

Abstract
Although ion channels are increasingly being discovered in cancer cells in vitro and in vivo, and shown to contribute to different aspects and stages of the cancer process, much less is known about the mechanisms controlling their expression. Here, we focus on voltage-gated Na+ channels (VGSCs) which are upregulated in many types of carcinomas where their activity potentiates cell behaviours integral to the metastatic cascade. Regulation of VGSCs occurs at a hierarchy of levels from transcription to post-translation. Importantly, mainstream cancer mechanisms, especially hormones and growth factors, play a significant role in the regulation. On the whole, in major hormone-sensitive cancers, such as breast and prostate cancer, there is a negative association between genomic steroid hormone sensitivity and functional VGSC expression. Activity-dependent regulation by positive feedback has been demonstrated in strongly metastatic cells whereby the VGSC is self-sustaining, with its activity promoting further functional channel expression. Such auto-regulation is unlike normal cells in which activity-dependent regulation occurs mostly via negative feedback. Throughout, we highlight the possible clinical implications of functional VGSC expression and regulation in cancer.
Keywords: voltage-gated sodium channel, metastasis, activity-dependent regulation, growth factor, hormone
1. Introduction
It is now well established that de novo expression of voltage-gated ion channels (VGICs) occurs in cancers in vitro and in vivo and plays a significant role in disease initiation and progression [1–6]. In particular, voltage-gated Na+ channels (VGSCs) are functionally expressed in many types of carcinomas (cancers of epithelial origin), including those of breast, cervix, colon, lung (small-cell, non-small-cell and mesothelioma), skin, ovary and prostate, where they promote disease progression, potentially leading to metastasis [7]. On the other hand, voltage-gated K+ channels (VGPCs) commonly control cellular proliferation in which VGSC activity plays no role [8,9]. In addition, VGPCs may be downregulated as cancer becomes more aggressive [2,8]. Such dichotomy in VGSC–VGPC expression in cancer is consistent with the notion that primary and secondary tumours are associated with expression of different genes and can even be controlled independently [10].
Expression and/or activity of VGICs can be regulated from transcription to post-translation. These include specific mechanisms of post-transcription/pre-translation, e.g. miRNAs [11] and post-translation, e.g. intracellular trafficking [1,2,6]. The primary regulators include hormones (mainly steroids and peptides) and growth factors which, in fact, are all intimately associated with various aspects and stages of the cancer process. In addition, some drugs used for cancer treatment may affect VGICs [12,13]. Here, we focus on the regulation of VGSCs which comprise a multi-gene family of at least nine different functional members (Nav1.1–1.9) coding for the pore-forming α-subunits (figure 1) [14]. There are also four auxiliary β-subunits of which one or two at a time can associate with an α-subunit and modulate channel expression and activity in plasma membrane [14].
Figure 1.

Schematic diagram of the structure and membrane topology of the voltage-gated sodium channel showing the main regulatory sites. Given α-subunits have four domains (DI–DIV) each composed of six transmembrane segments. Within the latter, segment four contains positively charged amino acids and this is the main voltage-sensitive region; the loop between transmembrane segments 5 and 6 is negatively charged and forms the pore region. Many types of modulatory sites exist for both α- and β-subunits as indicated by the key. The boxes adjacent to α- and β-subunits list the proteins known to interact with each subunit, respectively. PDZ, post-synaptic density protein (PSD95) and Drosophila disc large tumour suppressor (Dlg1) and zonula occludens-1 protein (zo-1); ER, endoplasmic reticulum; RXR, a motif which mediates retention of proteins in the ER. (Online version in colour.)
Several individual Nav isoforms are known to be functionally expressed in different human cancers. These include Nav1.5 in breast and colon cancers [8,15,16]; Nav1.6 in cervical cancer [17,18]; and Nav1.7 in breast, prostate and non-small cell lung cancers [8,19,20]. In addition, where studied (especially for Nav1.5 and Nav1.7), alternative splice variants of the isoforms have been found with sequence differences most notably in the extracellular loop between segments 3 and 4 of domain I [8,19,21]. At present, the rationale for such a phenomenon to occur in cancer is unclear, but consistent with channel expression being both (i) epigenetic [22] and (ii) oncofetal [23]. The mechanisms controlling alternative splicing also are not well characterized but could involve cAMP [24] and activity-dependence [25].
In the case of breast cancer (BCa), the oncofetal/neonatal isoform of Nav1.5 (nNav1.5) would allow greater Na+ entry into the cell, compared with the adult splice variant [26]. In turn, this could have implications for Na+–H+ exchange and intra/extracellular pH regulation [27]; enzyme activity, e.g. Na+/K+-ATPase and protein kinase A [27,28]; and Ca2+ homeostasis, e.g. Na+–Ca2+ exchange [29]. In addition, the charge-reversing aspartate (negative) to lysine (positive) amino acid change that occurs at position 211 could affect (i) response to extracellular (intrinsic or extrinsic) chemical factors, e.g. pH [30] and (ii) protein–protein interactions [31].
VGSC activity has been shown to contribute to many cell behaviours that may be important for metastasis. These include migration [32–34]; invasion [8,15,16,18,20,34–38]; colony formation in three-dimensional Matrigel [39]; process extension [40]; galvanotaxis; [8,41]; adhesion [42]; gene expression [43]; endocytic membrane activity [44–46]; vesicular patterning [47]; nitric oxide production [48]; and invadopodia formation [49]. Although the mechanisms underlying the involvement of VGSCs in such cell behaviours are not well characterized, several suggestions have been made. These include (i) interaction with cytoskeletal elements (and/or β-subunits) and (ii) modulation of ion fluxes/exchangers, gene expression and enzyme activity [8,18,32,39,49,50]. However, only a few specific mechanisms have been shown directly to be involved in the proposed role of VGSCs in metastasis-associated cellular behaviours. These mechanisms include PKA [51] and Na+/H+ exchanger [39,49]. Importantly, also, SCN5A (the gene that encodes Nav1.5) was deduced to be upstream of a network of genes/signalling cascades controlling human colon cancer invasiveness [16]. Thus, VGSCs would appear to play a significant role in cancer progression and are a potential novel therapeutic target against metastasis, the main cause of death in cancer patients [5,7,8,52].
Our overall approach here is to review, wherever possible, the available evidence for regulation of VGSC expression/activity in cancer cells. However, for completeness, we also cover some essential non-cancer examples.
2. Hormones
The development and/or progression of many cancers are well known to be hormone-dependent; hormone independence may develop during treatment, whereupon the cancer may become more aggressive [53]. Some cancers show particularly strong hormone sensitivity owing to the inherent nature of the native tissue, as for BCa and prostate cancer (PCa) for which oestrogen and androgen are key steroid hormones, respectively. Consequently, such cancers are commonly treated with hormone-based medication. A wide range of signalling mechanisms and cascades are associated with hormone action upon VGSCs, as illustrated in figure 2a.
Figure 2.

Summary diagrams showing possible regulatory pathways controlling voltage-gated sodium channel (VGSC) expression/activity in cells by (a) hormones and (b) growth factors. Some pathways mediate transcriptional activity; some actions are through the respective receptors, whereas some actions are directly on the channel. Some of the regulatory pathways have only been described in normal cells. Abbreviations are defined in text. VGSC↓ and VGSC↑ denote decreased and increased VGSC expression/activity, respectively. (Online version in colour.)
