

Abstract
MOZ-TIF2 is a leukemogenic fusion oncoprotein that confers self-renewal capability to hematopoietic progenitor cells and induces acute myelogenous leukemia (AML) with long latency in bone marrow transplantation assays. Here, we report that FLT3-ITD transforms hematopoietic cells in cooperation with MOZ-TIF2 in vitro and in vivo. Coexpression of FLT3-ITD confers growth factor independent survival/proliferation, shortens disease latency, and results in an increase in the number of leukemic stem cells (LSC). We show that STAT5, a major effector of aberrant FLT3-ITD signal transduction, is both necessary and sufficient for this cooperative effect. In addition, STAT5 signaling is essential for MOZ-TIF2–induced leukemic transformation itself. Lack of STAT5 in fetal liver cells caused rapid differentiation and loss of replating capacity of MOZ-TIF2–transduced cells enriched for LSCs. Furthermore, mice serially transplanted with Stat5−/− MOZ-TIF2 leukemic cells develop AML with longer disease latency and finally incomplete penetrance when compared with mice transplanted with Stat5+/+ MOZ-TIF2 leukemic cells. These data suggest that STAT5AB is required for the self-renewal of LSCs and represents a combined signaling node of FLT3-ITD and MOZ-TIF2 driven leukemogenesis. Therefore, targeting aberrantly activated STAT5 or rewired downstream signaling pathways may be a promising therapeutic option.
Introduction
Acute myelogenous leukemia (AML) is characterized by impaired differentiation of hematopoietic progenitors leading to an accumulation of more than 20% myeloid blast cells in the bone marrow (1). In murine bone marrow transplantation (BMT) models, AML can be transferred by transplanting a recipient mouse with leukemic cells derived from leukemic donor mice, possibly due to the presence of a leukemic stem cell (LSC). It is likely that the basis for both resistance and relapse of disease after induction of complete remission can be attributed to the persistence of the LSC population. Therefore, it is of central importance to understand the genetic mechanisms that support maintenance and integrity of LSC.
We and others have previously reported that certain fusion proteins associated with AML in humans, such as MOZ-TIF2 or MLL-AF9 (2, 3), can confer properties of LSC to committed hematopoietic progenitors, such as granulocyte-monocyte progenitors (GMP). However, in the case of MOZ-TIF2, mice transplanted with MOZ-TIF2–transduced GMPs or whole bone marrow cells developed monoclonal or oligoclonal leukemia with long median latency. These observations support a requirement for secondary mutations essential for the development a LSC phenotype. Interestingly, the occurrence of a FLT3-internal tandem duplication (ITD) has been described in a patient with AML harboring the MOZ-TIF2 fusion oncogene (4).
Monocytic leukemia zinc finger protein (MOZ) is a member of the MYST family of protein acetyltransferases. Beside its role in histone modification, MOZ associates with p53, PU.1, and AML1 and acts as a transcriptional coregulator (5). Gene targeting studies showed an essential function of MOZ in the development and maintenance of long-term hematopoietic stem cells (HSC; refs. 6, 7). In AML or MDS, often therapy-related, several MOZ-fusion genes have been detected, including MOZ-CBP, MOZ-p300, and MOZ-TIF2 (5). The MOZ-TIF2 fusion gene results from the pericentric inversion inv(8) (p12q13) and is composed of almost the entire MOZ protein and the C-terminal domain of TIF2, including the core-binding protein (CBP)–interacting domain in TIF2 (8, 9). MOZ-TIF2 (MT2) is thought to promote leukemogenesis through a dominant gain of MOZ function. Upon MOZ-dependent binding to nucleosomal components, TIF2-mediated recruitment of CBP results in histone acetylation, thereby enhancing chromatin accessibility and aberrantly maintaining MOZ target gene expression (10). In addition, MT2 has been shown to cause depletion of CBP from promyelocytic leukemia (PML) bodies and disrupts the normal function of CBP (11). FLT3 belongs to the class III receptor tyrosine kinase (RTK) family and represents one of the most frequently mutated genes in AML. FLT3-ITDs can be detected in 20% to 25% of patients with AML and are associated with poor clinical outcome (12). FLT3-ITDs confer aberrant signal transduction, factor independent growth in interleukin (IL)-3–dependent cell lines and cause a myeloproliferative disease in BMT and FLT3-ITD knockin mouse models (13–15). In stably transfected cell lines or primary AML cells, FLT3-ITD expression causes strong constitutive STAT5 activation (16–18). These data suggest that STAT5 may be a critical downstream target of FLT3-ITD signaling in leukemogenesis. This hypothesis is supported by data showing that tyrosine to phenylalanine mutation of residues 589 and 591 in the context of FLT3-ITD abolished STAT5 signaling and abrogated the development of a FLT3-ITD–induced myeloproliferative disease in mice (19). Interestingly, overexpression of constitutive STAT5A or FLT3 enhances self-renewal and alters differentiation in human hematopoietic progenitor cells (20–23). In myeloid leukemias, constitutive activation of STAT5 is widely observed (24, 25). In many cases, this activation can be attributed to gain-of-function mutations of upstream RTKs, such as FLT3-ITDs.
In the present study, we investigate the effect of constitutive FLT3-ITD expression on MT2-mediated leukemogenesis. In addition, we provide evidence that STAT5 is essential for the maintenance and initiation of solely MOZ-TIF2–driven AML.
Materials and Methods
Cell lines, plasmids, and reagents
MSCV-MOZ-TIF2, MSCV-FLT3-ITD, and MSCV-FLT3-ITD-589/591(Y->F)-GFP plasmids were generated as previously described (10, 19). The JAK1/JAK2-inhibitor ruxolitinib was obtained from Novartis Pharmaceuticals.
Animals
Four- to 6-week-old BALB/c mice were purchased from Taconic. Flt3ITD/+ and Flt3ITD/ITD knockin mice were generated as previously described (14). All mouse experiments were approved by the Institutional Animal Care and Use Committee.
Preparation of fetal liver cells
STAT5−/− livers from E14.5 fetuses derived from BALB/c STAT5+/− mating were isolated and dissociated in transplant medium by sequential aspiration through 21- and 27-gauge needles. Genotypes were determined by PCR as described (26).
Retroviral transduction
Retroviral supernatants were generated in 293T cells as previously described (13). Retroviral transduction procedure is described in detail in Supplementary Methods.
Bone marrow, serial, and limited dilution transplantation assays
BMT assays were carried out as described previously (13). For serial transplantation assays, secondary and tertiary recipient mice were sublethally irradiated (450 rads) and injected with 6 × 104–sorted GFP-positive cells from primary or secondary leukemic mice, respectively. For limiting dilution assays, bone marrow cells of 3 primary Flt3ITD/+-MOZ-TIF2-IRES-EGFP or C57BL/6-MOZ-TIF2-IRES-EGFP leukemic mice were pooled and 10,000, 5,000, 1,000, 100, and 20 cells were transplanted into sublethally irradiated recipient mice.
