

Abstract
Dicer is as an RNase III enzyme essential for the maturation of the majority of microRNAs. Recent studies have revealed down-regulation or hemizygous loss of Dicer in many tumor models and demonstrated that suppressing Dicer activity enhances tumorigenic activities of lung and breast cancer cells, which support Dicer as a haploinsufficient tumor suppressor in these cancer models. Surprisingly, we found that knocking down Dicer expression suppresses the growth and tumorigenic capacity of human prostate cancer cell lines, but enhances migratory capacities of some prostate cancer cell lines. Dicer is up-regulated in human prostate cancer specimens, but lower Dicer expression portends a shorter time to recurrence. Complete ablation of Dicer activity in a Pten null mouse model for prostate cancer significantly halts tumor growth and progression, demonstrating that microRNAs play a critical role in maintaining cancer cell fitness. In comparison, hemizygous loss of Dicer in the same model also reduces primary tumor burden, but induces a more locally invasive phenotype and causes seminal vesicle obstruction at high penetrance. Disrupting Dicer activity leads to an increase in apoptosis and senescence in these models, presumably through up-regulation of P16/INK4a and P27/Kip1. Collectively, these results highlight a pleotropic role of Dicer in tumorigenesis that is not only dosage-dependent but also tissue context-dependent.
Keywords: Prostate cancer, Dicer, Pten, Senescence, Apoptosis, Invasiveness
Introduction
Dicer is an RNase III enzyme essential for the maturation of almost all microRNAs (1). In addition, its C-terminal fragment also possesses DNase activity that is critical for DNA fragmentation during apoptosis (2). Numerous genetic studies using mouse models have been performed to specifically disrupt the RNase III activity of Dicer in various organs to determine the biological significance of functional microRNA biogenesis. The resulting biological consequences vary dramatically depending on the targeted organs. For examples, Dicer ablation severely affects limb morphogenesis (1), causes skin tissue disorganization (3, 4), impairs embryonic stem cell differentiation (5), and attenuates specified functions of the differentiated B lymphocytes (6). The most common cellular responses to Dicer ablation are increased apoptosis and senescence (7–9). In contrast, loss of function of Dicer in the vas deferens did not appear to cause major defects (10). Previously, we demonstrated that disrupting the RNase III function of Dicer in the prostate induces prostate epithelial apoptosis and causes prostate epithelial hypoplasia (11).
Array expression profiling analyses have revealed a global reduction of microRNA expression in various cancer models (12). This observation led to the hypothesis that the molecular machinery for microRNA maturation, such as Dicer, is deregulated in tumor tissues. Subsequently, lower Dicer expression was found to be associated with advanced tumor stages and poor clinical outcome in melanoma (13), neuroblastoma (14), breast (15–17), lung (18, 19) and ovarian cancers (20, 21). Recently, several lines of evidence have established Dicer as a haploinsufficient tumor suppressor in certain tumor models. Dicer hemizygous loss was frequently detected in lung, intestine and breast cancers etc. (22). In addition, suppressing Dicer expression in lung cancer and neuroblastoma enhanced their tumorigenicity (14, 23). Finally, studies using genetically engineered mouse models showed that Dicer haploinsufficiency promotes disease progression in mouse models for K-Ras-induced lung cancer and RB loss-induced retinoblastoma (22, 24). Of note, haploinsufficiency of Dicer had no effect on the progression of Myc induced B-cell lymphomagenesis in mice, suggesting that Dicer may not always function as a haploinsufficient tumor suppressor (25).
The role of Dicer in prostate carcinogenesis remains undetermined. Dicer hemizygous loss is not frequently observed in prostate cancer. Interestingly, global increase and decrease of microRNA expression in prostate adenocarcinoma have both been reported (12, 26, 27). Previously, two independent studies have shown that expression of many microRNA processing components including Drosha and all proteins in the RISC complex are transcriptionally upregulated in prostate cancer specimens as compared to that in control benign prostate tissues (28, 29). Chiosea et al. showed that Dicer upregulation is even correlated with advanced disease stage (29). These descriptive studies do not support Dicer as a haploinsufficient tumor suppressor in prostate cancer, and implicate that the role of Dicer in prostate carcinogenesis may not be exactly the same as that in other epithelial malignancies.
Herein, we sought to use cellular biological and genetic approaches to determine the role of Dicer in prostate carcinogenesis. Our results concur with previous studies that Dicer is required for efficient progression of tumorigenesis (25, 30, 31). Surprisingly, in contrast to previous studies showing that Dicer haploinsufficiency promotes (22, 24) or does not affect tumor progression (25), we found that haploinsufficiency of Dicer or partial suppression of Dicer expression impairs the proliferation and tumorigenesis of prostate cancer cells in vitro and in vivo. However, lowering Dicer activity can enhance the invasiveness of certain types of prostate cancer cell lines. Our study demonstrates a dosage-dependent pleiotropic role of Dicer in prostate tumorigenesis and highlights differential roles of Dicer in different organ systems.
Results
Dicer is upregulated in human prostate cancer specimens
We examined the expression level of Dicer in the primary human prostate epithelial cells (PrEC), two immortalized prostate epithelial cells (PNT1 and PNT2), and 5 cancerous human prostate cell lines using quantitative RT-PCR and Western blot analyses. Fig. 1A shows that although the expression level of Dicer transcript does not always correspond to that of the protein, all cancer cell lines express Dicer at comparable or higher levels than the primary (PrEC) and immortalized (PNT1 and PNT2) human prostate epithelial cells. We then compared the expression of Dicer in normal prostate specimens (N=26, samples from benign peripheral zone of prostate tissues) and prostate cancer specimens (N=37, 19 from non-recurrent tumor specimens and 18 from patients that developed early recurrent cancer). QRT-PCR analysis showed that Dicer expression in tumors is approximately 1.5–1.6 fold of that in normal tissues (Fig. 1B). This result, together with two previous studies (28, 29), shows that Dicer is upregulated in human prostate cancer tissues.
Fig. 1. Dicer is upregulated in prostate cancer, but relatively lower Dicer expression predicts poor clinical outcome.

(A) qRT-PCR and Western blot analyses of Dicer expression in normal primary (PrEC) and immortalized (PNT1, and PNT2) prostate epithelial cells, and cancerous human prostate epithelial cell lines. (B) qRT-PCR analysis of Dicer expression in normal and cancerous human prostate specimens. * p = 0.0008, one way ANOVA t test. (C) Kaplan-Meier survival plot for correlation of Dicer expression with prostate cancer recurrence in 5 years. Log-rank test: p = 0.0096.
