

Abstract
Inactivation of glycogen synthase kinase 3 (GSK3) has been shown to mediate axon growth during development and regeneration. Phosphorylation of GSK3 by the kinase Akt is well known to be the major mechanism by which GSK3 is inactivated. However, whether such regulatory mechanism of GSK3 inactivation is used in neurons to control axon growth has not been directly studied. Here by using GSK3 mutant mice, in which GSK3 is insensitive to Akt-mediated inactivation, we show that sensory axons regenerate normally in vitro and in vivo after peripheral axotomy. We also find that GSK3 in sensory neurons of the mutant mice is still inactivated in response to peripheral axotomy and such inactivation is required for sensory axon regeneration. Lastly, we provide evidence that GSK3 activity is negatively regulated by PI3K signaling in the mutant mice upon peripheral axotomy, and the PI3K-GSK3 pathway is functionally required for sensory axon regeneration. Together, these results indicate that in response to peripheral nerve injury GSK3 inactivation, regulated by an alternative mechanism independent of Akt-mediated phosphorylation, controls sensory axon regeneration.
Keywords: Axon regeneration, GSK3 signaling, PI3K signaling, In vivo electroporation
1. Introduction
It is well known that mature neurons in the mammalian central nervous system (CNS) cannot regenerate their axons after an injury. In contrast, mature CNS neurons in non-mammalian animals, such as C. elegans, Drosophila, and zebrafish, and mature neurons in the mammalian peripheral nervous system (PNS) regenerate their axons robustly after the nerve injury [1]. Thus, it is generally recognized that understanding the molecular mechanisms by which the naturally occurred axon regeneration is regulated will help to find ways to promote mammalian CNS axon regeneration. Our recent study has identified PI3K signaling as a key regulator of mammalian PNS axon regeneration [2]. Downstream of the PI3K signaling, we showed that inactivation of GSK3 controlled axon regeneration by regulation of the regeneration-associated transcription factor, Smad1. In addition to axon regeneration, previous studies, including ours, have also shown that GSK3 inactivation downstream of PI3K play important roles in regulation of neuronal polarization [3,4] and NGF-induced axon growth [5] during development of the nervous system.
Downstream of PI3K, GSK3 is believed to be inactivated via Akt-mediated phosphorylation of serine residues located at the N-terminus of GSK3 (serine 21 for GSK3α, serine 9 for GSK3β) [6,7]. However, when wild type GSK3s are replaced by mutant GSK3s, whose N-terminal serines are mutated to alanines and thus cannot be phosphorylated, the resulting mutant mice, GSK3α-S21A/GSK3β-S9A double knockin (DKI) mice, develop normally with no overt phenotype in the nervous system [8]. In addition, hippocampal neurons from the GSK3 DKI mice polarize normally in culture, although GSK3 is still spatially inactivated in the axon but not the dendrites [9]. These findings suggest that Akt-mediated phosphorylation might not be the major mechanism by which GSK3 is inactivated in neurons downstream of PI3K signaling.
Here we used the GSK3 DKI mice to investigate if Akt-mediated GSK3 phosphorylation is necessary for peripheral axotomy-induced GSK3 inactivation and axon regeneration. We found that adult sensory neurons from GSK3 DKI mice regenerated their axons normally both in vitro and in vivo. In addition, GSK3s were still inactivated in sensory neurons upon peripheral axotomy, and such inactivation was necessary for efficient axon regeneration. Lastly, we provided evidence that in the absence of Akt-mediated phosphorylation, GSK3 inactivation and axon regeneration of adult sensory neurons were still regulated downstream of the PI3K signaling. Our study indicates that an alternative pathway downstream of PI3K signaling functions to inactivate GSK3 and control axon regeneration.
2. Materials and methods
2.1. Animals
All experiments using animals were approved by the Institutional Animal Care and Use Committee of the Johns Hopkins University. The GSK3 double knockin mice were from Dario Alessi (University of Dundee, Scotland). The wild type littermates of the GSK3 knockin mice were used as the control group.
