

Abstract
Nicotine dependence is the leading cause of death in the United States. However, research on high rates of nicotine use in mental illness has primarily explained this co-morbidity as reflecting nicotine's therapeutic benefits, especially for cognitive symptoms, equating smoking with ‘self-medication’. We used a leading neurodevelopmental model of mental illness in rats to prospectively test the alternative possibility that nicotine dependence pervades mental illness because nicotine is simply more addictive in mentally ill brains that involve developmental hippocampal dysfunction. Neonatal ventral hippocampal lesions (NVHL) have previously been demonstrated to produce post-adolescent-onset, pharmacological, neurobiological and cognitive-deficit features of schizophrenia. Here, we show that NVHLs increase adult nicotine self-administration, potentiating acquisition-intake, total nicotine consumed and drug seeking. Behavioral sensitization to nicotine in adolescence prior to self-administration is not accentuated by NVHLs in contrast to increased nicotine self-administration and behavioral sensitization documented in adult NVHL rats, suggesting periadolescent neurodevelopmental onset of nicotine addiction vulnerability in the NVHL model. Delivering a nicotine regimen approximating the exposure used in the sensitization and self-administration experiments (i.e. as a treatment) to adult rats did not specifically reverse NVHL-induced cortical-hippocampal-dependent cognitive deficits and actually worsened cognitive efficiency after nicotine treatment stopped, generating deficits that resemble those due to NVHLs. These findings represent the first prospective evidence demonstrating a causal link between disease processes in schizophrenia and nicotine addiction. Developmental cortical-temporal limbic dysfunction in mental illness may thus amplify nicotine's reinforcing effects and addiction risk and severity, even while producing cognitive deficits that are not specifically or substantially reversible with nicotine.
Keywords: Addiction, dual diagnosis, hippocampus, nicotine dependence, schizophrenia, self-administration
Introduction
With pathogenic effects spanning brain and cardiopulmonary systems, nicotine dependence remains the single largest cause of death in the United States (Mokdad et al. 2004). As general population rates have fallen below 25%, smoking has become more concentrated in the mentally ill who now consume around half of all cigarettes sold (Lasser et al. 2000; Grant et al. 2004). Smoking rates exceeding 75% in schizophrenia populations are associated with decades cut from individual life spans, lower psychiatric treatment compliance and financial impoverishment as government assistance for the mentally ill is channeled into tobacco industry profits (Steinberg, Williams & Ziedonis 2004; Parks et al. 2006; Prochaska, Hall & Bero 2008).
Psychiatric research on nicotine use in mental illness has traditionally been guided by the hypothesis that this comorbidity reflects therapeutic effects of nicotine and/or tobacco, so that smoking in mental illness is widely accepted as synonymous with ‘self-medication’ (Dani & Harris 2005; DeHay et al. 2012). Human data encompassing genetic and histological analyses of nicotinic receptors and electrophysiological and cognitive responses to nicotine have been suggested to reflect schizophrenia-specific abnormalities that allow nicotine to function as a cognitive enhancer in this illness (Leonard et al. 2001; Dani & Harris 2005). Although this research has imparted neuroscientific credence to a medicinal value for nicotine use in schizophrenia, recently emerging data indicate that acute nicotine dosing has no cognitive therapeutic benefits for schizophrenic compared to non-schizophrenia smokers (Hahn 2013). Also, growing evidence suggests that chronic nicotine exposure may actually cause rather than treat cognitive and affective symptoms (Reitz et al. 2007; Slotkin 2008; Counotte et al. 2011; McDermott et al. 2013).
Generally, the self-medication hypothesis has not translated well into motivating clinicians or patients to treat nicotine addiction (DeHay et al. 2012), nor does it effectively explain why schizophrenia patients have increased addictions to other drugs like cocaine and alcohol, which are known for exacerbating rather than improving psychotic and/or cognitive symptoms (Volkow 2009). Given this larger picture, an alternative hypothesis becomes apparent: the connection between schizophrenia and nicotine dependence may reflect a more general, and involuntary biological process where one brain disease (i.e. schizophrenia) predisposes to and synergizes with another (i.e. addiction) (Chambers, Krystal & Self 2001).
Directly testing this alternative hypothesis necessitates pre-clinical approaches, not ethically possible in human subjects, where addictive drugs can be prospectively tested in well-controlled experiments using heuristic, drug-naive animal models of mental illness. For this purpose, we have applied the neonatal ventral hippocampal lesion (NVHL) model of schizophrenia. In this model, the axon-sparing neurotoxin ibotenic acid is delivered into the hippocampus of 7-day-old rats corresponding to the second trimester human fetal brain development when environmental and genetic risk factors are implicated in seeding schizophrenia (Lipska, Jaskiw & Weinberger 1993; Weinberger 1999). Similar to observations in humans with schizophrenia, NVHL rats have hippocampal atrophy that is proportional to overall syndrome severity (Chambers et al. 1996; Brambilla et al. 2013), and many secondary neurobiological and behavioral abnormalities involving prefrontal-cortical-striatal anatomy and function (Tseng, Chambers & Lipska 2009). Developmentally, NVHLs produce post-adolescent onset of ‘positive’ symptom-like behavioral abnormalities that are reducible with antipsychotic medications, superimposed on more insidiously presenting, earlier onset, cognitive and ‘negative’ symptoms that do not respond to anti-psychotics (Tseng et al. 2009).
Illustrative of a fundamental neurobiological connection between severe mental illness and addiction vulnerability, NVHLs also cause an involuntary amplification of short- and long-term behavioral sensitization to cocaine (Chambers & Taylor 2004), alcohol (Conroy, Rodd & Chambers 2007) and nicotine (Berg & Chambers 2008), corresponding to increased self-administration of cocaine (Chambers & Self 2002) and alcohol (Berg, Czachowski & Chambers 2011). The present study was designed to test whether this addiction vulnerability generalizes to nicotine, and to capture first proof of a causative relationship between early disruptions of hippocampal network development and adult-age nicotine addiction vulnerability, co-occurring with cognitive impairments that may or may not respond therapeutically to nicotine.
Methods and Materials
Subjects and neonatal surgeries
Subjects were born in our facility from Sprague-Dawley females arriving at 14–16 days gestation (Harlan, Indianapolis, IN, USA). Post-natal day (PD)-5 litters were culled to males in preparation for surgeries on PD-7. Pups weighing 16–19 g underwent surgeries performed under hypothermic anesthesia. Briefly, as described elsewhere (Lipska et al. 1993), stereotaxic-assisted Hamilton needle placement into the ventral hippocampus bilaterally (anterior-posterior –3.0 mm, medial-lateral ± 3.5 mm, anddorsal-ventral −5.0 mm from bregma) was followed by Ibotenic acid (3.0 μg; Sigma, St. Louis, MO, USA) delivery in 0.3 μl artificial cerebrospinal fluid (aCSF) to NVHL rats, or aCSF only to Sham-operated rats. Pups were returned to their awake mothers after 30 minutes of recovery on a heating pad, and thereafter reared under standard conditions until weaning on PD-21. At weaning, NVHL and Sham rats were housed in pairs (like lesion status) until adulthood (PD-56), when they were individually housed. Surgical and experimental procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Indiana University Institutional Animal Care and Use Committee.
Nicotine preparation
Nicotine hydrogen tartrate salt (Sigma) was dissolved in 0.9% sterile saline to a stock solution dose of 0.5 mg/ml [expressed in terms of the base of the salt (Matta et al. 2007)], adjusted to a pH 7.4. This solution was injected subcutaneously (sc) for adolescent sensitization and in the cognitive testing as pre-injections in volumes of 1 ml per kg of rat weight. For i.v. self-administration, doses were prepared daily on a per rat weight basis from stock solutions.
