

Abstract
Cell survival is conditional on the maintenance of a favourable acid–base balance (pH). Owing to intensive respiratory CO2 and lactic acid production, cancer cells are exposed continuously to large acid–base fluxes, which would disturb pH if uncorrected. The large cellular reservoir of H+-binding sites can buffer pH changes but, on its own, is inadequate to regulate intracellular pH. To stabilize intracellular pH at a favourable level, cells control trans-membrane traffic of H+-ions (or their chemical equivalents, e.g. 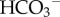 ) using specialized transporter proteins sensitive to pH. In poorly perfused tumours, additional diffusion-reaction mechanisms, involving carbonic anhydrase (CA) enzymes, fine-tune control extracellular pH. The ability of H+-ions to change the ionization state of proteins underlies the exquisite pH sensitivity of cellular behaviour, including key processes in cancer formation and metastasis (proliferation, cell cycle, transformation, migration). Elevated metabolism, weakened cell-to-capillary diffusive coupling, and adaptations involving H+/H+-equivalent transporters and extracellular-facing CAs give cancer cells the means to manipulate micro-environmental acidity, a cancer hallmark. Through genetic instability, the cellular apparatus for regulating and sensing pH is able to adapt to extracellular acidity, driving disease progression. The therapeutic potential of disturbing this sequence by targeting H+/H+-equivalent transporters, buffering or CAs is being investigated, using monoclonal antibodies and small-molecule inhibitors.
) using specialized transporter proteins sensitive to pH. In poorly perfused tumours, additional diffusion-reaction mechanisms, involving carbonic anhydrase (CA) enzymes, fine-tune control extracellular pH. The ability of H+-ions to change the ionization state of proteins underlies the exquisite pH sensitivity of cellular behaviour, including key processes in cancer formation and metastasis (proliferation, cell cycle, transformation, migration). Elevated metabolism, weakened cell-to-capillary diffusive coupling, and adaptations involving H+/H+-equivalent transporters and extracellular-facing CAs give cancer cells the means to manipulate micro-environmental acidity, a cancer hallmark. Through genetic instability, the cellular apparatus for regulating and sensing pH is able to adapt to extracellular acidity, driving disease progression. The therapeutic potential of disturbing this sequence by targeting H+/H+-equivalent transporters, buffering or CAs is being investigated, using monoclonal antibodies and small-molecule inhibitors.
Keywords: pH regulation, pH sensing, buffering, carbonic anhydrase, diffusion
1. Introduction
Hydrogen (H+) ions (or protons) are the smallest yet arguably the most reactive ions present in living organisms. All biological solutions have a certain concentration of H+ ions ([H+]) arising from the balance between deprotonation and protonation reactions of water, weak acids and weak bases. Equilibria between H+ ions and the unprotonated (A) and protonated (HA) forms of molecules is described by an acid-dissociation (Ka = [H+] × [A]/[HA]) (figure 1). Values of Ka span many orders of magnitude and, consequently, [H+] can vary greatly between different solutions. For this reason, [H+] is usually expressed on a logarithmic pH scale [1]. Complex solutes, including many biologically important molecules, are often ascribed with several Ka values, reflecting distinct proton-binding sites. At a given pH, protonatable sites with very high or very low pKa will be almost completely titrated or unbound, respectively. By contrast, the concentration (in a macroscopic sense) of protonated and unprotonated sites will be balanced if pKa is near the ambient pH. Such molecules are of major biological importance for two reasons. Firstly, the availability of HA and A protects (buffers) solutions from large pH changes in response to acid–base challenges. Secondly, a sustained change in pH alters the [HA]/[A] ratio, which could have secondary effects if the biological properties of HA and A differ substantially. The sensitivity of proteins to pH has exceptional bearing on cells because proteins act as pH buffers, and their function can change substantially if ionization state is altered by the binding or release of H+ ions (a form of post-translational modification) [2]. It is therefore not surprising that only a narrow range of pH is compatible with eukaryotic function.
Figure 1.
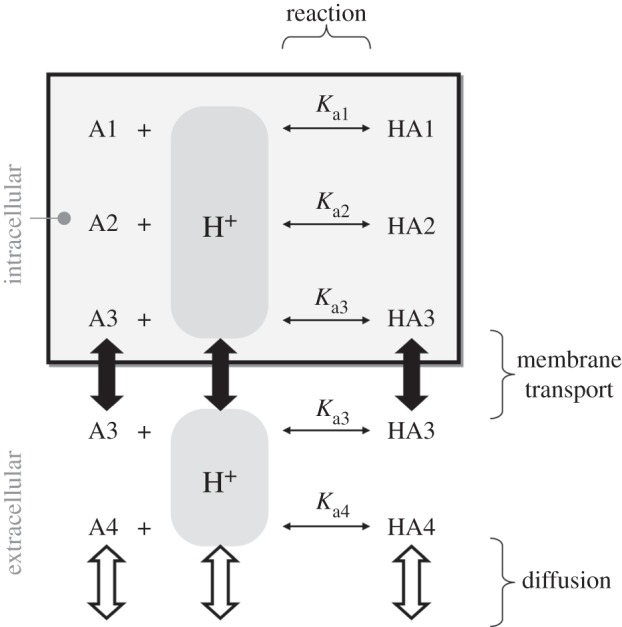
The dynamics of intra- and extracellular pH are determined by reaction, transport and diffusion fluxes. For illustrative purposes, two intracellular buffers (HA1/A1 and HA2/A2), one extracellular freely diffusible buffer (HA4/A4), and a buffer that can cross the cell membrane (HA3/A3) are shown.
Living tissue, unlike a simple salt solution, engages continually in the production or consumption of acids (or bases) through chemical reactions. Because of cellular respiration (yielding CO2 and lactic acid), most cells are net acid-producers hence intracellular pH (pHi) has a tendency to fall. A sustained and substantial acid–base challenge cannot be corrected by pH buffers alone because of their finite capacity (i.e. buffering reduces the amplitude of pH-changes but cannot, on its own, eliminate or reverse these). Also in contrast to a simple solution, living tissue is compartmentalized into intra- and extracellular spaces separated by the cell membrane (figure 1). The ability of biological membranes to allow the passage of selected molecules can give rise to pH differences between the compartments. Selective transport of H+-ions (or molecules that release or take-up H+ ions such as CO2 or 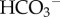 : the so-called H+-equivalents) across membranes is thus an effective means of changing pHi. As explained later, the usual pHi-regulatory strategy of cells is to balance the internal production of acid (or base) with an equal ‘corrective’ efflux of acid (or base) across the cell membrane.
: the so-called H+-equivalents) across membranes is thus an effective means of changing pHi. As explained later, the usual pHi-regulatory strategy of cells is to balance the internal production of acid (or base) with an equal ‘corrective’ efflux of acid (or base) across the cell membrane.
The biological potency and chemical omnipresence of H+ ions highlight the importance of regulating pH (where pH is controlled to suit protein function) and of adapting biology to a particular pH level (where gene products are selected or changed on the basis of ambient pH). As will be explained below, these processes are believed to play an important role in cancer disease progression.
2. Low micro-environmental O2 tension and pH as hallmarks of cancer
Histological studies in the 1950s by Thomlinson and Gray established that human tumours grow around blood vessels and that the outermost cells beyond a distance of approximately 200 µm from blood become necrotic [3]. A gradient of O2 tension develops across the layer of viable cells, driven by the high metabolic demand of cancer biochemistry and relatively long diffusion distances to the source [4]. O2 gradients have often been modelled by steady-state diffusion–reaction equations, where 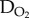 is the O2 diffusion coefficient and function R describes reactions
is the O2 diffusion coefficient and function R describes reactions
| 2.1 |
The presence of areas with low (<1%) O2 tension is associated with increased metastasis and poor patient survival [5], giving rise to the notion that hypoxia is a hallmark of malignant cancer. The discovery that hypoxia alters cell biology [6] (e.g. via hypoxia-inducible factor HIF1α [7]) offered a mechanism for adaptive changes, such as the switch-over to glycolytic metabolism (Warburg effect; [8]). Tumour hypoxia has since become a topic of considerable research, achieving promising outcomes with respect to understanding aetiology, improving diagnosis and developing treatments [6,9]. Among other micro-environmental factors specifically identified in tumours, extracellular acidity has emerged as another cancer hallmark [10–12]. Contrary to initial expectations, the intracellular compartment was shown to be alkaline [13] despite low extracellular pH (pHe). Other than in solid tumours, this trans-membrane [H+] distribution (acidic extracellularly/alkaline intracellularly) is not commonly observed in tissue. Two questions have emerged in response to these pioneering studies: firstly, how do solid tumours produce low pHe yet are able to maintain pHi within favourable limits, and, secondly, how does this trans-membrane pH-distribution affect disease progression?