2.2. Reagents and antibodies
LY294002 and 6-bromoindirubin-3′-oxime (BIO) were from Calbiochem (San Diego, CA). The βIII tubulin (TuJ1) was from Covance (Chantilly, VA). The antibodies against phospho-CRMP2(Thr514), GSK3α, Phospho-GSK3α (Ser21) and Phospho-GSK3β (Ser9) were from Cell Signaling (Beverly, MA). The GSK3β antibody was from BD Biosciences (Franklin Lakes, NJ).
2.3. Primary culture of adult DRG neurons
For in vitro regenerative axon growth experiments, the sciatic nerves of the adult mice were transected 1 week prior to the neuronal culture. Adult DRGs (L4 and L5) were then dissected out and digested at 37°C with collagenase A (1mg/ml, 90min) followed by trypLE express (1X, 20min). The digested DRG neurons were washed 3 times with the culture medium (MEM including 10% fetal calf serum, L-glutamine, and penicillin/streptomycin). The cells were then dissociated by trituration with 1 ml pipette tip, and the supernatant (containing dissociated single cells) was collected. The dissociated neurons were plated at low density onto glass coverslips coated with a mixture of 100 μg/ml poly-D-lysine and 10 μg/ml laminin for overnight culture in the absence of any growth factors. To transfect dissociated DRG neurons via electroporation before plating, the neurons were centrifuged to remove the culture medium and resuspended in 80 μl of Amaxa electroporation buffer (for mouse neurons), together with the DNA plasmid. Such cell mixture was then transferred to the electroporation cuvette and electroporated with the Amaxa Nucleofector. The cells were then immediately transferred to desired volume of prewarmed culture medium and plated. After 4–6 hours when neurons were attached to the coverslips, the culture medium was changed to remove the remnant electroporation buffer.
For experiments involving pharmacological drug treatment (PI3K and GSK3 inhibitors), the dissociated DRG neurons were first plated at high density in the presence of the indicated drug(s). After 3 days in culture, the drugs were washed out. The neurons were then resuspended and replated onto glass coverslips at lower density, followed by another overnight culture.
2.4. In vivo Electroporation of adult DRG neurons and sciatic nerve crush
The in vivo electroporation of adult DRG neurons were performed according to our previous published papers [10,11,12]. Briefly, the L4 and L5 DRGs on one side of the mouse were first surgically exposed. Solution of DNA plasmid encoding EGFP (3.0μg at 1μl) was injected into the DRGs using the capillary pipette connected to a Picospritzer II (Parker Ins.). The electroporation was then performed using a custom-made tweezer-like electrode and a BTX ECM830 Electro Square Porator (five 15 ms pulses at 35 V with 950 ms interval).
Two days after in vivo electroporation, the sciatic nerve on the side of the electroporated DRGs was crushed and the crush site was labeled with a size 10-0 nylon epineural suture. Three days after nerve crush, the mice were terminally anesthetized and perfused with ice-cold 4% paraformaldehyde (PFA). The whole sciatic nerve were dissected out and postfixed overnight in 4% PFA at 4°C.
2.5. Measurement of axon growth and regeneration
For in vitro axon growth experiments, the neurons were fixed and immunostained with the antibody against neuron specific tubulin (Tuj1). The fluorescence images of neurons were captured with an inverted fluorescence microscope controlled by the AxioVision 4.6 software (Carl Zeiss MicroImaging, Inc.). Only neurons with axons longer than twice the diameter of cell bodies were selected and the longest axon of each neuron was traced manually. About 50–100 neurons per experimental condition were measured to obtain the mean value, and at least 3 independent experiments were performed.
For in vivo axon regeneration experiments, the fluorescence images of the whole mount nerves were acquired using the MosaiX module of the AxioVision software. All identifiable EGFP-labeled axons in the whole mount sciatic nerve were manually traced from the crush site to the distal axonal tips and their lengths measured. At least 15 axons per mouse were measured and data from 6 mice for each experimental condition were used to calculate the mean axon length/mouse.
