SHIP, and a tyrosine-phosphorylated 100-kDa protein in Flt3 ligand-stimulated hematopoietic cells. Biochem Biophys Res Commun. 1999 Jan 19;254(2):440–445. doi: 10.1006/bbrc.1998.9959. [DOI] [PubMed] [Google Scholar]
- 39.Zhang S, Mantel C, Broxmeyer HE. Flt3 signaling involves tyrosyl-phosphorylation of SHP-2 and SHIP and their association with Grb2 and Shc in Baf3/Flt3 cells. J Leukoc Biol. 1999 Mar;65(3):372–380. doi: 10.1002/jlb.65.3.372. [DOI] [PubMed] [Google Scholar]
- 40.Chan RJ, Li Y, Hass MN, Walter A, Voorhorst CS, Shelley WC, et al. Shp-2 heterozygous hematopoietic stem cells have deficient repopulating ability due to diminished self-renewal. Exp Hematol. 2006 Sep;34(9):1230–1239. doi: 10.1016/j.exphem.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 41.Chan G, Cheung LS, Yang W, Milyavsky M, Sanders AD, Gu S, et al. Essential role for Ptpn11 in survival of hematopoietic stem and progenitor cells. Blood. Apr 21;117(16):4253–4261. doi: 10.1182/blood-2010-11-319517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu HH, Ji K, Alderson N, He Z, Li S, Liu W, et al. Kit-Shp2-Kit signaling acts to maintain a functional hematopoietic stem and progenitor cell pool. Blood. May 19;117(20):5350–5361. doi: 10.1182/blood-2011-01-333476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fukuda S, Broxmeyer HE, Pelus LM. Flt3 ligand and the Flt3 receptor regulate hematopoietic cell migration by modulating the SDF-1alpha(CXCL12)/CXCR4 axis. Blood. 2005 Apr 15;105(8):3117–3126. doi: 10.1182/blood-2004-04-1440. [DOI] [PubMed] [Google Scholar]
- 44.Hartman AD, Wilson-Weekes A, Suvannasankha A, Burgess GS, Phillips CA, Hincher KJ, et al. Constitutive c-jun N-terminal kinase activity in acute myeloid leukemia derives from Flt3 and affects survival and proliferation. Exp Hematol. 2006 Oct;34(10):1360–1376. doi: 10.1016/j.exphem.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 45.Zhang X, He Y, Liu S, Yu Z, Jiang ZX, Yang Z, et al. Salicylic acid based small molecule inhibitor for the oncogenic Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2) J Med Chem. Mar 25;53(6):2482–2493. doi: 10.1021/jm901645u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang EE, Chapeau E, Hagihara K, Feng GS. Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc Natl Acad Sci U S A. 2004 Nov 9;101(45):16064–16069. doi: 10.1073/pnas.0405041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan RJ, Leedy MB, Munugalavadla V, Voorhorst CS, Li Y, Yu M, et al. Human somatic PTPN11 mutations induce hematopoietic-cell hypersensitivity to granulocyte-macrophage colony-stimulating factor. Blood. 2005 May 1;105(9):3737–3742. doi: 10.1182/blood-2004-10-4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Munugalavadla V, Sims EC, Borneo J, Chan RJ, Kapur R. Genetic and pharmacologic evidence implicating the p85 alpha, but not p85 beta, regulatory subunit of PI3K and Rac2 GTPase in regulating oncogenic KIT-induced transformation in acute myeloid leukemia and systemic mastocytosis. Blood. 2007 Sep 1;110(5):1612–1620. doi: 10.1182/blood-2006-10-053058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Socolovsky M, Fallon AE, Wang S, Brugnara C, Lodish HF. Fetal anemia and apoptosis of red cell progenitors in Stat5a−/−5b−/− mice: a direct role for Stat5 in Bcl-X(L) induction. Cell. 1999 Jul 23;98(2):181–191. doi: 10.1016/s0092-8674(00)81013-2. [DOI] [PubMed] [Google Scholar]
- 50.Yang Z, Kondo T, Voorhorst CS, Nabinger SC, Ndong L, Yin F, et al. Increased c-Jun expression and reduced GATA2 expression promote aberrant monocytic differentiation induced by activating PTPN11 mutants. Mol Cell Biol. 2009 Aug;29(16):4376–4393. doi: 10.1128/MCB.01330-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mongomery DC. Design and Analysis of Experiments. 4th edn. John Wiley & Sons, Inc.; 1997. [Google Scholar]