Notably, the primary tumors that recurred expressed Dicer at a relatively lower level than the non-recurrent tumors (Fig. 1B). Consistently, analysis of a database from the Memorial Sloan Kettering Cancer Center (32) also shows that patients with relatively lower expression levels of Dicer (the lower 25% portion) in primary prostate cancer specimens are more likely to develop recurrent disease (Fig. 1C). Genomic analysis of the same database revealed that hemizygous loss of Dicer is very rare in primary human prostate cancer specimens (Supplemental Table 2). Collectively, our results showed that although a relatively lower expression level of Dicer predicts poor clinical outcome, Dicer is upregulated in prostate cancer specimens.
Ablation and attenuation of Dicer activity affect disease progression of a Pten null prostate cancer model
We sought to investigate whether the sustained and upregulated Dicer expression in prostate cancer cells reflects the functional necessity of Dicer for the survival and proliferation of prostate cancer cells. To address this issue, we reduced and ablated the RNase III activity of Dicer in a Pten null mouse model for prostate cancer, and evaluated the biological consequences. Briefly, ARR2PB-Cre (hereafter referred to as PB-Cre) (33), Ptenflox/flox (34), and Dicerflox/flox (1) mice were bred to generate a cohort of littermates that include PB-Cre;Ptenflox/flox, PB-Cre;Ptenflox/flox;Dicerflox/Wt, and PB-Cre;Ptenflox/flox;Dicerflox/flox mice (hereafter referred to as Pten−/−, Pten−/−Dicer−/+, and Pten−/−Dicer−/− mice). ARR2PB-Cre mice harbor the Cre transgene driven by a rat probasin promoter that is specifically active in prostate epithelia (33). Cre-LoxP mediated recombination disrupts the RNase III activity of Dicer by deleting its exon 24. The recombined allele encodes a truncated, functionally null Dicer.
In Dicerflox/flox mice, the ratio of genomic copy numbers of the floxed exon 24, and the non-floxed exon 21 is 1. Quantitative PCR analysis using genomic DNA showed that this ratio was reduced to 0.75 and 0.2 in 15 week-old Pten−/−Dicer−/+ and Pten−/−Dicer−/− mice, respectively, which confirms efficient deletion of exon 24 of Dicer (Fig. 2A). Taqman miRNA analysis shows that six representative prostate-expressed-microRNAs were downregulated in Pten−/−Dicer−/+ and Pten−/−Dicer−/− mice (Fig. 2B). In contrast the expression levels of the corresponding pre-microRNAs were not affected (Supplemental Fig. 1A). These results corroborate that Dicer activity is attenuated. Phospho-AKT staining confirmed that Pten was successfully disrupted in mice in all three groups (Supplemental Fig. 1B).
Fig. 2. Disrupting Dicer activity inhibits disease progression in the Pten null mouse model for prostate cancer.

(A) Quantitative PCR analyses of ratio of exon24/exon21 in genomic DNAs from 15-week-old Pten−/−, Pten−/−Dicer−/+, and Pten−/−Dicer−/− mice. (B) qRT-PCR analysis of 6 representative microRNAs in 15-week-old Pten−/−, Pten−/−Dicer−/+, and Pten−/−Dicer−/− mice. *: p < 0.05. (C) Images of prostates dissected from mice in different groups at 7, 15 and 32 weeks. Bar graphs show quantifications. Data represent means ± SD from 3–13 mice in individual groups. *: p < 0.05. Yellow bars = 5mm. (D–F) H&E staining of prostate tissues from the three groups at 7, 15 and 32 weeks. AP: anterior prostate; DLP: dorsolateral prostate; VP: ventral prostate. Black bars = 100 μm. (G) IHC analysis of smooth muscle actin (green). White bars = 100 μm.
Fig 2C shows representative images of the prostates dissected from 7-, 15- and 32-week-old Pten−/−, Pten−/−Dicer−/+, and Pten−/−Dicer−/− mice. The prostate tissues from Pten−/−Dicer−/−mice were significantly smaller and weighed less compared to those in the other two groups, which is in agreement with previous studies showing that complete ablation of Dicer activity negatively impacts tumor cell fitness (24, 25, 30, 31). Prostates from Pten−/−Dicer−/+ mice weighed similar to those from Pten−/− mice at 7 and 15 weeks of age, but weighed 50% less by 32 weeks.
Table 1 summarizes disease progression in all the examined experimental mice. Histological analysis revealed that despite the difference in tissue weight, disease progressed with similar dynamics in the three different groups at 7 weeks (Fig. 2D). All mice developed lesions at typical PIN III–IV stages based on the criteria established by Park et al (35). Glands with smooth or irregular outlines were surrounded by a fibromuscular sheath. Lumens of prostate lobes were filled with epithelial cells with nuclear pleomorphism and hyperchromasia displaying tufting and cribriform patterns. Figures 2E and 2F and Table 1 show that the majority of prostate lesions in Pten−/− and Pten−/−Dicer−/+ mice had progressed to at least microinvasive early cancer by 15 weeks and frank adenocarcinoma by 32 weeks. Immunostaining of smooth muscle actin reveals loss of the prostatic smooth muscle layer, corroborating the disease progression (Fig. 2G). In contrast, lesions in all of the Pten−/−Dicer−/− mice remained at the PIN III or IV stage by 15 weeks. Collectively, complete ablation of Dicer activity suppresses tumor growth and progression. In comparison, attenuating Dicer activity negatively impacts tumor growth, but does not affect disease progression.
Table 1.