3. Production and venting of metabolic acids
Cancer cells require a substantial input of energy to support their intensive programme of growth. This explains the high glucose utilization rate, measured to be most typically in the range 0.1–1 µmol (g tissue)−1 min−1 [14]. Under aerobic conditions, respiration of glucose to CO2 is coupled to the production of ATP, which consumes an H+ ion:
This acid–base disturbance is then cancelled out by ATP break-down elsewhere in the cell. As a result, the source of acidity from aerobic metabolism is CO2, once it hydrates to H+ and 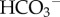 ions. Under anaerobic conditions, glycolytic ATP production is coupled to the chemical conversion of glucose to anionic lactate [15]:
ions. Under anaerobic conditions, glycolytic ATP production is coupled to the chemical conversion of glucose to anionic lactate [15]:
This reaction does not generate (or consume) H+ ions, indicating that glycolysis per se is pH neutral. However, subsequent ATP breakdown releases H+ ions, explaining how anaerobic metabolism yields acid. Depending on whether respiration is glycolytic or mitochondrial, cancer cells may be producing approximately 1–3 mmol · (l cell)−1 min−1 of acid (assuming an extracellular/intracellular volume ratio of 1/2; [16]). For a typical intracellular buffering capacity of approximately 30 mmol · (l cell)−1 · (pH unit)−1 [17], this magnitude of acid-loading would promptly and substantially alter pHi, if uncorrected. However, most cells have the capacity to remove respiratory end-products passively across the surface membrane. CO2 has a high lipid : water partition coefficient, allowing it to cross the lipid bilayer freely. In addition (although not without controversy [18]), specialized gas channels such as aquaporins (AQP) have been demonstrated to increase membrane permeability to CO2 [19]. Lactic acid, despite a much lower lipid : water partition coefficient, can cross the membrane as H+-lactate, translocated by H+-monocarboxylate transporters (MCT), including MCT1 and the hypoxia-inducible MCT4 [20] (according to the SoLute Carrier family naming convention, SLC16A1 and SLC16A3, respectively).
The rate of passive CO2 and H+-lactate venting from cells depends on trans-membrane concentration gradients. In well-perfused tissues, outwardly directed trans-membrane gradients are maintained by good diffusive coupling between the cell surface and capillary blood. By contrast, the often inadequate capillary perfusion of tumours gives rise to long diffusion distances and a considerable resistance to solute flux [21]. Extracellular build-up of CO2 or H+-lactate will reduce their venting, even across membranes with high permeability. However, CO2 and H+-lactate diffusion can be facilitated by biological adaptations that address the rate-limiting steps. Extracellular lactic acid (pKa = 3.8) remains almost completely ionized and the associated H+ ion is titrated by extracellular buffers. The overall rate of extracellular H+-lactate diffusion can be rate-limited by the effective mobility of H+ ions (DHapp). In highly buffered solutions, DHapp depends on the mobility of buffers [22], many of which are large proteins diffusing substantially slower than lactate. However, low molecular weight (mobile) buffers, such as amino acids, phosphates or CO2/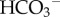 could facilitate H+ diffusion and improve H+-lactate venting (figure 2a). Extracellular CO2 also ionizes (pKa = 6.1) but to a much lesser degree than lactic acid. Although the spontaneous hydration reaction is very slow (time constant 5 s), it can be catalysed by exo-facial carbonic anhydrase (CA) enzymes [23–25], such as the tumour-associated isoforms CAIX and CAXII [26–30]. Catalysed conversion of CO2 to
could facilitate H+ diffusion and improve H+-lactate venting (figure 2a). Extracellular CO2 also ionizes (pKa = 6.1) but to a much lesser degree than lactic acid. Although the spontaneous hydration reaction is very slow (time constant 5 s), it can be catalysed by exo-facial carbonic anhydrase (CA) enzymes [23–25], such as the tumour-associated isoforms CAIX and CAXII [26–30]. Catalysed conversion of CO2 to 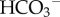 and H+ can facilitate overall CO2 diffusion by means of a parallel flux of H+ +
and H+ can facilitate overall CO2 diffusion by means of a parallel flux of H+ + 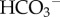 , a phenomenon first described in vitro by Gros & Moll [31]. Akin to the diffusion of other ionized weak acids, CA-facilitated CO2 diffusion also requires adequate mobile buffering to carry H+ ions in parallel to CO2 and
, a phenomenon first described in vitro by Gros & Moll [31]. Akin to the diffusion of other ionized weak acids, CA-facilitated CO2 diffusion also requires adequate mobile buffering to carry H+ ions in parallel to CO2 and 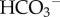 [32] (figure 2b). Net hydration by exofacial CAs will reduce steady-state pHe and contribute towards micro-environment acidity. Excessive extracellular acidification could become detrimental, but the extent of this may be curtailed by the inhibitory (i.e. negative feedback) effect of H+ ions on CA activity [33–35].
[32] (figure 2b). Net hydration by exofacial CAs will reduce steady-state pHe and contribute towards micro-environment acidity. Excessive extracellular acidification could become detrimental, but the extent of this may be curtailed by the inhibitory (i.e. negative feedback) effect of H+ ions on CA activity [33–35].
Figure 2.
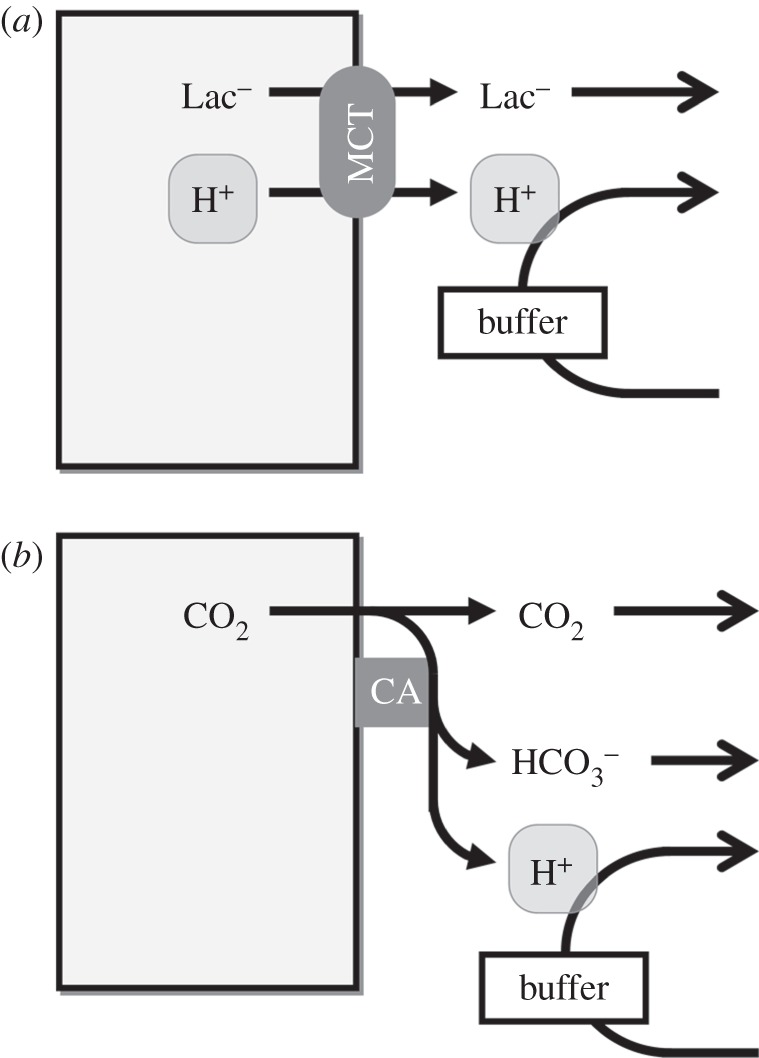
Venting of metabolically produced acids. (a) H+-lactate efflux across the membrane is facilitated by H+-monocarboxylate transport (MCT). Diffusion of H+ and lactate away from the cell-surface is necessary for sustained MCT activity. Mobile H+-buffers can facilitate H+ diffusion and support H+-lactate venting. (b) CO2 can permeate the cell membrane through the lipid bilayer or gas channels. Spontaneous CO2 hydration is slow, but can be accelerated by exo-facial carbonic anhydrase (CA) enzymes. Diffusion of the hydration products alongside CO2 represents a form of facilitated CO2 diffusion.