2.6. Western blot analysis
Dissociated DRG neurons or DRG tissues were collected and lysed using the RIPA buffer. The extracted proteins were analyzed with 4–12% gradient SDS/PAGE gel electrophoresis, and transferred onto PVDF membranes. After blocking with 5% non-fat milk, membranes were incubated with primary antibodies (4°C, overnight) followed by HRP linked secondary antibodies (room temperature, 1 hour). The densities of protein bands from 3 independent experiments were quantified using the Image J software.
2.7. Statistics
Data are presented as mean ± s.e.m. Two-tailed Student’s t-test was used to determine the statistical significance between different experimental groups, which was set at a value of p < 0.05.
3. Results
3.1. Akt mediated phosphorylation of GSK3α/β at Ser21/9 are not necessary for peripheral axotomy-induced axon regeneration in vitro and in vivo
We first examined if Akt-mediated phosphorylation of GSK3α/β at Ser21/9 were required for peripheral axotomy (conditioning lesion) induced regenerative axon growth by comparing adult sensory neurons from the GSK3 DKI mice with those from their wild type littermates. As shown in our previous studies, after a prior peripheral axotomy adult sensory neurons from wild type mice extended long and less branched axons in culture (Fig. 1). We found that peripheral axotomy induced similar regenerative axon growth of neurons from GSK3 DKI mice (Fig. 1). To confirm the in vitro result with an in vivo axon regeneration model, we examined sensory axon regeneration of GSK3 DKI mice in vivo using the sciatic nerve crush model. To accurately assess axon regeneration in vivo, we used our recently developed in vivo electroporation technique [12] to express EGFP in adult sensory neurons and their axons. Two days after electroporation, the sciatic nerves were subjected to a crush injury. Axon regeneration was then analyzed three days later by directly measuring the lengths of EGFP-labeled regenerating axons in whole mount nerves (Fig. 2). The result showed that there was no significant difference in sensory axon regeneration between GSK3 DKI and their wild type littermates (Fig. 2). Together, these results demonstrate clearly that phosphorylation of GSK3α/β at Ser21/9, presumably mediated by Akt, was not necessary for mouse sensory axon regeneration after peripheral nerve injury.
Figure 1.
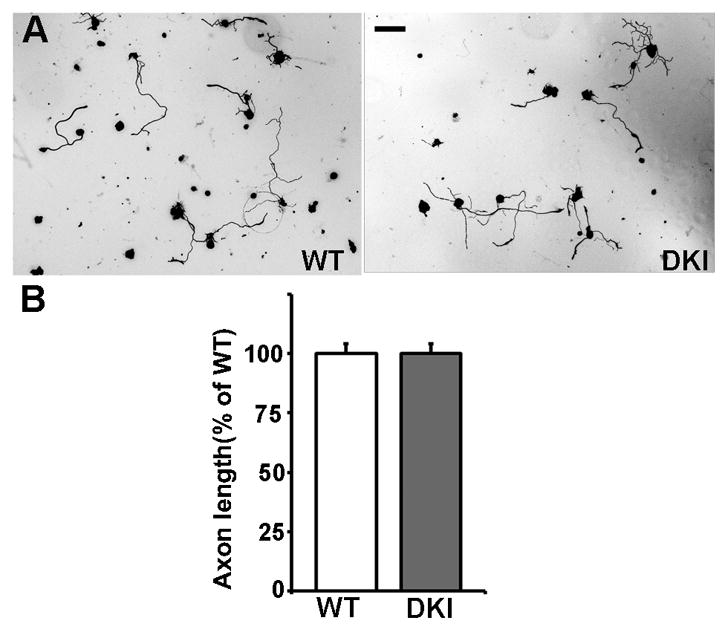
Adult sensory neurons from GSK3α-S21A/GSK3β-S9A double knockin mice show similar peripheral axotomy-induced regenerative axon growth to those from wild type mice. (A) Representative images of adult sensory neurons from double knockin (DKI) mice or wild type (WT) mice cultured overnight. Scale bar, 100 μm. (B) Quantification of axon length from at least 3 independent experiments. Note that there is no significant difference in axon length between neurons from wild type (WT) mice and neurons from double knockin mice (DKI). Error bars represent s.e.m.
Figure 2.