Histopathology
Peripheral blood was collected from the retroorbital plexus. Blood smears were stained with Wright–Giemsa. Total and differential blood counts were obtained with a Hemavet automated blood counter (Drew Scientific).
Spleen, liver, lymph nodes, and hind limb bones were collected in 10% neutral buffered formalin (Sigma). The bone was subsequently decalcified in EDTA. All specimens were embedded in paraffin. Histochemical stainings were conducted using hematoxylin and eosin (H&E) and the naphtanol-ASD-chloresterase reaction (ASD).
Flow cytometry
Single cell suspensions of spleen, bone marrow, and lymph nodes were prepared as described (27). Immunophenotypes of cells were analyzed with a FACSCalibur and BD Cell Quest 3.1 (Becton Dickinson). The analysis of intracellular phosphorylated STAT5 is described in Supplementary Methods. Staining and sorting for GMP or L-GMP population was conducted as described (2). The data were analyzed with BD FACSDiva software (Becton Dickinson) or FlowJo flow-cytometry analysis software.
Southern blot analysis
Genomic DNA (20 μg) digested overnight with NheI was fractionated through 1% agarose gels and transferred to nylon+ membrane (Amersham). Probes containing a GFP fragment were radiolabeled by using the Gene Images Random-Prime DNA Labeling Kit (Amersham). The blots were stripped with boiling in 0.5% SDS for 30 minutes and reprobed with radiolabeled neo probe.
Colony-forming unit assay and serial replating
Colony-forming unit (CFU) assays were conducted according to the Stem Cell Technology manual. Briefly, 5′-flurouracil primed bone marrow cells were transduced as described earlier and 2 × 105 to 2 × 106 cells were plated in either cytokine supplemented methylcellulose (M3434; Stem Cell Technologies) or in methylcellulose without cytokines (M3234) in the presence of antibiotics (G418 or puromycin in the first plate at 1 mg/mL or 2 μg/mL, respectively). After 7 days of incubation, colonies of greater than 50 cells were counted or collected for cytospin and Wright–Giemsa staining. Cells were pooled from duplicate plates and replated.
Statistics analyses
Statistical significance (P < 0.05) among groups was determined by Student t test (2-tailed, unpaired/unequal variances). Comparisons of survival curves were conducted using the log-rank test. All statistics were conducted using GraphPad Prism version 4.03. The frequency of LSCs was calculated according to Poisson statistics using L-Calc software (Stem Cell Technologies).
Results
FLT3-ITD decreases the latency of MOZ-TIF2–induced AML
The long latency and oligoclonal nature of MT2-mediated leukemia suggest that additional events are required to induce AML. To assess cooperation between FLT3-ITD and MT2 in vitro, we used a serial replating assay. Bone marrow cells derived from previously described Flt3-ITD knockin mice (14) or wild-type (WT) control mice were transduced with equivalent titers of MSCV-MOZ-TIF2-IRES-GFP or MSCV-IRES-GFP (hereafter MT2 or MIG, respectively) and plated in cytokine-supplemented methylcellulose. As expected, MT2 conferred serial replating activity independent of FLT3-ITD expression, whereas no colonies could be detected in Flt3ITD/+-MIG expressing cells after the third round of replating (Fig. 1A). In addition, no difference in colony numbers was observed between MT2-Flt3ITD/+ and MT2-WT cells. To address the question whether FLT3-ITD and MT2 cooperate in vivo, we conducted BMT assays. MT2-transduced WT or Flt3ITD/+ bone marrow cells were injected into lethally irradiated recipient mice and monitored for the onset of leukemia. As shown in Fig. 1B, Flt3ITD/+-MT2 mice developed disease with a significantly shorter median latency as compared with MT2 mice (P = 0.0036; log-rank test). In contrast, none of the mice transplanted with Flt3ITD/+-MIG or WT-MIG bone marrow cells developed disease up to day 120 posttransplantation (Fig. 1B). Leukemic mice exhibited splenomegaly, hepatomegaly, and infiltration of the submandibular gland, recapitulating the phenotype of human MT2-AML. Histopathology of various organs and peripheral blood smears showed infiltration with immature myeloid blasts consistent with AML (Fig. 1C and D). Fluorescence-activated cell sorting (FACS) analysis, gated on the GFP-positive leukemic population, showed the expression of Mac-1, Gr-1, and varying degrees of c-kit (Fig. 1E). No difference of disease phenotype about histopathologic findings, blood cell counts, or surface marker profile of leukemic blasts was seen between Flt3ITD/+-MT2 and WT-MT2-mice.
Figure 1.

FLT3-ITD decreases disease latency in cooperation with MOZ-TIF2. A, serial replating assay of FLT3ITD/+ or WT progenitor cells. Bone marrow (BM) cells transduced with MSCV-MT2-IRES-GFP were plated in M3434 at 2 × 104 cells per plate. Colonies were counted on day 7 and replated as indicated. Results shown represent duplicate measurements of at least 2 independent experiments. B, Kaplan–Meier survival curves of FLT3ITD/+-MT2 (n = 12), WT-MT2 (n =9), FLT3ITD/+-MIG (n =10), and WT-MIG (n = 8) mice. The log-rank test for comparison of cumulative incidence curves confirmed a significant (P < 0.001) decrease in disease latency in FLT3ITD/+-MT2 mice as compared with B6-MT2 mice. C, peripheral blood smears of moribund mice receiving FLT3ITD/+-MT2 (left) or WT-MT2 (right) bone marrow cells (May–Grünwald–Giemsa stain; 100×/1.4 oil). D, histopathology of transplant mice. Sections of bone marrow show positive (red) staining in maturing granulopoietic cells, whereas no or only faint staining in immature myeloid blasts (×100 and ×1,000, top 2 rows ASD-stainings); the following row shows the corresponding H&E stainings. Sections of spleen (H&E, ×100 and ×1,000): abundant blasts replacing splenic tissue. Sections of liver (H&E, ×100): portal infiltration of blasts. E, immunophenotype analysis of bone marrow and spleen cells derived from leukemic Flt3ITD/+-MT and WT-MT mice as indicated. Shown are dot plots gated on EGFP-positive cells.
Expression of FLT3-ITD enhances the number of LSCs in MOZ-TIF2–driven leukemogenesis
We next were interested, whether the shortened disease latency is caused by FLT3-ITD–induced hyperproliferation or whether the expression of FLT3-ITD results in an increase of LSC. Therefore, we conducted limiting dilution transplant assays using bone marrow cells derived from primary recipients. Mice developed phenotypically identical leukemia (data not shown) but with substantial differences in disease latency and incidence according to the transplanted cell numbers (Fig. 2). Calculation of the number of LSCs showed 2.3-fold enrichment in Flt3ITD/+-MT2 bone marrow cells as compared with MT2-MIG cells (Fig. 2).