Adolescent behavioral sensitization to nicotine
During mid-adolescence (PD-34 to 44), rats destined for adult nicotine self-administration were given 10 once daily injections (sc) of nicotine (0.5 mg/kg) or saline (1 ml/kg) during locomotor testing in 43 × 43 cm plexiglass arenas (Med Associates, St. Albans, VT, USA), equipped with 16 infrared beam arrays. Position, track, distance, speed and non-ambulatory movements were recorded over the 2-hour sessions. Rats were tested under red light and had injections delivered at the beginning of the second hour.
Adult intravenous cannulation and nicotine self-administration
On PD-56, subjects entering self-administration underwent jugular venous catheterization under sodium pentobarbital anesthesia. As detailed elsewhere (Chambers & Self 2002), Silastic tubing (Green Rubber, Woburn, MA, USA) placed into the animal's right vein coursed subcutaneously over the shoulder to exit the back via a 22-gauge cannula (Plastics One, Roanoke, VA, USA). Catheters were flushed daily with 0.3 ml heparinized saline (20 IU/ml) containing gentamicin sulfate (0.13 mg/ml). Catheter patency checks were conducted once a week on weekends using a 1 mg/0.1 ml i.v. push of methohexital sodium (Henry Schein, Indianapolis, IN, USA), which produces a 10–20-second loss of consciousness with patent catheters. Rats with failed or infected catheters were excluded from the experiment.
In preparation for nicotine self-administration on PD-59, to promote exploratory behavior, food restriction was started, maintaining body weight at 85% of pre-restriction weight (with 2–3 bricks/day of standard rat chow). This restriction continued for all rats until the fifth acquisition session. Self-administration sessions began on PD-60 in Med Associates chambers controlled by software that recorded instrumental activity. These units, housed inside sound-attenuating cubicles, were equipped with two non-retractable levers with cue lights, a house light and an infusion pump assembly (Razel Model A pump; Med Associates).
All self-administration sessions were 2 hours long beginning with house light on to signal nicotine availability and a single priming infusion of nicotine. Responses on the active lever (counterbalanced left/right between animals) resulted in house lights off and drug-paired lever cue light on for a 3-second infusion (0.015 mg/kg nicotine in 0.050 ml saline; FR1 schedule). A 17-second time-out followed with all lights out during which recorded lever presses produced no consequences. Rats progressed through three stages of sessions: acquisition, dose-response testing and extinction, with no days off between stages. For acquisition, rats were given a maximum of 35 once daily sessions, during which they were regarded as having acquired self-administration when they had accumulated (not necessarily consecutively) 20 days of > 20 active lever presses (nicotine hits) per day (i.e. resulting in a minimum exposure of 6 mg/kg nicotine during acquisition). These sessions occurred on a Monday–Friday (5 day) schedule with weekends (2 days) off so that rats remained in acquisition from 4 to 7 weeks depending on performance. Through acquisition, any rat that did not press once on the active lever in the prior session had a single sucrose pellet placed on the active lever for the subsequent session. At the end of acquisition upon reaching the 20 days/20 hits criteria or the 35th session, whichever came first, rats progressed on the very next day to the dose-response stage during which they had access to 7.5, 15, 30 and 15 μg nicotine/kg/infusion (one dose per session; not counter-balanced) over four daily sessions. The day after their last dose-response day, rats began once daily extinction sessions (2 hours; no nicotine pre-injections; house lights only on; lever presses producing no consequences), proceeding through these sessions until responding with ≤ 5 presses on the previously active lever and ≤ 10 presses on the inactive lever.
Adult radial arm maze (RAM) testing
Rats separate from those undergoing self-administration were prepared for cognitive testing on the RAM beginning on PD-60, with food restriction (to maintain weight at 85% of pre-restriction weight) starting on PD-59. Rats were fed regular chow after daily sessions. The RAM (Med Associates) was constructed in plexiglass and equipped with a central octagonal arena (29.5 cm diameter) with eight runways extending radially (61 × 9 cm with 17 cm high walls), standing 6.5 cm above the floor and surrounded by consistent visual landmarks.
In spatial learning and working memory testing based on the win-shift paradigm (Olton & Samuelson 2004), rats learned in daily sessions to efficiently enter all eight arms of the maze as reinforced by one-half of a Kellogs® Froot Loop® loaded at the end of each arm. Sessions lasted a maximum 300 seconds, or when animals had entered all eight arms, whichever came first. Three primary dependent measures were (1) entries-to-repeat (ETR: the total number of arms entered before the rat repeated an arm entry; (2) total session time and (3) Froot Loops consumed. Rats were tested over 24 (once daily) sessions organized into eight blocks of three sessions, spanning five sequential phases. In phase 1 (block 1), all animals received a pre-injection of saline (sc) 30 minutes prior to the session. In phase 2 (blocks 2–4), NVHL and Sham rats were randomized to receive saline or nicotine injections (0.5 mg/kg sc) 30 minutes prior. In phase 3 (blocks 5 and 6), all animals were only given saline pre-injections. Subsequently, animals were given a 2-week break in their home cages, then resumed testing for phase 4 (block 7) in which they were all given saline pre-injections followed by phase 5 (block 8) when they were all given nicotine (0.5 mg/kg) pre-injections.
Histology
After behavioral testing, rats were sacrificed by decapitation under brief isoflurane anesthesia. Brains were removed whole and cryostat cut into coronal sections (40 μm) through the rostral-caudal extent of the hippocampus. Mounted sections were fixed and thionin (0.5%) stained. Microscopic examination of sections for lesion verification was performed separate from, and blind to behavioral data; rats without appropriate lesions were excluded from the study. Appropriate lesions were identified as those showing bilateral evidence of tissue atrophy, paucity of nuclei and cellular disarray (with lateral ventricular enlargement) confined to the ventral hippocampus (Fig. 1). Brains with unilateral damage, damage encompassing the dorsal blades of the hippocampus, or direct damage to nearby structures (temporal cortex, amygdala, thalamus, basal ganglia) were excluded. From the self-administration experiment, 23 of 33 rats (70%) that underwent ibotenic acid delivery and had successful catheters, had appropriate hippocampal malformations and were included in the study. From the RAM experiment, 17 of 25 (68%) of ibotenic-exposed rats had acceptable NVHLs.
Figure 1.
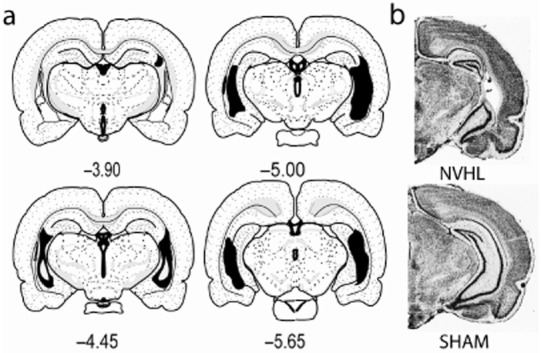
Mapping of hippocampal damage in neonatal ventral hippocampal lesions (NVHL) rats. (a) Coronal maps [from bregma (mm)] show the rostral-caudal extent of largest (black) to smallest (white inset) hippocampal damage among the 82 rats in the study. (b) Photomicrographs show typical NVHL histology versus a SHAM-operated control brain. [Maps are adapted from Swanson LW (2004) Brain Maps: Structure of the Rat Brain, 3rd edn. New York: Elsevier.]