Peripheral axon regeneration in GSK3α-S21A/GSK3β-S9A double knockin mice is comparable to that in wild type mice. (A) Representative images of EGFP labeled regenerating axons in mouse sciatic nerves from wild type (WT) or GSK3 double knockin (DKI) mice. Red arrowheads mark the crush sites. Scale bar, 500 μm. (B, C) Quantification of axon regeneration in vivo from at least 6 mice in each experimental group (wild type or double knockin) by measuring the lengths of all identifiable regenerating axons from the crush site (arrowheads) to the distal axon tips. Error bars represent s.e.m.
3.2. Akt-independent GSK3 inactivation upon peripheral axotomy is required for axon regeneration
Our previous studies have shown GSK3 inactivation upon peripheral nerve injury is necessary for axon regeneration [2,13]. These results seem to be contradictory to the above findings because phosphorylation of GSK3α/β at Ser21/9 is to date the most recognized mechanism by which GSK3 is inactivated. To solve this problem, we investigated how GSK3 activity in adult sensory neurons from GSK3 DKI mice was regulated in response to peripheral nerve injury. Because no phosphorylated GSK3 can be detected in GSK3 DKI mice (Fig. 3A), we examined the phosphorylation level of CRMP2. Previous studies, including ours, have shown that GSK3 is the major kinase that specifically phosphorylates CRMP2 at the residue Threonine 514 (Thr514), and as a result, and level of phosphorylated CRMP2 at Thr514 is often used as a marker for GSK3 activity in neurons [4,14,15]. To our surprise, we found that under control condition without nerve injury, the phospho-CRMP2 levels were similar in adult sensory neurons between GSK3 DKI mice and their wild type littermates (Fig. 3A), suggesting that mutation of GSK3s do not lead to enhanced GSK3 activities. Moreover, we found that peripheral nerve injury still resulted in reduced level of phospho-CRMP2 in sensory neurons of GSK3 DKI mice, similar to that of wild type mice (Fig. 3B, C). As a control, the total protein levels of CRMP2 were not affected by the peripheral nerve injury (Supplementary Fig. S1), indicating that peripheral nerve injury induces GSK3 inactivation in a manner that is independent of Akt-mediated GSK3 phosphorylation.
Figure 3.
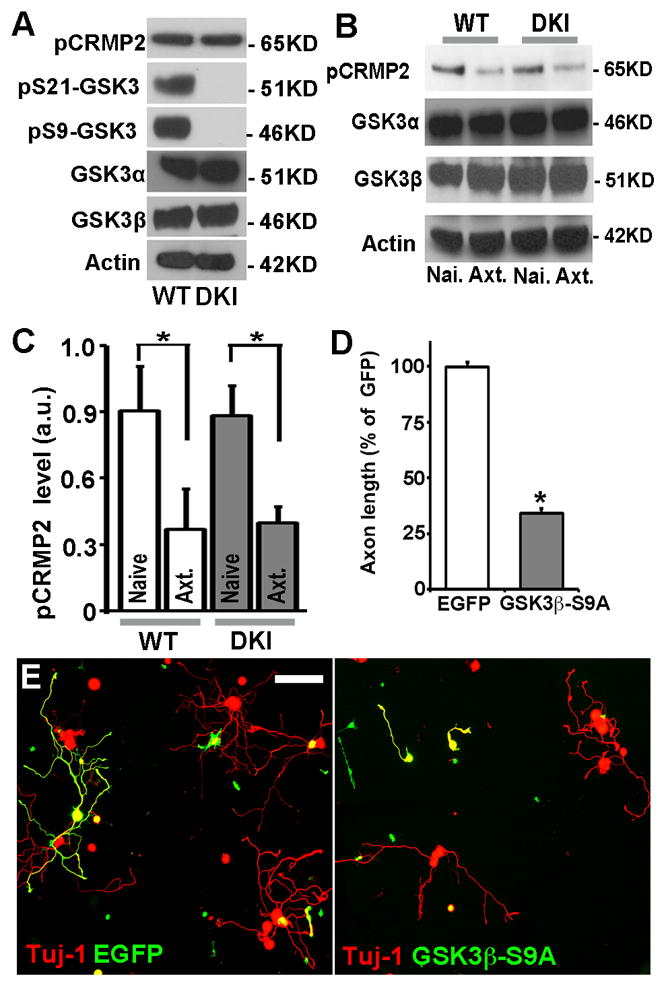
GSK3 inactivation in sensory neurons of GSK3α-S21A/GSK3β-S9A double knockin mice is necessary for regenerative axon growth. (A) Representative immunoblots of GSK3α, GSK3β and phosphorylated CRMP2 in adult sensory neurons from wild type (WT) or GSK3α-S21A/GSK3β-S9A double knockin (DKI) mice. Note the complete lack of phospho-GSK3α/β in the double knockin mice and similar levels of total GSK3α/β. (B) Representative immunoblots of GSK3α, GSK3β and phosphorylated CRMP2 in naïve (Nai.) adult sensory neurons or neurons with peripheral axotomy (Axt.) from wild type (WT) or double knockin (DKI) mice. Note that the levels of phosphorylated CRMP2 were reduced in adult sensory neurons from both wild type (WT) and double knockin (DKI) mice in response to peripheral axotomy. (C) Quantification of the levels of phosphorylated CRMP2 from 3 independent experiments. Error bars represent s.e.m. * indicates significant difference between indicated conditions. P<0.0001. (D) Quantification of axon growth from at least 3 independent experiments. Error bars represent s.e.m. * indicates significant difference from control (EGFP), P<0.0001. (E) Representative images of adult sensory neurons from GSK3α-S21A/GSK3β-S9A double knockin mice transfected with plasmids encoding EGFP or GSK3β-S9A. Scale bar, 100 μm.