Mobile buffers usually coexist with fixed buffers, therefore DHapp is typically lower than the diffusivity of CO2, 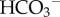 or lactate. Consequently, a relatively steep [H+]e gradient is needed to drive a diffusive H+ ion flux to match the flux of CO2, HCO3− or lactate. This may explain why it is important for [H+]e at the core of solid tumours to reach levels as high as 250 nM, i.e. pHe = 6.6 [4]. A mechanistic description of tumour pHe must account for diffusion–reaction processes involving H+ ions, CO2,
or lactate. Consequently, a relatively steep [H+]e gradient is needed to drive a diffusive H+ ion flux to match the flux of CO2, HCO3− or lactate. This may explain why it is important for [H+]e at the core of solid tumours to reach levels as high as 250 nM, i.e. pHe = 6.6 [4]. A mechanistic description of tumour pHe must account for diffusion–reaction processes involving H+ ions, CO2, 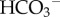 , lactate and buffers [25], and hence a single equation (such as equation (2.1) used for modelling hypoxia) is inadequate.
, lactate and buffers [25], and hence a single equation (such as equation (2.1) used for modelling hypoxia) is inadequate.
Facilitated CO2 and H+-lactate diffusion away from the surface of cells is expected to produce a more alkaline pHi that better supports cell proliferation. Indeed, facilitated CO2 venting appears to be a major role for CAIX and CAXII in tumour physiology [25,36], possibly explaining the faster growth rates measured in tumours expressing catalytically active enzyme [35,37]. As demonstrated in skeletal muscle [38], exo-facial CAs also improve H+-lactate venting by optimizing the ability of CO2/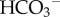 to neutralize H+ ions released by MCT. A similar MCT–CA interaction may be important in tumours.
to neutralize H+ ions released by MCT. A similar MCT–CA interaction may be important in tumours.
In the scheme developed so far, H+-lactate and CO2 are the principal sources of cellular acid, and their venting is rate-limited by resistances imposed by membranes (permeation) and the tortuous interstitial space (diffusion). Steady-state intracellular [H+] could be approximated by
In some cells, such as erythrocytes [39], steady-state pHi predicted by this equation is viable because plasma [H+], [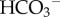 ] and [lactate−] are normally well controlled in the body. However, most cells do not have the privilege of direct access to a well-controlled milieu and, consequently, the predicted equilibrium pHi may not be compatible with biological needs. Due to its stoichiometry, MCT couples the transport of H+ ions with the transmembrane [lactate] gradient, and any further demand for H+ transport would have to be met by other means. In addition, many processes, such as cell division, require cells to manipulate pHi dynamically and independently of pHe. For these reasons, cells in most tissues, including tumours, have additional mechanisms for regulating pHi.
] and [lactate−] are normally well controlled in the body. However, most cells do not have the privilege of direct access to a well-controlled milieu and, consequently, the predicted equilibrium pHi may not be compatible with biological needs. Due to its stoichiometry, MCT couples the transport of H+ ions with the transmembrane [lactate] gradient, and any further demand for H+ transport would have to be met by other means. In addition, many processes, such as cell division, require cells to manipulate pHi dynamically and independently of pHe. For these reasons, cells in most tissues, including tumours, have additional mechanisms for regulating pHi.
4. pH regulation by membrane transport
In a solution where the concentrations and Ka of buffers are held constant, the only means of changing pH is by adding acid (or base). Whereas H+/H+-equivalent production (or consumption) by metabolism is not a feasible means of achieving pHi-homeostasis, cells can control their pHi by regulating the active transport of H+ ions or their chemical equivalents (OH−, 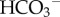 or CO32−) across the membrane [40,41]. Experimentally, it is possible to distinguish pHi-regulating proteins that translocate H+ (or OH−) ions from those that translocate
or CO32−) across the membrane [40,41]. Experimentally, it is possible to distinguish pHi-regulating proteins that translocate H+ (or OH−) ions from those that translocate 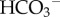 (or CO32−) ions by measuring fluxes in the presence and the absence of CO2/
(or CO32−) ions by measuring fluxes in the presence and the absence of CO2/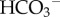 buffer [42] (figure 3).
buffer [42] (figure 3).
Figure 3.
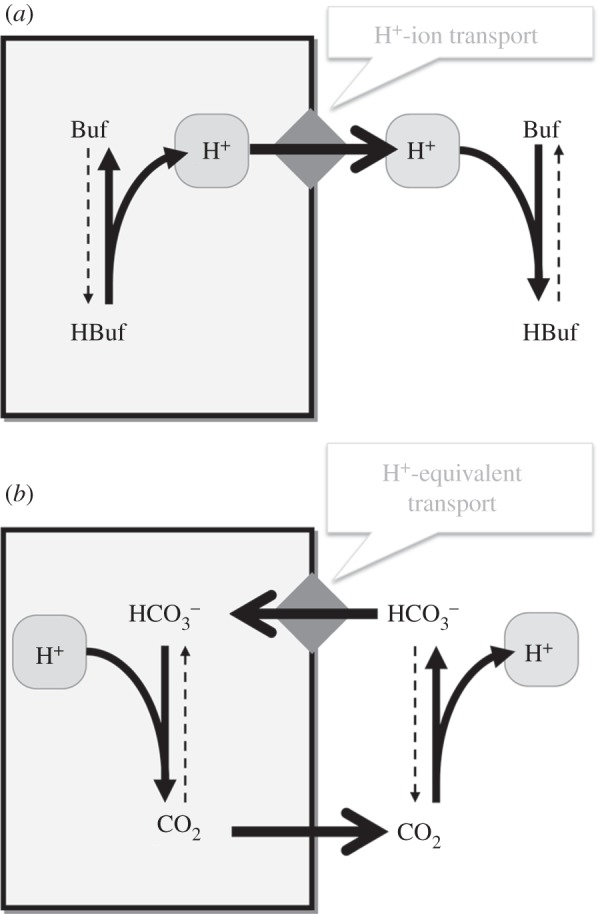
Regulation of intracellular pH by acid-extruding transporter-proteins (diamond symbol). (a) Extrusion of H+ ions alkalinizes the cell. This must also remove buffer-bound H+ ions to change pHi substantially. (b) Uptake of 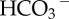 (or CO32−) ions also alkalinizes the cell because the subsequent reaction with intracellular H+ ions produces CO2, the passive venting of which completes the acid-extrusion process.
(or CO32−) ions also alkalinizes the cell because the subsequent reaction with intracellular H+ ions produces CO2, the passive venting of which completes the acid-extrusion process. 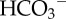 transport is considered to be H+-equivalent flux.
transport is considered to be H+-equivalent flux.
For a complete regulatory system, membrane transporters must ‘sense’ pHi and respond by producing net H+/H+-equivalent efflux or influx when pHi is either too low or too high, respectively. In practice, most cells express dedicated acid-loading and acid-extruding transporter proteins [40]. By working against each other, acid-extrusion and acid-loading fluxes can correct pHi disturbances and maintain pHi around a steady-state point (figure 4a). Acid-extruding transporter proteins include Na+/H+ exchangers (NHE) belonging to the SLC9 family [43], H+-ATPase pumps and transporters that produce net 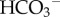 (or CO32−) influx, such as electroneutral Na+–
(or CO32−) influx, such as electroneutral Na+–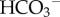 cotransport (NBCn1/SLC4A7), electrogenic Na+–2
cotransport (NBCn1/SLC4A7), electrogenic Na+–2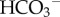 cotransport (NBCe1/SLC4A4) and electroneutral Na+-dependent Cl−/
cotransport (NBCe1/SLC4A4) and electroneutral Na+-dependent Cl−/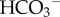 exchange (NDCBE/SLC4A8) [44]. Cl−/
exchange (NDCBE/SLC4A8) [44]. Cl−/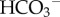 (or Cl−/OH−) exchangers of the SLC4A or SLC26 families are among acid-loading transporter proteins. Many cells use a combination of two or more acid-extruding and acid-loading transporter proteins. It may be speculated that this apparent redundancy is a demonstration of the fundamental importance of pHi control, particularly in the light of the fact that some nominally pHi-regulating proteins also service other regulatory systems (e.g. cell volume [45] or motility [46]), the transport demands of which may—at times—be at odds with pHi homeostasis. Different types of cancer have been shown to express various combinations of pHi-regulating transporters [17,47–50] (e.g. figure 4b). Despite the heterogeneity of pHi-regulatory mechanisms, some appear to be constitutive (e.g.