Summary of disease progression in analyzed experimental mice
| Phenotype | Pten−/− | Pten−/−Dicer+/− | Pten−/−Dicer−/− | ||||
|---|---|---|---|---|---|---|---|
| Age | Disease stages | N | Ear tag # | N | Ear tag # | N | Ear tag # |
| 7 weeks | PIN3/4 | 2 | 687, 860 | 3 | 372, 676, 732 | ||
| PIN4/early cancer | 4 | 208, 671, 696, 858 | 3 | 537, 669, 686 | |||
| Cancer | 2 | 713, 859 | |||||
| 14–18 weeks | PIN3 | 3 | 10, 731,984 | ||||
| PIN4 | 4 | 54, 482, 696,772 | 5 | 176, 720, 727, 729, 941 | |||
| Early cancer | 5 | 1, 157, 166, 556, 559 | 5 | 165, 215, 219, 431, 886 | |||
| Cancer | 4 | 128, 250, 750, 843 | |||||
| 32 weeks | Early cancer | 2 | 986,987 | 2 | 593, 861 | Not determined | |
| Cancer | 11 | 53, 200, 201, 202, 704, 708, 710, 711, 776, 986, 987, 999 | 11 | 52, 196, 204, 205, 206, 485, 522, 539, 593, 797, 798, 861, 990 | |||
Attenuating Dicer activity induces a more invasive phenotype
Despite smaller primary tumor burdens, Pten−/−Dicer−/+ mice developed seminal vesicle obstruction at high penetrance (69.2%) (9 out of 13 mice) by 32 weeks, which is in sharp contrast to 7.7% (1 out of 13 mice) observed in Pten−/− mice (p=0.004, Fisher exact test) (Fig. 3A). Histological analysis showed that there are more compact tumor tissues present at the region where seminal vesicles join the prostate in Pten−/−Dicer−/+ mice (arrow, Fig. 3B). However, we did not detect any micro-metastases in distal organs such as the liver and the lung in all the examined mice. These observations imply that tumors in Pten−/−Dicer−/+ mice are more locally invasive.
Fig. 3. Hemizygous loss of Dicer induces more invasive cancer.

(A) Images of urogenital organs dissected from Pten−/− and Pten−/−Dicer−/+ mice. Pie graphs quantify occurrence of seminal vesicle (SV) obstruction. (B) H&E staining of urogenital organs dissected from 32-week-old Pten−/− and Pten−/−Dicer−/+ mice. Pro: prostate; SV: seminal vesicle; VD: Vas deferens (C) Heatmap from microarray analysis shows changes of expression of 41 metastasis-associated genes in favor of metastasis in Pten−/−Dicer−/+ mice. (D) Validation of expression changes of representative genes in microarray analysis by qRT-PCR. Data represent means ± SD from 3 different mice. *: p < 0.05. (E–F) qRT-PCR analysis and Western blot analysis of expression of Vimentin and E-cadherin in prostate tissues from Pten−/−, Pten−/−Dicer−/+, and Pten−/−Dicer−/− mice at 15 weeks. Expression level of Vimentin (band intensity) is quantified using the Image J software. Individual lanes in Western Blot analysis represent specimens from different mice.
To understand the basis of this invasive phenotype, we examined global changes in gene expression in 15 week-old mouse prostates using Agilent 44k whole genome expression microarrays. A total of 544 genes were statistically significantly altered by at least 1.4 fold in the prostate tissues of either Pten−/−Dicer−/+ or Pten−/−Dicer−/− mice, or both mice, as compared with Pten−/− mice. Approximately 22% of genes were altered in the same trend in the prostate tissues of Pten−/−Dicer−/+ and Pten−/−Dicer−/− mice, while 70% of genes were only significantly affected in Pten−/−Dicer−/− mouse prostates. Very few genes were differentially regulated in the prostate tissues from Pten−/−Dicer−/+ and Pten−/−Dicer−/− mice or were only altered in those of Pten−/−Dicer−/+ mice. Data analysis revealed that a list of 41 genes (Fig. 3C) associated with epithelial-mesenchymal transition, cell adhesion, migration, and extracellular matrix deposition were significantly altered in favor of tumor metastasis in the prostate tissues of Pten−/−Dicer−/+ mice. QRT-PCR confirmed the changes in expression of some genes including growth factors (Tgfb1 and Ctgf), transcription factors (Snail, Twist2, and FoxC1), protease (Mmp7), and extracellular matrix components (Col5a and Col6a) (Fig. 3D). We also evaluated the expression of two other major players in EMT: Vimentin and E-Cadherin. Consistent to the invasive phenotype in Pten−/−Dicer−/+ mice, Western blot analysis showed that E-Cadherin and Vimentin were down-regulated and slightly up-regulated in Pten−/−Dicer−/+ mouse prostate tissues, respectively (Fig. 3F). Surprisingly, qRT-PCR analysis showed that they were both substantially down-regulated in prostate tissues of Pten−/−Dicer−/− mice, but were not altered in tissues from Pten−/−Dicer−/+ mice (Fig. 3E), suggesting the existence of important post-transcriptional regulatory mechanisms. Collectively, these findings corroborate the phenotypic invasiveness of the tumors in Pten−/−Dicer−/+ mice at a molecular level.
Abolishing Dicer activity enhances apoptosis and senescence in the Pten null prostate cancer model
TUNEL analysis reveals a 2.5 and 1.5 fold increase in apoptotic index in the prostates of 15 week-old Pten−/−Dicer−/− and Pten−/−Dicer−/+ mice, respectively, as compared to Pten−/−mice (Fig. 4A). Costaining of the cleaved caspase 3 (CC3) and the luminal cell marker cytokeratin 8 (K8) showed that almost all apoptotic cells are luminal cells, suggesting that attenuating Dicer activity negatively impacts the survival of luminal epithelial cells within the PIN lesions (Fig. 4B). Similar observations were made in the prostates of 7-week-old mice, though the increase in apoptosis does not reach statistical significance (Supplemental Figure 2). Pten-loss-induced cellular senescence (PICS) has been shown to impede rapid progression of fully developed prostate adenocarcinoma in the Pten null prostate cancer model (36, 37). Senescence-associated β-Galactosidase staining shows that tissues from Pten−/−Dicer−/− mice display a much stronger senescent phenotype than those from Pten−/− and Pten−/−Dicer−/+ mice (Fig. 4C). Collectively, these results demonstrate that impairing Dicer function induces luminal cell apoptosis while complete disruption of Dicer activity potentiates both apoptosis and senescence in the Pten null mouse model for prostate cancer.
Fig. 4. Suppressing Dicer activity enhances cellular apoptosis and senescence in the Pten null mouse model for prostate cancer.

(A) TUNEL assay and (B) IHC analysis of cleaved caspase 3 with luminal cell marker K8 in prostate tissues from Pten−/−, Pten−/−Dicer−/+, and Pten−/−Dicer−/− mice at 15 weeks. Bar graphs show quantifications. (C) β-galactosidase staining measuring cellular senescence in Pten−/−, Pten−/−Dicer−/+, and Pten−/−Dicer−/− mice. BGAV: β-galactosidase activity value. Bar graph quantifies the level of senescence. (D) Western blot analyses of expression of P16, P27, P15, P21, P53, and phosphor-γH2A.X in 15 week-old Pten−/−, Pten−/−Dicer−/+, and Pten−/−Dicer−/− mice. Individual lanes represent specimens from different mice. (E) qRT-PCR analysis of expression of three P53 target genes Bax, CD95 and Cyclin D2 in prostate tissues from Pten−/−, Pten−/−Dicer−/+, and Pten−/−Dicer−/− mice at 15 weeks. In all figures, data represent means ± SD. *: p < 0.05.