We next examined if such phosphorylation independent GSK3 inactivation was functionally required for axon regeneration by overexpression of the same GSK3β mutant (GSK3β-S9A) as that of endogenous protein in the GSK3 DKI mice. The result showed that overexpression of GSK3β-S9A drastically blocked regenerative axon growth of adult sensory neurons from GSK3 DKI mice (Fig. 3D, E), presumably by compensating the reduced activities of endogenous GSK3s. This result suggests that in the absence of Akt-mediated phosphorylation, peripheral axotomy-induced GSK3 inactivation is still necessary for axon regeneration.
3.3. PI3K signaling regulates axon regeneration and peripheral axotomy-induced GSK3 inactivation independent of Akt-mediated phoshorylation
Our latest study has shown that PI3K acts upstream of GSK3 inactivation to control peripheral nerve injury-induced axon regeneration [2]. We therefore tested if PI3K also regulated GSK3 activity in GSK3 DKI mice. We found that inhibition of PI3K with the specific inhibitor LY294002 in cultured adult sensory neurons from GSK3 DKI mice led to markedly increased level of phospho-CRMP2 (Fig. 4A, B). In contrast, the total protein level of CRMP2 was not affected (Supplementary Fig. S1), indicating PI3K inhibition resulted in drastically elevated GSK3 activity. Moreover, such increase in phospho-CRMP2 level could be fully reversed by adding the specific GSK3 inhibitor Bio, whereas Bio treatment alone had no effect on phospho-CRMP2 level, confirming GSK3 as the major kinase that phosphorylates CRMP2 in adult DRG neurons. Together these results indicate that in GSK3 DKI mice PI3K signaling regulates GSK3 activity in regenerating sensory neurons independent of Akt-mediated phosphorylation.
Figure 4.

PI3K regulates GSK3 activity and regenerative axon growth independent of Akt-mediated GSK3 phosphorylation. (A) Representative immunoblots of phosphorylated CRMP2 in adult sensory neurons of GSK3α-S21A/GSK3β-S9A double knockin mice treated with PI3K inhibitor LY294002 (LY) and/or GSK3 inhibitor 6-bromoindirubin-3′-oxime (Bio). Note that inhibition of PI3K leads to markedly increased level of phosphorylated CRMP2, which can be fully reverted by inhibition of GSK3. (B) Quantification of the levels of phosphorylated CRMP2 from 3 independent experiments. Error bars represent s.e.m. * indicates significant difference from control (DMSO), P<0.0001. # indicates significant difference from LY treated condition, P<0.0001. (C) Representative images of cultured adult sensory neurons with PI3K inhibitor LY294002 (LY) and/or GSK3 inhibitor 6-bromoindirubin-3′-oxime (Bio). Note that inhibition of PI3K drastically blocked axon growth, which can be fully rescued by inhibition of GSK3. Scale bar, 100 μm. (D) Quantification of axon growth from at least 3 independent experiments. Error bars represent s.e.m. * indicates significant difference from control (DMSO), P<0.001. # indicates significant difference from LY treated condition, P<0.001.