(a). Oestrogen
The effects of β-oestradiol (E2), the biologically active form of oestrogen, are classically mediated by two types of oestrogen receptor (ER): ERα and ERβ, which belong to the superfamily of ligand-activated transcription factors (‘nuclear receptors’). Like other members of this group, ERs function as dimers, which when bound to their ligand, undergo a translocation from the cytoplasm to the nucleus where they recognize specific sequences of DNA-response elements in the promoter regions of the target genes, ultimately resulting in their up- or downregulation [54]. However, non-transcription modes of ER action also occur. Thus, ERα can be localized at the plasma membrane in multi-protein complexes and mediate fast, non-genomic effects; several possible variants of such receptors exist including ER46, ERα-36. In addition, a novel transmembrane G-protein-coupled ER (GPER), previously called ‘GPR30’ also mediates fast, non-genomic effects [55].
A basal ‘negative’ association between expression of ERα and VGSC is apparent in human BCa cells. Thus, the strongly metastatic MDA-MB-231 cells are devoid of classic ERα, but express a functional VGSC, in particular nNav1.5 [8]. Conversely, the weakly/non-metastatic MCF-7 cells are ERα-positive and do not express any functional VGSC [8]. Additional data were obtained from MDA-MB-231 cells stably transfected with functional ERα (MDA-MB-231-ERα cells) [56]. In these cells, compared with controls expressing only the plasmid vector (VC5), there was a significant decrease in nNav1.5 mRNA levels by 96 ± 2% (figure 3a). Consistent with this, the proportion of cells expressing functional VGSCs (nNav1.5) was reduced from 71 to 43% (p < 0.05; n = 10–15 cells) [57]. Furthermore, treatment of the MDA-MB-231-ERα cells with the ER antagonist ICI-182 780 for more than 48 h produced the following significant effects: (i) the nNav1.5 mRNA level was increased by 211 ± 49% (figure 3b); (ii) the proportion of cells expressing functional VGSCs was increased by more than three-fold (figure 3c); and (iii) the lateral motility of the cells was increased by 23 ± 5% (figure 3d).
Figure 3.

Effects of oestrogen receptor α (ERα) on voltage-gated sodium channel (VGSC) expression and activity in MDA-MB-231 cells transfected with ERα (MDA-MB-231-ERα cells). (a) Basal levels of nNav1.5 mRNA were significantly lower in MDA-MB-231-ERα cells (ERα) compared with control cells expressing only the plasmid vector (VC5; p < 0.001; n = 4). (b) Treatment of MDA-MB-231-ERα cells with the ER antagonist ICI-182,780 (ICI; 1 µM) for more than 72 h significantly increased the nNav1.5 mRNA level, compared with non-treated cells (Cntl; p < 0.001; n = 4). (c) Similar treatment with ICI-182 780 for more than 48 h significantly increased the number of cells with VGSC activity, in comparison with those grown in normal medium (5% FBS–Cntl) as determined by patch-clamp recording (Fisher's exact test: p < 0.001; n = 73 cells). (d) Treatment of MDA-MB-231-ERα cells with ICI-182,780 for more than 72 h significantly increased the lateral motility of the cells (p < 0.01; n = 4). Further information is given in the supplementary Methods section.
Taken together, these results suggest that steady-state VGSC expression is upregulated transcriptionally in the absence of ERα expression/activity, in general agreement with ERα-negative (VGSC-expressing) cases of BCa being more aggressive [58]. Comparable data were obtained from ERα-knockout mice where mRNA expression of most Nav isoforms studied was upregulated [59].
However, MDA-MB-231 cells do express a ‘cell-surface’ ER. Thus, acute (10 s), extracellular application of E2 (10 nM) increased nNav1.5 current, via GPER coupled to PKA activation, leading to a reduction in cellular adhesiveness [51]. Acute application of E2 (10 nM) also increased VGSC currents in rat hypothalamus by a non-genomic mechanism [60]. By contrast, E2 inhibited VGSC currents in cultured N1E-115 mouse neuroblastoma cells [61]. Similarly, mouse dorsal root ganglion (DRG) neuronal VGSCs were inhibited by acute application of E2 and this occurred via a cell-surface ER-activated protein kinase C (PKC)–PKA pathway [62]. Thus, the quick effects of E2 on VGSCs may involve different intracellular signalling cascades, dependent on cell type.
The functional association of ERα/E2 with VGSCs is of clinical significance, because a significant body of evidence suggests that clinically prescribed ‘selective oestrogen receptor modulators’ (SERMs) can also affect VGSCs. Thus, raloxifene (‘Evista’) inhibited the VGSC current in guinea pig ventricular myocytes [12]. Tamoxifen also inhibited VGSC activity in the SHG-44 glioma cell line [13] and in rat cortical neurons [63]. Similar effects of SERMs were seen on rodent hypothalamic neurons [64] and ventricular myocytes [65].
In conclusion, (i) E2 has both non-genomic and genomic effects upon VGSC expression/activity; and (ii) transcriptionally, E2 (via ERα) downregulates functional VGSC (nNav1.5) expression in BCa cells. Some of these effects may manifest clinically in hormone-based treatment of patients and may impact upon the treatment itself. For example, drug-induced blockage of VGSCs may impair nerve and/or muscle activity. More work is needed (i) to generalize these notions; (ii) to improve our understanding of the role of ERβ; (iii) to elucidate whether the effects differ between cancer and corresponding normal cells; and (iv) to integrate the fast non-genomic effects with long-term genomic regulation [66].
(b). Androgen
Two isoforms of androgen receptor (AR) have been identified: AR-A, a 87-kDa protein with 187 amino acids cleaved from the amino-terminal domain and AR-B (110 kDa) which is considered the full-length receptor [54]. Little is known regarding the effects of androgen on VGSCs in cancer cells. As in the case of BCa cell lines, however, there is a negative association between basal expression of AR and VGSC in PCa cell lines. Thus, VGSC (Nav1.7) activity occurs in strongly metastatic, AR-devoid PC-3/M cells while AR-expressing, weakly/non-metastatic LNCaP cells do not possess functional VGSCs [19]. Application of (5α,17β)-17-hydroxy-androstan-3-one dihydrotestosterone (DHT) to androgen-deprived LNCaP cells decreased VGSC β1 mRNA expression over 24–48 h [67]. Surprisingly, DHT treatment also significantly (but transiently) increased both β1 and Nav1.7 mRNAs in PC-3 cells, presumably through a non-AR-dependent mechanism [67]. Such an effect could be mediated via a ‘cross-activated’ growth factor receptor signalling cascade [68]. In neuroblastoma ND7 cells, a nuclear interaction between the developmentally regulated transcription factor Brn-3a and AR resulted in a complex which bound to multiple elements within the promoter region of SCN9A (Nav1.7) and upregulated channel expression [69]. In differentiating mouse muscle C2 cells, VGSC currents were reduced by androgen treatment and abolished when AR was overexpressed; there was no change in VGSC mRNA level, suggesting that the inhibition was post-transcriptional [70].
In conclusion, from the limited available evidence, the effects of androgens on VGSC expression/activity appear complex. On the whole, however, the steady-state association is negative in PCa (as in BCa), again in line with hormone-insensitive (VGSC-expressing) cases of PCa being relatively more aggressive.