Disrupting Dicer activity induces senescence in MEF and primary skin cells by upregulating Arf and P53 (7). We investigated whether disrupting Dicer activity in the prostate may enhance apoptosis and senescence by upregulating cyclin-dependent kinase inhibitors and P53 by Western blot analyses. Expression levels of P16 and P27 were higher in Pten−/−Dicer−/+ and Pten−/−Dicer−/− mouse prostates than those in Pten−/− mice at 15 weeks (Fig. 4D). We were not able to determine the expression of P19 due to technical issues. The expressions of P15 and P21 were not altered (Fig. 4D). Surprisingly, P53 was down-regulated in Pten−/−Dicer−/− mouse prostates. In agreement with this, qRT-PCR analysis shows that the expression levels of three P53 target genes were also decreased in Pten−/−Dicer−/− mouse prostates. Collectively, these findings suggest that upregulation of P16/INK4a and P27/Kip1, but not P53, may contribute to the apoptotic and senescent phenotypes. Of note, senescence is not increased in the Pten−/−Dicer−/+ mice while the increase in apoptosis in Pten−/−Dicer−/+ mice is milder as compared to that in Pten−/−Dicer−/−mice. Therefore, further functional verifications are necessary to dissect whether upregulation of P16 and P27 plays a causative role or represents a consequence of impaired function of Dicer.
Inhibiting Dicer expression can elicit DNA damage in cultured cells (38). Recent studies also showed that Dicer and Drosha are involved in generating small RNAs like diRNAs and DDRNAs that are necessary for efficient DNA double strand repair (39, 40). We did not detect substantial increases in phosphorylation of γH2A.X by Western Blot in the prostate tissues of Pten−/−Dicer−/+ and Pten−/−Dicer−/− mice (Fig. 4D), suggesting that increased senescence and apoptosis in those mice may not be induced by deregulation of the DNA damage response.
Dicer downregulation affects proliferative and migratory capacities of human prostate cancer cells
To determine whether suppressing Dicer activity would affect the biology of human prostate cancer cells, we knocked down Dicer using two well-characterized Dicer shRNAs (23) in androgen receptor negative (PC3 and DU145) and androgen-receptor expressing prostate cancer cell lines (C4-2 and CWR22Rv1). Quantitative RT-PCR analysis showed that the maturation of some but not all prostate-expressed microRNAs that we tested were impaired when Dicer expression was knocked down (Supplemental Fig. 3). This suggests that knocking down the expression of Dicer in DU145 cells to the level comparable to that in normal or immortalized prostate epithelial cells (Figs. 1A and 5A) has an impact on the maturation of at least some microRNAs. Both shRNAs inhibited the in vitro growth of all the tested cell lines (Figs. 5A, E and S4). Knocking down Dicer in all four cell lines attenuated cell proliferation as determined by BrdU incorporation, and led to higher apoptotic indices as monitored by the expression of cleaved caspase 3 (Figs. 5B–C, F–G, and S4). Dicer suppression reduced the capacity of all four cell lines to form colonies in the soft agar assay (Figs. 5D, H, and S4). These studies demonstrate that reducing Dicer activity suppresses prostate cancer cell growth and tumorigenicity. In sharp contrast, knocking down Dicer expression in the breast cancer cell line MCF-7 did not affect cellular growth, apoptosis or BrdU incorporation (Fig. 5I–K), but instead enhanced the colony forming activity of MCF-7 cells in the soft agar assay, as has been reported previously (Fig. 5L) (23). These data suggest that cancer cells of different tissue origins may require distinct levels of Dicer activity for optimal survival and growth. Finally, transwell migration assay showed that reducing Dicer expression enhanced the migratory capacities of CWR22Rv1 cells (Fig. 6). The migration of C4-2 cells was also slightly increased when Dicer was knocked down, but did not reach statistical significance. In contrast, suppressing Dicer activity did not enhance the migratory capacities of the AR negative PC3 and DU145 cell lines (Data not shown).
Fig. 5. Suppressing Dicer activity attenuates proliferation and tumorigenesis of the human prostate cancer cells.

(A, E, I) In vitro proliferation curve of control and Dicer shRNA-expressing DU145 cells (A), C4-2 cells (E), and MCF-7 cells (I). Data represent means from three independent experiments. Insets: Western blot assay confirms knockdown of Dicer. (B, F, J) Bar graphs show quantification of BrdU positive proliferating cells in control and Dicer shRNA-expressing DU145 cells (B), C4-2 cells (F), and MCF-7 cells (J). (C, G, K) Bar graphs show quantification of cleaved caspase 3(CC3) positive apoptotic cells in control and Dicer shRNA-expressing DU145 cells (C), C4-2 cells (G), and MCF-7 cells (K). (D, H, L) Soft agar assay using control and Dicer shRNA-expressing DU145 cells (D), C4-2 cells (H), and MCF-7 cells (L). Data represent means ± SD. *: p < 0.05.
Fig. 6. Suppressing Dicer activity enhances in vitro migration of some prostate cancer cells.
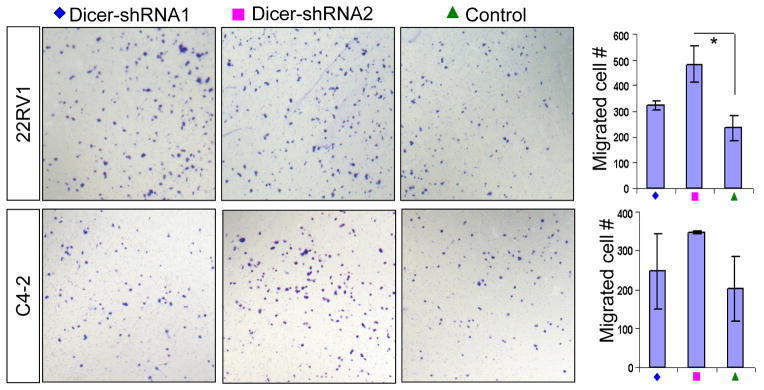
Images show transwell migration assay using control and Dicer shRNA-expressing CWR22Rv1 and C4-2 cells. Bar graphs show quantifications. Data represent means ± SD. *: p < 0.05.
