

Abstract
Synthetic nucleic acids are commonly used laboratory tools for modulating gene expression and have the potential to be widely used in the clinic. Progress towards nucleic acid drugs, however, has been slow and many challenges remain to be overcome before their full impact on patient care can be understood. Antisense oligonucleotides (ASOs) and small interfering RNAs (siRNAs) are the two most widely used strategies for silencing gene expression. We first describe these two approaches and contrast their relative strengths and weaknesses for laboratory applications. We then review the choices faced during development of clinical candidates and the current state of clinical trials. Attitudes towards clinical development of nucleic acid silencing strategies have repeatedly swung from optimism to depression during the past twenty years. Our goal is to provide the information needed to design robust studies with oligonucleotides, making use of the strengths of each oligonucleotide technology.
Keywords: antisense oligonucleotides, siRNA, mRNA, gene silencing, therapeutics
Overview
The ability to use synthetic agents to control gene expression facilitates many aspects of biological research and would have a transformative impact on the treatment of many diseases. Oligonucleotides are one promising class of synthetic agents. Such compounds can be designed to recognize any species of cellular DNA or RNA and, in theory, have the potential to modulate gene expression and affect the course of almost any disease.
This concept is not new. In 1978, Zamecnik first reported that a synthetic oligonucleotide (at that time a rare and difficult to obtain type of compound) complementary to Rous sarcoma virus 35S RNA acted as an efficient inhibitor of protein expression [1–3]. In spite of the obvious promise of this approach, progress has been slow because of the need to overcome many technical hurdles. Now, fueled by advances in antisense technology and the emergence of RNA interference, the modulation of gene expression by nucleic acids has become a routine tool for laboratory research. For patient care, the field has seen repeated disappointments that often mask the underlying steady progress being made.
The concept is simple: a target RNA is chosen based on a hypothesis about its physiological significance, a complementary oligonucleotide is synthesized, gene expression is assayed, and phenotypes examined. Reality is more complex. Here we describe strategies for using nucleic acids to control gene expression, with a focus on translating the technology into the clinic.
Basic principles of oligonucleotide-mediated gene silencing
Single-stranded antisense oligonucleotides (ASOs) and RNA interference (RNAi) share their fundamental principle: an oligonucleotide binds a target RNA through Watson-Crick base pairing. An ASO must survive and function as a single strand (Figure 1A). In contrast, during RNAi, a small RNA duplex associates with the RNA-induced silencing complex (RISC), one strand (the passenger strand) is lost, and the remaining strand (the guide strand) cooperates with RISC to bind complementary RNA. In contrast to ASOs, the guide strand is always associated with a complementary strand or a protein (Figure 1B). This difference between the two approaches leads to different strengths and weaknesses that affect drug development.
Figure 1.
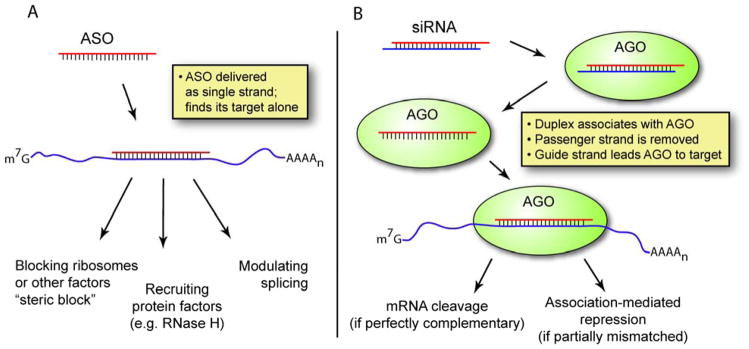
Comparison of the ASO and siRNA mechanisms. (A) ASOs must be stable as single-stranded oligonucleotides and find their target alone. (B) siRNAs are delivered as duplexes, then taken up by Argonaute (AGO), part of the RNA-induced silencing complex. Thus the “antisense oligonucleotide” that guides AGO to complementary mRNA is always present in the cell as a duplex or as part of a protein complex. Note that RISC also engages in complex biology with endogenous small RNAs [71].
Optimization strategies for antisense oligonucleotides: chemical modification
Unmodified single-stranded DNA or RNA oligonucleotides are too unstable to use in cells. The first type of optimization to be developed was, therefore, use of chemical modifications to increase the nuclease resistance. The earliest major breakthrough was the introduction of phosphorothioate (PS) linkages in place of the phosphodiester bond [4] (Figure 2A). This modification greatly improved stability towards digestion by nucleases. PS linkages also improved binding to serum proteins in vivo, increasing half-life and permitting greater delivery of active compound to tissues [5,6]. ASOs that only contain PS modifications were capable of producing antisense effects inside cells, but potencies were not always high nor were reliable results routine [7].
Figure 2.

Structures of some common chemically modified nucleotides. (A) Replacement of a nonbridging phosphate oxygen with sulfur gives the phosphorothioate (PS) linkage, which dramatically increases nuclease stability. (B) Various sugar modifications are compatible with unmodified DNA/RNA synthesis and increase binding affinity and nuclease stability. (C) PNA and PMO modifications have a neutral backbone structure that is dramatically different from the sugar-phosphate backbone of natural oligonucleotides.
Another obstacle was inadequate affinity for intended target sequences leading to low potencies. Poor potencies force use of high concentrations of oligonucleotides, which can lean to “off-target” effects – unintended phenotypes that are unrelated to inhibition of the intended target gene. Off-target effects can be due to recognition of other genes by binding to sequences that are similar to the intended target. Oligonucleotides can also bind directly to proteins and affect their function [8].
Chemical modifications can improve potency and selectivity by increasing binding affinity of oligonucleotides for their complementary sequences. Widely used modifications include 2′-O-methyl (2′-O-Me) [9], 2′-fluoro (2′-F) [10–13], and 2′-O-methoxyethyl (2′-MOE) [14,15] RNA (Figure 2B). Even more affinity can be gained using oligonucleotides modified with locked nucleic acid (LNA), which contains a methylene bridge between the 2′ and 4′ position of the ribose [16,17]. This bridge “locks” the ribose ring in a conformation that is ideal for binding, leading to high affinity for complementary sequences [18]. Related bridged nucleic acid (BNA) compounds have been developed and share these favorable properties [19–27]. Their high affinity has permitted the development of far shorter oligonucleotides than previously thought possible which nonetheless retain high potency [28].
The chemistry for introducing 2′-O-Me, 2′-MOE, 2′-F, or LNA into oligonucleotides is compatible with DNA or RNA synthesis, allowing chimeras with DNA or RNA bases to be easily obtained. This compatibility allows the properties of chemically modified oligonucleotides to be fine-tuned for specific applications – a major advantage for development that makes LNAs and other BNAs convenient tools for many applications.
Amplifying the effectiveness of ASOs: RNase H
The RNA strand of DNA•RNA hybrids is cleaved by the enzyme RNase H, an enzyme that exists in both the nucleus and cytoplasm of eukaryotic cells [29]. This catalytic cleavage can be exploited by synthetic oligonucleotides to increase potency [30,31]. “Gapmer” oligonucleotides contain 2–5 chemically modified nucleotides (e.g. LNA or 2′-MOE) on each terminus flanking a central 8–10 base “gap” of DNA [13,32]. The chemically modified oligonucleotides increase nuclease resistance and increase affinity for target sequences, while the DNA gap permits formation of a DNA•RNA hybrid that can be a good substrate for RNase H. Most of the ASOs currently in clinical development are gapmers of this type. It is also possible to use chemically modified oligonucleotides that mimic DNA structure and can recruit RNase H, yet bind strongly to complementary RNA. 2′-Fluoroarabinonucleic acid (2′F-ANA, Figure 2B) is the best example of this type of oligonucleotide [33,34].
Blocking translation
ASOs that lack a contiguous stretch of DNA (or DNA-like nucleotides) can also bind RNA and block gene expression. These oligomers are known as “steric blockers” because they act by blocking the ribosome rather than facilitating cleavage of RNA.
Besides the sugar-modified nucleotides discussed above, steric blockers can be made from oligomers that are quite different from DNA or RNA (Figure 2C). Peptide nucleic acid (PNAs) is an oligonucleotide mimic whose bases are linked by amide bonds and whose synthesis shares many features with peptide synthesis [35]. Because the amide backbone is uncharged, binding is characterized by high rates of association and high affinity [36,37]. Phosphorodiamidate morpholino oligomers (commonly called PMOs or “morpholinos”) are another uncharged DNA analogue [38]. PMOs do not bind complementary targets with the high affinities that characterize PNA binding, but have proven to be effective agents inside cells. PMOs are widely used as silencing agents in zebrafish [39,40].
Modulating splicing
mRNA is initially transcribed as a pre-mRNA containing intervening sequences or introns. These introns must be spliced out to form the mature mRNA. Up to 95% of multi-exon genes are subject to alternative splicing in which the pre-mRNA is spliced differently to form multiple variants of the mature mRNA [41]. Oligomers that target sequences within the pre-mRNA can affect splicing and increase production of desired isoforms [42–48]. In this case cleavage of the target RNA is not desired; these strategies use oligonucleotides that do not recruit RNase H, including LNA or 2′-modified oligonucleotides or uncharged analogues like PNA or PMO oligomers.
An impressive variety of genes and conditions can be treated by splice-switching oligonucleotides [42,43]. The most advanced target is the dystrophin gene, a large (2.6 Mb) gene containing 97 exons that encodes a protein essential for healthy muscle cells. Mutations in the dystrophin gene cause muscular dystrophy. In Duchenne muscular dystrophy (DMD), the most severe form of muscular dystrophy, mutations cause a frame shift resulting in no functional dystrophin protein. However, oligonucleotides have been used to remove the mutant exon and restore the proper reading frame, producing a truncated but partially functional dystrophin protein and potentially leading to milder symptoms [46–48]. Researchers have also successfully modulated the splicing of several other therapeutic targets, including β-globin (for β-thalassemia) [44,49,50], SMN2 (for spinal muscular atrophy) [45], HER2 or Bcl-x (for cancer) [51,52], and others [42,43].