(or Cl−/OH−) exchangers of the SLC4A or SLC26 families are among acid-loading transporter proteins. Many cells use a combination of two or more acid-extruding and acid-loading transporter proteins. It may be speculated that this apparent redundancy is a demonstration of the fundamental importance of pHi control, particularly in the light of the fact that some nominally pHi-regulating proteins also service other regulatory systems (e.g. cell volume [45] or motility [46]), the transport demands of which may—at times—be at odds with pHi homeostasis. Different types of cancer have been shown to express various combinations of pHi-regulating transporters [17,47–50] (e.g. figure 4b). Despite the heterogeneity of pHi-regulatory mechanisms, some appear to be constitutive (e.g. 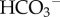 -dependent flux, [17,47]), whereas others (e.g. NHE1) emerge as highly dependent on the cell type and conditions.
-dependent flux, [17,47]), whereas others (e.g. NHE1) emerge as highly dependent on the cell type and conditions.
Figure 4.
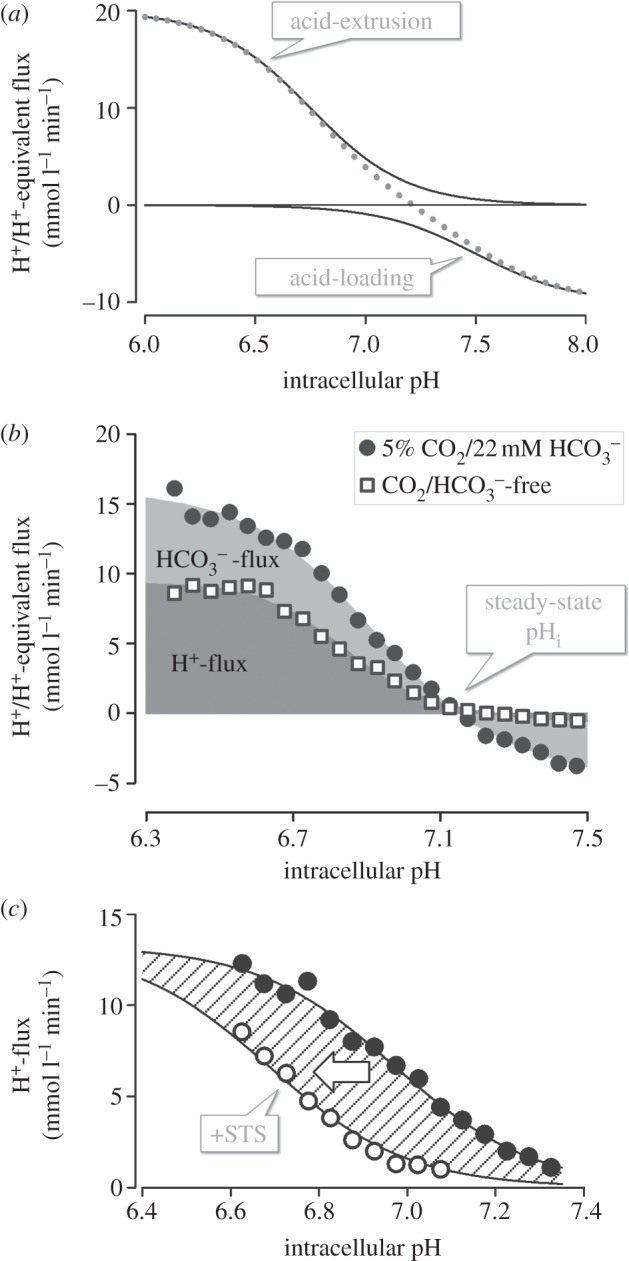
Flux analysis of H+/H+-equivalent membrane transport. (a) Hypothetical pHi dependence of flux by acid-extruding and acid-loading transporters: the shape of the pHi–flux relationship depends on the kinetics of transport, often described mathematically by a Hill equation. Acid extruders and acid loaders work against each other to produce net H+/H+-equivalent flux (dotted line). Intracellular pH stabilizes at a point of zero net flux. The angle at which the net flux curve crosses the pHi axis is a measure of the responsiveness of the pHi-regulating apparatus to pHi disturbances. (b) Example of flux analysis for colon HCT116 cancer cells cultured under normoxia (see [17]). Fluxes can be dissected into CO2/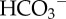 -dependent (i.e. involving
-dependent (i.e. involving 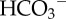 transport) and CO2/
transport) and CO2/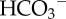 -independent (i.e. involving H+ transport). (c) Na+/H+ exchange in HCT116 cells is sensitive to kinases: 0.3 pHi unit acid-shift in pHi–flux relationship following inhibition of kinases by 20 nM staurosporin (STS) (see [17]).
-independent (i.e. involving H+ transport). (c) Na+/H+ exchange in HCT116 cells is sensitive to kinases: 0.3 pHi unit acid-shift in pHi–flux relationship following inhibition of kinases by 20 nM staurosporin (STS) (see [17]).
The energy for actively translocating H+/H+-equivalents across the membrane is ultimately financed by ATP consumption. In the case of primary active H+-ATPase pumps, the transport process is directly coupled to ATP hydrolysis. For the other pHi-regulating proteins, transport is driven by the energy stored in the electrochemical gradient of coupled ions (i.e. inward [Na+] or [Cl−] gradients). The free energy stored in ATP and trans-membrane [Na+] or [Cl−] gradients can affect the magnitude of H+/H+-equivalent flux produced by pHi regulators but a more physiologically important modulator of flux is the occupancy of the proteins’ pHi sensor. This can take the form of a binding site involved in the transport-cycle or a dedicated allosteric regulatory site. In the case of Cl−/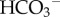 exchanger AE2 (SLC4A2), the allosteric pHi sensor has been described at the molecular level [51]. By coupling the activity of pHi-regulating proteins with signalling cascades, cells gain the ability to fine-tune the steady-state pHi in response to intrinsic (e.g. metabolic status [17,52]) or extrinsic (neural or hormonal [53–55]) influences. NHE1, for example, is sensitive to a wide range of signals [43,54,,55] (e.g. figure 4c). Factors associated with the tumour milieu, such as extracellular acidity [56], hypoxia [17] and limited
exchanger AE2 (SLC4A2), the allosteric pHi sensor has been described at the molecular level [51]. By coupling the activity of pHi-regulating proteins with signalling cascades, cells gain the ability to fine-tune the steady-state pHi in response to intrinsic (e.g. metabolic status [17,52]) or extrinsic (neural or hormonal [53–55]) influences. NHE1, for example, is sensitive to a wide range of signals [43,54,,55] (e.g. figure 4c). Factors associated with the tumour milieu, such as extracellular acidity [56], hypoxia [17] and limited 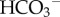 supply [48,56], can also greatly affect H+/H+-equivalent flux. These findings highlight the importance of investigating pHi regulation in the context of the tumour milieu.
supply [48,56], can also greatly affect H+/H+-equivalent flux. These findings highlight the importance of investigating pHi regulation in the context of the tumour milieu.
In summary, the resting pHi of a cell can be defined as the steady-state point at which net metabolic acid production is balanced by net membrane H+/H+-equivalent transport. These fluxes are likely to show considerable regional variation in solid tumours, resulting in the potential for large pHi gradients alongside pHe non-uniformity. This important aspect of tissue pH regulation cannot be investigated by measuring pH in suspensions or two-dimensional monolayers prepared from cultured cells. A more instructive approach to studying pH non-uniformity in tissue is to image cancer-derived multicellular three-dimensional spheroids for pHi and pHe (figure 5) [25,36,,56].