Discussion
A tissue context- and dosage-dependent role of Dicer in carcinogenesis
While most studies on epithelial malignancies concur that a relatively lower Dicer expression predicts poor clinical outcomes (13–21), it should be noted that cancerous tissues do not always express lower levels of Dicer than normal tissues do. For example, Dicer is expressed at a higher level in lung adenocarcinoma and cutaneous melanoma than in their respective benign counterparts (18, 41). These facts imply that an optimal level of Dicer is necessary for cancer cells to balance survival, proliferation and invasiveness to ensure efficient disease progression.
Our result that ablating Dicer activity significantly delays progression of prostate cancer is consistent with most of the previous studies showing that Dicer is essential for the maintenance of cancer cell fitness (22, 24, 25, 30, 31), although Kim et al showed that complete loss of Dicer synergizes with loss of function of Pten to stimulate progression of ovarian cancer (42). In contrast, hemizygous loss of Dicer accelerates progression of K-Ras induced lung cancer and Rb loss induced retinoblastoma (22, 24) but not that of Myc-induced lymphoma (25). We found that Dicer hemizygous loss or partial knockdown attenuates the proliferation, survival and tumorigenicity of prostate cancer cells but enhances the invasiveness. This result is consistent with the information obtained from human prostate specimens that Dicer expression is in general upregulated in cancerous tissues, but relatively lower Dicer expression predicts poor clinical outcome. In summary, Dicer plays a pleiotropic role in tumorigenesis that is not only dosage-dependent but also tissue context-dependent.
Mechanism by which loss of function of Dicer induces senescence and apoptosis
Dicer deletion can increase the expressions and activities of cyclin-dependent kinase inhibitors (7, 8). The molecular mechanisms underpinning such regulation have not been thoroughly understood. Suppressing Dicer activity can release gene silencing by removing localized promoter DNA methylation (43). Alternatively, Dicer deletion can up-regulate gene expression by releasing suppression from microRNAs (44).
The beta-galactosidase assay showed clearly that Pten−/−Dicer−/− mice displayed a more severe senescent phenotype, presumably through a synergy between Pten-loss induced-cellular senescence (37) and upregulation of P16 and P27 (45). Although P16 and P27 were also upregulated consistently in Pten−/−Dicer−/+ mice, we did not observe an increase in senescence as compared to Pten−/− mice. However, because of the semi-quantitative nature of the β-galactosidase assay, it is possible that a slight increase in senescence in Pten−/−Dicer−/+ mice that is below the detection threshold may also contribute to the decreased primary tumor burden in those mice.
Enhanced invasive phenotype in Pten−/−Dicer−/+ mice
Dicer was down-regulated in areas of invasive lung and breast cancers (16, 18). Lower Dicer expression level was also correlated with a mesenchymal phenotype of breast cancer cell lines (16). Several in vitro studies further showed that reducing Dicer activity enhances invasion of various human cancer cell lines (13, 17, 46). Our study showing that Dicer hemizygous loss promotes a more invasive phenotype provides additional in vivo genetic evidence that the level of Dicer activity regulates tumor invasiveness. This invasiveness may be caused by upregulation of master transcriptional factors of epithelial-mesenchymal transition, such as Twist and FoxC1, as shown in Fig. 3D. It is also proposed that suppressing Dicer activity may cause reduction of mature microRNAs that play critical roles in EMT, such as the miR-200 family members (47). Reducing Dicer expression only enhanced invasion of CWR22Rv1 and C4-2 cells but not PC3 and DU145 cells, suggesting that the mechanism through which Dicer regulates cell invasion can be interrupted in some cancer cell lines. Interestingly, PC3 and DU145 cells do not express the androgen receptor (AR). Dicer knockout has been shown to inhibit AR function in the prostate (48). Suppressing AR activity in human and rodent prostate cancer cells induces epithelial-mesenchymal transition (49). Therefore, it is possible that suppressing Dicer enhances prostate cancer cell migration partly by attenuating AR activity. Collectively, our study suggests that suppressing Dicer activity can suppress the growth of prostate tumor cells, but can lead to a more invasive phenotype.
Materials and Methods
Mouse and genotyping
Wild type C57BL/6 mice and Dicer1tm1bdh/J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). NOD/SCID beige mice were purchased from Charles River (Wilmington, MA). The ARR2PB-Cre transgenic mice (33) were from Dr. Fen Wang at the Institute of Bioscience and Technology, Texas A&M Health Science Center. The Ptenfl/fl mice (34) were from Dr. Hong Wu at the University of California Los Angeles (34). ARR2PB-Cre;Ptenfl/fl;Dicerfl/wt female mice and Ptenfl/fl;Dicerfl/wt male mice were mated to generated ARR2PB-Cre;Ptenfl/fl, ARR2PB-Cre;Ptenfl/fl;Dicerfl/wt, and ARR2PB-Cre;Ptenfl/fl;Dicerfl/fl mice. Prostate tissues were collected from 7-, 15-, or 32-week-old experimental mice. All mice received 80mg/kg BrdU 2.5 hours prior to euthanasia via I.P injection. All mice were housed and bred under the regulation of The Center for Comparative Medicine at the Baylor College of Medicine. Primers for genotyping are listed in Supplemental Table 1.
RNA from normal and cancerous human prostate specimens
All radical prostatectomy tissue samples were obtained from Baylor Prostate Specialized Programs of Research Excellence (SPORE) Tissue Core and collected from fresh radical prostatectomy specimens after obtaining informed consent under an institutional review board-approved protocol. Early recurrent and non-recurrent cancers are defined as PSA recurrence within one year and no PSA recurrence in 5 years, respectively. PSA recurrence is defined as having two successive follow-up PSA values >0.2 ng/mL >30 days following surgery. Cancer samples were at least 70% cancer.
RNA isolation and quantitative RT-PCR
Total RNA was isolated using the miRNA Kit (Qiagen, Gaithersburg, MD). Reverse transcription was performed using the Taqman microRNA RT kit (Applied Biosystems Incorporation, Foster City, CA) and superscript Kit (BioRad, Hercules, CA). Quantitative RT-PCR was performed using the Taqman microRNA assay (Applied Biosystems, Foster City, CA) and SYBR GreenER qPCR mix (BioRad, Hercules, CA) on a StepOne plus Real-Time PCR system (Applied Biosystems, Foster City, CA). The relative amount of specific mRNA and microRNA was normalized to Gapdh and U6, respectively. Primers for qRT-PCR are listed in Supplemental Table 1.