Data analysis
Parametric testing examined dependent variables of locomotor activity (cm/hour) in behavioral sensitization. Active and inactive lever pressing, nicotine intake and time-out pressing (on active and inactive levers) were examined in self-administration. ETR, session time and Froot Loops consumed, taken as the mean of three consecutive sessions for each block were dependent measures in RAM testing. Two-way analysis of variance (ANOVA) with independent variables lesion status and nicotine history was used with repeated measures testing across multiple sessions as appropriate. In sensitization/self-administration, the nicotine variable referred to adolescent nicotine versus saline exposure. In RAM testing, the nicotine variable reflected nicotine versus saline pre-injections during maze learning. Separate ANOVAs were applied to different phases of self-administration and RAM experiments. Significance was identified at P < 0.05 with mention of notable negative or marginal effects. Wherever significant interactions occurred between main effects and repeated time measures, secondary one-way ANOVAs were applied to specify when in the repeated measure the main effect was strongest.
Results
Adolescent nicotine sensitization and adult self-administration
In the first experiment, 45 rats first underwent daily experimenter-delivered injections of nicotine (0.5 mg/kg sc) (NVHL, n = 12; Sham, n = 10) or saline (1 ml/kg sc) (NVHL, n = 11; Sham, n = 12) for 10 days during mid-adolescence (PD 35–44) followed by adult self-administration (PD-60). This early nicotine exposure tested whether abnormal nicotine responsiveness occurs in NVHLs prior to the periadolescent onset of the full syndrome, and whether it interacts with NVHLs to alter adult self-administration. Although adolescent nicotine injections robustly sensitized locomotor activity (day × nicotine: F9,369 = 39.8, P < 0.001; nicotine: F1,41 = 261.5, P < 0.001), adolescent NVHL rats did not sensitize differently from Shams (Fig. 2).
Figure 2.
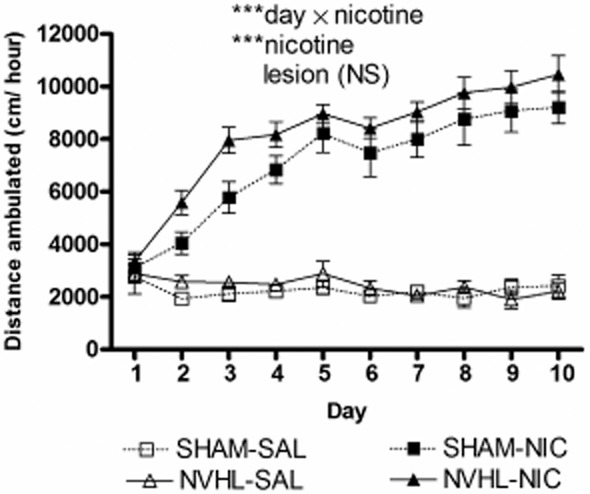
Adolescent nicotine behavioral sensitization is demonstrated as growth in post-injection locomotion due to nicotine injections (***P < 0.001) over 10 days. Neonatal ventral hippocampal lesions (NVHL) rats [nicotine (NIC) (n = 12); saline (SAL) (n = 11)] did not differ from SHAMS [nicotine (n = 10); saline (n = 12)]. Data depicted as means ± SEM
Upon reaching adulthood (PD-56), these 45 rats underwent jugular venous catheterization followed by i.v. nicotine self-administration 4 days later. Only the first 20 days of acquisition were analyzed (Fig. 3a–d) since over acquisition days 21–35, treatment group's numbers began to drop differentially as rats met acquisition criteria (20 days > 20 nicotine infusions/day) and progressed to dose-response testing. Over the first 20 days, nicotine was generally self-administered in increasing amounts via active lever presses (i.e. presses that delivered infusions) (days: F19,779 = 14.6, P < 0.001) (Fig. 3a), with no growth in inactive lever pressing (Fig. 3b). NVHL rats showed stronger acquisition in terms of active lever pressing (lesion: F1,41 = 16.0, P < 0.001) and shape of the acquisition curve (lesion × days: F19,779 = 2.05, P < 0.01). Post hoc ANOVAs (one way by lesion status on each day) detected significant increases in NVHL responding initially emerging on days 4 and 5, then becoming larger and more frequent over the next 3 weeks of acquisition. NVHLs did not differ from Shams on inactive lever pressing but they did show increased active lever time-out responding (lesion: F1,41 = 9.1, P < 0.01) (Fig. 3c). Time-out responding at the inactive level was flat and not different between groups (Fig. 3d).
Figure 3.
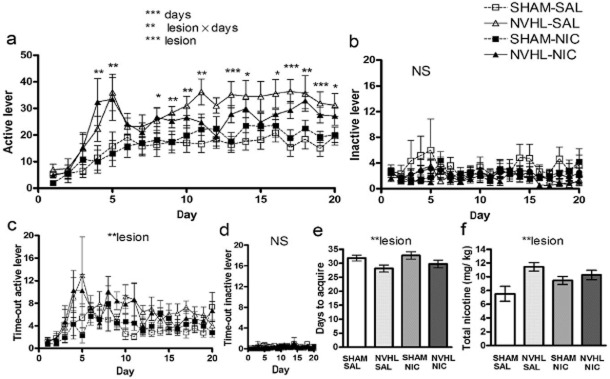
Adult acquisition of nicotine self-administration, same subjects (n = 45) as in Fig. 2. (a) Over the first 20 days, nicotine was self-administered in increasing amounts via active lever presses (***days: P < 0.001), (b) unaccompanied by growth in inactive lever pressing. Neonatal ventral hippocampal lesions (NVHL) rats showed stronger acquisition (in terms of overall active lever pressing (***lesion: P < 0.001) and shape of the acquisition curve (**lesion × days: P < 0.01), while not differing from SHAMS on inactive lever pressing. Significance levels of post hoc one-way ANOVAs on each day by lesion status are denoted directly above error bars (*P < 0.05; **P < 0.01; ***P < 0.001). (c) NVHLs increased active lever time-out responding (**P < 0.01) but not (d) time out-inactive lever pressing. Over acquisition, (e) NVHL rats achieved nicotine acquisition (20 days of ≥ 20 infusions/day) earlier than SHAMS (**P < 0.01), accumulating (f) greater total nicotine intake (**P < 0.01). Adolescent nicotine exposure (NIC groups) did not produce differential effects on these measures compared to adolescent saline exposure. Data depicted as means ± SEM
Based on the acquisition criteria in which all rats were given up to 35 days to achieve 20 days of ≥ 20 nicotine infusions/day, NVHL rats achieved acquisition criteria in fewer days than Shams (lesion: F1,41 = 7.3, P < 0.01) (Fig. 3e) and therefore entered the dose-response testing earlier. This provided the Shams with the opportunity of more acquisition sessions to catch up to the NVHLs in terms of cumulative nicotine intake. Even with this experimental design, NVHLs still had a greater total nicotine intake (lesion: F1,41 = 9.0, P < 0.01) (Fig. 3f) as calculated over all acquisition sessions.
Adolescent nicotine exposure had no effects or interactions on any measure during the acquisition stage of nicotine self-administration. However, during dose-response testing analyzed over 5 days, in which the last acquisition session was considered the first dose-response session, a dose-dependent effect of adolescent nicotine history on nicotine intake did emerge without lesion effects (day × nicotine history: F4,164 = 3.4, P < 0.05) (Fig. 4a). One-way (by nicotine history) post hoc ANOVAs performed across dosing days detected a nicotine-history-associated increase in nicotine intake at the 30 μg dose (F1,44 = 4.1, P < 0.05). As expected for dose-response testing, both daily nicotine intake (Fig. 4a) and active lever presses (Fig. 4b) varied significantly across days [(day: F4,164 = 40.3, P < 0.001) and (day: F4,164 = 10.1, P < 0.001), respectively]. However, inactive lever pressing did not differ across days or by lesion status or nicotine history. In parallel to active lever pressing, time-out active lever pressing did vary significantly across days (day: F4,164 = 9.2, P < 0.001) (Fig. 4d), also without lesion or nicotine history effects. Time-out inactive lever pressing (Fig. 4e) also varied across days (day: F4,164 = 3.1, P < 0.05) with a day × lesion interaction (F4,164 = 2.5, P < 0.05), in which Shams pressed more on the middle day (15 μg dose) (F1,44 = 5.7, P < 0.05). Despite this statistical significance, these effects in time-out inactive lever pressing were likely not meaningful due to the extremely low responding (averaging < 3 hits per 2 hours) observed over the course of dose-response testing.