Figure 5.
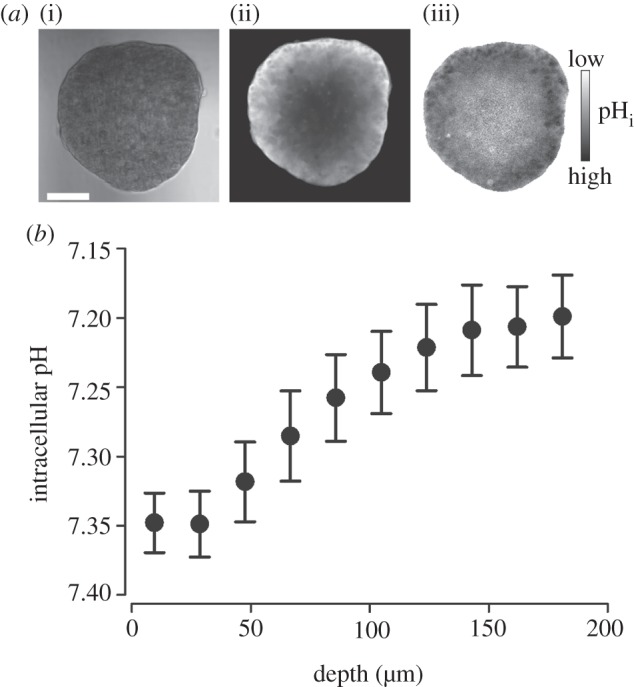
Intracellular pH gradients in multicellular spheroids grown from the ductal breast cancer line T47D. (a) (i) Transmission image (scale bar, 100 µm), (ii) fluorescence from intracellular carboxy-SNARF-1 (pH-sensitive reporter-dye), loaded into the intracellular compartment by passive entry of its acetoxymethyl ester; extracellular dye was washed away by perfusion in 5% CO2/22 mM 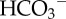 buffered normal Tyrode solution. (iii) Ratio of fluorescence at 580 and 640 nm is a measure of intracellular pH. (b) Intracellular pH at different depths (average of seven spheroids; mean radius 190 µm). pH-gradient results from restricted diffusion, depth-dependence of metabolic acid loading and H+/H+-equivalent membrane transport.
buffered normal Tyrode solution. (iii) Ratio of fluorescence at 580 and 640 nm is a measure of intracellular pH. (b) Intracellular pH at different depths (average of seven spheroids; mean radius 190 µm). pH-gradient results from restricted diffusion, depth-dependence of metabolic acid loading and H+/H+-equivalent membrane transport.
Active transport of H+/H+-equivalents is a means by which cancer cells can maintain an alkaline pHi, despite the substantial metabolic acid production and low pHe. However, the capacity of membrane transport to exercise full and autonomous control of pHi is limited by at least two factors. Firstly, active transport can place a substantial energetic burden on cells. The scale of this can be appreciated from the magnitude of fluxes typically produced by pHi regulators. To change pHi, membrane transporters must alter the concentration of free and buffer-bound H+ ions. In cytoplasm with a typical buffering capacity of 30 mmol. l−1 pH−1, for each free H+ ion there are approximately 105 buffer-bound H+ ions. Consequently, H+/H+-equivalent transport must be of the order of several mmol. l−1 min−1 to change pHi by a fraction of a unit per minute. Cancer cells are already challenged by a high demand for ATP and restricted respiratory substrate supply, and this may mean that pHi regulation cannot operate at full capacity. Indeed, Na+/H+ exchanger activity is reduced at low intracellular [ATP] [52].
A second limiting factor is the effect that pHi regulators have on extracellular pH. From a tissue point of view, membrane-bound pHi regulators neither produce nor consume H+/H+-equivalents, but change the distribution of H+ ions between the intracellular and extracellular compartments. During H+/H+-equivalent membrane transport, pHi and pHe will change in opposite directions. The magnitude of the pHe change will depend on extracellular buffering and H+ diffusion. In spheroids, which reproduce many aspects of the tissue microenvironment, the intrinsic extracellular buffering capacity is estimated to be equivalent to 5–10 mmol · (l interstitium)−1· (pH unit)−1 [21,56], i.e. lower than in the cytoplasm. As most fixed buffers reside on the surface of membranes, buffering capacity will depend on the degree of cell–cell packing (decreasing in ‘looser’ regions). Combined with weak diffusional coupling across the tumour interstitium, pHe changes may be considerable and add to the acidosis imposed by CO2 and H+-lactate venting (explaining why glycolysis-deficient tumours still generate low pHe [57,58]). Displacements of pHe can slow the transport cycle, either by means of trans inhibition or activation of allosteric sites [51,55,,59]. For instance, acid extrusion by Na+/H+ exchange is inhibited sharply at reduced pHe [60]. In multi-cellular spheroids, this inhibitory effect can be lessened by increasing extracellular mobile buffering capacity [56]. Since CO2/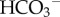 is the principal extracellular mobile buffer, the activity of exofacial CAs can have a substantial impact on the feedback between pHe and pHi-regulating transporters. The sensitivity of at least some pHi regulators to cellular energetics and to pHe may protect cancer cells from ATP depletion and excessive extracellular acidification beyond a point that is more damaging than a partially regulated pHi. In a growing tumour, where diffusion distances and metabolic rate change continuously, these safety checks can be important for the process of somatic evolution which seeks the most viable phenotype.
is the principal extracellular mobile buffer, the activity of exofacial CAs can have a substantial impact on the feedback between pHe and pHi-regulating transporters. The sensitivity of at least some pHi regulators to cellular energetics and to pHe may protect cancer cells from ATP depletion and excessive extracellular acidification beyond a point that is more damaging than a partially regulated pHi. In a growing tumour, where diffusion distances and metabolic rate change continuously, these safety checks can be important for the process of somatic evolution which seeks the most viable phenotype.
5. pH sensing, pH-driven selection and clinical perspectives
Cancer cells must be able to detect and respond to micro-environmental factors so that these can then guide somatic evolution. In the case of hypoxia, HIF is a transducer between O2 tension and cellular effects. The pH sensors involved in cancer disease progression have proved to be more challenging to identify, possibly because of difficulties in distinguishing bona fide pH sensors from the plethora of proteins that bind H+ ions [2]. Cells sensing an alkaline pHi have been shown to proliferate [61], enter the cell cycle [62,63], differentiate [64], migrate [65,66], reduce apoptosis [67] and clastogenesis [68], and undergo malignant transformation [69,70]—events that are critical in cancer formation and metastasis. Considering the complexity of these processes, the observed pHi sensitivity may involve a number of H+-binding molecular switches. pH sensing is not confined to the cytoplasm: H+ sensing G-protein-coupled receptors [71], H+ sensing ion channels (ASICs) [72] and the pH sensitivity of a number of ion channels [73] offer a means by which cells could respond to the pH of the tumour milieu.
Among the titratable sites on proteins, the imidazole group of histidine is an attractive candidate for pH-sensing moieties [74]. Although histidine makes up less than 3% of most proteins, it is commonly found in active or binding sites [75]. The reason for this can be traced to imidazole's pKa of 6.5, which means that even small changes in pHi can greatly affect its ionization state and ability to make salt bridges with other amino acids or prosthetic groups. With its unique chemical properties, histidine does not substitute well with any other amino acid [75]. Considering its prominence in active/binding sites, histidine mutations are expected to alter protein function. Among the three most common missense mutations in the tumour suppressor protein p53, two involve substitutions to histidine (Arg175 → His, Arg275 → His) [76]. A well-described Arg337 → His mutation destabilizes p53 tetramerization and hinders its interaction with DNA because a critical salt bridge with Asp352 is no longer stable at normal pHi [77].