We next investigated if the PI3K-GSK3 pathway functionally regulates regenerative axon growth of adult sensory neurons from GSK3 DKI mice. Similar to what we observed in wild type mice [2], we found that inhibition of PI3K signaling greatly impaired regenerative axon growth of adult sensory neurons from GSK3 DKI mice (Fig. 4C, D), which could be fully rescued when GSK3 activity was inhibited at the same time. These results demonstrate that the PI3K-GSK3 pathway functions independent of Akt-mediated GSK3 phosphorylation to control axon regeneration.
4. Discussion
Our previous studies have shown that GSK3 is inactivated in response to nerve growth factor (NGF) during development [5] and upon peripheral nerve injury during neural regeneration [2]. In both studies, we showed that GSK3 was inactivated downstream of the PI3K signaling, which is well known to inactivate GSK3 through phosphorylation of its N-terminal serine residue (serine 21 for GSK3α and serine 9 for GSK3β) by the kinase Akt. Thus, overexpression of a mutant GSK3β that cannot be phosphorylated at serine 9 residue (GSK3β-S9A) was able to block both developmental and regenerative axon growth [5,13], suggesting that inactivation of GSK3 by Akt-mediated phosphorylation is required for axon growth. However, to our knowledge no study has directly examined if Akt-mediated phosphorylation is necessary for GSK3 inactivation during axon growth. The GSK3 mutant mice, in which the serine residues of both GSK3α and GSK3β are replaced by alanine and therefore cannot be phosphorylated by Akt, provide us an opportunity to address this question.
Here we showed that in such GSK3 mutant mice peripheral nerve injury-induced sensory axon regeneration was not affected, indicating that Akt-mediated GSK3 phosphorylation is not necessary for axon regeneration. This result seems contradictory to our previous findings that overexpression of the same GSK3β mutant (GSK3β-S9A) as that in the mutant mice blocks axon growth of wild type neurons [13]. One likely explanation is that the level of GSK3 expression rather than its phosphorylation status determines its activity in neurons. Our previous studies have shown that the function of GSK3 depends on the levels of GSK3 activity [11,15]. In support of this idea, the expression levels of the mutated GSK3α and GSK3β in the GSK3 DKI mutant mice were similar to those in wild type mice. Importantly, the activity of GSK3β was also similar based on the phosphorylation status of its substrate CRMP2 (see Fig. 3A). Thus, it suggests that overexpression of GSK3β-S9A in wild type neurons inhibit axon growth via elevating the total GSK3 levels and the subsequent activity rather than competing with the endogenous protein to interfere with Akt-mediated phosphorylation. Indeed, we showed here that overexpression of GSK3β-S9A mutant in neurons from GSK3 DKI mice was also able to markedly blocked axon growth (see Fig. 3D, E). Under such condition, because the endogenous GSK3s and the ectopically expressed GSK3 are the same mutated proteins, the only explanation for the observed effect on axon growth is the increased level of GSK3 in neurons.
Moreover, here we also showed that peripheral axotomy still resulted in reduced GSK3 activity in GSK3 DKI mice, indicating an alternative mechanism acting to inactivate GSK3 independent of Akt-mediated phosphorylation (see Fig. 3B, C). In addition, we showed that such phosphorylation-independent GSK3 inactivation was functionally required for regenerative axon growth, as it could be blocked by overexpression of GSK3β-S9A, presumably via reversing peripheral axotomy-induced reduction in GSK3 activity. Finally, we provide evidence that in GSK3 DKI mice GSK3 is still inactivated downstream of PI3K signaling in the absence of Akt-mediated phosphorylation, and the PI3K-GSK3 pathway functions to control regenerative axon growth.