Targeting miRNAs
MicroRNAs (miRNAs) are endogenous small RNAs that can regulate normal physiological processes and affect disease [53–58]. miRNAs act through the RNAi pathway and recognize target mRNAs through complex patterns of recognition that are incompletely understood. An increasing number of miRNAs have been implicated in physiological processes that affect disease and interfering with these miRNAs might increase expression of their target genes and provide therapeutic lead compounds.
ASOs that are complementary to miRNAs can block their function [59–61]. For example, miR-122 is an abundant liver specific miRNA implicated in a variety of diseases including cancer and hepatitis C [62]. Oligonucleotides complementary to miR-122 have been shown to alter liver metabolism [63,64] and block hepatitis C virus replication [65,66]. Various chemistries have been shown to be active as inhibitors of miRNA function, including PNA [67], LNA [65–68], 2′-O-Me [69], 2′-MOE [64], and morpholino [70].
siRNAs
Over the past decade, double-stranded short interfering RNAs (siRNAs) have become widely used tools for silencing gene expression. When a duplex RNA enters cells it binds the protein machinery of the RNA induced silencing complex (RISC) [71]. Synthetic RNAs used for gene silencing are usually 19–22 bp duplexes. This length is sufficient to form a stable duplex and be recognized by RISC, but short enough to avoid most of the strong interferon response provoked by duplexes greater than 30 bp in length.
Since publication of the first report of gene silencing in mammalian cells in 2001 [72], siRNAs have been the subject of thousands of experimental studies aimed at examining function. While antisense oligonucleotides continue to be used for gene silencing, the robust nature of siRNAs and the relative ease of identifying active siRNAs have made them a favored silencing tool for many laboratories.
Chemical modifications and duplex RNAs
Unmodified duplex RNA is surprisingly stable and chemical modification of siRNAs is usually not essential for silencing gene expression in cultured cells (see Table 1). In vivo, however, unmodified siRNAs are not highly active and chemical modification can significantly improve their properties [73]. Chemically modified siRNAs can feature improved nuclease stability and an associated increase in duration of action [74–76]. Unmodified RNA is also rapidly cleared [77] and chemical modification, complexation with carrier agent, and local delivery to a disease target can help achieve improved in vivo results.
Table 1.
ASOs and siRNAs use different methods to overcome similar obstacles.
| goal | principal methods for achieving the goal
|
|
|---|---|---|
| ASOs | siRNA | |
| nuclease stability |
|
|
| binding affinity, disruption of existing target structure |
|
|
| overcoming immune stimulation |
|
|
| specificity of knockdown |
|
|
| cellular uptake |
|
|
Several patterns of chemical modification show increased potency across several sequences [76,78,79]. The introduction of double-stranded RNA into cells can activate the innate immune system [80–82]; this immune activation may be able to be harnessed for therapeutic benefit [83–87] but can also lead to dose-limiting toxicity when siRNAs are delivered systemically [88]. Chemical modifications can be used to reduce the immunogenicity of siRNAs [79,88,89]. Chemical modification becomes particularly important as an siRNA progresses towards clinical use [90,91].
The architecture of an siRNA duplex can also be modified. For example, longer RNA duplexes are recognized by Dicer, a component of the RISC machinery, and may have higher potency than their 21-mer counterparts [92,93].
Identifying a useful ASO or siRNA for gene silencing
The first step in any project is to identify a biology problem or disease state where manipulation of gene expression might produce a useful phenotype or physiologic effect. The second step is to identify one or more target genes to manipulate. Efficient agents may already have been identified in the literature. If no such data exist, commercial suppliers may offer duplex RNAs that have been validated for inhibition of the target gene. If commercial RNAs are unavailable, are too expensive, or are inadequate, investigators can experimentally identify ASOs or siRNAs themselves.
There are algorithms of siRNA design [94–96], but none are perfect and it will probably be necessary to test several duplex RNAs to find sequences that are sufficiently active and selective. Various groups have proposed computational [97–99] or experimental [99–104] methods for identifying potent ASOs. Aspects of mRNA secondary structure can be roughly predicted, but algorithms such as the popular mFOLD do not take into account factors like tertiary structure or the involvement of RNA binding proteins, and so they are of limited utility in practice [105]. Ultimately if a group is serious about finding a potent ASO, they should test as many oligonucleotides as time and budget permit [106,107].
Control experiments: you can never have too many
Regardless of whether ASOs or siRNAs are used for gene silencing, it is essential to use proper controls [108]. Understanding off-target effects can help investigators choose controls appropriately. ASOs and siRNAs are large synthetic molecules and can act in ways independent of Watson-Crick base pairing. For ASOs, this includes nonspecific binding to proteins both in serum and inside cells. Phosphorothioate backbone oligonucleotides are particularly liable to bind proteins – this is the source of their slower clearance from serum, but also the source of much of their toxicity [109]. Oligonucleotides can fold into complex secondary structures and bind proteins in a sequence-specific manner related to shape rather than pairing [110]. For siRNAs, cells can recognize double-stranded RNA and activate the innate immune system [80–82] and this immunostimulatory activity has been misinterpreted as RNA interference effects [111,112]. Finally, high concentrations of siRNA can saturate the RNAi machinery, leading to a global perturbation of miRNA-mediated regulation [113–115].
An ASO or siRNA will always have partial complementarity to non-target transcripts, and this can cause unintended gene repression and misleading phenotypes [116,117]. For siRNAs, one of the most common off-target effects occurs through 7–8 nucleotide complementarity at the 5′-end of the guide strand (the so-called “seed region”) to sites in the 3′-untranslated region of other genes. This type of base pairing is a prerequisite for miRNA-mediated gene repression, and partially complementary siRNA duplexes can enter the miRNA pathway and repress nontarget transcripts [117,118]. Careful use of chemistry and duplex design can help alleviate this type of off-target effects. For example, including two 2′-O-Me-RNA nucleotides at the 5′-end of the siRNA reduces its ability to enter the miRNA pathway [117,119]. siRNA designs that ensure loading of the correct guide strand into RISC can also help to reduce off-target effects [120–126].
It is impossible to avoid some level of off-target effects in oligonucleotide-mediated gene silencing – the goal is to minimize them by careful use of chemistry and to use multiple approaches with non-overlapping off-target effects so that an observed phenotype can be confidently ascribed to recognition of the desired target. For example, an investigator may be testing the hypothesis that inhibition of a protein will lead to decreased cell proliferation. How can one build a case that the observed protein knockdown and decreased cell proliferation are not indirect effects? Several types of controls are recommended for any oligonucleotide experiment:
When multiple ASOs or siRNAs target the same gene, they should have the same on-target effect but different off-target effects. Thus, possession of multiple ASOs or siRNAs that are complementary to the target mRNA and produce the same phenotype provides one piece of evidence supporting a relationship between recognition of the target mRNA, gene silencing, and phenotype.
Experiments should include negative control compounds containing mismatched bases relative to the target mRNA or having blocks of bases moved relative to the sequence of the parent oligomer (Figure 3). Sequences that are closely related to the oligonucleotide of interest are more likely to have similar immunogenic or other off-target effects, making them better controls than totally unrelated sequences. An ideal mismatched or scrambled control does not significantly perturb the levels of a gene of interest.
Protein and RNA levels of the target gene should both be tested where possible. If using siRNA or an RNase H competent ASO, both protein and RNA should decrease. A different result should raise suspicion about the mechanism of the silencing observed.
The dose-response of any knockdown should be tested and experiments should be carried out at the lowest possible concentration.
For gapmers or siRNAs, a technique called rapid amplification of 5′-cDNA ends (5′-RACE) can be used to verify that the targeted transcript is being cleaved at the predicted site [127–130]. While valuable, it is important to note that 5′-RACE merely detects cleavage, it is not quantitative and is not an indication of efficiency.
Finally, when possible, the ultimate control for gene silencing experiments is a functional rescue by an exogenous copy of the gene of interest containing silent mutations at the oligonucleotide’s target site. Nevertheless, these experiments can be quite challenging depending on the biological system being studied.
Figure 3.
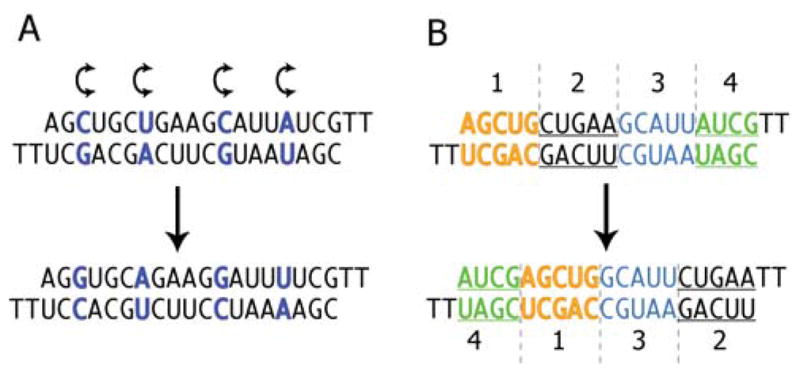
Typical mismatched and scrambled controls. (A) A duplex with several pairs of bases exchanged (from one strand to the other) should maintain very similar properties to the parent duplex but lose affinity for its target mRNA. (B) A scrambled control can be made by moving blocks of bases with respect to the parent oligomer.
Off-target effects are a significant concern for laboratory use and clinical development of nucleic acid acids. Unintended phenotypes are, however, a concern for development of any other type of drug including small molecules and proteins. The solution for minimizing off-target effects for nucleic acids is the same as for other classes of drug – iterative testing and rational optimization.