(c). Other hormones
Several other cancer-associated hormones also affect VGSC expression/activity. Insulin is a peptide hormone and its receptor (InsR), a tyrosine kinase, can occur in two alternatively spliced isoforms: InsR-A and InsR-B; the former is the predominant isoform in fetal life and is also the main subtype expressed in cancer as an ‘oncofetal’ phenomenon [71]. In InsR-expressing human BCa MDA-MB-231 cells, addition of insulin (in serum-free medium) increased cellular migration [72]. Interestingly, blocking VGSC activity by applying tetrodotoxin (TTX) in the presence of insulin increased, rather than decreased, migration implying that insulin additionally controlled the functional ‘coupling’ between VGSC activity and the cells’ motility [72]. Insulin has been shown to affect VGSC expression/activity also in non-cancer cells. In cultured bovine adrenal chromaffin cells, insulin increased cell-surface expression of VGSC (Nav1.7) and β1 via PI3K; the transcriptional pathway involved phosphorylation of glycogen synthase kinase-3β (GSK-3β), and downregulation of InsR substrates 1 and 2 and Akt [73]. In cardiomyocytes, the transcription factor ‘NForkhead box O 1’, which shares conserved DNA sequences with the insulin responsive element, regulated Nav1.5 expression by directly binding to the SCN5A promoter and inhibiting transcriptional activity [74]. Ion channels may also be modulated by glucocorticoids [75]. As regards VGSCs, serum- and glucocorticoid-inducible kinase 1 (SGK1) upregulated Nav1.5 expressed in frog oocytes [76]. The effect could involve channel phosphorylation and ubiquitin ligase Nedd4 [76,77]. In addition, although progesterone was shown to inhibit VGSC currents in cultured N1E-115 mouse neuroblastoma cells, the high IC50 of this effect (17 µM) would make the result rather uncertain [61]. The VGSC current in rat striatal neurons was also inhibited by microM progesterone, and this was thought to occur via a plasma membrane receptor [78]. It would be worthwhile repeating and extending these experiments, because progesterone is known to be involved in many cancers [79].
3. Growth factors
Growth factors (GFs) are well known to be involved in cancer initiation and progression [80]. Consequently, GF receptors and their associated signalling mechanisms are major targets for cancer therapy [81]. In general, GFs signal via their respective receptor tyrosine kinases (RTKs) which, in humans, comprise some 20 subfamilies [80]. Binding of GF activates an RTK by inducing receptor dimerization, but some may form oligomers in the absence of the activating ligand [80]. Dimerization results in the activation of the intracellular tyrosine kinase domains, in turn, triggering signalling pathways that can include JAK/STAT, MAP kinase and PI3 kinase [80]. The signalling mechanisms and cascades specifically associated with GF effects upon VGSCs are summarized in figure 2b.
(a). Epidermal growth factor
Epidermal growth factor (EGF) commonly regulates ion channel, including VGSC, expression in neurons and muscles [82–85]. In rat and human PCa cells, EGF upregulated functional VGSC (Nav1.7) expression which, in turn, promoted motility, endocytic membrane activity and invasion [86,87]. Similarly, in human BCa cells, EGF upregulated functional Nav1.5 expression through a PI3K-dependent signalling cascade [88]. In an extensive study on the human non-small-cell lung carcinoma (NSCLC) cell line H460, EGF upregulated functional Nav1.7 expression transcriptionally via ERK1/2 and increased Matrigel invasiveness (figure 4) [89]. Importantly, the EGF-induced increase in the invasiveness was blocked completely by silencing Nav1.7 expression, i.e. the effect of EGF on invasion was mediated solely via the channel upregulation [89].
Figure 4.

Upregulation of functional expression of Nav1.7 in human non-small-cell lung cancer H460 cells and consequent increase in invasiveness via ERK1/2 signalling. (a) Current–voltage (I–V) plots for control/untreated cells (open squares) and cells treated in the presence of serum for 24 h with 100 ng ml−1 EGF (closed squares), 1 μM gefitinib/Gef (open circles) or 10 μg ml−1 EGF receptor blocking antibody (filled circles). Currents were evoked using 30 ms depolarizing steps in 5 mV intervals (−90 to +70 mV) from a holding potential of −90 mV. (b) I–V plots for control/untreated cells (open squares) and cells treated with 10 μM U0126 (closed squares). (c) Relative Nav1.7 mRNA expression showing effect of serum starvation for 48 h and treatment for 24 h with EGF (100 ng ml−1), Gef (1 μM) and co-application of EGF + Gef. (d) Matrigel invasion measured after 48 h in control medium (CTL), 0.5 μM TTX, 100 ng ml−1 EGF, 1 μM Gef and EGF + TTX. (e) Effects of treatment with 10 μM U0126 or 100 nM wortmannin (WORT) for 24 h on relative Nav1.7 mRNA expression, compared with control/untreated (CTL) cells. (f) Matrigel invasion measured over 48 h in control/untreated cells (CTL), and following treatment with 10 μM U0126, 10 μM U0126/1 μM TTX, 100 nM WORT, and WORT + TTX. (g) Proposed model for EGF-mediated upregulation of Nav1.7 and consequent invasiveness of H460 cells. Stimulation of EGFR with EGF results in increased functional expression of Nav1.7 via ERK1/2. Following transcription and translation, the mature Nav1.7 protein is trafficked to the cell surface where it becomes functional. At the resting membrane potential, VGSCs are partially activated but not fully inactivated, resulting in a basal influx of Na+. This increase in [Na+]i then drives cell invasion through an, as yet, unknown mechanism. All data are presented as means ± s.e. (n = 6–13). Statistical analyses were with Student's t-test or one-way ANOVA and Student–Newman–Keuls correction, as appropriate; significance: *p < 0.05, **p < 0.01, ***p < 0.001. Adapted from [89]. (Online version in colour.)
In conclusion, the effects of EGF on carcinoma cell lines studied to date are consistent with the following basic scheme: EGF → VGSC upregulation (transcriptional and functional) → increased metastatic cell behaviours.
(b). Insulin-like growth factor
Incubation of MDA-MB-231 cells for 24 h with AG1024, an inhibitor of the insulin-like growth factor-1 receptor (IGF-1R) had no effect on the VGSC (nNav1.5) peak current density [90]. In addition, in cultured rat hippocampal neurons, IGF-1 had no effect on the amplitude of the VGSC currents [91]. However, in cultured bovine adrenal chromaffin cells, chronic application of IGF-1 upregulated plasma membrane expression of Nav1.7; this was a transcriptional effect involving the PI3K-Akt pathway and GSK-3β inhibition [92]. Taken together, these findings suggested a positive feedback loop between Nav1.7 expression and GSK-3β inhibition. Further work is required (i) to determine the possible role of IGF-1R signalling in VGSC expression/activity in cancer cells, and (ii) to elucidate the possible overlap between the IGF-1R and InsR signalling in this.
(c). Nerve growth factor
Long-term (more than 24 h) treatment with nerve growth factor (NGF) upregulated Nav1.7 functional expression in the strongly metastatic MAT-LyLu rat PCa cell line; acute application had no effect [93]. The action of NGF was suppressed by both the pan-tropomyosin-related receptor tyrosine kinases (Trk) antagonist K252a, and the PKA inhibitor KT5720, suggesting (i) that it was receptor mediated, and (ii) that PKA was a signalling intermediate [93]. Indeed, the SCN9A promoter in human PCa PC-3 cells was shown to be activated by NGF [94]. While it appears that NGF can affect both VGSC expression and cancer cell motility, further work is required to clarify the connection between the two effects and the nature of the associated receptor and downstream signalling mechanisms. The in vivo relevance of these findings also remains to be determined. Interestingly, it has recently been reported that progression of PCa in mice is accelerated by activity in the impinging autonomic nerves [95]. An intriguing question is whether such activity would release NGF from the nerves and thus impact upon the cancer VGSCs [96].
(d). Vascular endothelial growth factor
Vascular endothelial growth factor (VEGF) plays a key role in angiogenesis [97] which also involves invasive cell behaviour [98]. Not surprisingly, therefore, VEGF has been shown to affect VGSC expression/activity in several cell types. Thus, VEGF increased the invasiveness of the cervical cancer cell line, ME180, by upregulating Nav1.6 expression via p38 MAPK signalling [99]. In human umbilical vein endothelial cells (HUVECs), VGSC (Nav1.5 and Nav1.7) activity promoted several kinds of angiogenic cell behaviour, including chemotaxis and tubular differentiation; Nav1.5 potentiated VEGF-induced ERK1/2 activation through the PKCα-B-Raf signalling axis, possibly through membrane depolarization, influx of Na+, reverse-mode Na+–Ca2+ exchange and rise in intracellular Ca2+ [100,101]. In cultured rat hippocampal neurons, VEGF decreased VGSC availability [102], whereas in bladder DRG neurons, VEGF upregulated VGSC expression leading to increased excitability [103]. Thus, VEGF can have mixed effects on VGSC expression/activity in various cell types, and it would be worthwhile to perform further studies on cancer cells and cancer-associated endothelia.