Figure 4.
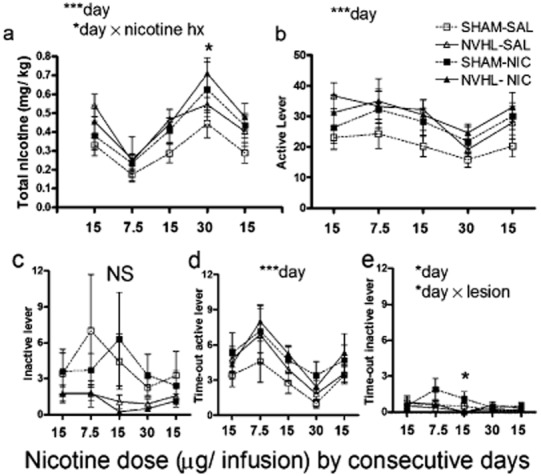
Dose-response testing after acquisition of nicotine self-administration, same subjects (n = 45) as in Figs 2 and 3. (a) Total nicotine intake varied significantly between days as the doses of nicotine infusions changed (***day: P < 0.001), interacting with nicotine history in adolescence (*day × nicotine hx: P < 0.05). This effect was carried by nicotine history-related increases in intake at the highest nicotine dose (one-way ANOVA by nicotine history: *P < 0.05). (b) Active lever presses differed across days (***P < 0.001). (c) Inactive lever pressing did not vary significantly whereas (d) Time-out active lever responding did vary by day (***P < 0.001) as did (e) time-out inactive lever pressing (P < 0.05) where sham rats showed greater responding in a dose dependent manner (*day × lesion: P < 0.05) with shams showing greater responding specifically at the middle 15 μg dose (post hoc ANOVA by nicotine history: *P < 0.05). Data are depicted as means ± SEM throughout all figures
Having established that total nicotine intake and active lever presses did not differ over the five dose-response sessions according to lesion status, we also determined more specifically that NVHL and Sham rats also did not differ on active or inactive lever pressing on their very last day of nicotine intake (dose-response day 5; 15 μg dose). This confirmed that despite robust lesion-based differences in active lever responding and nicotine intake over the acquisition stage, the added acquisition sessions allotted to Sham rats did allow NVHL and Sham rats to arrive at comparable levels of nicotine reinforcement and exposure by the time of (and measured over) the dose-response days just before extinction testing. Thus, lesion-based differences in subsequent extinction responding can be interpreted as signifying persistent changes in nicotine-seeking behavior due to NVHLs, independent from possible effects of very recent drug-taking behavior, although lesion-based differences in nicotine intake earlier in acquisition might be still be predictive of, or contribute to, later extinction differences. Indeed, in the first extinction session when rats pursued nicotine in daily 2-hour sessions, but without nicotine reinforcement, NVHL rats again demonstrated greater active lever responses (lesion: F1,41 = 7.5, P < 0.01) (Fig. 5a). When dividing the first extinction session into 4 × 30-minute segments, this main effect was accompanied by a significant within-session tapering of active lever pressing from an initial extinction burst (segment: F3,123 = 94, P < 0.001). Compared to their overall average of 8.1 presses per 30 minutes recorded in their final dose-response session when they were receiving nicotine, active lever pressing was higher for all rat groups in the first 30 minutes of the first extinction session, dropping below the 8.1 average across the 30- to 120- minute segments. A lesion × segment interaction (F3,123 = 8.2, P < 0.001) indicated that NVHLs amplified the magnitude of the extinction burst as confirmed by a post hoc one-way (lesion status) ANOVA testing at each time segment, which showed a significant increase for NVHLs in the first 30-minute segment (F1,44 = 8.2, P < 0.001). In analysis of extinction responding over a longer time span encompassing the first 3 days of extinction testing, NVHL rats also showed persistent elevations in drug seeking on the previously nicotine-paired lever (lesion: F1,36 = 8.6, P < 0.01), superimposed on an extinguishing pattern of responding across days (day: F2,72 = 30.2, P < 0.001) (Fig. 5b). On the inactive lever, where responding was still much less overall than at the previously nicotine-paired lever, an overall extinction pattern was also observed (day: F2,72 = 8.7, P < 0.001) with Sham rats tending to have higher responding than NVHLs on day 1 that habituated significantly more compared to NVHLs over the 3 days (day × lesion: F2,72 = 5.5, P < 0.01) (Fig. 5c).
Figure 5.
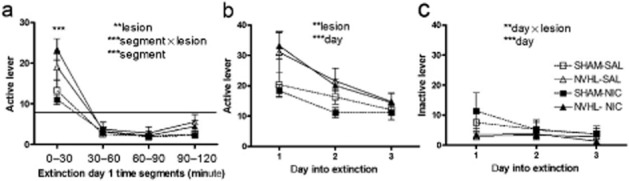
Drug seeking measured by extinction phase lever pressing. For extinction day 1, same subjects (n = 45) are included as in Figs 4. Subsequent extinction day analyses exclude rats that had met extinction criteria on previous days [day 2: n = 2 sham; n = 1 neonatal ventral hippocampal lesions (NVHL); day 3: n = 3 sham; n = 2 NVHL] (a) During extinction day 1, divided into 4 × 30-minute segments, an extinction burst marked by increased rates of active lever pressing in the first 30 minutes [compared to the 8.1 active lever presses per 30 minutes recorded in the prior nicotine reinforced session (horizontal line)] extinguished significantly over the next 90 minutes (***segment: P < 0.001). Overall lesion effects (**lesion: P < 0.01 and ***segment × lesion: P < 0.001) to increase drug seeking were carried most prominently by increased pressing during the initial extinction burst (one-way by lesion status: P < 0.001). (b) Over the first three extinction days on the previously nicotine-paired lever, NVHLs again showed elevated drug seeking (**P < 0.01) superimposed on an extinguishing pattern (***day; P < 0.001), while on the inactive lever (c), SHAM rats tended to have higher responding that habituated more significantly (**day × lesion: P < 0.01; ***day: P < 0.001). Unlike the dose-response data, adolescent nicotine history effects had no effects on nicotine seeking during extinction
Adult learning and working memory deficits and response to nicotine
Different sets of NVHL and Sham rats (n = 37) entered a second experiment beginning in adulthood (PD- 60) that tested the effects of nicotine on learning and working memory performance on the eight-arm RAM. This testing measures prefrontal-cortical-hippocampal network dysfunction analogous to that underlying the contextual-spatial working memory deficits in human schizophrenia (Fuller et al. 2009; Gold et al. 2010) and is sensitive to nicotinic receptor manipulation and the NVHL model (Levin 1988; Chambers et al. 1996). All rats received saline pre-injections 30 minutes before testing across blocks 1, 5, 6 and 7 but were randomized to receive nine (once daily) nicotine pre-injections (0.5 mg/kg sc) (NVHL, n = 9; Sham, N = 10) or saline (NVHL, n = 8; Sham, n = 10) before testing across blocks 2–4. By design, this dosing regimen closely approximated that used in the first experiment. In nicotine sensitization, the 10 × 0.5 mg/kg doses (5 mg/kg total exposure) were behaviorally activating (compared to saline) from 10 to 60 minutes post-injection. In nicotine self-administration, the acquisition criteria of a minimum of 20 infusions/day × 20 days would produce a total exposure of approximately 6 mg/kg within 20 to 35 sessions.