Introducing ASOs or siRNAs into cultured cells
The most common method for promoting uptake of ASOs and siRNAs in cell culture involves use of cationic lipids to transfect nucleic acids. Mixing cationic lipid with negatively charged nucleic acids yields a complex that can cross cell membranes and release active oligonucleotide into the cytoplasm of cells.
Many different cationic lipids are available and activity depends on the cell line used. Even for a given cell line, the preferred lipid may vary depending on whether an ASO or siRNA is being transfected. Not all cell lines can be transfected using cationic lipid, and if literature precedent is unavailable it may be necessary to experimentally test different cationic lipids to find one that can successfully transfect a cell line of interest. It is also possible to electroporate oligonucleotides into cells [131–133]. This method can be highly effective and useful for cell lines that cannot be readily transfected by lipid, but requires specialized equipment and expertise.
Recently, it has become apparent that active ASOs can freely enter some cell lines without the need to add lipid [134,135]. The transfection protocol is thus simplified and any off-target effects due to exposure of cells to lipid are avoided. The method can also be used with cell lines that are not compatible with lipid-mediated transfection. However, higher concentrations of ASO are needed relative to the amounts used in lipid-mediated transfections.
Delivery of ASOs and siRNAs in vivo
As gene silencing technologies move from cultured cells into animal models and ultimately clinical application, the challenge of delivery increases. Delivering oligonucleotides in whole organisms requires crossing many barriers [136,137]. Degradation by serum nucleases, clearance by the kidney, or inappropriate biodistribution can prevent the oligonucleotide from ever reaching its target organ. The oligonucleotide must pass through the blood vessel wall and navigate the interstitial space and extracellular matrix. Finally, if the oligonucleotide succeeds in reaching the appropriate cell membrane, it will usually be taken up into an endosome, from which it must escape to be active.
ASOs are usually delivered in saline and rely on chemical modifications to enable uptake. Their phosphorothioate backbone binds to serum proteins, slowing excretion by the kidney [109]. The aromatic nucleobases also interact with other hydrophobic molecules in serum and on cell surfaces. Many types of cells in vivo express surface receptors that actively take up oligonucleotides; these are often lost when cells are cultured, which explains why lipid seems more important for delivering ASOs in culture than in vivo [138].
Delivery is even more challenging for duplex RNAs than single-stranded oligonucleotides. In an siRNA, all of the aromatic nucleobases are on the inside, leaving only heavily hydrated phosphates on the outside of the duplex. This hydrated surface interacts poorly with cell surfaces and is rapidly excreted in the urine. Thus researchers have invested heavily in the development of delivery vehicles for siRNAs [137,139–142]. The predominant technologies for delivering siRNAs involve complexing the RNA with cationic and neutral lipids [139,143,144], although encouraging results have also been obtained using peptide transduction domains [145] and cationic polymers [130]. Including PEGylated lipids in the formulation prolongs the circulating half-life of the particles [146]. Conjugation of cholesterol to one strand of the siRNA gave effective knockdown in the liver of mice [147], but the quantities of material required (50 mg/kg) were several orders of magnitude higher than current lipid-based formulations (as low as 0.01 mg/kg [148]).
Lipid-based formulations are ideal for targeting the liver, since lipid nanoparticles are readily taken up by liver cells. For targeting other organs following systemic administration, researchers have conjugated various ligands to the siRNA itself or included them as part of a formulation. Promising strategies include the use of aptamers [149], antibodies/fragments [150], vitamins [151] and other targeting ligands [130,152].
siRNA or ASO?
For cell culture experiments siRNAs will often be the better choice. It is relatively simpler to discover potent siRNAs and it may be easier to obtain siRNAs since unmodified RNA works with high potency. ASOs, on the other hand, must contain chemical modifications to be active inside cells. For experiments where the goal is to develop compounds for testing in animals or investigate therapeutic development the choice is more complex.
Several studies have directly compared the activity of various ASOs and siRNAs [105,153–157]. These studies can be misleading if one of the compounds contains suboptimal chemistry or sequence selection (e.g. first generation PS-DNA ASOs); both potent and effective antisense oligonucleotides or siRNAs can generally be found if researchers are willing to invest in optimizing each technology [158]. An ideal target sequence for an ASO is not necessarily ideal for an siRNA, and vice versa.
In vivo, the choice of ASO versus siRNA is unsettled and will continue to evolve over the next decade. The challenges involved in delivering oligonucleotides to a given target tissue should be considered before choosing between them. For example, in animal models of Huntington’s disease, antisense oligonucleotides or siRNAs have been infused directly into the central nervous system. In the case of single-stranded oligonucleotides, researchers observed wide distribution throughout the mouse CNS including deep-brain penetration [159]. In contrast, others found that siRNA infused into the monkey brain penetrated into brain tissue only up to about 12 mm from the site of infusion [159].
ASOs and siRNAs share important similarities as drug candidates. Both platforms are intended to modulate gene expression. Both are nucleic acids and contain an antisense strand intended to recognize a target mRNA. They also have important differences. ASOs have one strand while siRNAs have two, a basic fact that may lower cost and simplify delivery. On the other hand, siRNAs have proven to be a more robust technology in cell culture in the hands of most users. It is not clear whether this will be true in vivo, but the possibility that siRNAs might have superior potency for at least some applications is a major driving force for their continued development.
Clinical progress
The clinical progress of oligonucleotide drugs has been slow because realizing the potential for ASOs and duplex RNAs requires inventing a new model for pharmaceutical development that allows large, highly charged molecules to be synthesized economically, distribute to target tissues, enter cells, and function within acceptable limits for toxicity. Oligonucleotides are unlike traditional small molecule drugs (<500–700 molecular weight) and much effort has been required to understand their properties and optimize them. Antibody therapeutics provide a useful comparison. This class of molecules is now a major source of new drugs, but they also required many years to develop. Oligonucleotides may eventually have similar success. One ASO has been approved by the FDA and at least 22 oligonucleotide drugs are in Phase II or III clinical trials (Table 2). Many more are earlier in the process of clinical development [91,158,160,161].
Table 2.
Oligonucleotide drug candidates in advanced clinical trials (Phase II or higher). Information taken from company websites and from references cited in the text.
| drug | company | phase | target gene and disease | notes and references |
|---|---|---|---|---|
| mRNA1-targeted Antisense oligonucleotide drug candidates | ||||
|
| ||||
| Mipomersen | ISIS/Genzyme | III | ApoB for hypercholeseterolemia | PS-MOE gapmer, intravenous delivery; met all endpoints in four Phase III trials |
| Genasense (Oblimersen) | Genta | III | Bcl-2 for melanoma, leukemia, lymphoma | PS-DNA, intravenous delivery of naked ASO |
| OGX-011 (Custirsen) | ISIS/Teva/OncoGenex | III | Clusterin for prostate, NSCLC and breast cancer | PS-MOE gapmer |
| AP 12009 (Trabedersen) | Antisense Pharma | III | TGF-β2 for brain cancer | PS-DNA |
| GS-101 (Aganirsen) | Gene Signal | III | Insulin receptor substrate-1 for corneal neovascularization | PS-DNA, topical delivery (eyedrops) |
| LOR-2040 | Lorus | II | Ribonucleotide reductase for cancer | PS-DNA |
| ASM8 | Pharmaxis | II | multiple targets for allergic asthma | two PS-DNA ASOs, delivered by inhalation |
| Archexin | Rexahn | II | AKT-1 for cancer | PS-DNA |
| LY2181308 | ISIS/Lilly | II | Survivin for cancer | PS-MOE gapmer |
| ISIS-EIF4ERx | ISIS/Lilly | II | eIF4E for cancer | PS-MOE gapmer |
| OGX-427 | ISIS/OncoGenex | II | Hsp27 for cancer | PS-MOE gapmer |
| Alicaforsen | ISIS/Atlantic | II | ICAM-1 for ulcerative colitis | available when requested by physicians for patients with inflammatory bowel disease |
| AEG35156 | Aegera | II | XIAP for cancer | PS 2′-O-Me gapmer |
|
| ||||
| Splice-switching oligonucleotide drug candidates | ||||
|
| ||||
| PRO051 (GSK2402968) | Prosenza/GSK | III | Dystrophin (exon 51 skipping) for DMD | 20-mer PS, fully-2′-O-Me ASO |
| AVI-4658 | AVI BioPharma | II | Dystrophin (exon 51 skipping) for DMD | 30-mer morpholino |
| PRO044 | Prosenza | I/II | Dystrophin (exon 44 skipping) for DMD | PS, fully-2′-O-Me ASO |
|
| ||||
| anti-miR oligonucleotide drug candidate | ||||
|
| ||||
| Miravirsen (SPC3649) | Santaris | II | miR-122 for hepatitis C virus | LNA-modified anti-miR oligonucleotide, delivered intravenously |
|
| ||||
| siRNA drug candidates | ||||
|
| ||||
| ALN-RSV01 | Alnylam/Cubist/Kyowa Kirin | II | RSV (viral nucleocapsid) | unmodified siRNA, intranasal or inhaled |
| PF-655 | Quark/Pfizer/Silence | II | RTP801 for wet AMD and diabetic macular edema | chemically modified, intraocular |
| QPI-1002 | Quark/Novartis/Silence | II | p53 for acute renal failure | chemically modified, intravenous |
| Excellair | ZaBeCor | II | SYK kinase for athsma | inhaled |
|
| ||||
| other advanced oligonucleotide drug candidates | ||||
|
| ||||
| IMO-2055 | Idera/Merck | II | TLR-9 activation for cancer treatment | CpG-rich oligonucleotide causes activation of TLR-9 |
| GRN163L (Imetelstat) | Geron | II | Telomerase inhibitor for cancer treatment | 13-mer N3′ thiophosphoramidate oligonucleotide (lipid conjugate); inhibits telomerase by direct binding, not an antisense effect |
ASO-mediated gene inhibition in the clinic
ASOs began clinical development in the 1990s with first generation compounds consisting of phosphorothioate DNA. One program from ISIS Pharmaceuticals succeeded, leading to FDA approval of Fomivirsen for treatment of CMV retinitis [162,163]. Development was facilitated by the location of the disease target and mode of administration. Fomivirsen is administered directly into the eye, reducing the amount of material needed and decreasing concerns about systemic side effects. One lesson for future work from these studies was that local delivery of oligonucleotides can simplify clinical studies and contribute to efficient trial design.