Expression microarray
Expression microarray assays were performed using 4×44K Whole Mouse Genome Oligo Microarray chip (Agilent Technologies, Santa Clara, CA) and the Feature Extraction Software v9.1.3.1 (Agilent Technologies) was used to extract and analyze the signals. Array data have been deposited on GEO (GSE42820).
Western Blot analysis
Tissue and cells were lysed in RIPA buffer containing protease inhibitors (Sigma, St. Louis, MO) and PhosSTOP Phosphatase Inhibitor Cocktail (Roche, Pleasanton, CA). Western Blot analysis was performed as described previously (50) using primary antibodies against Dicer (13D6, Clonegene, Hartford, CT), P15, P16, P19 (Santa Cruz Biotechnology, Santa Cruz, CA), P21, P27, P53, phospho-γH2A.X (Cell signaling Technologies, Danvers, MA), Vimentin (Fitzgerald, Germany), E-Cadherin and β-actin (Sigma, St. Louis, MO) and peroxidase-conjugated goat anti-mouse IgG or goat anti-rabbit IgG (Jackson ImmunoResearch, Inc., West Grove, PA). Results were quantified using the NIH Image J software.
Lentivirus production and transduction
The Dicer shRNA lentiviral vectors and the control pSicoR vector were made by the laboratory of Dr. Tyler Jacks (23), and were purchased from Addgene (Cambridge, MA). Lentivirus preparation, titering, and infection of dissociated prostate cells were performed as described previously (50).
Cell culture, and in vitro and in vivo cellular assays
The primary PrEC cells were cultured in the PrEGM media and both were purchased from Lonza (Walkersville, MD). PNT1a and PNT2 cells were from the European Collection of Cell Cultures and were obtained from the laboratory of Michael Ittmann at the Baylor College of Medicine. LNCaP, PC-3, DU145, CWR22Rv1, and MCF-7 cells were from American Type Culture Collection (Manassas, VA). C4-2 cells were from UroCor, Inc. (Oklahoma City, OK). These cells were maintained in RPMI 1640 with 10% FBS with penicillin and streptomycin (Invitrogen, Carlsbad, CA). All cell lines were maintained at 37 °C in a humid atmosphere containing 5% CO2.
For in vitro proliferation assay, 1.5–3 ×105 cells were seeded in 6 well-plates. Cells were counted using a VI-Cell Cell Viability Analyzer (Bechman-Coulter, Brea, CA) for 4 sequential days. Cell proliferation was also measured by the MTT assay according to the manufacturer’s instruction (Promega, Fitchburg, WI). Soft agar assays were performed using 6-well plates as described previously (22). 5000 cells were plated in each well in triplicates. For BrdU labeling assays, BrdU (Sigma, St. Louis, MO) was added in cell culture media to a final concentration of 50 μM and incubated for 1 hour at 37 °C with 5% CO2. The in vitro cell migration assay was performed using the BioCoat Matrigel Invasion Chamber (BD Biosciences, San Jose, CA). Briefly, 1–6 ×104 cells were put in each chamber and incubated for 12–24 hours before invaded cells were quantified according to the manufacturer’s instruction.
Histology, immunohistochemical and senescence analyses
Histological and IHC analyses were performed as described previously (50). Slides were made from formalin-fixed and paraffin-embedded prostate tissues or from frozen tissues. Tissue sections were either stained with H&E or the following antibodies: mouse anti-cytokeratin 8 (Covance, Berkeley, California), mouse anti-BrdU (DSHB, Iowa City, Iowa), mouse anti-smooth muscle actin (Sigma-Aldrich, St. Louis, MO), and rabbit anti-pAKT and anti-cleaved caspase 3 (Cell Signaling Technologies, Danvers, MA). For fluorescence visualization, sections were stained with the Alexa Fluor 594 goat anti-rabbit IgG (H+L) and Alexa Fluor 594 goat anti-mouse IgG (H+L) (Invitrogen, Carlsbad, California) secondary antibodies. Sections were counterstained with DAPI in mounting medium (Vector Laboratories, Burlingame, CA) and analyzed by regular or confocal fluorescent microscopy. Senescence associated beta-galactosidase staining was performed using the Senescence β-Galactosidase (SABG) Staining Kit (Cell Signaling Technologies, Danvers, MA) according to the manufacturer’s instruction. Staining was performed for 7 hours at 37 °C in dark. The intensity of SABG staining was quantified as described previously (51).
Statistics
All experiments were performed using 3–13 mice in independent experiments. P values were calculated using the Student’s t test and One-Way ANOVA analysis with Excel software and Graph-Pad Prism. The 0.05 level of confidence was accepted for statistical significance. For microarray data analysis, two-sided t-test and fold changes (using log-transformed values), were used to assess differential expression. Expression heat maps were generated using JavaTreeview (PMID: 15180930).
Supplementary Material
Acknowledgments
Source of support: National Cancer Institute, Cancer Prevention and Research Institute of Texas.