Figure 2.
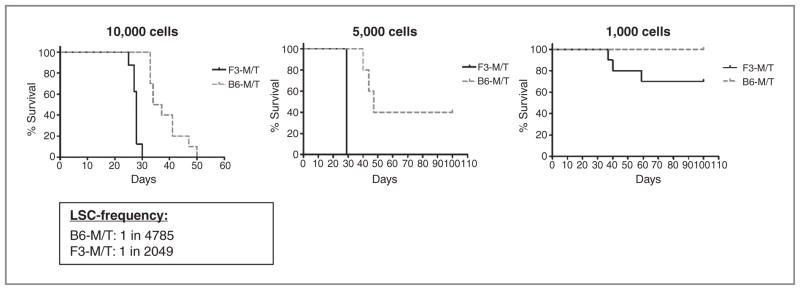
Expression of FLT3-ITD enhances the number of LSCs. Bone marrow cells derived from leukemic FLT3ITD/+-MT2 or WT-MT2 mice were transplanted through limiting dilution into sublethally irradiated recipients. Shown are Kaplan–Meier survival curves of mice transplanted with the indicated cell numbers. LCS frequency for the indicated genotype was calculated by applying Poisson statistics using the L-Calc program.
Taken together, these data suggest that FLT3-ITD has no effect on MOZ-TIF2–induced disease phenotype but enhances the development of leukemogenesis and increases the number of LSCs.
FLT3-ITD–mediated cooperative effects are STAT5-dependent in vitro
To further analyze the FLT3-ITD–mediated cooperative effect on MT2-induced leukemogenesis, we investigated whether constitutively active FLT3 can compensate for external cytokine and growth factor signaling. As shown in Fig. 1A, expression of MT2 confers serial replating capability to hematopoietic progenitors in methylcellulose in the presence of cytokines. However, MT2-transduced progenitors could only be grown and replated in the presence of either IL-3, granulocyte macrophage colony-stimulating factor (GM-CSF), or combinations of cytokines (Supplementary Fig. S1), indicating that MT2-mediated transformation of progenitor cells in vitro is growth factor dependent and signaling downstream of IL-3 or GM-CSF is required for cell survival.
FLT3-ITD has been shown to confer IL-3–independent cell growth to BaF/3 and 32D cells (13, 17). To investigate whether FLT3-ITD could abrogate growth factor dependence of MT2, bone marrow cells of Flt3+/ITD, Flt3ITD/ITD, or control mice were transduced with MT2 and plated in methylcellulose without cytokines (M3234). No replating activity was seen in progenitor cells expressing FLT3-ITD, MT2, or MIG alone (Fig. 3A). In contrast, MT2-transduced bone marrow cells derived from homozygous Flt3-ITD mice could be replated for several rounds, whereas the colony number of Flt3ITD/+-MT2 cells was diminished in the third plate (Fig. 3A). These data suggest that FLT3-ITD expression is able to substitute for external growth factor signaling in a dose-dependent manner. As already mentioned, constitutive activation of STAT5 plays an important role in FLT3-ITD–mediated leukemogenesis. To address the question whether STAT5 contributes to FLT3-ITD–induced cytokine-independent growth in MT2-positive leukemia, we cotransduced primary bone marrow cells with vectors expressing MT2 and FLT3-ITD or the FLT3ITD-589/591 Y->F mutant that selectively abrogates STAT5 signaling (19), and plated them in M3234 (Fig. 3B). Transduced cells from the first plate were pooled and replated in M3234. MT2 and FLT3-ITD coexpressing bone marrow progenitors produced significant numbers of colonies in the second and third rounds of plating (Fig. 3B), again indicating that FLT3-ITD supported serial replating activity of MT2 in the absence of growth factors. In contrast, progenitors transduced with FLT3-WT, FLT3-ITD, or MT2 (Fig. 3B) alone failed to yield colonies in the second round of replating. In addition, STAT5 signaling was required for cooperative serial replating activity in vitro, as no colonies were observed upon transduction with MT2 and FLT3-ITD-589/591. The data suggested that STAT5 activity significantly contributed to cytokine-independent replating capacity of progenitor cells transformed by MT2. To test this hypothesis, a constitutively active STAT5 mutant [STAT5(1*6*)] was used to transduce bone marrow cells, that were first plated in methylcellulose with cytokines (data not shown). Positively selected clones were then transduced with a MT2 retroviral vector and replated in M3234. The colonies derived from single STAT5 (1*6*)-transduced cells were very small and diffuse (Fig. 3C, bottom left) and exhibited limited replating capability (Fig. 3C). In contrast, progenitors coexpressing both STAT5(1*6*) and MT2 formed colonies with clonogenic activities to at least 6 rounds of plating (Fig. 3C). These colonies were consistent with blast-like colonies, with round and compact morphology (Fig. 3C, bottom middle), and Wright–Giemsa staining showed a predominance of immature myelomonocytic blasts (Fig. 3C, bottom right).
Figure 3.

FLT3-ITD requires STAT5 to confer growth factor–independent serial replating activity to MOZ-TIF2–transformed cells. A, serial replating assay of FLT3ITD/+ or WT progenitor cells. Bone marrow cells transduced with MSCV-MT2-IRES-GFP were plated in M3234 without cytokines at 1 × 105 cells per plate. Colonies were counted on day 7 and replated as indicated. Results shown represent duplicate measurements of at least 2 independent experiments. B, serial replating assay of progenitor cells transduced with MT2, FLT3-WT, FLT3-ITD, and mutants. Transduced bone marrow cells were plated in M3234 at 1 × 106 cells without cytokines. Puromycin and neomycin were added to select the positively transduced clones on the first plate. Results shown represent duplicate measurements of at least 2 independent experiments. C, serial replating assay of progenitor cells transduced with STAT5(1*6*)–constitutive active mutant or MT2+STAT5(1*6*). Bone marrow cells transduced with STAT5(1*6*) retroviral expression vector were plated in M3234 with no cytokines in the presence of puromycin to select for the positively transduced clones. For MT2 and STAT5(1*6*) coexpressing colonies, bone marrow cells were first transduced with STAT5(1*6*) and selected with puromycin on M3434 plates. Three days later, cells were pooled, transduced with MOZ-TIF2 retroviral vector, and plated in M3234 with neomycin. Results shown are representative of at least 2 independent experiments in duplicate. The bottom shows colonies of bone marrow cells transduced with STAT5(1*6*) [left] and MT2+STAT5(1*6*) [middle]. Bottom right, Wright–Giemsa staining of cells from a MT2 and STAT5(1*6*) cotransduced colonies. D, serial replating assay of progenitor cells transduced with STAT5-WT or STAT5-DN in the presence of MT2 and FLT3-ITD. Transductions were done sequentially: MOZ-TIF2-neo transduced cells were first selected in M3434 with 1 mg/mL neomycin, then transduced with FLT3-ITD-GFP, and selected in M3234. After transduction with STAT5 expression plasmids, cells were plated in M3234 with puromycin. Results shown are representative of at least 3 independent experiments in duplicate. The bottom shows colonies from bone marrow cells expressing MT2/FLT3-ITD (left), MT2/FLT3-ITD/STAT5-WT (middle), and MT2/FLT3-ITD/STAT5-DN (right) in M3234 without cytokine. E, flow cytometric analysis of cells pooled from first M3234 methylcellulose culture plate. Cells cotransduced with MT2, FLT3-ITD, and STAT5-DN (bottom right) show more differentiated granulocytes (Mac-1+Gr-1hi) than cells without STAT5-DN (bottom left).