(e). Other growth factors
Other GFs are also associated with cancer development and/or VGSC expression/activity, and two are worthy of highlighting. Fibroblast growth factors (FGFs) can be classified as secretory (FGF1–10 and FGF15–23) or intracellular/non-secretory (FGF11–14) [104]. At present, there are no published data on the possible effect of FGF(s) on VGSC(s) expressed in cancer cells. However, work on other cell types suggests that intracellular FGFs co-localize and interact directly with VGSCαs to produce a range of modulatory effects [105,106]. Transforming growth factor-β1 (TGF-β1) is a member of a superfamily of proteins that includes bone morphogenetic proteins, activins and inhibins. Activin A increased VGSC currents in the neuroblastoma Neuro 2a cell line [107]. In adult cardiomyocytes, TGF-β1 increased VGSC (Nav1.5) activity and, concomitantly, reduced the outward current [108]. This observation is reminiscent of the ‘cellular excitability’ hypothesis of cancer progression, i.e. concurrent upregulation of VGSC and downregulation of VGPC activities [2]. However, nearly opposite effects were seen on neonatal cardiomyocytes [109]. Such contrasting effects may be caused, at least partially, by changes in gene expression during development and may relate to the oncofetal nature of VGSC expression in cancer. It would be interesting to test the effects of TGF-β1 on expression/activity of VGSCs and VGPCs in cancer cells.
In overall conclusion, several GFs can affect VGSC expression and activity in a variety of cell types, including cancer and cancer-associated cells. Importantly, the multiplicity of GF regulation of VGSCs raises the possibility that it may be clinically more advantageous to block the VGSC (the ‘hub’) rather than the individual GF pathways because the latter can cross-talk (even with hormonal pathways) and compensate for each other (figure 5) [110,111].
Figure 5.
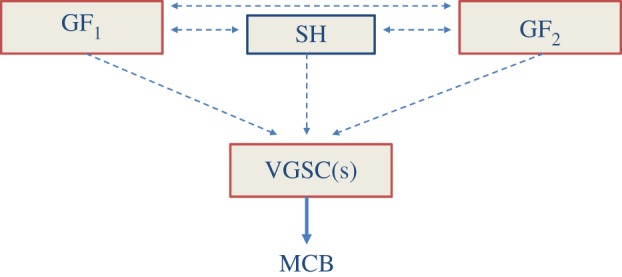
A ‘conceptual’ scheme showing how growth factors (GF1, GF2, etc.) and steroid hormone (SH) signalling systems can feed through and compensate for each other in regulating expression/activity of the VGSC(s). In turn, VGSC activity enhances metastatic cell behaviour (MCB). Dotted, horizontal lines denote the interactive pathways, involving mostly the intracellular signalling cascades. (Online version in colour.)
4. Auto-regulation
Auto-regulation of VGSCs may occur in either of two ways: dependent upon channel activity and association with β1 subunits.
(a). Activity-dependent regulation
Activity-dependent regulation of ion channel function is well known to occur in the central nervous system and is critical for correct neuronal development, wiring and plasticity [112]. In particular, expression of neuronal VGSCα is often regulated by negative feedback, such that patterned activity or chronic treatment with VGSC openers leads to reduction of mRNA/protein expression and change in the balance of intracellular trafficking in favour of channel internalization [113–115]. Conversely, treatment with VGSC blockers increases functional protein expression at the plasma membrane [116]. Thus, steady-state VGSC expression is normally tightly regulated in order to optimize activity and avoid hyper-excitability.
Notably, however, a different situation appears to exist in cancer cells. In strongly metastatic rat PCa MAT-LyLu cells, expression of Nav1.7 was found to be maintained by positive feedback [28]. Thus, chronic pre-treatment of MAT-LyLu cells with TTX inhibited the VGSC current (INa) which persisted after drug washout; this suppression could be reversed with the Na+ ionophore monensin, implying that a rise in intracellular Na+ was involved. In addition, TTX reduced PKA phosphorylation, and the PKA inhibitor KT5720 also reduced INa, whereas the adenylate cyclase (AC) activator forskolin increased INa, suggesting that the positive feedback mechanism is dependent on AC/PKA (figure 6a) [28,118,119]. A similar mechanism of auto-regulation was found for nNav1.5 expression in human metastatic BCa MDA-MB-231 cells [34]. Importantly, pre-treatment with TTX eliminated the VGSC-dependent migration in MAT-LyLu cells [28], and suppressed both basal and PKA-induced migration and invasion in MDA-MB-231 cells [34]. These results suggested that inhibition of INa would collapse the positive feedback loop, which would, in turn, reduce the cells’ migratory and invasive capacity. Indeed, it is well established that it is the influx of Na+ through VGSC that is essential for invasion [8,20,39]. In both BCa and NSCLC cell lines, the partial opening of VGSC at the resting membrane potential, Vm, allows tonic Na+ influx, which in turn keeps Vm sufficiently depolarized to maintain a small but continuous influx of Na+ [20,39,89]. Thus, under the ensuing pathophysiological conditions, Na+ influx is sufficient to maintain the positive feedback and the invasive capability of cancer cells.
Figure 6.
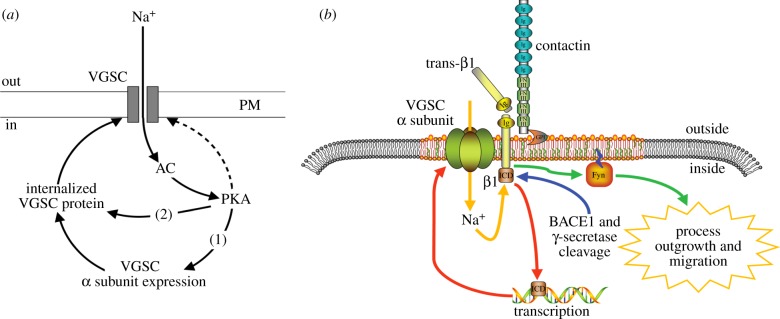
Activity-dependent regulation of VGSC expression and VGSC-dependent migration. (a) In Mat-LyLu [28] and MDA-MB-231 [34] cells, INa activates adenylate cyclase (AC) and protein kinase A (PKA), which in turn (i) potentiates α-subunit mRNA expression and (ii) increases channel expression at the plasma membrane, without affecting total cellular α-subunit protein level. PKA also directly phosphorylates surface-expressed VGSCs, although this may be independent of PKA (dashed line). Adapted from [28]. (b) Interplay between α and β1 subunits in transcription and process outgrowth, modelled from cerebellar granule neurons. Trans adhesion between β1 on an adjacent cell and a VGSC signalling complex (comprising α, β1 subunits and contactin), initiates a signalling cascade via FYN kinase that enhances process outgrowth and migration. Proteolytic processing of β1 by BACE1 and γ-secretase is proposed to release the soluble intracellular domain of β1, which may in turn enhance transcription of α subunit genes. Nav1.6 activity is required for β1-mediated process outgrowth, and in turn, β1 is required for normal localization of α-subunits. Thus, INa may fine-tune the dual processes of gene expression and migration. Adapted from [117]. (Online version in colour.)