Other trials, however, were not successful. Drugs from Genta, Hybridon, and ISIS Pharmaceuticals failed in Phase III clinical trials. Reasons that contribute to the lack of success include i) incomplete understanding of the biological target and the consequences of its repression, ii) use of relatively inefficient first generation chemically-modified PS-DNA oligonucleotides, and iii) targeting disease tissues that are not prime locations for oligonucleotide biodistribution.
More recent trials have begun to revive optimism. Gapmer designs with optimized chemistry have led to improved potencies [28,164]. Biodistribution of oligonucleotides is better understood and some of the more promising trials involve inhibiting expression of genes in the liver, an organ known to accumulate ASO [165]. The importance of convenient markers of activity has been recognized. Such markers allow an ASO-mediated down-regulation of target gene expression to be demonstrated early in clinical trials, permitting resources to be devoted to the most promising drug candidates.
An example of the new wave of more promising ASOs is Mipomersen, an ASO from ISIS Pharmaceuticals designed to inhibit expression of Apo-B (Figure 4) [166]. Mipomersen is a gapmer containing phosphorothioate-modified DNA and 2′-O-MOE-RNA. Data from animal models show a robust and prolonged repression of apo-B expression [167]. In patients, the desired physiologic response upon systemic administration was demonstrated by monitoring serum LDL-cholesterol: all primary, secondary and tertiary endpoints have been met in four separate Phase III clinical trials [166]. Some toxic effects have been noted, and while these have been relatively mild they may (at least initially) limit the patient population to patients who are at the most severe risk for atherosclerosis [168].
Figure 4.
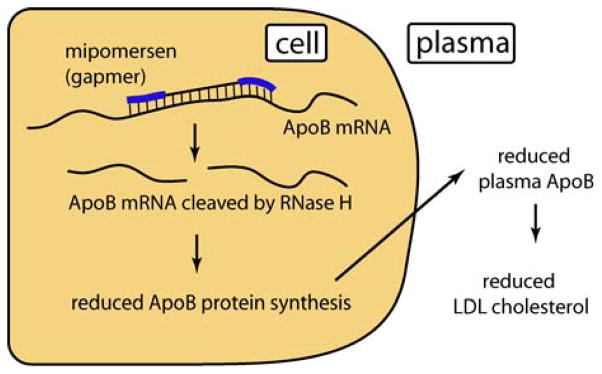
Mechanism of action of Mipomersen.
Eleven other traditional antisense oligonucleotides are in advanced clinical trials (Table 2). Targets relevant to cancer are the most highly represented, but there are also ASOs in trials against asthma, corneal neovascularization and ulcerative colitis. Many of these ASOs contain optimized chemistry and are taking advantage of the lessons learned over the past two decades in terms of delivery.
Modulating splicing in the clinic
Three splice-switching ASOs are in Phase II or III clinical trials, all of them for treatment of Duchenne muscular dystrophy (Figure 5). Prosenza has developed 2′-OMe phosphorothioate oligonucleotides [46] while AVI BioPharma has favored development of morpholino oligomers [48]. Both drugs show promise in clinical development, but two particular challenges face splice switching oligomers against DMD – the first is that of delivery. For significant clinical benefit, the ASOs would need to enter muscle tissue [144], including heart tissue. Currently all of the splice-switching ASOs are delivered naked and are not efficiently taken up into heart muscle cells (cardiomyocytes) in particular. Delivery is aided to some degree by the weakened, “leaky” nature of dystrophic muscle cells – but as the drug begins to take effect and the muscle cells recover, they are not such easy targets for further drug uptake. One possible solution is to use a targeted delivery system such as a cell-penetrating peptide [169,170]. Peptide-conjugated PMOs (PPMOs) take advantage of an active cell-internalization process and are taken up far more efficiently than unconjugated PMOs [171–177]. Even at lower doses, they distribute to more muscle cells bodywide, including healthy muscle cells and cardiomyocytes.
Figure 5.

Mode of action of drug candidates PRO051 and AVI-4568. (A) This DMD patient is missing dystrophin exon 50. Splicing of the pre-mRNA gives mature mRNA that is out-of-frame, and so no functional dystrophin can be produced. (B) In the presence of a splice-switching ASO that favors exclusion of exon 51, the cell splices exon 49 to exon 52, which restores the reading frame and causes translation of a shorter but partially functional dystrophin protein.
The second challenge is that DMD is caused by a large family of mutations. Exon 51 skipping would in principle be helpful for ~13% of DMD patients, including those with deletions of exon 50, 52, 45–50, 48–50 or 49–50. Exon 44 skipping could help another ~6% of DMD patients. Further splice-switching ASOs could be developed that would help other patients, but the populations become increasingly small. It is unclear whether the regulatory process could one day be adjusted to approve, for example, personalized dystrophin-targeted splice-switching ASOs as a class [170]. Extensive clinical trials for a sequence with a small target population might be prohibitively expensive.
Clinical trials for ASOs targeting microRNA
The first anti-miR oligonucleotide to be tested in humans is being developed by Santaris Pharmaceuticals. Miravirsen is a 15-mer phosphorothioate oligonucleotide containing 8 LNA modifications. It is complementary to mir-122 and designed to inhibit replication of hepatitis C virus [65,66] (Figure 6). Since miravirsen was the first anti-miR to be tested in humans, no one could predict the effect of inhibiting a miRNA, thus simultaneously de-repressing a family of mRNA targets in humans. Accordingly, Phase I testing started conservatively at 0.2 mg/kg delivered intravenously or subcutaneously. However, the drug was so well tolerated at the planned endpoint dose of 6 mg/kg that the trial was extended to a 12 mg/kg upper dose.
Figure 6.
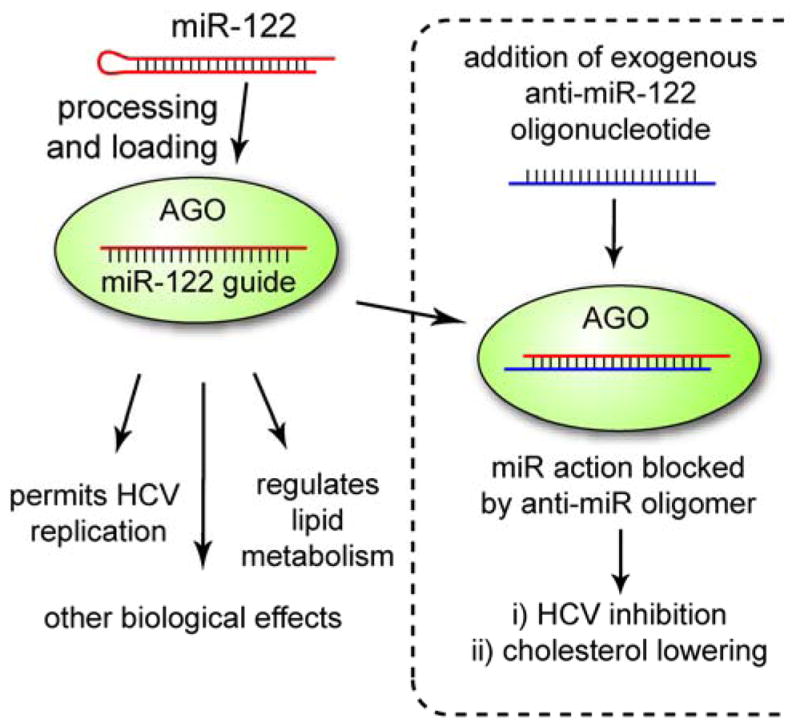
miR-122 is a liver-specific miRNA that regulates multiple pathways. Therapeutic inhibition of miR-122 by Miravirsen blocks HCV replication and lowers plasma cholesterol.
While miravirsen is designed to treat HCV, inhibition of miR-122 also lowers plasma cholesterol (Figure 6). Researchers at Santaris made use of this fact to demonstrate dose-dependent pharmacology in their Phase I trial in spite of the fact that the trial enrolled healthy volunteers. Phase II clinical trials on HCV patients began in September 2010.
siRNAs in the clinic
A decade after the first siRNA experiments [72,178], a dozen siRNA drugs are in clinical development [91]. The four most advanced are in Phase II trials (Table 2). As with ASOs, some of the earliest drugs to enter trials were very simple “first generation” siRNAs containing no chemical modifications. Two of these drugs, both targeting the VEGF pathway in the eye, had reached advanced clinical trials (Phase II and Phase III) but were withdrawn [91]. The therapeutic siRNA field is still young, however, many valuable lessons have been learned from 20 years of clinical work with ASOs that can now be applied to siRNA clinical development [179].
While cationic lipid-based formulations are clearly dominant in terms of potency, they deliver siRNAs into the endosome, the part of the cell where innate immune receptors are most intensely displayed. This means that siRNAs delivered by cationic lipid-based formulations are particularly vulnerable to immune stimulation. As such, a Phase I drug candidate targeting ApoB from Tekmira Pharmaceuticals was withdrawn from clinical trials after one patient at the highest dose level showed severe flu-like symptoms typical of an immune response [180]. Tekmira halted the trial in autumn 2009 and optimized both the lipid delivery agent and the siRNA cargo, decreasing the immunogenicity of both, before returning to clinical testing of the improved candidate.
Conclusions and future prospects
The field of oligonucleotide therapeutics has often swung from irrational optimism to irrational despair. In the laboratory as well, gene knockdown experiments have fallen in and out of favor with researchers. In reality, oligonucleotides are useful tools with strengths and weaknesses. Furthermore, different oligonucleotide technologies have different strengths, and many of the pendulum swings in the field have caused a switch from one oligonucleotide technology to another [181–184]. If researchers approach the oligonucleotide toolbox with careful experimentation and the broadest possible understanding, we believe they will continue to find useful tools for both laboratory experiments and therapeutic development.