To assess whether STAT5 signaling was essential for the immortalization of myeloid progenitor cells transformed by MT2, we used a dominant-negative STAT5 mutant (STAT5-DN) that lacks the STAT5-transactivation domain. Cotransduction of the STAT5-DN mutant in bone marrow progenitor cells expressing MOZ-TIF2 and FLT3-ITD greatly reduced serial-replating activity as compared with controls (Fig. 3D). The few colonies were small and diffuse (Fig. 3D, bottom right), in contrast to the large condensed colonies from the controls (Fig. 3D, bottom left and middle). Of note, more Mac1+Gr-1hi/int cells were observed when STAT5-DN was coexpressed (Fig. 3E, right), as revealed by flow-cytometry analysis. This observation suggests the possibility that the STAT5-DN mutant may have reversed the differentiation block in myeloid blast cells mediated by MT2.
FLT3-ITD requires STAT5 for cooperation with MOZ-TIF2 in vivo
We next hypothesized that STAT5, as an important downstream event of FLT3-ITD signaling, might also serve as a crucial cooperating effector in MT2-induced leukemogenesis. To test this possibility, we conducted a BMT study using bone marrow cells transduced with MT2-neo, together with equivalent titers of MIG, MSCV-FLT3-ITD-GFP, or MSCV-FLT3-589/591-GFP. While all transplanted mice eventually developed AML, they did so with different median latencies (Table 1). As shown in Fig. 4A, mice receiving MT2-neo plus FLT3-ITD–transduced bone marrow cells showed a significantly shorter median survival (69 days) than those expressing only MT2-neo (158 days; P < 0.0001; log-rank test). Statistically significant cooperativity between MT2 and FLT3-ITD was abrogated when FLT3-ITD residues 589 and 591 were mutated, as indicated by the increase in median survival to 152 days in FLT3-ITD589/891/MT2-neo cotransplanted mice (Fig. 4A).
Table 1.
Summary of bone marrow transplant experiments with MOZ-TIF2 (MT2) alone or in the presence of FLT3-ITD and various mutants
| Constructs | No. of diseased mice/BMT mice | Disease Latency days (median) | Spleen wt. mg (median) | Liver wt. mg (median) | WBC × 106 cells/mL (median) |
|---|---|---|---|---|---|
| MT2-neo + GFP | 8/8 | 125–179 (158) | 180–810 (390) n = 7 | 860–1,860 (1,170) n = 7 | 7.3–269.2 (94.4) n = 7 |
| MT2-neo + FLT3-ITD-GFP | 8/8 | 53–82 (69) | 130–689 (313) n = 6 | 740–2,119 (1,100) n = 6 | 5.4–110.6 (38.7) n = 6 |
| MT2-neo + FLT3-ITD-589/591-GFP | 8/8 | 89–197 (136) | 270–630 (380) n = 7 | 1,070–1,430 (1,160) n = 7 | 8.4–134.4 (33.4) n = 6 |
NOTE: The median survival times are taken from the Kaplan–Meier survival plots shown in Fig. 4A. The n values in each field represent the number of animals from which the particular analysis was done.
Figure 4.

FLT3-ITD requires STAT5 for cooperation with MOZ-TIF2 to induce AML in a murine BMT model. A, Kaplan–Meier survival analysis of transplanted mice. Mice were transplanted with 1 × 106 bone marrow cells transduced with retroviral vectors expressing MT2 alone (n = 8) or together with an equivalent titer of retroviral vectors expressing FLT3-ITD-589/591 (n = 8), or FLT3-ITD (n = 8). Median survival of mice expressing MT2, FLT3-ITD-589/591, and FLT3ITD was 158, 152, and 69 days, respectively. The cumulative survival of MT2-mice was significantly different from mice transduced with FLT3-ITD (P < 0.0001; log-rank test). B, Southern blot analysis of transplanted mice. Genomic DNA was isolated from the splenic cells of leukemic mice in which bone marrow cells were transduced with retroviral vectors MSCV-MT2-neo + MSCV-GFP (lanes 1–4); MSCV-MT2-neo + MSCV-FLT3-ITD-GFP (lanes 5–8); MSCV-MT2-neo + MSCV-FLT3-ITD-589/591 (lanes 9–13). Top, DNA was digested with NheI, which cuts once in the long terminal repeat site of the retroviral vectors, transferred to nylon membrane and hybridized with radioactive GFP probe to show the provirus integration of FLT3-ITD and various mutants. The blot was then stripped and hybridized with radioactive neo probe to detect MT2 provirus integration (bottom). C, flow cytometric staining of intracellular phosphorylated STAT5 of transplanted mice. A representative diagram of flow analysis of intracellular phosphorylated STAT5 at 80 days posttransplantation of splenic cells (left) and bone marrow (right) cells is shown.
Impaired cooperativity between MT2 and the FLT3ITD-589/591 mutant was also shown by immunophenotypic studies (Supplementary Fig. S2A). All transplant recipients showed markedly elevated populations of Mac-1+Gr-1−/low immature myeloid and blast cells (Supplementary Fig. S2A, first and second panels), consistent with the histopathologic findings of various organs including spleen, liver, and bone marrow derived from diseased animals (Supplementary Fig. S2B). However, the contribution of GFP-transduced cells to this immature myeloid phenotype differed among transplant groups. Only mice transplanted with FLT3-ITD-GFP + MT2-neo had GFP-positive Mac-1+Gr-1−/low cells, whereas in FLT3-ITD-589/591-GFP + MT2-neo animals no GFP-positive Mac-1+Gr-1−/low cells were detected (Supplementary Fig. S2A, third panel). This observation provides strong support for the hypothesis that coexpression of FLT3-ITD with MT2 causes a survival or proliferative advantage. Southern blot analysis of DNA isolated from the spleens of diseased mice showed the presence of intact FLT3-ITD and FLT3-ITD-589/591 provirus (Fig. 4B, top lane), as well as MT2 provirus (Fig. 4B, bottom lane). These data argue against the possibility that the absence of GFP-positive immature myeloid populations in MT2 and FLT3-ITD-589/591 transplant mice was due to the lack of provirus integration. Finally, FACS analysis of phosphorylated STAT5 in spleen cells (Fig. 4C, left) and bone marrow cells (Fig. 4C, right) showed that STAT5 was activated only in MT2-neo/FLT3-ITD mice, again supporting the hypothesis that STAT5 signaling is crucial for cooperation with MT2-induced leukemogenesis.