These data therefore suggest that in metastatic carcinoma cells, VGSCα expression is at minimum self-sustaining, and thus may potentiate metastasis in persistent fashion. The results also raise the possibility that chronic blockage of VGSCs would suppress both channel activity and expression and, consequently, provide a dual advantage in any clinical anti-metastatic treatment.
(b). VGSC β1 subunit
Downstream functions of the VGSC β1 subunit, which is both a channel modulator and a cell adhesion molecule, may also be regulated in part by INa and this interaction may be reciprocal [120]. In BCa MCF-7 cells, β1 downregulation with siRNA reduced cellular adhesion to substrate and increased VGSC-dependent migration [121]. However, TTX had no effect on basal β1 subunit mRNA levels, consistent with lack of functional VGSC activity in these weakly/non-metastatic cells [121]. By contrast, the reverse could occur. Thus, downregulating SCN1B with siRNA upregulated nNav1.5 mRNA and protein expression [121]. It appeared that putting the cells into a higher metastatic mode by reducing their substrate adhesion automatically upregulated the VGSC expression. Such reciprocal expression of endogenous β1 and Nav1.7 has also been seen in NSCLC cells [89]. Moreover, downregulating β1 alone by siRNA was sufficient to cause a modest (approx. 30%) enhancement of A549 cell invasion, whereas overexpression of exogenous β1 reduced H460 cell invasion to a similar extent [89]. Thus, the VGSC β1 subunit has a multi-functional role in cancer and appears to be involved in a complex feedback loop that regulates channel expression/function [117]. In turn, this would modulate adhesion-mediated functions of the β1 subunit, including cellular process extension and invasiveness (figure 6b).
5. Conclusion and future perspectives
The idea that VGSCs are expressed during cancer progression and that VGSC activity enhances cell behaviours linked to metastasis, such as motility, invasion and adhesion, is now well-established. There is increasing evidence, as highlighted here, that such VGSC expression is under the control of ‘mainstream’ cancer mechanisms, principally hormones and growth factors, thus placing VGSCs as key regulators in cancer progression. However, other messenger molecules, including immune modulators, can also affect channel expression/activity [122]. Although regulation of VGSCs in cancer occurs clearly at a hierarchy of levels from transcription to post-translation, much work remains to determine the precise mechanisms involved. Regarding the former, developmentally regulated transcription factors, such as REST (‘neuron-specific silencing factor’), are also known to affect both the cancer process and channel expression [84].
It may be possible in the future, therefore, to combine conventional (e.g. hormone-based) therapies with clinically viable VGSC blockers. This may even help alleviate some of the problems associated with hormone-based therapies such as the insensitivity that frequently ensues in such treatments. Importantly, there are some indications that VGSC regulation may differ between ‘cancer’ and ‘normal’ cells. If so, understanding the signalling pathways that regulate VGSC expression/activity in cancer may provide additional avenues for preventing or suppressing metastatic disease. Furthermore, a newly discovered ‘non-canonical’ role of intracellular VGSCs in cell behaviour warrants investigation in the context of cancer [123]. In fact, a systematic analysis of the whole interactive network of ion channels and transporters is required ultimately in order to gain an overall and precise understanding of the role of ionic activity in cancer and to exploit this knowledge clinically.
Funding statement
We acknowledge continued support from the Pro Cancer Research Fund (PCRF)—rolling grant (M.B.A.D., S.P.F.), and the Medical Research Council—Fellowship no. G1000508(95657) to W.J.B.
References
- 1.Brackenbury WJ. 2012. Voltage-gated sodium channels and metastatic disease. Channels 6, 352–361. ( 10.4161/chan.21910) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Djamgoz MBA. 2011. Bioelectricity of cancer: voltage-gated ion channels and direct-current electric fields. In The physiology of bioelectricity in development, tissue regeneration, and cancer (ed. Pullar C.), pp. 269–294. London, UK: Taylor & Francis. [Google Scholar]
- 3.Fraser SP, Pardo LA. 2008. Ion channels: functional expression and therapeutic potential in cancer. Colloquium on ion channels and cancer. EMBO Rep. 9, 512–515. ( 10.1038/embor.2008.75) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pedersen SF, Stock C. 2013. Ion channels and transporters in cancer: pathophysiology, regulation and clinical potential. Cancer Res. 73, 1658–1661. ( 10.1158/0008-5472.CAN-12-4188) [DOI] [PubMed] [Google Scholar]
- 5.Djamgoz MBA, Onkal R. 2013. Persistent current blockers of voltage-gated sodium channels: a clinical opportunity for controlling metastatic disease. Recent Patents Anti-Cancer Drug Discov. 8, 66–84. [DOI] [PubMed] [Google Scholar]
- 6.Huber SM. 2013. Oncochannels. Cell Calcium 53, 241–255. ( 10.1016/j.ceca.2013.01.001) [DOI] [PubMed] [Google Scholar]
- 7.Yildirim S, Altun S, Gumushan H, Patel A, Djamgoz MBA. 2012. Voltage-gated sodium channel activity promotes prostate cancer metastasis in vivo. Cancer Lett. 323, 58–61. ( 10.1016/j.canlet.2012.03.036) [DOI] [PubMed] [Google Scholar]
- 8.Fraser SP, et al. 2005. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin. Cancer Res. 11, 5381–5389. ( 10.1158/1078-0432.CCR-05-0327) [DOI] [PubMed] [Google Scholar]
- 9.Wulff H, Castle NA, Pardo LA. 2009. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Disc. 8, 982–1001. ( 10.1038/nrd2983) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poplawski AB, et al. 2010. Frequent genetic differences between matched primary and metastatic breast cancer provide an approach to identification of biomarkers for disease progression. Eur. J. Hum. Genet. 18, 560–568. ( 10.1038/ejhg.2009.230) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao J, et al. 2010. Small RNAs control sodium channel expression, nociceptor excitability, and pain thresholds. J. Neurosci. 30, 10 860–10 871. ( 10.1523/JNEUROSCI.1980-10.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu H, Yang L, Jin MW, Sun HY, Huang Y, Li GR. 2010. The selective estrogen receptor modulator raloxifene inhibits cardiac delayed rectifier potassium currents and voltage-gated sodium current without QTc interval prolongation. Pharmacol. Res. 62, 384–390. ( 10.1016/j.phrs.2010.07.008) [DOI] [PubMed] [Google Scholar]
- 13.Wang S, Jiao B-H. 2009. The inhibition of tamoxifen on sodium channel in SHG-44 glioma cell-line. Chin. J. Appl. Physiol. 25, 207–210. [PubMed] [Google Scholar]