Over the long course of cancer disease progression, cells accumulate genetic changes that are retained if selected positively by the micro-environment [78–80]. If extracellular acidity (a complex derivative of diffusion distance, buffering, metabolic rate and membrane transport) were a major selection pressure (as hinted by its prominence as a cancer hallmark) then at least some mutations are likely to relate to genes or gene regulators for H+/H+-equivalent transporters, pH sensors or proteins involved in acid-yielding metabolic pathways [79]. Poor prognosis for tumours with low pHe may indicate that acidity has identified a population of cells with the appropriate pH sensing and regulatory apparatus necessary to thrive and even become resistant to drugs (e.g. weakly basic drugs such as doxorubicin [81,82]). As cancer cells (and possibly stromal cells [83]) have ultimate control over pHe, the direction and rate of change of this selection pressure can be adapted to optimise disease progression. The higher acid-per-ATP yield of glycolysis, when compared with mitochondrial respiration, may explain the prominence of the Warburg effect in cancer [84]. In summary, the plasticity of pHi regulation and versatility of protein pH sensitivity offer a mechanism for cancer cells to exploit pH as a selection pressure. By contrast, the genetic stability of normal host cells would hinder their adaptability to the micro-environment.
Growing evidence for the importance of pH in cancer biology has solicited many ambitious ideas for therapy targeting pH-handling proteins with low molecular weight inhibitors or monoclonal antibodies [85–87]. The strategy of blocking acid extrusion from cancer cells and allowing intracellular acid to accumulate to lethal levels may be achieved readily in vitro, but efficacy in vivo would need to overcome major obstacles. Firstly, acid–base handling proteins are also important for normal cells and inhibition could lead to unacceptable systemic toxicity. It is therefore essential to identify, at the molecular level, acid–base handling mechanisms in cancer that differ substantially from normal cells. For instance, hypoxia-induced MCT4 and CAIX are associated with tumours. Alternatively, drugs could be tailored chemically to become more efficacious in the tumour milieu, e.g. through chemical activation at low pH and/or O2 tension. A second obstacle is the redundancy in mechanisms for acid extrusion that could compensate for the targeted protein, particularly in cancer cells which have the means to change and adapt dynamically. This raises the question of whether therapy targeted at manipulating pHi will have the desired clinical efficacy. The hypothesis of acid-driven disease progression has highlighted the importance of extracellular pH as a target for therapy. Unlike interventions that alter intracellular pH by targeting one (or more) of many membrane-bound transporter proteins, extracellular pH could be manipulated by altering H+ diffusivity or buffering capacity. Raising mobile buffering capacity with systemic bicarbonate offers an attractive means of changing the course of acid-driven somatic evolution by ablating the underlying selection pressure [88]. The steady flow of new data in support for the prominent role played by H+ ions in cancer will keep pH in the spotlight for novel therapeutic approaches for years to come.
6. Concluding remarks
Tissues can regulate and adapt to a particular pH distribution through a two-way interaction between pH and proteins. By continuously evolving superior phenotypes, cancer cells can exploit this interaction to out-compete host cells and metastasize. As the common denominator of a vast array of chemical reactions and transport processes, it is challenging to understand how the concentration of H+ ions is regulated and sensed. However, the combination of physiological, biochemical, genetic and computational approaches is supplying new ideas on how to exploit the pH/biology interaction in the management of cancer.
Acknowledgements
The authors acknowledge Jian Ping Jen for assistance in performing experiments on T47D spheroid.
Funding statement
Our research is supported by the Royal Society (P.S.), Association for International Cancer Research (P.S., A.L.H.), European Union (FP7 ‘IonTrac’, P.S., R. D.V.J.), British Heart Foundation (R.D.V.J.) and Cancer Research UK (A.L.H.).
References
- 1.Roos A, Boron WF. 1981. Intracellular pH. Physiol. Rev. 61, 296–434. [DOI] [PubMed] [Google Scholar]
- 2.Schonichen A, Webb BA, Jacobson MP, Barber DL. 2013. Considering protonation as a posttranslational modification regulating protein structure and function. Annu. Rev. Biophys. 42, 289–314. ( 10.1146/annurev-biophys-050511-102349) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomlinson RH, Gray LH. 1955. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br. J. Cancer 9, 539–549. ( 10.1038/bjc.1955.55) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vaupel P, Kallinowski F, Okunieff P. 1989. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 49, 6449–6465. [PubMed] [Google Scholar]
- 5.Hockel S, Schlenger K, Vaupel P, Hockel M. 2001. Association between host tissue vascularity and the prognostically relevant tumor vascularity in human cervical cancer. Int. J. Oncol. 19, 827–832. [PubMed] [Google Scholar]
- 6.Harris AL. 2002. Hypoxia—a key regulatory factor in tumour growth. Nat. Rev. Cancer 2, 38–47. ( 10.1038/nrc704) [DOI] [PubMed] [Google Scholar]
- 7.Wang GL, Jiang BH, Rue EA, Semenza GL. 1995. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl Acad. Sci. USA 92, 5510–5514. ( 10.1073/pnas.92.12.5510) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warburg O. 1930. The metabolism of tumours. London, UK: Arnold Constable. [Google Scholar]
- 9.Semenza GL. 2003. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732. ( 10.1038/nrc1187) [DOI] [PubMed] [Google Scholar]
- 10.Wike-Hooley JL, Haveman J, Reinhold HS. 1984. The relevance of tumour pH to the treatment of malignant disease. Radiother. Oncol. 2, 343–366. ( 10.1016/S0167-8140(84)80077-8) [DOI] [PubMed] [Google Scholar]
- 11.Gillies RJ, Liu Z, Bhujwalla Z. 1994. 31P-MRS measurements of extracellular pH of tumors using 3-aminopropylphosphonate. Am. J. Physiol. 267, C195–C203. [DOI] [PubMed] [Google Scholar]
- 12.Tannock IF, Rotin D. 1989. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 49, 4373–4384. [PubMed] [Google Scholar]
- 13.Griffiths JR, Stevens AN, Iles RA, Gordon RE, Shaw D. 1981. 31P-NMR investigation of solid tumours in the living rat. Biosci. Rep. 1, 319–325. ( 10.1007/BF01114871) [DOI] [PubMed] [Google Scholar]
- 14.Kallinowski F, Schlenger KH, Runkel S, Kloes M, Stohrer M, Okunieff P, Vaupel P. 1989. Blood flow, metabolism, cellular microenvironment, and growth rate of human tumor xenografts. Cancer Res. 49, 3759–3764. [PubMed] [Google Scholar]
- 15.Moll W, Gros G. 2008. Combined glycolytic production of lactate(−) and ATP(4−) derived protons (= dissociated lactic acid) is the only cause of metabolic acidosis of exercise—a note on the OH(−) absorbing function of lactate(−) production. J. Appl. Physiol. 105, 365. [DOI] [PubMed] [Google Scholar]
- 16.Clauss MA, Jain RK. 1990. Interstitial transport of rabbit and sheep antibodies in normal and neoplastic tissues. Cancer Res. 50, 3487–3492. [PubMed] [Google Scholar]
- 17.Hulikova A, Harris AL, Vaughan-Jones RD, Swietach P. 2013. Regulation of intracellular pH in cancer cell lines under normoxia and hypoxia. J. Cell Physiol. 228, 743–752. ( 10.1002/jcp.24221) [DOI] [PubMed] [Google Scholar]