We thank Drs. Lawrence Donehower and Jeffrey Rosen for critical comments, Dr. Xiongbin Lu for sharing reagents. This work is supported by NIH R00 CA125937 (L.X.), Cancer Prevention Research Institute of Texas RP110005 (L.X.), NIH U01 CA141497 (M.M.I.), and NIH P30 CA125123 (PI: Kent Osborne).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Harfe BD, McManus MT, Mansfield JH, Hornstein E, Tabin CJ. The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc Natl Acad Sci U S A. 2005;102(31):10898–903. doi: 10.1073/pnas.0504834102. Epub 2005/07/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakagawa A, Shi Y, Kage-Nakadai E, Mitani S, Xue D. Caspase-dependent conversion of Dicer ribonuclease into a death-promoting deoxyribonuclease. Science. 2010;328(5976):327–34. doi: 10.1126/science.1182374. Epub 2010/03/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yi R, O’Carroll D, Pasolli HA, Zhang Z, Dietrich FS, Tarakhovsky A, et al. Morphogenesis in skin is governed by discrete sets of differentially expressed microRNAs. Nature genetics. 2006;38(3):356–62. doi: 10.1038/ng1744. Epub 2006/02/08. [DOI] [PubMed] [Google Scholar]
- 4.Andl T, Murchison EP, Liu F, Zhang Y, Yunta-Gonzalez M, Tobias JW, et al. The miRNA-processing enzyme dicer is essential for the morphogenesis and maintenance of hair follicles. Current biology: CB. 2006;16(10):1041–9. doi: 10.1016/j.cub.2006.04.005. Epub 2006/05/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, et al. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes & development. 2005;19(4):489–501. doi: 10.1101/gad.1248505. Epub 2005/02/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koralov SB, Muljo SA, Galler GR, Krek A, Chakraborty T, Kanellopoulou C, et al. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell. 2008;132(5):860–74. doi: 10.1016/j.cell.2008.02.020. Epub 2008/03/11. [DOI] [PubMed] [Google Scholar]
- 7.Mudhasani R, Zhu Z, Hutvagner G, Eischen CM, Lyle S, Hall LL, et al. Loss of miRNA biogenesis induces p19Arf-p53 signaling and senescence in primary cells. J Cell Biol. 2008;181(7):1055–63. doi: 10.1083/jcb.200802105. Epub 2008/07/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu S, Guo K, Zeng Q, Huo J, Lam KP. The RNase III enzyme Dicer is essential for germinal center B-cell formation. Blood. 2012;119(3):767–76. doi: 10.1182/blood-2011-05-355412. Epub 2011/11/26. [DOI] [PubMed] [Google Scholar]
- 9.O’Rourke JR, Georges SA, Seay HR, Tapscott SJ, McManus MT, Goldhamer DJ, et al. Essential role for Dicer during skeletal muscle development. Developmental biology. 2007;311(2):359–68. doi: 10.1016/j.ydbio.2007.08.032. Epub 2007/10/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pastorelli LM, Wells S, Fray M, Smith A, Hough T, Harfe BD, et al. Genetic analyses reveal a requirement for Dicer1 in the mouse urogenital tract. Mamm Genome. 2009;20(3):140–51. doi: 10.1007/s00335-008-9169-y. Epub 2009/01/27. [DOI] [PubMed] [Google Scholar]
- 11.Zhang L, Zhang B, Valdez JM, Wang F, Ittmann M, Xin L. Dicer Ablation Impairs Prostate Stem Cell Activity and Causes Prostate Atrophy. Stem Cells. 2010 doi: 10.1002/stem.455. Epub 2010/06/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–8. doi: 10.1038/nature03702. Epub 2005/06/10. [DOI] [PubMed] [Google Scholar]
- 13.Jafarnejad SM, Ardekani GS, Ghaffari M, Martinka M, Li G. Sox4-mediated Dicer expression is critical for suppression of melanoma cell invasion. Oncogene. 2012 doi: 10.1038/onc.2012.239. Epub 2012/06/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin RJ, Lin YC, Chen J, Kuo HH, Chen YY, Diccianni MB, et al. microRNA signature and expression of Dicer and Drosha can predict prognosis and delineate risk groups in neuroblastoma. Cancer Res. 2010;70(20):7841–50. doi: 10.1158/0008-5472.CAN-10-0970. Epub 2010/09/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khoshnaw SM, Rakha EA, Abdel-Fatah TM, Nolan CC, Hodi Z, Macmillan DR, et al. Loss of Dicer expression is associated with breast cancer progression and recurrence. Breast cancer research and treatment. 2012;135(2):403–13. doi: 10.1007/s10549-012-2169-3. Epub 2012/07/24. [DOI] [PubMed] [Google Scholar]
- 16.Grelier G, Voirin N, Ay AS, Cox DG, Chabaud S, Treilleux I, et al. Prognostic value of Dicer expression in human breast cancers and association with the mesenchymal phenotype. British journal of cancer. 2009;101(4):673–83. doi: 10.1038/sj.bjc.6605193. Epub 2009/08/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noh H, Hong S, Dong Z, Pan ZK, Jing Q, Huang S. Impaired MicroRNA Processing Facilitates Breast Cancer Cell Invasion by Upregulating Urokinase-Type Plasminogen Activator Expression. Genes & cancer. 2011;2(2):140–50. doi: 10.1177/1947601911408888. Epub 2011/07/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiosea S, Jelezcova E, Chandran U, Luo J, Mantha G, Sobol RW, et al. Overexpression of Dicer in precursor lesions of lung adenocarcinoma. Cancer Res. 2007;67(5):2345–50. doi: 10.1158/0008-5472.CAN-06-3533. Epub 2007/03/03. [DOI] [PubMed] [Google Scholar]
- 19.Karube Y, Tanaka H, Osada H, Tomida S, Tatematsu Y, Yanagisawa K, et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer science. 2005;96(2):111–5. doi: 10.1111/j.1349-7006.2005.00015.x. Epub 2005/02/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faggad A, Budczies J, Tchernitsa O, Darb-Esfahani S, Sehouli J, Muller BM, et al. Prognostic significance of Dicer expression in ovarian cancer-link to global microRNA changes and oestrogen receptor expression. The Journal of pathology. 2010;220(3):382–91. doi: 10.1002/path.2658. Epub 2009/12/05. [DOI] [PubMed] [Google Scholar]
- 21.Merritt WM, Lin YG, Han LY, Kamat AA, Spannuth WA, Schmandt R, et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med. 2008;359(25):2641–50. doi: 10.1056/NEJMoa0803785. Epub 2008/12/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar MS, Pester RE, Chen CY, Lane K, Chin C, Lu J, et al. Dicer1 functions as a haploinsufficient tumor suppressor. Genes & development. 2009;23(23):2700–4. doi: 10.1101/gad.1848209. Epub 2009/11/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nature genetics. 2007;39(5):673–7. doi: 10.1038/ng2003. Epub 2007/04/03. [DOI] [PubMed] [Google Scholar]