STAT5-deficient LSCs fail to serially replate and have a higher propensity to differentiate in vitro
Activation of STAT5 seems to be essential to mediate the cooperative effect of FLT3-ITD and MT2. We next asked, whether STAT5 is also important for the maintenance of MT2-LSCs itself. To investigate this issue, we used fetal liver cells derived from Stat5ab−/− mice or WT littermates (26). Fetal liver cells were transduced with MT2-GFP and transplanted into lethally irradiated primary recipients. L-GMP (lin−c-KithiFcγRhi) were sorted from secondary recipients transplanted with fetal liver cells expressing MT2-GFP in a Stat5ab−/− or Stat5ab+/+ background (Supplementary Fig. S3). The sorted cells were incubated in liquid culture media containing IL-3, IL-6, and stem cell factor, or in M3434 methylcellulose. After 3 days in liquid culture, Stat5−/− L-GMP rapidly upregulated Mac-1 and downregulated the immature cell surface marker c-Kit (Fig. 5A, top). After a 7-day culture in M3434 methylcellulose, there was a 3-fold decrease in the percentage of cells expressing c-Kit in Stat5−/− L-GMP when compared with Stat5+/+ L-GMP (Fig. 5A, bottom). Moreover, we observed a rapid upregulation of the myeloid differentiation marker Gr1 in Stat5−/− L-GMP (data not shown). This phenotype was partially rescued by reexpression of STAT5A in Stat5−/− L-GMP (Fig. 5B). Upon transduction with MSCV-STAT5a-pgk-puro and selection for 3 days in M3434 media, sorted L-GMP showed increased expression of c-Kit (12.7% vs. 3.95%) as compared with empty vector control cells. These observations suggest that the lack of STAT5 prompts L-GMP to differentiate in vitro. To further investigate whether this property of Stat5−/− L-GMP affects the serial replating potential of L-GMP, sorted cells were plated in M3434 for 7 days, then pooled and replated for 3 more rounds (Fig. 5C). While Stat5+/+ L-GMP maintained serial replating activity to the fourth round of replating, Stat5−/− L-GMP failed to replate (Fig. 5C). To further elaborate whether activation of STAT5 or the mere presence of STAT5 confers self-renewal activity, we treated MT2 leukemic blast with ruxolitinib, a potent JAK1/JAK2-inhibitor. Treatment with ruxolitinib is expected to partially abolish JAK2-mediated STAT5-phosphorylation without significantly affecting expression levels of STAT5. In vitro, ruxolitinib treatment had no obvious effect on cell viability of MT2 or FLT3+/ITD-MT2 bone marrow blasts maintained in liquid culture in the absence of cytokines or in M3434 supplemented with cytokines (Supplementary Fig. S4A and S4B, respectively). Western blot analyses confirmed a slight decrease of STAT5-phosphorylation in MT2 blasts cultured in M3434 for 7 days (Supplementary Fig. S4C), indicating effective but not complete inhibition of JAK2 signaling. To investigate whether ruxolitinib treatment affects the serial replating potential of MT2 or FLT3+/ITD-MT2 blasts, cells were replated for 2 more rounds (Supplementary Fig. S4D). As expected, replating activity of MT2 cells coexpressing FLT3-ITD was not diminished upon inhibition of JAK2-signaling. In contrast, we observed a substantial decrease in colony numbers of ruxolitinib-treated MT2 cells in rounds 2 and 3, however, still more than 200 colonies were recovered in plate 3. Taken together with the results from STAT5−/− L-GMP (Fig. 5), the incomplete inhibition of self-renewal activity by ruxolitinib suggests that both STAT5 expression and its phosphorylation play a role in self-renewal activity.
Figure 5.

STAT5-deficient LSCs fail to serially replate and have a higher propensity to differentiate in vitro. A, leukemic-GMPs were sorted 21 days posttransplantation from secondary recipients initially transplanted with leukemic cells derived from MT2-GFP Stat5−/− or Stat5+/+ fetal liver cells. The cells were analyzed by flow cytometry either after incubation for 3 days in liquid transplant medium containing 10 ng/mL IL-3, 10 ng/mL IL-6, and 50 ng/mL stem cell factor (top), or after incubation for 7 days in M3434. Results shown are representative of 2 independent experiments. B, expression of STAT5A upregulates the immature surface marker c-Kit in STAT5-deficient L-GMP. L-GMP populations were sorted from mice transplanted with MT2-GFP in a Stat5-deficient background or Stat5-WT background. The Stat5+/+ L-GMP cells were infected with MSCV-pgk-puro retroviral vector (second panel). Stat5−/− L-GMP cells were infected with MSCV-pgk-puro retroviral vector (third panel) or a STAT5A expressing construct (fourth panel). The infected cells were selected for 3 days in M3434 with puromycin and then analyzed by flow cytometry. C, L-GMPs were serially replated in M3434 methylcellulose at 2 × 103 cells per plate in the first and second plates or at 2 × 104 cells per plate in the third and fourth plates. The results shown are representative of duplicate measurements of 2 independent experiments.
STAT5 is required for the self-renewal properties of MOZ-TIF2 LSC in an in vivo serial transplantation assay
To further investigate the role of STAT5 signaling in the maintenance of LSCs in vivo, we used serial transplantation assays. Leukemic Stat5−/− or Stat5+/+ cells (2 × 105 or 6 × 104), sorted for the expression of GFP upon transduction with the MT2-GFP vector, were serially injected into sublethally irradiated recipient mice. We observed that mice transplanted with Stat5−/− leukemic cells developed AML with significantly prolonged latency compared with the Stat5+/+ control group (log-rank test; P < 0.0001; Fig. 6A; Supplementary Table S1). Moreover, median survival in the mice transplanted with Stat5−/− leukemic cells progressively increased from 57 days in the first recipients (Fig. 6A, left), to 80 days in the second serial transplant (Fig. 6A, middle), and to 100 days (Fig. 6A, right) in the third serial transplant as compared with 35 days in mice with Stat5+/+ leukemic cells in all serial transplants (Fig. 6A). Interestingly, only 7 of 8 mice transplanted with Stat5−/− leukemic cells developed AML in the tertiary transplant experiment, suggesting that LSCs might become exhausted during serial transplantation in the absence of STAT5. The difference in disease latency was likely not due to differences in abilities of homing or engraftment between Stat5−/− or Stat5+/+ leukemic cells (Fig. 6B). All mice receiving either Stat5−/− or Stat5+/+ cells developed AML with similar disease phenotypes (data not shown). These data indicate that STAT5 is important for the maintenance of LSCs generated by MT2-induced transformation in vitro and in vivo.