- 18.Boron WF, Endeward V, Gros G, Musa-Aziz R, Pohl P. 2011. Intrinsic CO2 permeability of cell membranes and potential biological relevance of CO2 channels. ChemPhysChem 12, 1017–1019. ( 10.1002/cphc.201100034) [DOI] [PubMed] [Google Scholar]
- 19.Boron WF. 2010. Sharpey–Schafer lecture: gas channels. Exp. Physiol. 95, 1107–1130. ( 10.1113/expphysiol.2010.055244) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halestrap AP, Price NT. 1999. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem. J. 343, 281–299. ( 10.1042/0264-6021:3430281) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hulikova A, Harris AL, Vaughan-Jones RD, Swietach P. 2012. Acid-extrusion from tissue: the interplay between membrane transporters and pH buffers. Curr. Pharm. Des. 18, 1331–1337. ( 10.2174/138161212799504920) [DOI] [PubMed] [Google Scholar]
- 22.Junge W, McLaughlin S. 1987. The role of fixed and mobile buffers in the kinetics of proton movement. Biochim. Biophys. Acta 890, 1–5. ( 10.1016/0005-2728(87)90061-2) [DOI] [PubMed] [Google Scholar]
- 23.Maren TH. 1967. Carbonic anhydrase: chemistry, physiology, and inhibition. Physiol. Rev. 47, 595–781. [DOI] [PubMed] [Google Scholar]
- 24.Svastova E, et al. 2004. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 577, 439–445. ( 10.1016/j.febslet.2004.10.043) [DOI] [PubMed] [Google Scholar]
- 25.Swietach P, Patiar S, Supuran CT, Harris AL, Vaughan-Jones RD. 2009. The role of carbonic anhydrase 9 in regulating extracellular and intracellular pH in three-dimensional tumor cell growths. J. Biol. Chem. 284, 20 299–20 310. ( 10.1074/jbc.M109.006478) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Potter CP, Harris AL. 2003. Diagnostic, prognostic and therapeutic implications of carbonic anhydrases in cancer. Br. J. Cancer 89, 2–7. ( 10.1038/sj.bjc.6600936) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pastorekova S, Parkkila S, Zavada J. 2006. Tumor-associated carbonic anhydrases and their clinical significance. Adv. Clin. Chem. 42, 167–216. ( 10.1016/s0065-2423(06)42005-9) [DOI] [PubMed] [Google Scholar]
- 28.Opavsky R, Pastorekova S, Zelnik V, Gibadulinova A, Stanbridge EJ, Zavada J, Kettmann R, Pastorek J. 1996. Human MN/CA9 gene, a novel member of the carbonic anhydrase family: structure and exon to protein domain relationships. Genomics 33, 480–487. ( 10.1006/geno.1996.0223) [DOI] [PubMed] [Google Scholar]
- 29.Pastorek J, et al. 1994. Cloning and characterization of MN, a human tumor-associated protein with a domain homologous to carbonic anhydrase and a putative helix-loop-helix DNA binding segment. Oncogene 9, 2877–2888. [PubMed] [Google Scholar]
- 30.Tureci O, et al. 1998. Human carbonic anhydrase XII: cDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. Proc. Natl Acad. Sci. USA 95, 7608–7613. ( 10.1073/pnas.95.13.7608) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gros G, Moll W. 1974. Facilitated diffusion of CO2 across albumin solutions. J. Gen. Physiol. 64, 356–371. ( 10.1085/jgp.64.3.356) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geers C, Gros G. 2000. Carbon dioxide transport and carbonic anhydrase in blood and muscle. Physiol. Rev. 80, 681–715. [DOI] [PubMed] [Google Scholar]
- 33.Khalifah RG, Edsall JT. 1972. Carbon dioxide hydration activity of carbonic anhydrase: kinetics of alkylated anhydrases B and C from humans (metalloenzymes-isoenzymes-active sites-mechanism). Proc. Natl Acad. Sci. USA 69, 172–176. ( 10.1073/pnas.69.1.172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alterio V, et al. 2009. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl Acad. Sci. USA 106, 16 233–16 238. ( 10.1073/pnas.0908301106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McIntyre A, et al. 2012. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clin. Cancer Res. 18, 3100–3111. ( 10.1158/1078-0432.CCR-11-1877) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swietach P, Wigfield S, Cobden P, Supuran CT, Harris AL, Vaughan-Jones RD. 2008. Tumor-associated carbonic anhydrase 9 spatially coordinates intracellular pH in three-dimensional multicellular growths. J. Biol. Chem. 283, 20 473–20 483. ( 10.1074/jbc.M801330200) [DOI] [PubMed] [Google Scholar]
- 37.Chiche J, Ilc K, Laferriere J, Trottier E, Dayan F, Mazure NM, Brahimi-Horn MC, Pouyssegur J. 2009. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 69, 358–368. ( 10.1158/0008-5472.CAN-08-2470) [DOI] [PubMed] [Google Scholar]
- 38.Hallerdei J, Scheibe RJ, Parkkila S, Waheed A, Sly WS, Gros G, Wetzel P, Endeward V. 2010. T tubules and surface membranes provide equally effective pathways of carbonic anhydrase-facilitated lactic acid transport in skeletal muscle. PLoS ONE 5, e15137 ( 10.1371/journal.pone.0015137) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Swietach P, Tiffert T, Mauritz JM, Seear R, Esposito A, Kaminski CF, Lew VL, Vaughan-Jones RD. 2010. Hydrogen ion dynamics in human red blood cells. J. Physiol. 588, 4995–5014. ( 10.1113/jphysiol.2010.197392) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boron WF. 2004. Regulation of intracellular pH. Adv. Physiol. Educ. 28, 160–179. ( 10.1152/advan.00045.2004) [DOI] [PubMed] [Google Scholar]
- 41.Thomas RC. 1976. Ionic mechanism of the H+ pump in a snail neurone. Nature 262, 54–55. ( 10.1038/262054a0) [DOI] [PubMed] [Google Scholar]
- 42.Thomas RC. 1976. The effect of carbon dioxide on the intracellular pH and buffering power of snail neurones. J. Physiol. 255, 715–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wakabayashi S, Shigekawa M, Pouyssegur J. 1997. Molecular physiology of vertebrate Na+/H+ exchangers. Physiol. Rev. 77, 51–74. [DOI] [PubMed] [Google Scholar]
- 44.Romero MF, Fulton CM, Boron WF. 2004. The SLC4 family of HCO3− transporters. Pflugers Arch. 447, 495–509. ( 10.1007/s00424-003-1180-2) [DOI] [PubMed] [Google Scholar]
- 45.Hoffmann EK, Lambert IH, Pedersen SF. 2009. Physiology of cell volume regulation in vertebrates. Physiol. Rev. 89, 193–277. ( 10.1152/physrev.00037.2007) [DOI] [PubMed] [Google Scholar]
- 46.Lauritzen G, et al. 2012. The Na+/H+ exchanger NHE1, but not the Na+, HCO3− cotransporter NBCn1, regulates motility of MCF7 breast cancer cells expressing constitutively active ErbB2. Cancer Lett. 317, 172–183. ( 10.1016/j.canlet.2011.11.023) [DOI] [PubMed] [Google Scholar]
- 47.Lee AH, Tannock IF. 1998. Heterogeneity of intracellular pH and of mechanisms that regulate intracellular pH in populations of cultured cells. Cancer Res. 58, 1901–1908. [PubMed] [Google Scholar]
- 48.Boedtkjer E, et al. 2013. Contribution of Na+, HCO3−-cotransport to cellular pH control in human breast cancer: a role for the breast cancer susceptibility locus NBCn1 (SLC4A7). Int. J. Cancer 132, 1288–1299. ( 10.1002/ijc.27782) [DOI] [PubMed] [Google Scholar]
- 49.Cardone RA, Casavola V, Reshkin SJ. 2005. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 5, 786–795. ( 10.1038/nrc1713) [DOI] [PubMed] [Google Scholar]
- 50.Martinez-Zaguilan R, Lynch RM, Martinez GM, Gillies RJ. 1993. Vacuolar-type H+-ATPases are functionally expressed in plasma membranes of human tumor cells. Am. J. Physiol. 265, C1015–C1029. [DOI] [PubMed] [Google Scholar]
- 51.Stewart AK, Kurschat CE, Vaughan-Jones RD, Alper SL. 2009. Putative re-entrant loop 1 of AE2 transmembrane domain has a major role in acute regulation of anion exchange by pH. J. Biol. Chem. 284, 6126–6139. ( 10.1074/jbc.M802051200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cassel D, Katz M, Rotman M. 1986. Depletion of cellular ATP inhibits Na+/H+ antiport in cultured human cells. Modulation of the regulatory effect of intracellular protons on the antiporter activity. J. Biol. Chem. 261, 5460–5466. [PubMed] [Google Scholar]