- 14.Catterall WA, Goldin AL, Waxman SG. 2005. International Union of Pharmacology. XLVII. Nomenclature and structure–function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409. ( 10.1124/pr.57.4.4) [DOI] [PubMed] [Google Scholar]
- 15.Roger S, Besson P, Le Guennec JY. 2003. Involvement of a novel fast inward sodium current in the invasion capacity of a breast cancer cell line. Biochim. Biophys. Acta 1616, 107–111. ( 10.1016/j.bbamem.2003.07.001) [DOI] [PubMed] [Google Scholar]
- 16.House CD, et al. 2010. Voltage-gated Na+ channel SCN5A is a key regulator of a gene transcriptional network that controls colon cancer invasion. Cancer Res. 70, 6957–6967. ( 10.1158/0008-5472.CAN-10-1169) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diaz D, Delgadillo DM, Hernandez-Gallegos E, Ramirez-Dominguez ME, Hinojosa LM, Ortiz CS, Berumen J, Camacho J, Gomora JC. 2007. Functional expression of voltage-gated sodium channels in primary cultures of human cervical cancer. J. Cell. Physiol. 210, 469–478. ( 10.1002/jcp.20871) [DOI] [PubMed] [Google Scholar]
- 18.Hernandez-Plata E, Ortiz CS, Marquina-Castillo B, Medina-Martinez I, Alfaro A, Berumen J, Rivera M, Gomora JC. 2012. Overexpression of Nav1.6 channels is associated with the invasion capacity of human cervical cancer. Int. J. Cancer 130, 2013–2023. ( 10.1002/ijc.26210) [DOI] [PubMed] [Google Scholar]
- 19.Diss JKJ, Archer SN, Hirano J, Fraser SP, Djamgoz MBA. 2001. Expression profiles of voltage-gated Na+ channel α-subunit genes in rat and human prostate cancer cell lines. Prostate 48, 1–14. ( 10.1002/pros.1095) [DOI] [PubMed] [Google Scholar]
- 20.Roger S, et al. 2007. Voltage-gated sodium channels potentiate the invasive capacities of human non-small-cell lung cancer cell lines. Int. J. Biochem. Cell Biol. 39, 774–786. ( 10.1016/j.biocel.2006.12.007) [DOI] [PubMed] [Google Scholar]
- 21.Ou SW, Kameyama A, Hao LY, Horiuchi M, Minobe E, Wang WY, Makita N, Kameyama M. 2005. Tetrodotoxin-resistant Na+ channels in human neuroblastoma cells are encoded by new variants of Nav1.5/SCN5A. Eur. J. Neurosci. 22, 793–801. ( 10.1111/j.1460-9568.2005.04280.x) [DOI] [PubMed] [Google Scholar]
- 22.Sharma S, Kelly TK, Jones PA. 2010. Epigenetics in cancer. Carcinogenesis 31, 27–36. ( 10.1093/carcin/bgp220) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monk M, Holding C. 2001. Human embryonic genes re-expressed in cancer cells. Oncogene 20, 8085–8091. ( 10.1038/sj.onc.1205088) [DOI] [PubMed] [Google Scholar]
- 24.Oh Y, Waxman SG. 1998. Novel splice variants of the voltage-sensitive sodium channel alpha subunit. Neuroreport 9, 1267–1272. ( 10.1097/00001756-199805110-00002) [DOI] [PubMed] [Google Scholar]
- 25.Lin W-H, Guenay C, Marley R, Prinz A, Baines RA. 2012. Activity-dependent alternative splicing increases persistent sodium current and promotes seizure. J. Neurosci. 32, 7267–7277. ( 10.1523/JNEUROSCI.6042-11.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Onkal R, Mattis JH, Fraser SP, Diss JKJ, Shao D, Okuse K, Djamgoz MBA. 2008. Alternative splicing of Nav1.5: an electrophysiological comparison of ‘neonatal’ and ‘adult’ isoforms, and critical involvement of a lysine residue. J. Cell. Physiol. 216, 716–726. ( 10.1002/jcp.21451) [DOI] [PubMed] [Google Scholar]
- 27.Bers DM, Barry WH, Despa S. 2003. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc. Res. 57, 897–912. ( 10.1016/S0008-6363(02)00656-9) [DOI] [PubMed] [Google Scholar]
- 28.Brackenbury WJ, Djamgoz MBA. 2006. Activity-dependent regulation of voltage-gated Na+ channel expression in Mat-LyLu rat prostate cancer cell line. J. Physiol. 573, 343–356. ( 10.1113/jphysiol.2006.106906) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pieske B, Houser SR. 2003. [Na+]i handling in the failing human heart. Cardiovasc. Res. 57, 874–886. ( 10.1016/S0008-6363(02)00841-6) [DOI] [PubMed] [Google Scholar]
- 30.Khan A, Kyle JW, Hanck DA, Lipkind GM, Fozzard HA. 2006. Isoform-dependent interaction of voltage-gated sodium channels with protons. J. Physiol. 576, 493–501. ( 10.1113/jphysiol.2006.115659) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Onkal R. 2010. Neonatal Nav1.5 voltage-gated Na+ channel: Regulation, electrophysiology and pharmacology. PhD thesis, Imperial College London, London, UK. [Google Scholar]
- 32.Fraser SP, Salvador V, Manning EA, Mizal J, Altun S, Raza M, Berridge RJ, Djamgoz MBA. 2003. Contribution of functional voltage-gated Na+ channel expression to cell behaviors involved in the metastatic cascade in rat prostate cancer. I. Lateral motility. J. Cell. Physiol. 195, 479–487. ( 10.1002/jcp.10312) [DOI] [PubMed] [Google Scholar]
- 33.Fulgenzi G, Graciotti L, Faronato M, Soldovieri MV, Miceli F, Amoroso S, Annunziato L, Procopio A, Taglialatela M. 2006. Human neoplastic mesothelial cells express voltage-gated sodium channels involved in cell motility. Int. J. Biochem. Cell Biol. 38, 1146–1159. ( 10.1016/j.biocel.2005.12.003) [DOI] [PubMed] [Google Scholar]
- 34.Chioni AM, Shao D, Grose R, Djamgoz MBA. 2010. Protein kinase A and regulation of neonatal Nav1.5 expression in human breast cancer cells: activity-dependent positive feedback and cellular migration. Int. J. Biochem. Cell Biol. 42, 346–358. ( 10.1016/j.biocel.2009.11.021) [DOI] [PubMed] [Google Scholar]
- 35.Grimes J, Fraser SP, Stephens GJ, Downing JEG, Laniado ME, Foster CS, Abel PD, Djamgoz MBA. 1995. Differential expression of voltage-activated Na+ currents in two prostatic tumour cell lines: contribution to invasiveness in vitro. FEBS Lett. 369, 290–294. ( 10.1016/0014-5793(95)00772-2) [DOI] [PubMed] [Google Scholar]
- 36.Laniado ME, Lalani E-N, Fraser SP, Grimes JA, Bhangal G, Djamgoz MBA, Abel PD. 1997. Expression and functional analysis of voltage-activated Na+ channels in human prostate cancer cell lines and their contribution to invasion in vitro. Am. J. Pathol. 150, 1213–1221. [PMC free article] [PubMed] [Google Scholar]
- 37.Bennett ES, Smith BA, Harper JM. 2004. Voltage-gated Na+ channels confer invasive properties on human prostate cancer cells. Pflügers Arch. 447, 908–914. ( 10.1007/s00424-003-1205-x) [DOI] [PubMed] [Google Scholar]
- 38.Smith P, Rhodes NP, Shortland AP, Fraser SP, Djamgoz MBA, Ke Y, Foster CS. 1998. Sodium channel protein expression enhances the invasiveness of rat and human prostate cancer cells. FEBS Lett. 423, 19–24. ( 10.1016/S0014-5793(98)00050-7) [DOI] [PubMed] [Google Scholar]
- 39.Gillet L, Roger S, Besson P, Lecaille F, Gore J, Bougnoux P, Lalmanach G, Le Guennec JY. 2009. Voltage-gated sodium channel activity promotes cysteine cathepsin-dependent invasiveness and colony growth of human cancer cells. J. Biol. Chem. 284, 8680–8691. ( 10.1074/jbc.M806891200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fraser SP, Ding Y, Liu A, Foster CS, Djamgoz MBA. 1999. Tetrodotoxin suppresses morphological enhancement of the metastatic Mat-LyLu rat prostate cancer cell line. Cell Tissue Res. 295, 505–512. ( 10.1007/s004410051256) [DOI] [PubMed] [Google Scholar]