Acknowledgments
This work was supported by the National Institutes of Health (NIGMS 77253 and 73042 to DRC), the Robert A Welch Foundation (I-1244 to DRC), and by the Natural Sciences and Engineering Research Council of Canada (postdoctoral fellowship to JKW).
Footnotes
The authors declare no conflicts of interest.
Author Contributions
JKW and DRC wrote the manuscript.
Teaching Materials
PowerPoint slides of the figures from this review are supplied as supporting information in the online version of this article.
References
- 1.Stephenson ML, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S A. 1978;75:285–288. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zamecnik PC, Stephenson ML. Inhibition of Rous sarcoma Virus Replication and Cell Transformation by a Specific Oligodeoxynucleotide. Proc Natl Acad Sci USA. 1978;75:280–284. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agrawal S. Remembering Paul C. Zamecnik, M.D. “Father of Antisense” (1912–2009) Oligonucleotides. 2010;20:47–50. doi: 10.1089/oli.2009.2001.obz. [DOI] [PubMed] [Google Scholar]
- 4.Eckstein F. Developments in RNA chemistry, a personal view. Biochimie. 2002;84:841–848. doi: 10.1016/s0300-9084(02)01459-1. [DOI] [PubMed] [Google Scholar]
- 5.Geary RS, Yu RZ, Levin AA. Pharmacokinetics of phosphorothioate antisense oligodeoxynucleotides. Curr Opin Investig Drugs. 2001;2:562–573. [PubMed] [Google Scholar]
- 6.Watanabe TA, Geary RS, Levin AA. Plasma protein binding of an antisense oligonucleotide targeting human ICAM-1 (ISIS 2302) Oligonucleotides. 2006;16:169–180. doi: 10.1089/oli.2006.16.169. [DOI] [PubMed] [Google Scholar]
- 7.Stein CA, Krieg AM. Problems in interpretation of data derived from in vitro and in vivo use of antisense oligodeoxynucleotides. Antisense Res Dev. 1994;4:67–69. doi: 10.1089/ard.1994.4.67. [DOI] [PubMed] [Google Scholar]
- 8.While direct binding to proteins is generally considered an unwanted off-target effect in the gene silencing field it has spawned a field of its own. See for example Thiel KW, Giangrande PH. Therapeutic applications of DNA and RNA aptamers. Oligonucleotides. 2009;19:209–222. doi: 10.1089/oli.2009.0199.
- 9.Chiang MY, Chan H, Zounes MA, et al. Antisense oligonucleotides inhibit intercellular adhesion molecule 1 expression by two distinct mechanisms. J Biol Chem. 1991;266:18162–18171. [PubMed] [Google Scholar]
- 10.Williams DM, Benseler F, Eckstein F. Properties of 2′-fluorothymidine-containing oligonucleotides: interaction with restriction endonuclease EcoRV. Biochemistry. 1991;30:4001–4009. doi: 10.1021/bi00230a027. [DOI] [PubMed] [Google Scholar]
- 11.Pieken WA, Olsen DB, Aurup H, et al. Structure-function relationship of hammerhead ribozymes as probed by 2′-modifications. Nucleic Acids Symp Ser. 1991:51–53. [PubMed] [Google Scholar]
- 12.Kawasaki AM, Casper MD, Freier SM, et al. Uniformly modified 2′-deoxy-2′-fluoro-phosphorothioate oligonucleotides as nuclease-resistant antisense compounds with high affinity and specificity for RNA targets. J Med Chem. 1993;36:831–841. doi: 10.1021/jm00059a007. [DOI] [PubMed] [Google Scholar]
- 13.Monia BP, Lesnik EA, Gonzalez C, et al. Evaluation of 2′-modified oligonucleotides containing 2′-deoxy gaps as antisense inhibitors of gene expression. J Biol Chem. 1993;268:14514–14522. [PubMed] [Google Scholar]
- 14.Freier SM, Altmann KH. The ups and downs of nucleic acid duplex stability: structure-stability studies on chemically-modified DNA:RNA duplexes. Nucl Acids Res. 1997;25:4429–4443. doi: 10.1093/nar/25.22.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin P. A New Access to 2′-O-Alkylated Ribonucleosides and Properties of 2′-O-Alkylated Oligoribonucleotides. Helv Chim Acta. 1995;78:486–504. [Google Scholar]
- 16.Koshkin AA, Singh SK, Nielsen P, et al. LNA (Locked Nucleic Acids): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron. 1998;54:3607–3630. [Google Scholar]
- 17.Obika S, Nanbu D, Hari Y, et al. Stability and structural features of the duplexes containing nucleoside analogues with a fixed N-type conformation, 2′-O,4′-C-methyleneribonucleosides. Tetrahedron Lett. 1998;39:5401–5404. [Google Scholar]
- 18.Braasch DA, Corey DR. Locked nucleic acid (LNA): Fine-tuning the recognition of DNA and RNA. Chem Biol. 2001;8:1–7. doi: 10.1016/s1074-5521(00)00058-2. [DOI] [PubMed] [Google Scholar]
- 19.Leumann CJ. DNA analogues: From supramolecular principles to biological properties. Bioorg Med Chem. 2002;10:841–854. doi: 10.1016/s0968-0896(01)00348-0. [DOI] [PubMed] [Google Scholar]
- 20.Obika S. Development of bridged nucleic acid analogues for antigene technology. Chem Pharm Bull (Tokyo) 2004;52:1399–1404. doi: 10.1248/cpb.52.1399. [DOI] [PubMed] [Google Scholar]
- 21.Christensen NK, Petersen M, Nielsen P, et al. A Novel Class of Oligonucleotide Analogues Containing 2′-O,3′-C-Linked [3.2. 0]Bicycloarabinonucleoside Monomers: Synthesis, Thermal Affinity Studies, and Molecular Modeling. J Am Chem Soc. 1998;120:5458–5463. [Google Scholar]
- 22.Rajwanshi VK, Hakansson AE, Sorensen MD, et al. The Eight Stereoisomers of LNA (Locked Nucleic Acid): A Remarkable Family of Strong RNA Binding Molecules. Angew Chem Int Ed Engl. 2000;39:1656–1659. doi: 10.1002/(sici)1521-3773(20000502)39:9<1656::aid-anie1656>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 23.Morita K, Hasegawa C, Kaneko M, et al. 2′-O,4′-C-ethylene-bridged nucleic acids (ENA): highly nuclease-resistant and thermodynamically stable oligonucleotides for antisense drug. Bioorg Med Chem Lett. 2002;12:73–76. doi: 10.1016/s0960-894x(01)00683-7. [DOI] [PubMed] [Google Scholar]
- 24.Renneberg D, Bouliong E, Reber U, et al. Antisense properties of tricyclo-DNA. Nucl Acids Res. 2002;30:2751–2757. doi: 10.1093/nar/gkf412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hari Y, Obika S, Ohnishi R, et al. Synthesis and properties of 2′-O,4′-C-methyleneoxymethylene bridged nucleic acid. Bioorg Med Chem. 2006;14:1029–1038. doi: 10.1016/j.bmc.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 26.Srivastava P, Barman J, Pathmasiri W, et al. Five- and six-membered conformationally locked 2′,4′-carbocyclic ribo-thymidines: synthesis, structure, and biochemical studies. J Am Chem Soc. 2007;129:8362–8379. doi: 10.1021/ja071106y. [DOI] [PubMed] [Google Scholar]
- 27.Bramsen JB, Laursen MB, Nielsen AF, et al. A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucl Acids Res. 2009;37:2867–2881. doi: 10.1093/nar/gkp106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Straarup EM, Fisker N, Hedtjarn M, et al. Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucl Acids Res. 2010;38:7100–7111. doi: 10.1093/nar/gkq457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu H, Lima WF, Zhang H, et al. Determination of the Role of the Human RNase H1 in the Pharmacology of DNA-like Antisense Drugs. J Biol Chem. 2004;279:17181–17189. doi: 10.1074/jbc.M311683200. [DOI] [PubMed] [Google Scholar]
- 30.Minshull J, Hunt T. The use of single-stranded DNA and RNase H to promote quantitative ‘hybrid arrest of translation’ of mRNA/DNA Hybrids in reticulocyte lysate cell-free translations. Nucl Acids Res. 1986;14:6433–6451. doi: 10.1093/nar/14.16.6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamura H, Oda Y, Iwai S, et al. How does RNase H recognize a DNA.RNA hybrid? Proc Natl Acad Sci U S A. 1991;88:11535–11539. doi: 10.1073/pnas.88.24.11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wahlestedt C, Salmi P, Good L, et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc Natl Acad Sci U S A. 2000;97:5633–5638. doi: 10.1073/pnas.97.10.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Damha MJ, Wilds CJ, Noronha A, et al. Hybrids of RNA and arabinonucleic acids (ANA and 2′F-ANA) are substrates of Ribonuclease H. J Am Chem Soc. 1998;120:12976–12977. [Google Scholar]
- 34.Watts JK, Damha MJ. 2′F-Arabinonucleic acids (2′F-ANA) -- History, properties, and new frontiers. Can J Chem. 2008;86:641–656. [Google Scholar]
- 35.Nielsen PE, Egholm M, Berg RH, et al. Sequence-Selective Recognition of DNA by Strand Displacement with a Thymine-Substituted Polyamide. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- 36.Bentin T, Nielsen PE. Enhanced peptide nucleic acid binding to supercoiled DNA: possible implications for DNA “breathing” dynamics. Biochemistry. 1996;35:8863–8869. doi: 10.1021/bi960436k. [DOI] [PubMed] [Google Scholar]
- 37.Smulevitch SV, Simmons CG, Norton JC, et al. Enhancement of strand invasion by oligonucleotides through manipulation of backbone charge. Nat Biotech. 1996;14:1700–1704. doi: 10.1038/nbt1296-1700. [DOI] [PubMed] [Google Scholar]
- 38.Summerton J, Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997;7:187–195. doi: 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]