Taken together, our study provides clear evidence that during neural regeneration neuronal GSK3 is inactivated downstream of PI3K signaling by a mechanism independent of Akt-mediated phosphorylation. Importantly, GSK3 inactivation is functionally necessary for regenerative axon growth, consistent with our previous findings obtained using wild type mice [2]. Future studies are needed to identify the molecular mechanism by which GSK3 is inactivated in neurons to control axon growth.
Supplementary Material
Axon regeneration is normal in GSK3α-S21A/GSK3β-S9A mice in vitro and in vivo.
GSK3 inactivation in mutant mice was necessary for axon regeneration.
GSK3 acted downstream of PI3K signaling in mutant mice to control axon regeneration.
Acknowledgments
Authors would like to thank Drs. Dario Alessi and Le Ma for the GSK3α-S21A/GSK3β-S9A double knockin mice. This work was supported by grants (to F.Z.) from NIH (R01NS064288,), Alzheimer’s Association (NIRG-10-56491), and the Craig H. Neilsen Foundation. B.Z. was supported by China Scholarship Council (CSC-201206170117).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Saijilafu, Zhang BY, Zhou FQ. Signaling pathways that regulate axon regeneration. Neurosci Bull. 2013 doi: 10.1007/s12264-013-1357-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saijilafu EM, Hur CM, Liu ZX, Jiao WL, Xu FQ, Zhou PI3K–GSK3 signalling regulates mammalian axon regeneration by inducing the expression of Smad1. Nat Commun. 2013;4:2690. doi: 10.1038/ncomms3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. 2005;120:123–135. doi: 10.1016/j.cell.2004.12.033. [DOI] [PubMed] [Google Scholar]
- 4.Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 5.Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron. 2004;42:897–912. doi: 10.1016/j.neuron.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 6.Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci. 2010;11:539–551. doi: 10.1038/nrn2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim YT, Hur EM, Snider WD, Zhou FQ. Role of GSK3 Signaling in Neuronal Morphogenesis. Front Mol Neurosci. 2011;4:48. doi: 10.3389/fnmol.2011.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gartner A, Huang X, Hall A. Neuronal polarity is regulated by glycogen synthase kinase-3 (GSK-3beta) independently of Akt/PKB serine phosphorylation. J Cell Sci. 2006;119:3927–3934. doi: 10.1242/jcs.03159. [DOI] [PubMed] [Google Scholar]
- 10.Liu CM, Wang Saijilafu RY, Jiao ZX, Zhang BY, Zhou FQ. MicroRNA-138 and SIRT1 form a mutual negative feedback loop to regulate mammalian axon regeneration. Genes Dev. 2013;27:1473–1483. doi: 10.1101/gad.209619.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hur Saijilafu EM, Lee BD, Kim SJ, Xu WL, Zhou FQ. GSK3 controls axon growth via CLASP-mediated regulation of growth cone microtubules. Genes Dev. 2011;25:1968–1981. doi: 10.1101/gad.17015911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saijilafu, Hur EM, Zhou FQ. Genetic dissection of axon regeneration via in vivo electroporation of adult mouse sensory neurons. Nat Commun. 2011;2:543. doi: 10.1038/ncomms1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou FQ, Walzer M, Wu YH, Zhou J, Dedhar S, Snider WD. Neurotrophins support regenerative axon assembly over CSPGs by an ECM-integrin-independent mechanism. J Cell Sci. 2006;119:2787–2796. doi: 10.1242/jcs.03016. [DOI] [PubMed] [Google Scholar]
- 14.Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ, Connolly CN, Sutherland C. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J Biol Chem. 2004;279:50176–50180. doi: 10.1074/jbc.C400412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim WY, Zhou FQ, Zhou J, Yokota Y, Wang YM, Yoshimura T, Kaibuchi K, Woodgett JR, Anton ES, Snider WD. Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron. 2006;52:981–996. doi: 10.1016/j.neuron.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.