- 24.Lambertz I, Nittner D, Mestdagh P, Denecker G, Vandesompele J, Dyer MA, et al. Monoallelic but not biallelic loss of Dicer1 promotes tumorigenesis in vivo. Cell death and differentiation. 2010;17(4):633–41. doi: 10.1038/cdd.2009.202. Epub 2009/12/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arrate MP, Vincent T, Odvody J, Kar R, Jones SN, Eischen CM. MicroRNA biogenesis is required for Myc-induced B-cell lymphoma development and survival. Cancer Res. 2010;70(14):6083–92. doi: 10.1158/0008-5472.CAN-09-4736. Epub 2010/07/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103(7):2257–61. doi: 10.1073/pnas.0510565103. Epub 2006/02/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ozen M, Creighton CJ, Ozdemir M, Ittmann M. Widespread deregulation of microRNA expression in human prostate cancer. Oncogene. 2008;27(12):1788–93. doi: 10.1038/sj.onc.1210809. Epub 2007/09/25. [DOI] [PubMed] [Google Scholar]
- 28.Ambs S, Prueitt RL, Yi M, Hudson RS, Howe TM, Petrocca F, et al. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res. 2008;68(15):6162–70. doi: 10.1158/0008-5472.CAN-08-0144. Epub 2008/08/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiosea S, Jelezcova E, Chandran U, Acquafondata M, McHale T, Sobol RW, et al. Up-regulation of dicer, a component of the MicroRNA machinery, in prostate adenocarcinoma. Am J Pathol. 2006;169(5):1812–20. doi: 10.2353/ajpath.2006.060480. Epub 2006/10/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nittner D, Lambertz I, Clermont F, Mestdagh P, Kohler C, Nielsen SJ, et al. Synthetic lethality between Rb, p53 and Dicer or miR-17-92 in retinal progenitors suppresses retinoblastoma formation. Nature cell biology. 2012;14(9):958–65. doi: 10.1038/ncb2556. Epub 2012/08/07. [DOI] [PubMed] [Google Scholar]
- 31.Ravi A, Gurtan AM, Kumar MS, Bhutkar A, Chin C, Lu V, et al. Proliferation and tumorigenesis of a murine sarcoma cell line in the absence of DICER1. Cancer Cell. 2012;21(6):848–55. doi: 10.1016/j.ccr.2012.04.037. Epub 2012/06/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. Epub 2010/06/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin C, McKeehan K, Wang F. Transgenic mouse with high Cre recombinase activity in all prostate lobes, seminal vesicle, and ductus deferens. Prostate. 2003;57(2):160–4. doi: 10.1002/pros.10283. Epub 2003/09/02. [DOI] [PubMed] [Google Scholar]
- 34.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4(3):209–21. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 35.Park JH, Walls JE, Galvez JJ, Kim M, Abate-Shen C, Shen MM, et al. Prostatic intraepithelial neoplasia in genetically engineered mice. Am J Pathol. 2002;161(2):727–35. doi: 10.1016/S0002-9440(10)64228-9. Epub 2002/08/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–30. doi: 10.1038/nature03918. Epub 2005/08/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alimonti A, Nardella C, Chen Z, Clohessy JG, Carracedo A, Trotman LC, et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. The Journal of clinical investigation. 2010;120(3):681–93. doi: 10.1172/JCI40535. Epub 2010/03/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang KF, Ren H, Cao J, Zeng GL, Xie J, Chen M, et al. Decreased Dicer expression elicits DNA damage and up-regulation of MICA and MICB. J Cell Biol. 2008;182(2):233–9. doi: 10.1083/jcb.200801169. Epub 2008/07/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu L, Mao L, Qi Y. Roles of DICER-LIKE and ARGONAUTE Proteins in TAS-Derived Small Interfering RNA-Triggered DNA Methylation. Plant physiology. 2012;160(2):990–9. doi: 10.1104/pp.112.200279. Epub 2012/08/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Francia S, Michelini F, Saxena A, Tang D, de Hoon M, Anelli V, et al. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature. 2012;488(7410):231–5. doi: 10.1038/nature11179. Epub 2012/06/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma Z, Swede H, Cassarino D, Fleming E, Fire A, Dadras SS. Up-regulated Dicer expression in patients with cutaneous melanoma. PLoS One. 2011;6(6):e20494. doi: 10.1371/journal.pone.0020494. Epub 2011/06/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim J, Coffey DM, Creighton CJ, Yu Z, Hawkins SM, Matzuk MM. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc Natl Acad Sci U S A. 2012;109(10):3921–6. doi: 10.1073/pnas.1117135109. Epub 2012/02/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ting AH, Suzuki H, Cope L, Schuebel KE, Lee BH, Toyota M, et al. A requirement for DICER to maintain full promoter CpG island hypermethylation in human cancer cells. Cancer Res. 2008;68(8):2570–5. doi: 10.1158/0008-5472.CAN-07-6405. Epub 2008/04/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu JY, Reynolds SH, Hatfield SD, Shcherbata HR, Fischer KA, Ward EJ, et al. Dicer-1-dependent Dacapo suppression acts downstream of Insulin receptor in regulating cell division of Drosophila germline stem cells. Development. 2009;136(9):1497–507. doi: 10.1242/dev.025999. Epub 2009/04/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Majumder PK, Grisanzio C, O’Connell F, Barry M, Brito JM, Xu Q, et al. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell. 2008;14(2):146–55. doi: 10.1016/j.ccr.2008.06.00. Epub 2008/08/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han L, Zhang A, Zhou X, Xu P, Wang GX, Pu PY, et al. Downregulation of Dicer enhances tumor cell proliferation and invasion. International journal of oncology. 2010;37(2):299–305. doi: 10.3892/ijo_00000678. Epub 2010/07/03. [DOI] [PubMed] [Google Scholar]
- 47.Martello G, Rosato A, Ferrari F, Manfrin A, Cordenonsi M, Dupont S, et al. A MicroRNA targeting dicer for metastasis control. Cell. 2010;141(7):1195–207. doi: 10.1016/j.cell.2010.05.017. Epub 2010/07/07. [DOI] [PubMed] [Google Scholar]
- 48.Narayanan R, Jiang J, Gusev Y, Jones A, Kearbey JD, Miller DD, et al. MicroRNAs are mediators of androgen action in prostate and muscle. PLoS One. 2010;5(10):e13637. doi: 10.1371/journal.pone.0013637. Epub 2010/11/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S, et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res. 2012;72(2):527–36. doi: 10.1158/0008-5472.CAN-11-3004. Epub 2011/11/24. [DOI] [PubMed] [Google Scholar]
- 50.Xin L, Ide H, Kim Y, Dubey P, Witte ON. In vivo regeneration of murine prostate from dissociated cell populations of postnatal epithelia and urogenital sinus mesenchyme. Proc Natl Acad Sci U S A. 2003;100 (Suppl 1):11896–903. doi: 10.1073/pnas.1734139100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shlush LI, Itzkovitz S, Cohen A, Rutenberg A, Berkovitz R, Yehezkel S, et al. Quantitative digital in situ senescence-associated beta-galactosidase assay. BMC cell biology. 2011;12:16. doi: 10.1186/1471-2121-12-16. Epub 2011/04/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.