Over the first block of RAM testing, NVHL rats showed no impairments in entries-to-repeat (ETR) (Fig. 6a) but were significantly slower in session time (lesion: F1,33 = 2.1, P < 0.05) (Fig. 6b) while showing no differences in Froot Loops eaten (Fig. 6c). Across blocks 2–4, all rats demonstrated learning with increased ETRs (blocks: F2,66 = 13.6, P < 0.001), decreased session times (blocks: F2,66 = 63.5, P < 0.001) and more Froot Loops eaten (blocks: F2,66 = 38.5, P < 0.001). Now, NVHL rats did show impaired cognition with lower ETRs (lesion: F1,33 = 26.7, P < 0.001) that could not be due to differences in food reward motivation since there were no lesion-based differences in Froot Loops consumed. Efficiency in completing the maze was also again impaired by NVHLs (lesion: F1,33 = 4.6, P < 0.05), but enhanced by nicotine pre-injections (nicotine: F1,33 = 5.5, P < 0.05). Nicotine pre-injections did not improve ETR however, and did not interact with NVHLs to specifically reverse NVHL deficits in ETR or session time.
Figure 6.
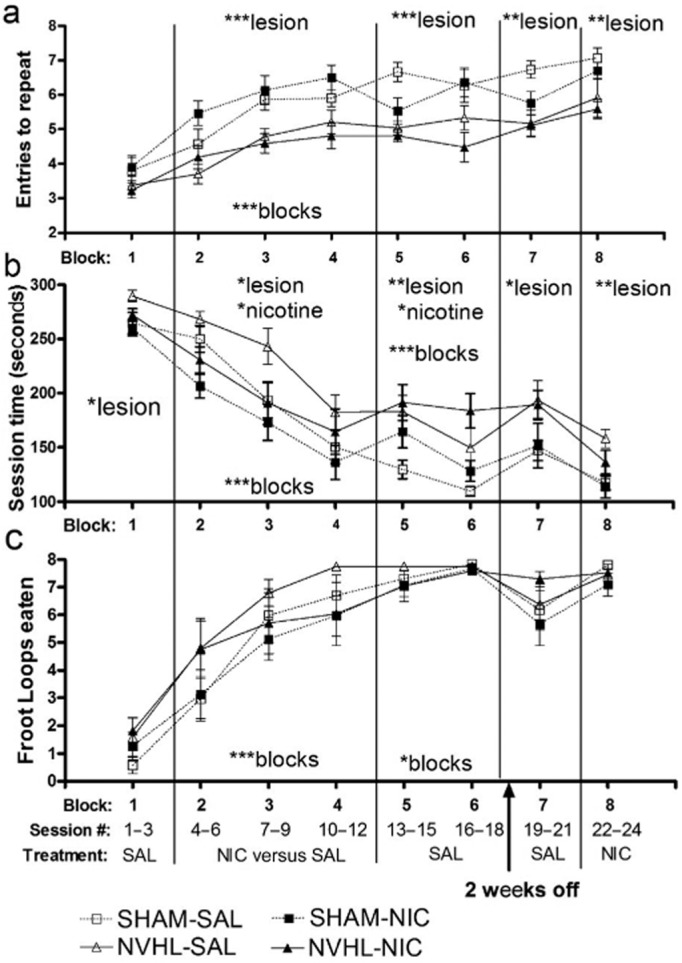
Radial arm maze testing of spatial learning and working memory. Rats received nine once daily nicotine (neonatal ventral hippocampal lesions; (NVHL, n = 9; SHAM, n = 10) versus saline pre-injections (NVHL, n = 8; SHAM, n = 10) across blocks 2–4 with all receiving saline elsewhere except for the last block where they all received nicotine. In block 1, NVHL rats showed no impairments in (a) entries-to-repeat (ETR) but were (b) significantly slower in session time (*P < 0.05). Across blocks 2–4, rats demonstrated learning with increased ETRs (***P < 0.001), decreased session times (***P < 0.001), and more (c) Froot Loops eaten (***P < 0.001), but NVHL rats showed impaired cognition with lower ETRs (***P < 0.001). Nicotine pre-injections produced no effects on ETR where the cognitive deficits due to NVHLs were most robust, but did improve cognitive efficiency in terms of reduced session time (*P < 0.05). This improvement did not specifically reverse NVHL deficits on session time (*lesion: P < 0.05). Over blocks 5–6, the recent nicotine exposure produced new cognitive deficits in terms of session time (*P < 0.05) with NVHL deficits in ETR (*** P < 0.001) and session time persisting (**P < 0.01). In long-term recall ending with nicotine pre-injections (block 7–8), NVHL deficits in ETR (block #7; **P < 0.01; block #8:** P < 0.01) and session time (block #7:* P < 0.05; block #8: **P < 0.01) also persisted, with no group differences in Fruit Loops consumed and no effects or interactions with prior nicotine pre-injections (blocks 2–4)
Over blocks 5 and 6, when all rats were again receiving saline pre-injections, the recent nicotine exposure produced new cognitive deficits in terms of marginally worsening ETR (nicotine: F1,33 = 3.6, P = 0.06) and significantly worsening session time (nicotine: F1,33 = 4.4, P < 0.05). NVHL deficits in ETR (lesion: F1,33 = 21.8, P < 0.001) and session time (lesion: F1,33 = 14.8, P < 0.01) persisted across these blocks and were not interactive with the effects of prior nicotine exposure. Figure 7 depicts the session data covering the transition from nicotine pre-injections back to saline pre-injections (bins 4 through 6). These groupings plotted according to nicotine exposure (Fig. 7a) and lesion status (Fig. 7b) allow a more clear view of the effects of nicotine withdrawal (and NVHLs) on cognition.
Figure 7.
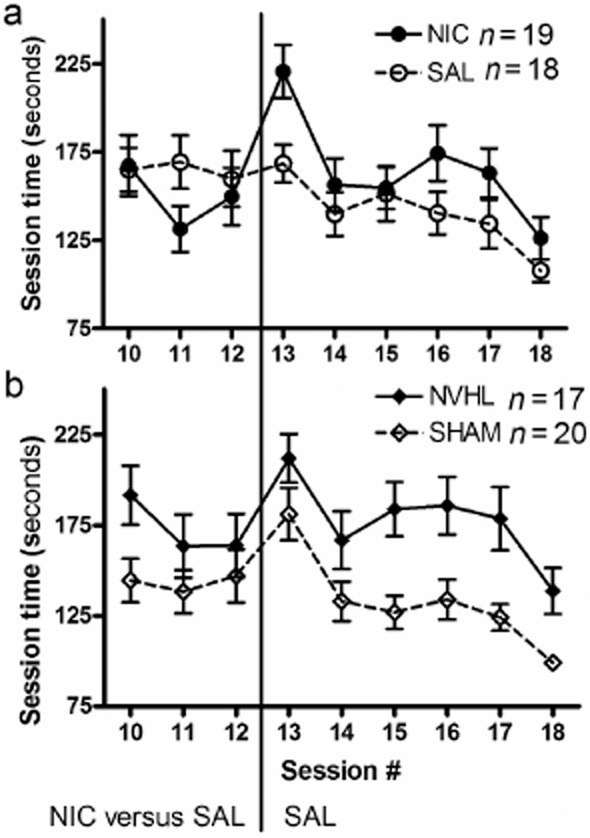
Radial arm maze session times covering the transition from nicotine to saline pre-injections, same subjects (n = 37) as in Fig. 6 with blocks 4, 5 and 6 decomposed into individual sessions for visual clarity. Comparison of rats according to (a) nicotine versus saline exposure (during sessions 4–12) depicts the detrimental cognitive effects of nicotine withdrawal. The comparison by (b) lesion status suggests how nicotine withdrawal impacts NVHL and Sham rats similarly
After block 6, all animals had 2 weeks off from RAM testing so that subsequent blocks would serve as measures of long-term recall. Nicotine pre-injections were given in the final (eighth) block to all rats to test for potential ‘nicotine rescue’ effects of any cognitive deficits. Across blocks 7 and 8, NVHL deficits in ETR (lesion: block #7: F1,33 = 12.2, P < 0.01; block #8: F1,33 = 10.6, P < 0.01) (Fig. 6a) and session time (lesion: block #7: F1,33 = 7.0, P < 0.05; block #8: F1,33 = 11.7, P < 0.01) (Fig. 6b) persisted, with no group differences in Froot Loops consumed (Fig. 6c) and no effects of prior nicotine (or nicotine history interactions with NVHLs) on any measures.