- 39.Corey DR, Abrams JM. Morpholino antisense oligonucleotides: tools for investigating vertebrate development. Genome Biol. 2001;2:REVIEWS1015. doi: 10.1186/gb-2001-2-5-reviews1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bill BR, Petzold AM, Clark KJ, et al. A primer for morpholino use in zebrafish. Zebrafish. 2009;6:69–77. doi: 10.1089/zeb.2008.0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pan Q, Shai O, Lee LJ, et al. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 42.Aartsma-Rus A, van Ommen GJ. Antisense-mediated exon skipping: a versatile tool with therapeutic and research applications. RNA. 2007;13:1609–1624. doi: 10.1261/rna.653607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bauman J, Jearawiriyapaisarn N, Kole R. Therapeutic potential of splice-switching oligonucleotides. Oligonucleotides. 2009;19:1–13. doi: 10.1089/oli.2008.0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dominski Z, Kole R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc Natl Acad Sci U S A. 1993;90:8673–8677. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hua Y, Sahashi K, Hung G, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goemans NM, Tulinius M, van den Akker JT, et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med. 2011;364:1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- 47.Aartsma-Rus A, van Ommen GJ. Less is more: therapeutic exon skipping for Duchenne muscular dystrophy. Lancet Neurol. 2009;8:873–875. doi: 10.1016/S1474-4422(09)70229-7. [DOI] [PubMed] [Google Scholar]
- 48.Alter J, Lou F, Rabinowitz A, et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med. 2006;12:175–177. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- 49.Suwanmanee T, Sierakowska H, Fucharoen S, et al. Repair of a splicing defect in erythroid cells from patients with beta-thalassemia/HbE disorder. Mol Ther. 2002;6:718–726. doi: 10.1006/mthe.2002.0805. [DOI] [PubMed] [Google Scholar]
- 50.Suwanmanee T, Sierakowska H, Lacerra G, et al. Restoration of human beta-globin gene expression in murine and human IVS2-654 thalassemic erythroid cells by free uptake of antisense oligonucleotides. Mol Pharmacol. 2002;62:545–553. doi: 10.1124/mol.62.3.545. [DOI] [PubMed] [Google Scholar]
- 51.Wan J, Sazani P, Kole R. Modification of HER2 pre-mRNA alternative splicing and its effects on breast cancer cells. Int J Cancer. 2009;124:772–777. doi: 10.1002/ijc.24052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bauman JA, Li SD, Yang A, et al. Anti-tumor activity of splice-switching oligonucleotides. Nucl Acids Res. 2010;38:8348–8356. doi: 10.1093/nar/gkq731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bartel DP. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 54.Farazi TA, Spitzer JI, Morozov P, et al. miRNAs in human cancer. J Pathol. 2011;223:102–115. doi: 10.1002/path.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9:775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soifer HS, Rossi JJ, Saetrom P. MicroRNAs in disease and potential therapeutic applications. Mol Ther. 2007;15:2070–2079. doi: 10.1038/sj.mt.6300311. [DOI] [PubMed] [Google Scholar]
- 57.Lu M, Zhang Q, Deng M, et al. An analysis of human microRNA and disease associations. PLoS One. 2008;3:e3420. doi: 10.1371/journal.pone.0003420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang Q, Wang Y, Hao Y, et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucl Acids Res. 2009;37:D98–104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Esau CC. Inhibition of microRNA with antisense oligonucleotides. Methods. 2008;44:55–60. doi: 10.1016/j.ymeth.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 60.Davis S, Propp S, Freier SM, et al. Potent inhibition of microRNA in vivo without degradation. Nucl Acids Res. 2009;37:70–77. doi: 10.1093/nar/gkn904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Montgomery RL, van Rooij E. Therapeutic advances in MicroRNA targeting. J Cardiovasc Pharmacol. 2011;57:1–7. doi: 10.1097/FJC.0b013e3181f603d0. [DOI] [PubMed] [Google Scholar]
- 62.Girard M, Jacquemin E, Munnich A, et al. miR-122, a paradigm for the role of microRNAs in the liver. J Hepatol. 2008;48:648–656. doi: 10.1016/j.jhep.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 63.Krutzfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005:438. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 64.Esau C, Davis S, Murray SF, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metabolism. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 65.Elmen J, Lindow M, Schutz S, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–U810. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 66.Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fabani MM, Gait MJ. miR-122 targeting with LNA/2′-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA-peptide conjugates. RNA. 2008;14:336–346. doi: 10.1261/rna.844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Orom UA, Kauppinen S, Lund AH. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene. 2006;372:137–141. doi: 10.1016/j.gene.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 69.Meister G, Landthaler M, Dorsett Y, et al. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10:544–550. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kloosterman WP, Lagendijk AK, Ketting RF, et al. Targeted inhibition of miRNA maturation with morpholinos reveals a role for miR-375 in pancreatic islet development. PLoS Biol. 2007;5:e203. doi: 10.1371/journal.pbio.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Siomi H, Siomi MC. On the road to reading the RNA-interference code. Nature. 2009;457:396–404. doi: 10.1038/nature07754. [DOI] [PubMed] [Google Scholar]
- 72.Elbashir SM, Harborth J, Lendeckel W, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 73.Watts JK, Deleavey GF, Damha MJ. Chemically modified siRNA: tools and applications. Drug Discov Today. 2008;13:842–855. doi: 10.1016/j.drudis.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 74.Morrissey DV, Lockridge JA, Shaw L, et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat Biotech. 2005;23:1002–1007. doi: 10.1038/nbt1122. [DOI] [PubMed] [Google Scholar]
- 75.Morrissey DV, Blanchard K, Shaw L, et al. Activity of stabilized short interfering RNA in a mouse model of hepatitis B virus replication. Hepatology. 2005;41:1349–1356. doi: 10.1002/hep.20702. [DOI] [PubMed] [Google Scholar]
- 76.Allerson CR, Sioufi N, Jarres R, et al. Fully 2′-Modified Oligonucleotide Duplexes with Improved in Vitro Potency and Stability Compared to Unmodified Small Interfering RNA. J Med Chem. 2005;48:901–904. doi: 10.1021/jm049167j. [DOI] [PubMed] [Google Scholar]
- 77.Braasch DA, Paroo Z, Constantinescu A, et al. Biodistribution of phosphodiester and phosphorothioate siRNA. Bioorg Med Chem Lett. 2004;14:1139–1143. doi: 10.1016/j.bmcl.2003.12.074. [DOI] [PubMed] [Google Scholar]
- 78.Koller E, Propp S, Murray H, et al. Competition for RISC binding predicts in vitro potency of siRNA. Nucl Acids Res. 2006;34:4467–4476. doi: 10.1093/nar/gkl589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Deleavey GF, Watts JK, Alain T, et al. Synergistic effects between analogs of DNA and RNA improve the potency of siRNA-mediated gene silencing. Nucl Acids Res. 2010;38:4547–4557. doi: 10.1093/nar/gkq181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Marques JT, Williams BRG. Activation of the mammalian immune system by siRNAs. Nat Biotech. 2005;23:1399–1405. doi: 10.1038/nbt1161. [DOI] [PubMed] [Google Scholar]
- 81.Robbins M, Judge A, MacLachlan I. siRNA and Innate Immunity. Oligonucleotides. 2009;19:89–102. doi: 10.1089/oli.2009.0180. [DOI] [PubMed] [Google Scholar]
- 82.Olejniczak M, Galka P, Krzyzosiak WJ. Sequence-non-specific effects of RNA interference triggers and microRNA regulators. Nucl Acids Res. 2010;38:1–16. doi: 10.1093/nar/gkp829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schlee M, Hornung V, Hartmann G. siRNA and isRNA: Two Edges of One Sword. Mol Ther. 2006;14:463–470. doi: 10.1016/j.ymthe.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 84.Poeck H, Besch R, Maihoefer C, et al. 5′-triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med. 2008;14:1256–1263. doi: 10.1038/nm.1887. [DOI] [PubMed] [Google Scholar]
- 85.Kortylewski M, Swiderski P, Herrmann A, et al. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat Biotechnol. 2009;27:925–932. doi: 10.1038/nbt.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gantier MP, Tong S, Behlke MA, et al. Rational design of immunostimulatory siRNAs. Mol Ther. 2010;18:785–795. doi: 10.1038/mt.2010.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Khairuddin N, Gantier MP, Blake SJ, et al. siRNA-induced immunostimulation through TLR7 promotes antitumoral activity against HPV-driven tumors in vivo. Immunol Cell Biol. 2011 doi: 10.1038/icb.2011.1019. in press. [DOI] [PubMed] [Google Scholar]
- 88.Judge A, MacLachlan I. Overcoming the innate immune response to small interfering RNA. Hum Gene Ther. 2008;19:111–124. doi: 10.1089/hum.2007.179. [DOI] [PubMed] [Google Scholar]
- 89.Judge AD, Bola G, Lee ACH, et al. Design of Noninflammatory Synthetic siRNA Mediating Potent Gene Silencing in Vivo. Mol Ther. 2006;13:494–505. doi: 10.1016/j.ymthe.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 90.Corey DR. Chemical modification: the key to clinical application of RNA interference? J Clin Invest. 2007;117:3615–3622. doi: 10.1172/JCI33483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Watts JK, Corey DR. Clinical status of duplex RNA. Bioorg Med Chem Lett. 2010;20:3203–3207. doi: 10.1016/j.bmcl.2010.03.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim DH, Behlke MA, Rose SD, et al. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotech. 2005;23:222–226. doi: 10.1038/nbt1051. [DOI] [PubMed] [Google Scholar]
- 93.Collingwood MA, Rose SD, Huang L, et al. Chemical modification patterns compatible with high potency dicer-substrate small interfering RNAs. Oligonucleotides. 2008;18:187–200. doi: 10.1089/oli.2008.0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yuan B, Latek R, Hossbach M, et al. siRNA Selection Server: an automated siRNA oligonucleotide prediction server. Nucl Acids Res. 2004;32:W130–134. doi: 10.1093/nar/gkh366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boese Q, Leake D, Reynolds A, et al. Mechanistic insights aid computational short interfering RNA design. Methods Enzymol. 2005;392:73–96. doi: 10.1016/S0076-6879(04)92005-8. [DOI] [PubMed] [Google Scholar]