- 52.Lee BH, Williams IR, Anastasiadou E, Boulton CL, Joseph SW, Amaral SM, et al. FLT3 internal tandem duplication mutations induce myeloproliferative or lymphoid disease in a transgenic mouse model. Oncogene. 2005 Nov 24;24(53):7882–7892. doi: 10.1038/sj.onc.1208933. [DOI] [PubMed] [Google Scholar]
- 53.Beverly LJ, Varmus HE. MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene. 2009 Mar 5;28(9):1274–1279. doi: 10.1038/onc.2008.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mizukawa B, Wei J, Shrestha M, Wunderlich M, Chou FS, Griesinger A, et al. Inhibition of Rac GTPase signaling and downstream prosurvival Bcl-2 proteins as combination targeted therapy in MLL-AF9 leukemia. Blood. Nov 10;118(19):5235–5245. doi: 10.1182/blood-2011-04-351817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schuringa JJ, Chung KY, Morrone G, Moore MA. Constitutive activation of STAT5A promotes human hematopoietic stem cell self-renewal and erythroid differentiation. J Exp Med. 2004 Sep 6;200(5):623–635. doi: 10.1084/jem.20041024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li L, Modi H, McDonald T, Rossi J, Yee JK, Bhatia R. A critical role for SHP2 in STAT5 activation and growth factor-mediated proliferation, survival, and differentiation of human CD34+ cells. Blood. Aug 11;118(6):1504–1515. doi: 10.1182/blood-2010-06-288910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muller JP, Schonherr C, Markova B, Bauer R, Stocking C, Bohmer FD. Role of SHP2 for FLT3-dependent proliferation and transformation in 32D cells. Leukemia. 2008 Oct;22(10):1945–1948. doi: 10.1038/leu.2008.73. [DOI] [PubMed] [Google Scholar]
- 58.Masson K, Liu T, Khan R, Sun J, Ronnstrand L. A role of Gab2 association in Flt3 ITD mediated Stat5 phosphorylation and cell survival. Br J Haematol. 2009 Jul;146(2):193–202. doi: 10.1111/j.1365-2141.2009.07725.x. [DOI] [PubMed] [Google Scholar]
- 59.Harir N, Pecquet C, Kerenyi M, Sonneck K, Kovacic B, Nyga R, et al. Constitutive activation of Stat5 promotes its cytoplasmic localization and association with PI3-kinase in myeloid leukemias. Blood. 2007 Feb 15;109(4):1678–1686. doi: 10.1182/blood-2006-01-029918. [DOI] [PubMed] [Google Scholar]
- 60.Chan RJ, Feng GS. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood. 2007 Feb 1;109(3):862–867. doi: 10.1182/blood-2006-07-028829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Feng GS. Shp-2 tyrosine phosphatase: signaling one cell or many. Exp Cell Res. 1999 Nov 25;253(1):47–54. doi: 10.1006/excr.1999.4668. [DOI] [PubMed] [Google Scholar]
- 62.Leischner H, Albers C, Grundler R, Razumovskaya E, Spiekermann K, Bohlander S, et al. SRC is a signaling mediator in FLT3-ITD− but not in FLT3-TKD-positive AML. Blood. 2012 Apr 26;119(17):4026–4033. doi: 10.1182/blood-2011-07-365726. [DOI] [PubMed] [Google Scholar]
- 63.Okamoto M, Hayakawa F, Miyata Y, Watamoto K, Emi N, Abe A, et al. Lyn is an important component of the signal transduction pathway specific to FLT3/ITD and can be a therapeutic target in the treatment of AML with FLT3/ITD. Leukemia. 2007 Mar;21(3):403–410. doi: 10.1038/sj.leu.2404547. [DOI] [PubMed] [Google Scholar]
- 64.Robinson LJ, Xue J, Corey SJ. Src family tyrosine kinases are activated by Flt3 and are involved in the proliferative effects of leukemia-associated Flt3 mutations. Exp Hematol. 2005 Apr;33(4):469–479. doi: 10.1016/j.exphem.2005.01.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.