Discussion
This study demonstrates that early developmental hippocampal damage increases the reinforcing effects of nicotine in adulthood while also producing cognitive impairments that are not specifically treated by nicotine. This modeling accurately simulates clinical phenomenology of greater severities of nicotine dependence in mentally ill people, including observations of schizophrenia patients consuming more nicotine than non-schizophrenic, nicotine-dependent subjects (Williams et al. 2005). While contributing to mounting evidence pointing to the importance of hippocampal function in the pathogenesis of addictive disorders (Chambers et al. 2001; Sudai et al. 2011; Chambers 2013), these findings replicate, and begin to biologically explain, enhanced nicotine addiction vulnerability in the absence of specific cognitive therapeutic effects of nicotine in schizophrenia subjects (Hahn 2013).
NVHLs impact the maturation and function of prefrontal cortical-ventral striatal circuits to which the ventral hippocampus directly projects, in multiple ways that correspond to neural and behavioral findings in human schizophrenia and subjects with addictions (Liu et al. 1998; Chambers et al. 2001; Heerey, Bell-Warren & Gold 2008; Tseng et al. 2009). Behaviorally, NVHL rats show baseline cognitive impulsivity in their approach to natural rewards that is worsened by prior cocaine history (Chambers et al. 2005), mirroring impulsivity found in populations with nicotine dependence and other addictions (Bickel, Odum & Madden 1999). The present study identified impulsive and perseverative styles of nicotine seeking in the NVHL model like those previously shown in cocaine self-administration (Chambers & Self 2002). Specifically, NVHL rats showed increased active lever time-out responding during acquisition of nicotine self-administration and increased nicotine seeking in extinction, that mirror the same abnormalities they show in cocaine self-administration. Together, these findings confirm that NVHLs produce a failure in inhibitory control over motivated behavior associated with multiple addictive drugs abused at particularly high rates in schizophrenia.
Both NVHLs and human smokers show prefrontal cortical regional atrophy (Chambers et al. 2010a; Durazzo et al. 2013). In NVHL rats, this prefrontal atrophy corresponds to pyramidal cell neuronal atrophy and derangements in excitatory and inhibitory neurotransmission, occurring on top of neostriatal supersensitivity to dopamine signaling (Tseng et al. 2007; Chambers et al. 2010a; Chambers, Sentir & Engleman 2010b). Together, these abnormalities may contribute to enhanced recruitment of striatal activation patterns associated with addictive drug-induced behavioral adaptation in NVHL rats (Tseng et al. 2007; Chambers et al. 2010a) resulting in augmented behavioral sensitization and self-administration with multiple addictive drugs (Chambers & Self 2002; Conroy et al. 2007; Chambers et al. 2010a; Berg et al. 2011).
Given that the NVHL model increases behavioral sensitization to nicotine in adulthood (Berg & Chambers 2008) but not in adolescence as shown here, we can surmise that heightened nicotine responsiveness due to early hippocampal perturbation is involuntary, and emerges developmentally in phase with the post-adolescent onset of the full schizophrenia syndrome of the model. These findings comport with emerging clinical and neuroimaging data suggestive of a developmental coincidence, and neurobiological connection between nicotine addiction vulnerability and schizophrenic pathology in the brain, in which altered maturation of prefrontal-cortical striatal circuits plays a major role (Chambers, Taylor & Potenza 2003; Compton et al. 2009; Zhang, Stein & Hong 2010). Abnormalities in cortical-hippocampal network architecture and function, due to a large variety of genetic and early environmental backgrounds are implicated across mental illnesses other than schizophrenia—including post-traumatic stress disorder, borderline personality and primary mood disorders (Bremner et al. 2000; Teicher, Anderson & Polcari 2012)—which also encompass elevated rates of nicotine addiction (Lasser et al. 2000; Grant et al. 2004; Pulay et al. 2010). Accordingly, the present findings may also illustrate a more general role of hippocampal malfunction in generating addiction vulnerability leading to many ‘dual diagnosis’ combinations that involve nicotine.
Adolescent nicotine exposure in rats similar to what we employed can produce adult prefrontal cortical physiological abnormalities with cognitive deficits that resemble those characterized in schizophrenia (Counotte et al. 2009, 2011). Adolescent nicotine injections also amplify nicotine sensitization tested in adulthood (Bracken et al. 2011). Our study design did not test these effects, and we did not see adolescent nicotine sensitization impact adult self-administration in the same way that the NVHL model did. The relative lack of effect of the adolescent nicotine exposure on subsequent adult nicotine self-administration may have been due to several concurrent factors: The adolescent exposure was not self-administered, it was not delivered i.v., and it was delivered in a different context from the adult self-administration. Further, as hinted by the dose-response testing, where only the 30 μg dose revealed a significant adolescent nicotine-history effect, the 15 μg dose we used in acquisition may have been too low to reflect prior nicotine exposure effects. Finally, it is possible that NVHLs pushed nicotine reinforcement to a ceiling where relatively weaker nicotine dose history effects were largely obscured.
In cognitive testing, adult nicotine exposure produced mild learning benefits at least in terms of time efficiency in completing the RAM. However, this beneficial effect was not specific to NVHL rats, improving Sham performance as well. Nicotine also did not at all ameliorate the primary cognitive deficit measure of ETR that is actually the most robustly impaired dimension of cognition in NVHLs measured on the RAM. Finally, the non-specific mild cognitive benefit of nicotine came at a price in terms of actually impairing performance once the nicotine stopped, producing new deficits that resembled a mild form of what the NVHL model does to cognition even without nicotine exposure. Again, this nicotine withdrawal effect occurred non-specifically in NVHL and Sham rats alike, indicating that the net short-term cost-benefits of nicotine on cognition in the NVHL are not different from Shams. These observations are consistent with rigorously controlled human experimentation demonstrating a lack of differential cognitive benefits of nicotine in healthy versus schizophrenia nicotine users, and detrimental cognitive effects of nicotine withdrawal (Hahn 2013). Together with emerging animal and human data suggesting that chronic nicotine exposure can actually worsen cognition and other psychiatric symptoms (Reitz et al. 2007; Slotkin 2008; Counotte et al. 2011; McDermott et al. 2013), our findings suggest the feasibility of two, non-contradictory, bidirectional causal dynamics underlying the link between schizophrenia and nicotine addiction, in which each disease worsens the severity of the other. We did not test every possible manner in which nicotine could work as a medicine for abnormalities in the NVHL model or schizophrenia, and therefore cannot rule out the possibility that nicotine could still be therapeutic in some way. However, our results showing that early developmental perturbation of the hippocampus amplifies the reinforcing action of nicotine, while also producing cognitive problems that are not specifically or differentially treatable with nicotine, calls into question the self-medication hypothesis as the most widely espoused explanation for high rates of nicotine dependence in schizophrenia.