- 41.Djamgoz MBA, Mycielska M, Madeja Z, Fraser SP, Korohoda W. 2001. Directional movement of rat prostatic cancer cells in direct-current electric field: involvement of voltage-gated Na+ channel activity. J. Cell Sci. 114, 2697–2705. [DOI] [PubMed] [Google Scholar]
- 42.Palmer CP, et al. 2008. Single cell adhesion measuring apparatus (SCAMA): application to cancer cell lines of different metastatic potential and voltage-gated Na+ channel expression. Eur. Biophys. J. 37, 359–368. ( 10.1007/s00249-007-0219-2) [DOI] [PubMed] [Google Scholar]
- 43.Mycielska ME, Palmer CP, Brackenbury WJ, Djamgoz MBA. 2005. Expression of Na+-dependent citrate transport in a strongly metastatic human prostate cancer PC-3M cell line: regulation by voltage-gated Na+ channel activity. J. Physiol. 563, 393–408. ( 10.1113/jphysiol.2004.079491) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mycielska ME, Fraser SP, Szatkowski M, Djamgoz MBA. 2003. Contribution of functional voltage-gated Na+ channel expression to cell behaviors involved in the metastatic cascade in rat prostate cancer. II. Secretory membrane activity. J. Cell. Physiol. 195, 461–469. ( 10.1002/jcp.10265) [DOI] [PubMed] [Google Scholar]
- 45.Onganer PU, Djamgoz MBA. 2005. Small-cell lung cancer (human): potentiation of endocytic membrane activity by voltage-gated Na+ channel expression in vitro. J. Membr. Biol. 204, 67–75. ( 10.1007/s00232-005-0747-6) [DOI] [PubMed] [Google Scholar]
- 46.Nakajima T, et al. 2009. Eicosapentaenoic acid inhibits voltage-gated sodium channels and invasiveness in prostate cancer cells. Br. J. Pharmacol. 56, 420–431. ( 10.1111/j.1476-5381.2008.00059.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krasowska M, Grzywna ZJ, Mycielska ME, Djamgoz MBA. 2004. Patterning of endocytic vesicles and its control by voltage-gated Na+ channel activity in rat prostate cancer cells: fractal analyses. Eur. Biophys. J. 33, 535–542. ( 10.1007/s00249-004-0394-3) [DOI] [PubMed] [Google Scholar]
- 48.Williams EL, Djamgoz MBA. 2005. Nitric oxide and metastatic cell behaviour. Bioessays 27, 1228–1238. ( 10.1002/bies.20324) [DOI] [PubMed] [Google Scholar]
- 49.Brisson L, et al. 2013. Nav1.5 Na+ channels allosterically regulate the NHE-1 exchanger and promote the activity of breast cancer cell invadopodia. J. Cell Sci. 126, 4835–4842. ( 10.1242/jcs.123901) [DOI] [PubMed] [Google Scholar]
- 50.Diss JKJ, Fraser SP, Djamgoz MBA. 2004. Voltage-gated Na+ channels: functional consequences of multiple subtypes and isoforms for physiology and pathophysiology. Eur. Biophys. J. 33, 180–193. ( 10.1007/s00249-004-0389-0) [DOI] [PubMed] [Google Scholar]
- 51.Fraser SP, Özerlat-Gunduz I, Onkal R, Diss JKJ, Latchman DS, Djamgoz MBA. 2010. Estrogen and non-genomic upregulation of voltage-gated Na+ channel activity in MDA-MB-231 human breast cancer cells: role in adhesion. J. Cell. Physiol. 224, 527–539. ( 10.1002/jcp.22154) [DOI] [PubMed] [Google Scholar]
- 52.Onkal R, Djamgoz MBA. 2009. Molecular pharmacology of voltage-gated sodium channel expression in metastatic disease: clinical potential of neonatal Nav1.5 in breast cancer. Eur. J. Pharmacol. 625, 206–219. ( 10.1016/j.ejphar.2009.08.040) [DOI] [PubMed] [Google Scholar]
- 53.Manavathi B, Dey O, Gajulapalli V, Narsihma R, Bhatia RS, Bugide S, Kumar R. 2013. Derailed estrogen signaling and breast cancer: an authentic couple. Endocr. Rev. 34, 1–32. ( 10.1210/er.2011-1057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burris TP, et al. 2013. Nuclear receptors and their selective pharmacologic modulators. Pharmacol. Rev. 65, 710–778. ( 10.1124/pr.112.006833) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Renoir J-M, Marsaud V, Lazennec G. 2013. Estrogen receptor signaling as a target for novel breast cancer therapeutics. Biochem. Pharmacol. 85, 449–465. ( 10.1016/j.bcp.2012.10.018) [DOI] [PubMed] [Google Scholar]
- 56.Jiang SY, Jordan VC. 1992. Growth regulation of estrogen receptor-negative breast cancer cells transfected with complementary DNAs for estrogen receptor. J. Natl Cancer Inst. 84, 580–591. ( 10.1093/jnci/84.8.580) [DOI] [PubMed] [Google Scholar]
- 57.Özerlat I. 2009. Effects of estrogen on voltage-gated sodium channel expression and function in human breast cancer cell lines. PhD thesis, University of London, London, UK. [Google Scholar]
- 58.Sheikh MS, Garcia M, Pujol P, Fontana JA, Rochefort H. 1994. Why are estrogen-receptor-negative breast cancers more aggressive than the estrogen-receptor-positive breast cancers? Invasion Metastasis 14, 329–336. [PubMed] [Google Scholar]
- 59.Hu F, Wang Q, Wang P, Wang W, Qian WY, Xiao H, Wang L. 2012. 17 Beta-estradiol regulates the gene expression of voltage-gated sodium channels: role of estrogen receptor alpha and estrogen receptor beta. Endocrine 41, 274–280. ( 10.1007/s12020-011-9573-z) [DOI] [PubMed] [Google Scholar]
- 60.Kow LM, Devidze N, Pataky S, Shibuya I, Pfaff DW. 2006. Acute estradiol application increases inward and decreases outward whole-cell currents of neurons in rat hypothalamic ventromedial nucleus. Brain Res. 1116, 1–11. ( 10.1016/j.brainres.2006.07.104) [DOI] [PubMed] [Google Scholar]
- 61.Barann M, Gothert M, Bruss M, Bonisch H. 1999. Inhibition by steroids of [14C]-guanidinium flux through the voltage-gated sodium channel and the cation channel of the 5-HT3 receptor of N1E-115 neuroblastoma cells. Naunyn Schmiedeberg's Arch. Pharmacol. 360, 234–241. ( 10.1007/s002109900089) [DOI] [PubMed] [Google Scholar]
- 62.Wang Q, Cao J, Hu F, Lu R, Wang J, Ding H, Gao R, Xiao H. 2013. Effects of estradiol on voltage-gated sodium channels in mouse dorsal root ganglion neurons. Brain Res. 1512, 1–8. ( 10.1016/j.brainres.2013.02.047) [DOI] [PubMed] [Google Scholar]
- 63.Smitherman KA, Sontheimer H. 2001. Inhibition of glial Na+ and K+ currents by tamoxifen. J. Membr. Biol. 181, 125–135. [DOI] [PubMed] [Google Scholar]
- 64.Hardy SP, deFelipe C, Valverde MA. 1998. Inhibition of voltage-gated cationic channels in rat embryonic hypothalamic neurones and C1300 neuroblastoma cells by triphenylethylene antioestrogens. FEBS Lett. 434, 236–240. ( 10.1016/S0014-5793(98)00974-0) [DOI] [PubMed] [Google Scholar]
- 65.Borg JJ, Yuill KH, Hancox JC, Spencer IC, Kozlowski RZ. 2002. Inhibitory effects of the antiestrogen agent clomiphene on cardiac sarcolemmal anionic and cationic currents. J. Pharmacol. Exp. Therapeut. 303, 282–292. ( 10.1124/jpet.102.038901) [DOI] [PubMed] [Google Scholar]