Figure 6.

STAT5 is required for self-renewal of MOZ-TIF2 LSC in an in vivo serial transplantation assay. A, Kaplan–Meier survival analysis of serial transplant Stat5−/− or in a Stat5+/+ background is statistically significant (P < 0.0001; log-rank mice. The difference in cumulative survival of mice expressing MT2 in a test) in all 3 serial transplantations. B, Stat5−/− or Stat5+/+ leukemic cells have similar homing and engraftment capacities. Homing of the injected GFP-positive cells was analyzed 18 hours postinjection, while engraftment of the injected GFP-positive cells was analyzed 21 days after transplantation. Results shown are the percentage of GFP-positive cells in spleen (first and second row) or bone marrow (third row) of 2 representative mice from the first serial transplantation. Similar results with no significant difference were obtained in the second or third serial transplant (data not shown). FITC, fluorescein isothiocyanate.
Discussion
There is accumulating evidence that alterations in transcription factors and tyrosine kinases cooperate in the process of leukemia development. This idea is supported by clinical studies of human AML and by several murine models (e.g., AML1-ETO and c-KIT, MLL-leukemias and RAS mutations; refs. 28, 29). FLT3-ITD mutations have been shown to cooperate with PML-RARα, AML1-ETO, and MLL-AF9 (30–33). In these models, the expression of FLT3-ITD is either necessary to induce AML at all (AML1-ETO), to induce a short-latency APL-like disease with complete penetrance (PML-RARα) or to shorten disease latency (MLL-AF9). Here, we show that physiologic expression of FLT3-ITD from its endogenous promoter or supraphysiologic expression of FLT3-ITD upon retroviral infection reduces disease latency in a MOZ-TIF2–induced leukemia model. Interestingly, both models displayed the same leukemia phenotype indistinguishable in FACS and histopathology analyses. To address the question whether the expression of FLT3-ITD simply provides a survival/proliferation advantage to LSCs, we conducted limiting dilution BMT assays. We show that the coexpression of FLT3-ITD not only shortens disease latency for all cell numbers transplanted, but also increases the number of LSCs 2.3-fold. These data correlate with the observation that expression of FLT3-ITD has been observed in LSCs in human disease and predicts poor clinical outcome (34, 35). In contrast, no increase of LSCs was seen in a MLL-AF9–mediated leukemia model upon FLT3-ITD expression (32). However, the expression of MLL-AF9 itself is able to induce a strong leukemia self-renewal and maintenance program (36, 37), whereas mice transplanted with MOZ-TIF2–transduced hematopoietic progenitors developed monoclonal or oligoclonal AML with long disease latencies indicating that a second cooperative event is required for MOZ-TIF2–induced transformation.
In vitro, MOZ-TIF2 confers serial replating activities to murine progenitor cells in methylcellulose assays, however, MOZ-TIF2 transformed progenitors still require cytokines. Because FLT3-ITD can substitute for IL-3 or GM-CSF in supporting serial replating activities, we speculated that downstream pathways common to FLT3-ITD, IL-3 and GM-CSF were essential for the process. Interestingly, normal FLT3 ligand–mediated FLT3 signaling leads to only weak phosphorylation of STAT5 in BaF/3 cells (18). In contrast, FLT3-ITD expression results in constitutive STAT5 activation in stably transfected cell lines as well as primary AML cells (16, 17). We tested whether STAT5 might be a key cooperative signal downstream of FLT3-ITD with divergent but complementary genetic approaches. We observed that constitutively active STAT5 is sufficient in cooperating with MOZ-TIF2 to confer cytokine-independent serial replating activities to progenitor cells but not a STAT5 dominant-negative mutant or a FLT3-ITD589/591–mutant deficient in STAT5 signaling (19). Moreover, FLT3-ITD589/591 and MT2 transplant mice showed disease latencies similar to mice transplanted with MT2 alone. These results indicate that STAT5 signaling is a key cooperative signal downstream of FLT3-ITD.
To further assess the role of STAT5 in MT2-driven AML, we transduced fetal liver cells isolated from Stat5−/− mice or Stat5+/+ littermates with a retrovirus expressing MT2-IRES-GFP and sorted the L-GMP population. Interestingly, L-GMPs depleted of STAT5 rapidly differentiated into a more mature population (c-KitloMac1hiGr1hi) and failed to serially replate in methylcellulose. Moreover, MT2 leukemic cells without STAT5 induced AML with significantly longer disease latencies along serial transplantations and incomplete penetrance in tertiary recipients. Although we provide evidence that homing and engraftment is similar in mice transplanted with Stat5−/−- or Stat5+/+-MOZ-TIF2 cells (Fig. 6B), the observed STAT5-dependent exhaustion may still be caused by transplantation-associated factors in our serial transplantation experiments and needs to be further addressed by secondary depletion of STAT5 in already leukemic mice.
In previous work, Ito and colleagues showed that PML−/− LSC cells or LSC cells depleted of PML with A2O3 could migrate out of the G0 phase, thus decreasing their serial transplant-ability (38). To investigate if STAT5 deficiency drives more LSCs to cycle, we conducted cell-cycle analysis in L-GMPs, however, no significant differences were seen in LSC with or without STAT5 (data not shown).
Recently, the role of Stat5b was investigated in a 2-oncogene leukemia model, namely MN1 and HOXA9 (39). Depletion of Stat5b inhibited the MN1/HOXA9–mediated expansion of LSCs by 86-fold, indicating a critical role of Stat5b in the maintenance of the LSC population. Furthermore, in a BCR–ABL–driven murine leukemia model, expression of Stat5ab was essential for the initiation of leukemic disease and depletion of Stat5ab caused induction of apoptotic cell death followed by effective elimination of both, myeloid and lymphoid leukemia (40). Of note, the mere reexpression of selected STAT5 target genes could not rescue the disease phenotype.
In the leukemia model presented here, we propose a dual function for STAT5. First, STAT5 confers growth factor–independent growth, an increase in the number of LSCs, and a more aggressive disease, however, indistinguishable about disease phenotype. This effect is likely to be dependent on STAT5-phosphorlyation by virtue of FLT3-ITD mutations. Second, STAT5, at least in the context of MOZ-TIF2 leukemia, blocks differentiation and shifts the balance toward maintenance of self-renewal activity. This function might be partially dependent on the sole expression of STAT5 as LSCs lacking STAT5 expression rapidly differentiate into a mature ckitloMac1+Gr1hi population in vitro and exhibit an exhaustion phenotype in vivo. However, as mice transplanted with Stat5−/− MT2 cells still develop leukemia, alternative pathways such as STAT3, PI3K-Akt, or Ras-Raf-MAPK-Erk may partially compensate the loss of STAT5. Interestingly, in Drosophila melanogaster a noncanonical JAK-STAT signaling pathway has been identified (41). Unphosphorylated STAT proteins have been found to colocalize to heterochromatin protein 1 (HP1) and are required to stabilize HP1 localization, H3K9-methylation, and transcriptional repression. Loss of STAT was followed by HP1 displacement and heterochromatin destabilization. Whether similar mechanisms are active in mammalian cells and which STAT proteins are involved, needs further investigation.