These findings highlight what could be a central pitfall in the self-medication explanation in that it focuses on, and promotes, only a therapeutic value to nicotine, while ignoring its highly addictive activity, and the likelihood that it is this activity that is pathologically amplified by the biology of mental illness. In circumventing this issue, these data provide a new view on neurodevelopmental mechanisms that predispose the mentally ill to nicotine addiction that should be studied further in this model and in human subjects for discovery of new prevention and treatment strategies. Perhaps more immediately, these findings provide a neuroscientific demonstration accessible to clinicians and patients alike that identifies nicotine dependence, not as a medication-modality for mental illness, but as a co-morbid addiction to which the mentally ill are highly biologically vulnerable.
Acknowledgments
This study was funded by a National Science Foundation GK-12 Doctoral Training Program grant to S.A.B. (as part of her PHD dissertation) and National Institute of Alcoholism and Alcohol Abuse grants (1RC2 AA019366 and R01 AA020396) (R.A.C.; E.A.E.). Support for translating this science to the clinical community via open access is provided by Indiana CTSI (NIH grant TR000006). [Correction added on 2 September 2013, after first online publication: The full funding information for this paper is now included.]
Authors Contribution
SAB and RAC were involved in all components of the experimental design, execution, data analysis and manuscript preparation. AMS and BSC played critical roles in the management of rats through the longitudinal experiment and the hands on data collection involving self-administration and RAM testing. RAC and EAE co-supervised the project and were integral to manuscript preparation.
References
- Berg SA, Chambers RA. Accentuated behavioral sensitization to nicotine in the neonatal ventral hippocampal lesion model of schizophrenia. Neuropharmacology. 2008;54:1201–1207. doi: 10.1016/j.neuropharm.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg SA, Czachowski CL, Chambers RA. Alcohol seeking and consumption in the NVHL neurodevelopmental rat model of schizophrenia. Behav Brain Res. 2011;218:346–349. doi: 10.1016/j.bbr.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickel WK, Odum AL, Madden GJ. Impulsivity and cigarrette smoking: delay discounting in current, never, and ex-smokers. Psychopharmacology (Berl) 1999;146:447–454. doi: 10.1007/pl00005490. [DOI] [PubMed] [Google Scholar]
- Bracken AL, Chambers RA, Berg SA, Rodd ZA, McBride WJ. Nicotine exposure during adolescence enhances behavioral sensitivity to nicotine during adulthood in Wistar rats. Pharmacol Biochem Behav. 2011;99:87–93. doi: 10.1016/j.pbb.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla P, Perlini C, Rajagopalan P, Saharan P, Rambaldelli G, Bellani M, Dusi N, Cerini R, Pozzi Mucelli R, Tansella M, Thompson PM. Schizophrenia severity, social functioning and hippocampal neuroanatomy: three-dimensional mapping study. Br J Psychiatry. 2013;202:50–55. doi: 10.1192/bjp.bp.111.105700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS. Hippocampal volume reduction in major depression. Am J Psychiatry. 2000;157:115–118. doi: 10.1176/ajp.157.1.115. [DOI] [PubMed] [Google Scholar]
- Chambers RA. Adult hippocampal neurogenesis in the pathogenesis of addiction and dual diagnosis disorders. Drug Alcohol Depend. 2013;130:1–12. doi: 10.1016/j.drugalcdep.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers RA, Jones RM, Brown S, Taylor JR. Natural reward related learning in rats with neonatal ventral hippocampal lesions and prior cocaine exposure. Psychopharmacology (Berl) 2005;179:470–478. doi: 10.1007/s00213-004-2042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers RA, Krystal JK, Self DW. A neurobiological basis for substance abuse comorbidity in schizophrenia. Biol Psychiatry. 2001;50:71–83. doi: 10.1016/s0006-3223(01)01134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers RA, Moore J, McEvoy JP, Levin ED. Cognitive effects of neonatal hippocampal lesions in a rat model of schizophrenia. Neuropsychopharmacology. 1996;15:587–594. doi: 10.1016/S0893-133X(96)00132-7. [DOI] [PubMed] [Google Scholar]
- Chambers RA, Self DW. Motivational responses to natural and drug rewards in rats with neonatal ventral hippocampal lesions: an animal model of dual diagnosis schizophrenia. Neuropsychopharmacology. 2002;27:889–905. doi: 10.1016/S0893-133X(02)00365-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers RA, Sentir AM, Conroy SK, Truitt WA, Shekhar A. Cortical-striatal integration of cocaine history and prefrontal dysfunction in animal modeling of dual diagnosis. Biol Psychiatry. 2010a;67:788–792. doi: 10.1016/j.biopsych.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers RA, Sentir AM, Engleman EA. Ventral and dorsal striatal dopamine efflux and behavior in rats with simple vs. co-morbid histories of cocaine sensitization and neonatal ventral hippocampal lesions. Psychopharmacology (Berl) 2010b;212:73–83. doi: 10.1007/s00213-010-1929-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers RA, Taylor JR. Animal modeling dual diagnosis schizophrenia: sensitization to cocaine in rats with neonatal ventral hippocampal lesions. Biol Psychiatry. 2004;56:308–316. doi: 10.1016/j.biopsych.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Chambers RA, Taylor JR, Potenza MN. Developmental neurocircuitry of motivation in adolescence: a critical period of addiction vulnerability. Am J Psychiatry. 2003;160:1041–1052. doi: 10.1176/appi.ajp.160.6.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton MT, Kelley ME, Ramsay CE, Pringle M, Goulding SM, Esterberg ML, Stewart T, Walker EF. Association of pre-onset cannabis, alcohol, and tobacco use with age at onset of prodrome and age at onset of psychosis in first-episode patients. Am J Psychiatry. 2009;166:1251–1257. doi: 10.1176/appi.ajp.2009.09030311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy SK, Rodd Z, Chambers RA. Ethanol sensitization in a neurodevelopmental lesion model of schizophrenia in rats. Pharmacol Biochem Behav. 2007;86:386–394. doi: 10.1016/j.pbb.2006.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counotte DS, Goriounova NA, Li KW, Loos M, van der Schors RC, Schetters D, Schoffelmeer AN, Smit AB, Mansvelder HD, Pattij T, Spijker S. Lasting synaptic changes underlie attention deficits caused by nicotine exposure during adolescence. Nat Neurosci. 2011;14:417–419. doi: 10.1038/nn.2770. [DOI] [PubMed] [Google Scholar]
- Counotte DS, Spijker S, Van de Burgwal LH, Hogenboom F, Schoffelmeer AN, De Vries TJ, Smit AB, Pattij T. Long-lasting cognitive deficits resulting from adolescent nicotine exposure in rats. Neuropsychopharmacology. 2009;34:299–306. doi: 10.1038/npp.2008.96. [DOI] [PubMed] [Google Scholar]
- Dani JA, Harris RA. Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat Neurosci. 2005;8:1465–1470. doi: 10.1038/nn1580. [DOI] [PubMed] [Google Scholar]
- DeHay T, Morris C, May MG, Devine K, Waxmonsky J. Tobacco use in youth with mental illnesses. J Behav Med. 2012;35:139–148. doi: 10.1007/s10865-011-9336-6. [DOI] [PubMed] [Google Scholar]