- 96.Huesken D, Lange J, Mickanin C, et al. Design of a genome-wide siRNA library using an artificial neural network. Nat Biotech. 2005;23:995–1001. doi: 10.1038/nbt1118. [DOI] [PubMed] [Google Scholar]
- 97.Sczakiel G, Homann M, Rittner K. Computer-aided search for effective antisense RNA target sequences of the human immunodeficiency virus type 1. Antisense Res Dev. 1993;3:45–52. doi: 10.1089/ard.1993.3.45. [DOI] [PubMed] [Google Scholar]
- 98.Stull RA, Taylor LA, Szoka FC., Jr Predicting antisense oligonucleotide inhibitory efficacy: a computational approach using histograms and thermodynamic indices. Nucl Acids Res. 1992;20:3501–3508. doi: 10.1093/nar/20.13.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sczakiel G. Theoretical and experimental approaches to design effective antisense oligonucleotides. Front Biosci. 2000;5:D194–201. doi: 10.2741/sczakiel. [DOI] [PubMed] [Google Scholar]
- 100.Gifford LK, Opalinska JB, Jordan D, et al. Identification of antisense nucleic acid hybridization sites in mRNA molecules with self-quenching fluorescent reporter molecules. Nucl Acids Res. 2005;33:e28. doi: 10.1093/nar/gni024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rittner K, Burmester C, Sczakiel G. In vitro selection of fast-hybridizing and effective antisense RNAs directed against the human immunodeficiency virus type 1. Nucl Acids Res. 1993;21:1381–1387. doi: 10.1093/nar/21.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ho SP, Bao Y, Lesher T, et al. Mapping of RNA accessible sites for antisense experiments with oligonucleotide libraries. Nat Biotechnol. 1998;16:59–63. doi: 10.1038/nbt0198-59. [DOI] [PubMed] [Google Scholar]
- 103.Kronenwett R, Haas R, Sczakiel G. Kinetic selectivity of complementary nucleic acids: bcr-abl-directed antisense RNA and ribozymes. J Mol Biol. 1996;259:632–644. doi: 10.1006/jmbi.1996.0345. [DOI] [PubMed] [Google Scholar]
- 104.Milner N, Mir KU, Southern EM. Selecting effective antisense reagents on combinatorial oligonucleotide arrays. Nat Biotechnol. 1997;15:537–541. doi: 10.1038/nbt0697-537. [DOI] [PubMed] [Google Scholar]
- 105.Rudnick SI, Swaminathan J, Sumaroka M, et al. Effects of local mRNA structure on posttranscriptional gene silencing. Proc Natl Acad Sci U S A. 2008;105:13787–13792. doi: 10.1073/pnas.0805781105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bacon TA, Wickstrom E. Walking along human c-myc mRNA with antisense oligodeoxynucleotides: maximum efficacy at the 5′ cap region. Oncogene Res. 1991;6:13–19. [PubMed] [Google Scholar]
- 107.Stewart AJ, Canitrot Y, Baracchini E, et al. Reduction of expression of the multidrug resistance protein (MRP) in human tumor cells by antisense phosphorothioate oligonucleotides. Biochem Pharmacol. 1996;51:461–469. doi: 10.1016/0006-2952(95)02220-1. [DOI] [PubMed] [Google Scholar]
- 108.Whither RNAi? Nat Cell Biol. 2003;5:489–490. doi: 10.1038/ncb0603-490. [DOI] [PubMed] [Google Scholar]
- 109.Levin AA. A review of issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim Biophys Acta. 1999;1489:69–84. doi: 10.1016/s0167-4781(99)00140-2. [DOI] [PubMed] [Google Scholar]
- 110.While direct binding to proteins is generally considered an unwanted off-target effect in the gene silencing field it has spawned a field of its own. See for example Thiel KW, Giangrande PH. Therapeutic applications of DNA and RNA aptamers. Oligonucleotides. 2009;19:209–222. doi: 10.1089/oli.2009.0199.
- 111.Robbins M, Judge A, Ambegia E, et al. Misinterpreting the therapeutic effects of small interfering RNA caused by immune stimulation. Hum Gene Ther. 2008;19:991–999. doi: 10.1089/hum.2008.131. [DOI] [PubMed] [Google Scholar]
- 112.Kleinman ME, Yamada K, Takeda A, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591–597. doi: 10.1038/nature06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yi R, Doehle BP, Qin Y, et al. Overexpression of exportin 5 enhances RNA interference mediated by short hairpin RNAs and microRNAs. RNA. 2005;11:220–226. doi: 10.1261/rna.7233305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Grimm D, Streetz KL, Jopling CL, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 115.Khan AA, Betel D, Miller ML, et al. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat Biotech. 2009;27:549–555. doi: 10.1038/nbt.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jackson AL, Bartz SR, Schelter J, et al. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotech. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 117.Fedorov Y, Anderson EM, Birmingham A, et al. Off-target effects by siRNA can induce toxic phenotype. RNA. 2006;12:1188–1196. doi: 10.1261/rna.28106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jackson AL, Burchard J, Schelter J, et al. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA. 2006;12:1179–1187. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jackson AL, Burchard J, Leake D, et al. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA. 2006;12:1197–1205. doi: 10.1261/rna.30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bramsen JB, Laursen MB, Damgaard CK, et al. Improved silencing properties using small internally segmented interfering RNAs. Nucl Acids Res. 2007;35:5886–5897. doi: 10.1093/nar/gkm548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen PY, Weinmann L, Gaidatzis D, et al. Strand-specific 5′-O-methylation of siRNA duplexes controls guide strand selection and targeting specificity. RNA. 2008;14:263–274. doi: 10.1261/rna.789808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sano M, Sierant M, Miyagishi M, et al. Effect of asymmetric terminal structures of short RNA duplexes on the RNA interference activity and strand selection. Nucl Acids Res. 2008;36:5812–5821. doi: 10.1093/nar/gkn584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sun X, Rogoff HA, Li CJ. Asymmetric RNA duplexes mediate RNA interference in mammalian cells. Nat Biotech. 2008;26:1379–1382. doi: 10.1038/nbt.1512. [DOI] [PubMed] [Google Scholar]
- 124.Ui-Tei K, Naito Y, Zenno S, et al. Functional dissection of siRNA sequence by systematic DNA substitution: modified siRNA with a DNA seed arm is a powerful tool for mammalian gene silencing with significantly reduced off-target effect. Nucl Acids Res. 2008;36:2136–2151. doi: 10.1093/nar/gkn042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chang CI, Yoo JW, Hong SW, et al. Asymmetric shorter-duplex siRNA structures trigger efficient gene silencing with reduced nonspecific effects. Mol Ther. 2009;17:725–732. doi: 10.1038/mt.2008.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lapierre J, Salomon W, Cardia J, et al. Potent and systematic RNAi mediated silencing with single oligonucleotide compounds. RNA. 2011 doi: 10.1261/rna.2399411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lasham A, Herbert M, Coppieters ‘t, Wallant N, et al. A rapid and sensitive method to detect siRNA-mediated mRNA cleavage in vivo using 5′ RACE and a molecular beacon probe. Nucl Acids Res. 2009 doi: 10.1093/nar/gkp1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Alvarez R, Elbashir S, Borland T, et al. RNA interference-mediated silencing of the respiratory syncytial virus nucleocapsid defines a potent antiviral strategy. Antimicrob Agents Chemother. 2009;53:3952–3962. doi: 10.1128/AAC.00014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Judge AD, Robbins M, Tavakoli I, et al. Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. J Clin Invest. 2009;119:661–673. doi: 10.1172/JCI37515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Davis ME, Zuckerman JE, Choi CHJ, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bergan R, Hakim F, Schwartz GN, et al. Electroporation of synthetic oligodeoxynucleotides: a novel technique for ex vivo bone marrow purging. Blood. 1996;88:731–741. [PubMed] [Google Scholar]
- 132.Spiller DG, Giles RV, Grzybowski J, et al. Improving the intracellular delivery and molecular efficacy of antisense oligonucleotides in chronic myeloid leukemia cells: a comparison of streptolysin-O permeabilization, electroporation, and lipophilic conjugation. Blood. 1998;91:4738–4746. [PubMed] [Google Scholar]
- 133.Li S, editor. Electroporation Protocols: Preclinical and Clinical Gene Medicine (Methods in Molecular Biology. Vol. 423. Humana Press; New York: 2008. [Google Scholar]