These data combined with the fact that STAT5 activation represents a common signaling node of FLT3-ITD and MOZ-TIF2 oncogenes indicate that targeting STAT5 might represent a promising therapeutic option. The feasibility of such an approach has been shown by Schepers and colleagues RNA interference (RNAi)–mediated reduction of STAT5 expression and concomitant inhibition of STAT5 phosphorylation significantly decreased the expansion of primary CD34+ AML blasts in vitro (42). However, in this experimental setting, knockdown of STAT5 also caused reduction of primary CD34+ cord blood progenitor cells. These observations are in line with data showing that conditional deletion of Stat5ab in adult mice caused a decrease of LT-HSC number, loss of HSC quiescence, and a decline in total white blood cell counts at 4 months (43). Therefore, specific targeting of STAT5 is likely restricted to temporary treatment, thus avoiding long-term adverse effects. Alternatively, aberrant STAT5 activation might rewire downstream pathways and render LSCs dependent on critical players in these networks. Targeting these so called nononcogenes might selectively kill LSCs while sparing their normal counterparts (44). Nevertheless, in the context of aberrant STAT5 activation, this hypothesis needs to be addressed by further experiments.
Supplementary Material
Acknowledgments
The authors are thankful for the valuable discussions with members of the Gilliland laboratory and Birgit Enders for scientific input and technical assistance.
Grant Support
D.G. Gilliland received funding support from the Howard Hughes Medical Institute (Boston, MA), L. Hennighausen and G.W. Robinson received funding support from the intramural program of the National Institutes of Diabetes, Digestive and Kidney Diseases (NIDDK)/NIH and the DFG Mercator Visiting Professor program.
Footnotes
Disclosure of Potential Conflicts of Interest
T. Kindler is a consultant/advisory board member of Novartis Advisory Board. No potential Conflicts of interest were disclosed by the other authors.
Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors’ Contributions
Conception and design: W.F. Tam, S. Pante, E. Bockamp, L. Hennighausen, T. Kindler
Development of methodology: W.F. Tam, D. Sasca, D.G. Gilliland, T. Kindler
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): W.F. Tam, B.H. Lee, S. Pante, G. Wernig, A. Kreft, G.W. Robinson, L. Hennighausen, T. Kindler
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): W.F. Tam, P.S. Hähnel, A. Schüler, B.H. Lee, N. Zhu, G. Raffel, T. Mercher, E. Bockamp, D. Sasca, T. Kindler
Writing, review, and/or revision of the manuscript: W.F. Tam, P.S. Hähnel, B.H. Lee, N. Zhu, S. Pante, G. Raffel, G.W. Robinson, L. Hennighausen, D.G. Gilliland, T. Kindler
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): W.F. Tam, R. Okabe, S. Pante
Study supervision: W.F. Tam, T. Kindler
Performance of experiments: A. Schüler
References
- 1.Estey E, Dohner H. Acute myeloid leukaemia. Lancet. 2006;368:1894–907. doi: 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- 2.Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6:587–96. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 3.Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006;10:257–68. doi: 10.1016/j.ccr.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 4.Billio A, Steer EJ, Pianezze G, Svaldi M, Casin M, Amato B, et al. A further case of acute myeloid leukaemia with inv(8)(p11q13) and MOZ-TIF2 fusion. Haematologica. 2002;87:ECR15. [PubMed] [Google Scholar]
- 5.Yang XJ, Ullah M. MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene. 2007;26:5408–19. doi: 10.1038/sj.onc.1210609. [DOI] [PubMed] [Google Scholar]
- 6.Thomas T, Corcoran LM, Gugasyan R, Dixon MP, Brodnicki T, Nutt SL, et al. Monocytic leukemia zinc finger protein is essential for the development of long-term reconstituting hematopoietic stem cells. Genes Dev. 2006;20:1175–86. doi: 10.1101/gad.1382606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katsumoto T, Aikawa Y, Iwama A, Ueda S, Ichikawa H, Ochiya T, et al. MOZ is essential for maintenance of hematopoietic stem cells. Genes Dev. 2006;20:1321–30. doi: 10.1101/gad.1393106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carapeti M, Aguiar RC, Goldman JM, Cross NC. A novel fusion between MOZ and the nuclear receptor coactivator TIF2 in acute myeloid leukemia. Blood. 1998;91:3127–33. [PubMed] [Google Scholar]
- 9.Liang J, Prouty L, Williams BJ, Dayton MA, Blanchard KL. Acute mixed lineage leukemia with an inv(8)(p11q13) resulting in fusion of the genes for MOZ and TIF2. Blood. 1998;92:2118–22. [PubMed] [Google Scholar]
- 10.Deguchi K, Ayton PM, Carapeti M, Kutok JL, Snyder CS, Williams IR, et al. MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell. 2003;3:259–71. doi: 10.1016/s1535-6108(03)00051-5. [DOI] [PubMed] [Google Scholar]
- 11.Kindle KB, Troke PJ, Collins HM, Matsuda S, Bossi D, Bellodi C, et al. MOZ-TIF2 inhibits transcription by nuclear receptors and p53 by impairment of CBP function. Mol Cell Biol. 2005;25:988–1002. doi: 10.1128/MCB.25.3.988-1002.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010;116:5089–102. doi: 10.1182/blood-2010-04-261867. [DOI] [PubMed] [Google Scholar]
- 13.Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99:310–8. doi: 10.1182/blood.v99.1.310. [DOI] [PubMed] [Google Scholar]
- 14.Lee BH, Tothova Z, Levine RL, Anderson K, Buza-Vidas N, Cullen DE, et al. FLT3 mutations confer enhanced proliferation and survival properties to multipotent progenitors in a murine model of chronic myelomonocytic leukemia. Cancer Cell. 2007;12:367–80. doi: 10.1016/j.ccr.2007.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li L, Piloto O, Nguyen HB, Greenberg K, Takamiya K, Racke F, et al. Knockin of an internal tandem duplication mutation into murine FLT3 confers myeloproliferative disease in a mouse model. Blood. 2008;111:3849–58. doi: 10.1182/blood-2007-08-109942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tse KF, Mukherjee G, Small D. Constitutive activation of FLT3 stimulates multiple intracellular signal transducers and results in transformation. Leukemia. 2000;14:1766–76. doi: 10.1038/sj.leu.2401905. [DOI] [PubMed] [Google Scholar]
- 17.Mizuki M, Fenski R, Halfter H, Matsumura I, Schmidt R, Muller C, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. 2000;96:3907–14. [PubMed] [Google Scholar]