- Durazzo TC, Mon A, Gazdzinski S, Meyerhoff DJ. Chronic cigarette smoking in alcohol dependence: associations with cortical thickness and N-acetylaspartate levels in the extended brain reward system. Addict Biol. 2013;18:379–391. doi: 10.1111/j.1369-1600.2011.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller RL, Luck SJ, Braun EL, Robinson BM, McMahon RP, Gold JM. Impaired visual working memory consolidation in schizophrenia. Neuropsychology. 2009;23:71–80. doi: 10.1037/a0013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold JM, Hahn B, Zhang WW, Robinson BM, Kappenman ES, Beck VM, Luck SJ. Reduced capacity but spared precision and maintenance of working memory representations in schizophrenia. Arch Gen Psychiatry. 2010;67:570–577. doi: 10.1001/archgenpsychiatry.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BF, Hasin DS, Chou SP, Stinson FS, Dawson DA. Nicotine dependence and psychiatric disorders in the United States: results from the National Epidemiologic Survey on alcohol and related conditions. Arch Gen Psychiatry. 2004;61:1107–1115. doi: 10.1001/archpsyc.61.11.1107. [DOI] [PubMed] [Google Scholar]
- Hahn B. A test of the cognitive self-medication hypothesis of tobacco smoking in schizophrenia. Biol Psychiatry. 2013 doi: 10.1016/j.biopsych.2013.03.017. doi: 10.1016/j.biopsych.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerey EA, Bell-Warren KR, Gold JM. Decision-making impairments in the context of intact reward sensitivity in schizophrenia. Biol Psychiatry. 2008;64:62–69. doi: 10.1016/j.biopsych.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasser K, Boyd JW, Woolhandler S, Heimmerlstein D, McCormick D, Bor D. Smoking in mental illness: a population-based prevalence study. JAMA. 2000;284:2606–2610. doi: 10.1001/jama.284.20.2606. [DOI] [PubMed] [Google Scholar]
- Leonard S, Adler L, Benhammou K, Berger R, Breese CR, Drebring C, Gault J, Lee M, Logel J, Olincy A, Ross RG, Stevens K, Sullivan B, Vianzon R, Virnich DE, Waldo M, Walton K, Freedman R. Smoking and mental illness. Pharmacol Biochem Behav. 2001;70:561–570. doi: 10.1016/s0091-3057(01)00677-3. [DOI] [PubMed] [Google Scholar]
- Levin ED. Psychopharmacological effects in the radial-arm maze. Neurosci Biobehav Rev. 1988;12:169–175. doi: 10.1016/s0149-7634(88)80008-3. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Jaskiw GE, Weinberger DR. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: a potential animal model of schizophrenia. Neuropsychopharmacology. 1993;9:67–75. doi: 10.1038/npp.1993.44. [DOI] [PubMed] [Google Scholar]
- Liu X, Matochik JA, Cadet JL, London ED. Smaller volume of prefrontal lobe in polysubstance abusers: a magnetic resonance imaging study. Neuropsychopharmacology. 1998;18:243–252. doi: 10.1016/S0893-133X(97)00143-7. [DOI] [PubMed] [Google Scholar]
- Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, Gardiner PS, Grady SR, Heberlein U, Leonard SS, Levin ED, Lukas RJ, Markou A, Marks MJ, McCallum SE, Parameswaran N, Perkins KA, Picciotto MR, Quik M, Rose JE, Rothenfluh A, Schafer WR, Stolerman IP, Tyndale RF, Wehner JM, Zirger JM. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl) 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- McDermott MS, Marteau TM, Hollands GJ, Hankins M, Aveyard P. Change in anxiety following successful and unsuccessful attempts at smoking cessation: cohort study. Br J Psychiatry. 2013;202:62–67. doi: 10.1192/bjp.bp.112.114389. [DOI] [PubMed] [Google Scholar]
- Mokdad AH, Marks JS, Stroup JS, Gerberding JL. Actual causes of death in the United States, 2000. JAMA. 2004;291:1238–1245. doi: 10.1001/jama.291.10.1238. [DOI] [PubMed] [Google Scholar]
- Olton D, Samuelson RJ. Rememberances of places passed: spatial memory in rats. J Exp Psychol Anim Behav Process. 1976;2:97–116. [Google Scholar]
- Parks J, Svendsen D, Singer P, Foti ME. Morbidity and mortaloty in people with serious mental illness. Alexandria, VA: National Associatry of State Mental Health Program Directors; 2006. [Google Scholar]
- Prochaska JJ, Hall SM, Bero LA. Tobacco use among individuals with schizophrenia: what role has the tobacco industry played. Schizophr Bull. 2008;34:555–567. doi: 10.1093/schbul/sbm117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulay AJ, Stinson FS, Ruan WJ, Smith SM, Pickering RP, Dawson DA, Grant BF. The relationship of DSM-IV personality disorders to nicotine dependence-results from a national survey. Drug Alcohol Depend. 2010;108:141–145. doi: 10.1016/j.drugalcdep.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, den Heijer T, van Duijn C, Hofman A, Breteler MMB. Relation between smoking and risk of dementia and Alzheimer disease: the Rotterdam study. Neurology. 2007;69:998–1005. doi: 10.1212/01.wnl.0000271395.29695.9a. [DOI] [PubMed] [Google Scholar]
- Slotkin TA. If nicotiine is a developmental neurotoxicant in animal studies, dare we recommend nicotine replacement therapy in pregnant women and adolescence? Neurotoxicol Teratol. 2008;30:1–19. doi: 10.1016/j.ntt.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Steinberg ML, Williams JM, Ziedonis DM. Financial implications of cigarette smoking among individuals with schizophrenia. Tob Control. 2004;13:206. [PMC free article] [PubMed] [Google Scholar]
- Sudai E, Croitoru O, Shaldubina A, Abraham L, Gispan I, Flaumenhaft Y, Roth-Deri I, Kinor N, Aharoni S, Ben-Tzion M, Yadid G. High cocaine dosage decreases neurogenesis in the hippocampus and impairs working memory. Addict Biol. 2011;16:251–260. doi: 10.1111/j.1369-1600.2010.00241.x. [DOI] [PubMed] [Google Scholar]
- Teicher MH, Anderson CM, Polcari A. Childhood maltreatment is associated with reduced volume in the hippocampal subfields CA3, dentate gyrus, and subiculum. Proc Natl Acad Sci U S A. 2012;109:E563–E572. doi: 10.1073/pnas.1115396109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Chambers RA, Lipska BK. The neonatal ventral hippocampal lesion as a heuristic neurodevelopmental model of schizophrenia. Behav Brain Res. 2009;204:295–305. doi: 10.1016/j.bbr.2008.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Lewis BL, Lipska BK, O'Donnell P. Post-pubertal disruption of medial prefrontal cortical dopamine-glutamate interactions in a developmental animal model of schizophrenia. Biol Psychiatry. 2007;62:730–738. doi: 10.1016/j.biopsych.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND. Substance use disorders in schizophrenia—clinical implications of comorbidity. Schizophr Bull. 2009;35:469–472. doi: 10.1093/schbul/sbp016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger DR. Cell biology of the hippocampal formation in schizophrenia. Biol Psychiatry. 1999;45:395–402. doi: 10.1016/s0006-3223(98)00331-x. [DOI] [PubMed] [Google Scholar]
- Williams JM, Ziedonis DM, Abanyie F, Steinberg ML, Foulds J, Benowitz NL. Increased nicotine and cotinine levels in smokers with schizophrenia and schizoaffective disorder is not a metabolic effect. Schizophr Res. 2005;79:323–335. doi: 10.1016/j.schres.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Zhang X, Stein EA, Hong LE. Smoking and schizophrenia independently and additively reduce white matter integrity between striatum and frontal cortex. Biol Psychiatry. 2010;68:674–677. doi: 10.1016/j.biopsych.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]