- 134.Stein CA, Hansen JB, Lai J, et al. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucl Acids Res. 2009 doi: 10.1093/nar/gkp841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang Y, Qu Z, Kim S, et al. Down-modulation of cancer targets using locked nucleic acid (LNA)-based antisense oligonucleotides without transfection. Gene Ther. 18:326–333. doi: 10.1038/gt.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang J, Lu Z, Wientjes MG, et al. Delivery of siRNA therapeutics: barriers and carriers. Aaps J. 2010;12:492–503. doi: 10.1208/s12248-010-9210-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Juliano R, Alam MR, Dixit V, et al. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucl Acids Res. 2008;36:4158–4171. doi: 10.1093/nar/gkn342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Koller E, Vincent TM, Chappell A, et al. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucl Acids Res. 2011;39:4795–4807. doi: 10.1093/nar/gkr089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Grimm D, Kay MA. Therapeutic application of RNAi: is mRNA targeting finally ready for prime time? J Clin Invest. 2007;117:3633–3641. doi: 10.1172/JCI34129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.De Paula D, Bentley MVLB, Mahato RI. Hydrophobization and bioconjugation for enhanced siRNA delivery and targeting. RNA. 2007;13:431–456. doi: 10.1261/rna.459807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Akhtar S, Benter IF. Nonviral delivery of synthetic siRNAs in vivo. J Clin Invest. 2007;117:3623–3632. doi: 10.1172/JCI33494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li W, Szoka FC., Jr Lipid-based Nanoparticles for Nucleic Acid Delivery. Pharm Res. 2007;24:438–449. doi: 10.1007/s11095-006-9180-5. [DOI] [PubMed] [Google Scholar]
- 144.Tseng YC, Mozumdar S, Huang L. Lipid-based systemic delivery of siRNA. Adv Drug Deliv Rev. 2009;61:721–731. doi: 10.1016/j.addr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Eguchi A, Meade BR, Chang YC, et al. Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nat Biotech. 2009;27:567–571. doi: 10.1038/nbt.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Papahadjopoulos D, Allen TM, Gabizon A, et al. Sterically stabilized liposomes: improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc Natl Acad Sci U S A. 1991;88:11460–11464. doi: 10.1073/pnas.88.24.11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Soutschek J, Akinc A, Bramlage B, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 148.Semple SC, Akinc A, Chen J, et al. Rational design of cationic lipids for siRNA delivery. Nat Biotech. 2010;28:172–176. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 149.Dassie JP, Liu XY, Thomas GS, et al. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotech. 2009;27:839–849. doi: 10.1038/nbt.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Song E, Zhu P, Lee SK, et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotech. 2005;23:709–717. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 151.Sato Y, Murase K, Kato J, et al. Resolution of liver cirrhosis using vitamin A–coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat Biotech. 2008;26:431–442. doi: 10.1038/nbt1396. [DOI] [PubMed] [Google Scholar]
- 152.Kim SS, Ye C, Kumar P, et al. Targeted delivery of siRNA to macrophages for anti-inflammatory treatment. Mol Ther. 2010;18:993–1001. doi: 10.1038/mt.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ferrari N, Bergeron D, Tedeschi AL, et al. Characterization of Antisense Oligonucleotides Comprising 2′-Deoxy-2′-Fluoro-beta-D-Arabinonucleic Acid (FANA): Specificity, Potency, and Duration of Activity. Ann N Y Acad Sci. 2006;1082:91–102. doi: 10.1196/annals.1348.032. [DOI] [PubMed] [Google Scholar]
- 154.Stein D, Foster E, Huang SB, et al. A specificity comparison of four antisense types: morpholino, 2′-O-methyl RNA, DNA, and phosphorothioate DNA. Antisense Nucleic Acid Drug Dev. 1997;7:151–157. doi: 10.1089/oli.1.1997.7.151. [DOI] [PubMed] [Google Scholar]
- 155.Summerton JE. Morpholino, siRNA, and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Curr Top Med Chem. 2007;7:651–660. doi: 10.2174/156802607780487740. [DOI] [PubMed] [Google Scholar]
- 156.Gruenweller A, Wyszko E, Bieber B, et al. Comparison of different antisense strategies in mammalian cells using locked nucleic acids, 2′-O-methyl RNA, phosphorothioates and small interfering RNA. Nucl Acids Res. 2003;31:3185–3193. doi: 10.1093/nar/gkg409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kretschmer-Kazemi Far R, Sczakiel G. The activity of siRNA in mammalian cells is related to structural target accessibility: a comparison with antisense oligonucleotides. Nucl Acids Res. 2003;31:4417–4424. doi: 10.1093/nar/gkg649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 159.Gagnon KT. HD Therapeutics - CHDI Fifth Annual Conference (Meeting Report) IDrugs. 2010;13:219–223. [PubMed] [Google Scholar]
- 160.Kunze D, Kraemer K, Fuessel S. Antisense Oligonucleotides: Insights from Preclinical Studies and Clinical Trials. In: Erdmann VA, Barciszewski J, editors. RNA Technologies and Their Applications. Springer-Verlag; Berlin: 2010. pp. 285–303. [Google Scholar]
- 161.Haussecker D. The Business of RNAi Therapeutics. Hum Gene Ther. 2008;19:451–462. doi: 10.1089/hum.2008.007. [DOI] [PubMed] [Google Scholar]
- 162.Azad RF, Driver VB, Tanaka K, et al. Antiviral activity of a phosphorothioate oligonucleotide complementary to RNA of the human cytomegalovirus major immediate-early region. Antimicrob Agents Chemother. 1993;37:1945–1954. doi: 10.1128/aac.37.9.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Grillone LR, Lanz R. Fomivirsen. Drugs Today (Barc) 2001;37:245–255. doi: 10.1358/dot.2001.37.4.620590. [DOI] [PubMed] [Google Scholar]
- 164.Bennett CF. Pharmacological Properties of 2′-O-Methoxyethyl-modified Oligonucleotides. In: Crooke ST, editor. Antisense Drug Technology: Principles, Strategies, and Applications. 2. CRC Press; Boca Raton: 2008. pp. 273–303. [Google Scholar]
- 165.Levin AA, Yu RZ, Geary RS. Basic Principles of the Pharmacokinetics of Antisense Oligonucleotide Drugs. In: Crooke ST, editor. Antisense Drug Technology: Principles, Strategies, and Applications. 2. CRC Press; Boca Raton: 2008. pp. 183–215. [Google Scholar]
- 166.Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. The Lancet. 2010;375:998–1006. doi: 10.1016/S0140-6736(10)60284-X. [DOI] [PubMed] [Google Scholar]
- 167.Crooke RM, Graham MJ, Lemonidis KM, et al. An apolipoprotein B antisense oligonucleotide lowers LDL cholesterol in hyperlipidemic mice without causing hepatic steatosis. J Lipid Res. 2005;46:872–884. doi: 10.1194/jlr.M400492-JLR200. [DOI] [PubMed] [Google Scholar]
- 168.Kling J. Safety signal dampens reception for mipomersen antisense. Nat Biotech. 2010;28:295–297. doi: 10.1038/nbt0410-295. [DOI] [PubMed] [Google Scholar]
- 169.Moulton HM, Moulton JD. Morpholinos and their peptide conjugates: therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim Biophys Acta. 2010;1798:2296–2303. doi: 10.1016/j.bbamem.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 170.Goyenvalle A, Davies KE. Challenges to oligonucleotides-based therapeutics for Duchenne muscular dystrophy. Skeletal Muscle. 2011;1:8. doi: 10.1186/2044-5040-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wu B, Moulton HM, Iversen PL, et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc Natl Acad Sci U S A. 2008;105:14814–14819. doi: 10.1073/pnas.0805676105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yin H, Moulton HM, Betts C, et al. Functional rescue of dystrophin-deficient mdx mice by a chimeric peptide-PMO. Mol Ther. 2010;18:1822–1829. doi: 10.1038/mt.2010.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yin H, Moulton HM, Betts C, et al. A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum Mol Genet. 2009;18:4405–4414. doi: 10.1093/hmg/ddp395. [DOI] [PubMed] [Google Scholar]
- 174.Yin H, Moulton HM, Seow Y, et al. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum Mol Genet. 2008;17:3909–3918. doi: 10.1093/hmg/ddn293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Jearawiriyapaisarn N, Moulton HM, Sazani P, et al. Long-term improvement in mdx cardiomyopathy after therapy with peptide-conjugated morpholino oligomers. Cardiovasc Res. 2010;85:444–453. doi: 10.1093/cvr/cvp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Jearawiriyapaisarn N, Moulton HM, Buckley B, et al. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol Ther. 2008;16:1624–1629. doi: 10.1038/mt.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Amantana A, Moulton HM, Cate ML, et al. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug Chem. 2007;18:1325–1331. doi: 10.1021/bc070060v. [DOI] [PubMed] [Google Scholar]
- 178.Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Corey DR. RNAi learns from antisense. Nat Chem Biol. 2007;3:8–11. doi: 10.1038/nchembio0107-8. [DOI] [PubMed] [Google Scholar]
- 180.www.tekmirapharm.com
- 181.Silence is Golden. Nat Biotech. 2006;24:306–308. [Google Scholar]
- 182.Frederickson RM. RNAi gets vote of confidence from big pharma. Mol Ther. 2007;15:221–222. doi: 10.1038/sj.mt.6300099. [DOI] [PubMed] [Google Scholar]
- 183.Potera C. Antisense--down, but not out. Nat Biotech. 2007;25:497–499. doi: 10.1038/nbt0507-497. [DOI] [PubMed] [Google Scholar]
- 184.Schmidt C. RNAi momentum fizzles as pharma shifts priorities. Nat Biotech. 2011;29:93–94. doi: 10.1038/nbt0211-93. [DOI] [PubMed] [Google Scholar]
- 185.Zoubeidi A, Chi K, Gleave M. Targeting the cytoprotective chaperone, clusterin, for treatment of advanced cancer. Clin Cancer Res. 2010;16:1088–1093. doi: 10.1158/1078-0432.CCR-09-2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Agrawal S, Kandimalla ER. Synthetic agonists of Toll-like receptors 7, 8 and 9. Biochem Soc Trans. 2007;35:1461–1467. doi: 10.1042/BST0351461. [DOI] [PubMed] [Google Scholar]
- 187.Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer. 2008;8:167–179. doi: 10.1038/nrc2275. [DOI] [PubMed] [Google Scholar]
- 188.Joseph I, Tressler R, Bassett E, et al. The telomerase inhibitor imetelstat depletes cancer stem cells in breast and pancreatic cancer cell lines. Cancer Res. 2010;70:9494–9504. doi: 10.1158/0008-5472.CAN-10-0233. [DOI] [PubMed] [Google Scholar]