- 18.Zhang S, Fukuda S, Lee Y, Hangoc G, Cooper S, Spolski R, et al. Essential role of signal transducer and activator of transcription (Stat) 5a but not Stat5b for Flt3-dependent signaling. J Exp Med. 2000;192:719–28. doi: 10.1084/jem.192.5.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rocnik JL, Okabe R, Yu JC, Lee BH, Giese N, Schenkein DP, et al. Roles of tyrosine 589 and 591 in STAT5 activation and transformation mediated by FLT3-ITD. Blood. 2006;108:1339–45. doi: 10.1182/blood-2005-11-011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore MA, Dorn DC, Schuringa JJ, Chung KY, Morrone G. Constitutive activation of Flt3 and STAT5A enhances self-renewal and alters differentiation of hematopoietic stem cells. Exp Hematol. 2007;35:105–16. doi: 10.1016/j.exphem.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 21.Schuringa JJ, Chung KY, Morrone G, Moore MA. Constitutive activation of STAT5A promotes human hematopoietic stem cell self-renewal and erythroid differentiation. J Exp Med. 2004;200:623–35. doi: 10.1084/jem.20041024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung KY, Morrone G, Schuringa JJ, Wong B, Dorn DC, Moore MA. Enforced expression of an Flt3 internal tandem duplication in human CD34+ cells confers properties of self-renewal and enhanced erythropoiesis. Blood. 2005;105:77–84. doi: 10.1182/blood-2003-12-4445. [DOI] [PubMed] [Google Scholar]
- 23.Wierenga AT, Vellenga E, Schuringa JJ. Maximal STAT5-induced proliferation and self-renewal at intermediate STAT5 activity levels. Mol Cell Biol. 2008;28:6668–80. doi: 10.1128/MCB.01025-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gouilleux-Gruart V, Gouilleux F, Desaint C, Claisse JF, Capiod JC, Delobel J, et al. STAT-related transcription factors are constitutively activated in peripheral blood cells from acute leukemia patients. Blood. 1996;87:1692–7. [PubMed] [Google Scholar]
- 25.Birkenkamp KU, Geugien M, Lemmink HH, Kruijer W, Vellenga E. Regulation of constitutive STAT5 phosphorylation in acute myeloid leukemia blasts. Leukemia. 2001;15:1923–31. doi: 10.1038/sj.leu.2402317. [DOI] [PubMed] [Google Scholar]
- 26.Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–47. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kindler T, Cornejo MG, Scholl C, Liu J, Leeman DS, Haydu JE, et al. K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to gamma-secretase inhibitors. Blood. 2008;112:3373–82. doi: 10.1182/blood-2008-03-147587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang YY, Zhou GB, Yin T, Chen B, Shi JY, Liang WX, et al. AML1-ETO and C-KIT mutation/overexpression in t(8;21) leukemia: implication in stepwise leukemogenesis and response to Gleevec. Proc Natl Acad Sci U S A. 2005;102:1104–9. doi: 10.1073/pnas.0408831102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ono R, Kumagai H, Nakajima H, Hishiya A, Taki T, Horikawa K, et al. Mixed-lineage-leukemia (MLL) fusion protein collaborates with Ras to induce acute leukemia through aberrant Hox expression and Raf activation. Leukemia. 2009;23:2197–209. doi: 10.1038/leu.2009.177. [DOI] [PubMed] [Google Scholar]
- 30.Kelly LM, Kutok JL, Williams IR, Boulton CL, Amaral SM, Curley DP, et al. PML/RARalpha and FLT3-ITD induce an APL-like disease in a mouse model. Proc Natl Acad Sci U S A. 2002;99:8283–8. doi: 10.1073/pnas.122233699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schessl C, Rawat VP, Cusan M, Deshpande A, Kohl TM, Rosten PM, et al. The AML1-ETO fusion gene and the FLT3 length mutation collaborate in inducing acute leukemia in mice. J Clin Invest. 2005;115:2159–68. doi: 10.1172/JCI24225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stubbs MC, Kim YM, Krivtsov AV, Wright RD, Feng Z, Agarwal J, et al. MLL-AF9 and FLT3 cooperation in acute myelogenous leukemia: development of a model for rapid therapeutic assessment. Leukemia. 2008;22:66–77. doi: 10.1038/sj.leu.2404951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ono R, Nakajima H, Ozaki K, Kumagai H, Kawashima T, Taki T, et al. Dimerization of MLL fusion proteins and FLT3 activation synergize to induce multiple-lineage leukemogenesis. J Clin Invest. 2005;115:919–29. doi: 10.1172/JCI22725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levis M, Murphy KM, Pham R, Kim KT, Stine A, Li L, et al. Internal tandem duplications of the FLT3 gene are present in leukemia stem cells. Blood. 2005;106:673–80. doi: 10.1182/blood-2004-05-1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pollard JA, Alonzo TA, Gerbing RB, Woods WG, Lange BJ, Sweetser DA, et al. FLT3 internal tandem duplication in CD34+/CD33− precursors predicts poor outcome in acute myeloid leukemia. Blood. 2006;108:2764–9. doi: 10.1182/blood-2006-04-012260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–22. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 37.Somervaille TC, Matheny CJ, Spencer GJ, Iwasaki M, Rinn JL, Witten DM, et al. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell. 2009;4:129–40. doi: 10.1016/j.stem.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453:1072–8. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heuser M, Sly LM, Argiropoulos B, Kuchenbauer F, Lai C, Weng A, et al. Modeling the functional heterogeneity of leukemia stem cells: role of STAT5 in leukemia stem cell self-renewal. Blood. 2009;114:3983–93. doi: 10.1182/blood-2009-06-227603. [DOI] [PubMed] [Google Scholar]
- 40.Hoelbl A, Schuster C, Kovacic B, Zhu B, Wickre M, Hoelzl MA, et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol Med. 2010;2:98–110. doi: 10.1002/emmm.201000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi S, Larson K, Guo D, Lim SJ, Dutta P, Yan SJ, et al. Drosophila STAT is required for directly maintaining HP1 localization and heterochromatin stability. Nat Cell Biol. 2008;10:489–96. doi: 10.1038/ncb1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schepers H, van Gosliga D, Wierenga AT, Eggen BJ, Schuringa JJ, Vellenga E. STAT5 is required for long-term maintenance of normal and leukemic human stem/progenitor cells. Blood. 2007;110:2880–8. doi: 10.1182/blood-2006-08-039073. [DOI] [PubMed] [Google Scholar]
- 43.Wang Z, Li G, Tse W, Bunting KD. Conditional deletion of STAT5 in adult mouse hematopoietic stem cells causes loss of quiescence and permits efficient nonablative stem cell replacement. Blood. 2009;113:4856–65. doi: 10.1182/blood-2008-09-181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.