- 53.Moolenaar WH, Tsien RY, van der Saag PT, de Laat SW. 1983. Na+/H+ exchange and cytoplasmic pH in the action of growth factors in human fibroblasts. Nature 304, 645–648. ( 10.1038/304645a0) [DOI] [PubMed] [Google Scholar]
- 54.Puceat M, Vassort G. 1995. Neurohumoral modulation of intracellular pH in the heart. Cardiovasc. Res. 29, 178–183. [DOI] [PubMed] [Google Scholar]
- 55.Vaughan-Jones RD, Spitzer KW, Swietach P. 2009. Intracellular pH regulation in heart. J. Mol. Cell Cardiol. 46, 318–331. ( 10.1016/j.yjmcc.2008.10.024) [DOI] [PubMed] [Google Scholar]
- 56.Hulikova A, Vaughan-Jones RD, Swietach P. 2011. Dual role of CO2/HCO3− buffer in the regulation of intracellular pH of three-dimensional tumor growths. J. Biol. Chem. 286, 13 815–13 826. ( 10.1074/jbc.M111.219899) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Helmlinger G, Sckell A, Dellian M, Forbes NS, Jain RK. 2002. Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin. Cancer Res. 8, 1284–1291. [PubMed] [Google Scholar]
- 58.Newell K, Franchi A, Pouyssegur J, Tannock I. 1993. Studies with glycolysis-deficient cells suggest that production of lactic acid is not the only cause of tumor acidity. Proc. Natl Acad. Sci. USA 90, 1127–1131. ( 10.1073/pnas.90.3.1127) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Niederer SA, Swietach P, Wilson DA, Smith NP, Vaughan-Jones RD. 2008. Measuring and modeling chloride-hydroxyl exchange in the Guinea-pig ventricular myocyte. Biophys. J. 94, 2385–2403. ( 10.1529/biophysj.107.118885) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vaughan-Jones RD, Wu ML. 1990. Extracellular H+ inactivation of Na+-H+ exchange in the sheep cardiac Purkinje fibre. J. Physiol. 428, 441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pouyssegur J, Chambard JC, Franchi A, Paris S, Van Obberghen-Schilling E. 1982. Growth factor activation of an amiloride-sensitive Na+/H+ exchange system in quiescent fibroblasts: coupling to ribosomal protein S6 phosphorylation. Proc. Natl Acad. Sci. USA 79, 3935–3939. ( 10.1073/pnas.79.13.3935) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Putney LK, Barber DL. 2003. Na-H exchange-dependent increase in intracellular pH times G2/M entry and transition. J. Biol. Chem. 278, 44 645–44 649. ( 10.1074/jbc.M308099200) [DOI] [PubMed] [Google Scholar]
- 63.Turchi L, Loubat A, Rochet N, Rossi B, Ponzio G. 2000. Evidence for a direct correlation between c-Jun NH2 terminal kinase 1 activation, cyclin D2 expression, and G(1)/S phase transition in the murine hybridoma 7TD1 cells. Exp. Cell Res. 261, 220–228. ( 10.1006/excr.2000.5060) [DOI] [PubMed] [Google Scholar]
- 64.Wang H, Singh D, Fliegel L. 1997. The Na+/H+ antiporter potentiates growth and retinoic acid-induced differentiation of P19 embryonal carcinoma cells. J. Biol. Chem. 272, 26 545–26 549. ( 10.1074/jbc.272.42.26545) [DOI] [PubMed] [Google Scholar]
- 65.Klein M, Seeger P, Schuricht B, Alper SL, Schwab A. 2000. Polarization of Na+/H+ and Cl−/HCO3− exchangers in migrating renal epithelial cells. J. Gen. Physiol. 115, 599–608. ( 10.1085/jgp.115.5.599) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lagana A, Vadnais J, Le PU, Nguyen TN, Laprade R, Nabi IR, Noel J. 2000. Regulation of the formation of tumor cell pseudopodia by the Na+/H+ exchanger NHE1. J. Cell Sci. 113, 3649–3662. [DOI] [PubMed] [Google Scholar]
- 67.McConkey DJ, Orrenius S. 1996. Signal transduction pathways in apoptosis. Stem Cells 14, 619–631. ( 10.1002/stem.140619) [DOI] [PubMed] [Google Scholar]
- 68.Morita T, Nagaki T, Fukuda I, Okumura K. 1992. Clastogenicity of low pH to various cultured mammalian cells. Mutat. Res. 268, 297–305. ( 10.1016/0027-5107(92)90235-T) [DOI] [PubMed] [Google Scholar]
- 69.Reshkin SJ, et al. 2000. Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypes. FASEB J. 14, 2185–2197. ( 10.1096/fj.00-0029com) [DOI] [PubMed] [Google Scholar]
- 70.Gillies RJ, Martinez-Zaguilan R, Martinez GM, Serrano R, Perona R. 1990. Tumorigenic 3T3 cells maintain an alkaline intracellular pH under physiological conditions. Proc. Natl Acad. Sci. USA 87, 7414–7418. ( 10.1073/pnas.87.19.7414) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ludwig MG, Vanek M, Guerini D, Gasser JA, Jones CE, Junker U, Hofstetter H, Wolf RM, Seuwen K. 2003. Proton-sensing G-protein-coupled receptors. Nature 425, 93–98. ( 10.1038/nature01905) [DOI] [PubMed] [Google Scholar]
- 72.Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. 1997. A proton-gated cation channel involved in acid-sensing. Nature 386, 173–177. ( 10.1038/386173a0) [DOI] [PubMed] [Google Scholar]
- 73.Glitsch M. 2011. Protons and Ca2+: ionic allies in tumor progression? Physiology (Bethesda) 26, 252–265. ( 10.1152/physiol.00005.2011) [DOI] [PubMed] [Google Scholar]
- 74.Srivastava J, Barber DL, Jacobson MP. 2007. Intracellular pH sensors: design principles and functional significance. Physiology (Bethesda) 22, 30–39. ( 10.1152/physiol.00035.2006) [DOI] [PubMed] [Google Scholar]
- 75.Betts MJ, Barnes MR, Gray IC. 2013. Amino acid properties and consequences of substitition. In Bioinformatics for geneticists (eds Barnes MR, Gray IC.), pp. 289–316. Chichester, UK: John Wiley & Sons. [Google Scholar]
- 76.Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. 2007. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene 26, 2157–2165. ( 10.1038/sj.onc.1210302) [DOI] [PubMed] [Google Scholar]
- 77.DiGiammarino EL, Lee AS, Cadwell C, Zhang W, Bothner B, Ribeiro RC, Zambetti G, Kriwacki RW. 2002. A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nat. Struct. Biol. 9, 12–16. ( 10.1038/nsb730) [DOI] [PubMed] [Google Scholar]
- 78.Nowell PC. 1976. The clonal evolution of tumor cell populations. Science 194, 23–28. ( 10.1126/science.959840) [DOI] [PubMed] [Google Scholar]
- 79.Fang JS, Gillies RD, Gatenby RA. 2008. Adaptation to hypoxia and acidosis in carcinogenesis and tumor progression. Semin. Cancer Biol. 18, 330–337. ( 10.1016/j.semcancer.2008.03.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gillies RJ, Verduzco D, Gatenby RA. 2012. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat. Rev. Cancer 12, 487–493. ( 10.1038/nrc3298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Raghunand N, et al. 1999. Enhancement of chemotherapy by manipulation of tumour pH. Br. J. Cancer 80, 1005–1011. ( 10.1038/sj.bjc.6690455) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gerweck LE, Kozin SV, Stocks SJ. 1999. The pH partition theory predicts the accumulation and toxicity of doxorubicin in normal and low-pH-adapted cells. Br. J. Cancer 79, 838–842. ( 10.1038/sj.bjc.6690134) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Koukourakis MI, Giatromanolaki A, Harris AL, Sivridis E. 2006. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor-associated stroma. Cancer Res. 66, 632–637. ( 10.1158/0008-5472.CAN-05-3260) [DOI] [PubMed] [Google Scholar]
- 84.Gatenby RA, Gillies RJ. 2004. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 4, 891–899. ( 10.1038/nrc1478) [DOI] [PubMed] [Google Scholar]
- 85.Le Floch R, et al. 2011. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc. Natl Acad. Sci. USA 108, 16 663–16 668. ( 10.1073/pnas.1106123108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fais S, De Milito A, You H, Qin W. 2007. Targeting vacuolar H+-ATPases as a new strategy against cancer. Cancer Res. 67, 10 627–10 630. ( 10.1158/0008-5472.CAN-07-1805) [DOI] [PubMed] [Google Scholar]
- 87.Neri D, Supuran CT. 2011. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 10, 767–777. ( 10.1038/nrd3554) [DOI] [PubMed] [Google Scholar]
- 88.Estrella V, et al. 2013. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 73, 1524–1535. ( 10.1158/0008-5472.CAN-12-2796) [DOI] [PMC free article] [PubMed] [Google Scholar]