- 66.Hammes S, Kelly M, Slingerland J. 2013. Integration of genomic and non-genomic steroid receptor actions. Steroids 78, 529–529. ( 10.1016/j.steroids.2013.01.001) [DOI] [PubMed] [Google Scholar]
- 67.Diss JKJ, Fraser SP, Walker MM, Patel A, Latchman DS, Djamgoz MBA. 2008. β-subunits of voltage-gated sodium channels in human prostate cancer: quantitative in vitro and in vivo analyses of mRNA expression. Prostate Cancer Prostatic Dis. 11, 325–333. ( 10.1038/sj.pcan.4501012) [DOI] [PubMed] [Google Scholar]
- 68.Oliver VL, Poulios K, Ventura S, Haynes JM. 2013. A novel androgen signalling pathway uses dihydrotestosterone, but not testosterone, to activate the epidermal growth factor receptor signalling cascade in prostate stromal cells. Br. J. Pharmacol. 170, 592–601. ( 10.1111/bph.12307) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berwick DC, Diss JKJ, Budhram-Mahadeo VS, Latchman DS. 2010. A simple technique for the prediction of interacting proteins reveals a direct brn-3a-androgen receptor interaction. J. Biol. Chem. 285, 15 286–15 295. ( 10.1074/jbc.M109.071456) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tabb JS, Fanger GR, Wilson EM, Maue RA, Henderson LP. 1994. Suppression of sodium-channel function in differentiating C2 muscle-cells stably overexpressing rat androgen receptors. J. Neurosci. 14, 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frasca F, Pandini C, Scalia P, Mineo R, Costantino A, Goldfine ID, Belfiore A, Vigneri R. 1999. Insulin receptor isoform A, a newly recognized, high affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 19, 3278–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pan H, Djamgoz MBA. 2008. Biochemical constitution of extracellular medium is critical for control of human breast cancer MDA-MB-231 cell motility. J. Membr. Biol. 223, 27–36. ( 10.1007/s00232-008-9110-z) [DOI] [PubMed] [Google Scholar]
- 73.Nemoto T, Yanagita T, Kanai T, Wada A. 2009. Drug development targeting the glycogen synthase kinase-3β (GSK-3β)-mediated signal transduction pathway: the role of GSK-3β in the maintenance of steady-state levels of insulin receptor signaling molecules and Nav1.7 sodium channel in adrenal chromaffin cells. J. Pharmacol. Sci. 109, 157–161. ( 10.1254/jphs.08R20FM) [DOI] [PubMed] [Google Scholar]
- 74.Mao W, You T, Ye B, Li X, Dong HH, Hill JA, Li FQ, Xu HD. 2012. Reactive oxygen species suppress cardiac Nav1.5 expression through Foxo1. PLoS ONE 7, e32738 ( 10.1371/journal.pone.0032738) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lang F, Shumilina E. 2013. Regulation of ion channels by the serum- and glucocorticoid-inducible kinase SGK1. FASEB J. 27, 3–12. ( 10.1096/fj.12-218230) [DOI] [PubMed] [Google Scholar]
- 76.Boehmer C, Wilhelm V, Palmada M, Wallisch S, Henke G, Brinkmeier H, Cohen P, Pieske B, Lang F. 2003. Serum and glucocorticoid inducible kinases in the regulation of the cardiac sodium channel SCN5A. Cardiovasc. Res. 57, 1079–1084. ( 10.1016/S0008-6363(02)00837-4) [DOI] [PubMed] [Google Scholar]
- 77.Abriel H. 2010. Cardiac sodium channel Nav1.5 and interacting proteins: physiology and pathophysiology. J. Mol. Cell. Cardiol. 48, 2–11. ( 10.1016/j.yjmcc.2009.08.025) [DOI] [PubMed] [Google Scholar]
- 78.Kelley BG, Mermelstein PG. 2011. Progesterone blocks multiple routes of ion flux. Mol. Cell. Neurosci. 48, 137–141. ( 10.1016/j.mcn.2011.07.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim JJ, Kurita T, Bulun SE. 2013. Progesterone action in endometrial cancer, endometriosis, uterine fibroids, and breast cancer. Endocr. Rev. 34, 130–162. ( 10.1210/er.2012-1043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lemmon MA, Schlessinger J. 2010. Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134. ( 10.1016/j.cell.2010.06.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gao J, Chang YS, Jallal B, Viner J. 2012. Targeting the insulin-like growth factor axis for the development of novel therapeutics in oncology. Cancer Res. 72, 3–12. ( 10.1158/0008-5472.CAN-11-0550) [DOI] [PubMed] [Google Scholar]
- 82.Ahern CA, Zhang JF, Wookalis MJ, Horn R. 2005. Modulation of the cardiac sodium channel Nav.5 by Fyn, a Src family tyrosine kinase. Circ. Res. 96, 991–998. ( 10.1161/01.RES.0000166324.00524.dd) [DOI] [PubMed] [Google Scholar]
- 83.Jespersen T, Gavillet B, van Bemmelen MX, Cordonier S, Thomas MA, Staub O, Abriel H. 2006. Cardiac sodium channel Nav1.5 interacts with and is regulated by the protein tyrosine phosphatase PTPH1. Biochem. Biophys. Res. Commun. 348, 1455–1462. ( 10.1016/j.bbrc.2006.08.014) [DOI] [PubMed] [Google Scholar]
- 84.Toledo-Aral JJ, Brehm P, Halegoua S, Mandel G. 1995. A single pulse of nerve growth factor triggers long-term neuronal excitability through sodium channel gene induction. Neuron 14, 607–611. ( 10.1016/0896-6273(95)90317-8) [DOI] [PubMed] [Google Scholar]
- 85.Liu H, Sun H-Y, Lau C-P, Li GR. 2007. Regulation of voltage-gated cardiac sodium current by epidermal growth factor receptor kinase in guinea pig ventricular myocytes. J. Mol. Cell. Cardiol. 42, 760–768. ( 10.1016/j.yjmcc.2006.10.013) [DOI] [PubMed] [Google Scholar]
- 86.Ding Y, Brackenbury WJ, Onganer PU, Montano X, Porter LM, Bates LF, Djamgoz MBA. 2008. Epidermal growth factor upregulates motility of Mat-LyLu rat prostate cancer cells partially via voltage-gated Na+ channel activity. J. Cell. Physiol. 215, 77–81. ( 10.1002/jcp.21289) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Uysal-Onganer P, Djamgoz MBA. 2007. Epidermal growth factor potentiates in vitro metastatic behaviour of human prostate cancer PC-3M cells: involvement of voltage-gated sodium channel. Mol. Cancer 6, 76 ( 10.1186/1476-4598-6-76) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pan H, Zhao L, Dai Y, Chen H, Huang B. 2011. Epidermal growth factor upregulates voltage-gated sodium channel/Nav1.5 expression and human breast cancer cell invasion. Basic Clin. Med. 31, 388–393. [Google Scholar]
- 89.Campbell TM, Main MJ, Fitzgerald EM. 2013. Functional expression of the voltage-gated sodium channel, Nav1.7, underlies epidermal growth factor-mediated invasion in human non-small cell lung cancer cells. J. Cell. Sci. 126, 4939–4949. ( 10.1242/jcs.130013) [DOI] [PubMed] [Google Scholar]
- 90.Güzel R. 2012. Studies of ionic mechanisms associated with human cancers. PhD thesis, Imperial College London, London, UK. [Google Scholar]
- 91.Xing CH, Yin YL, He XP, Xie ZP. 2006. Effects of insulin-like growth factor 1 on voltage-gated ion channels in cultured rat hippocampal neurons. Brain Res. 1072, 30–35. ( 10.1016/j.brainres.2005.10.091) [DOI] [PubMed] [Google Scholar]
- 92.Yanagita T, et al. 2011. Transcriptional up-regulation of cell surface Nav1.7 sodium channels by insulin-like growth factor-1 via inhibition of glycogen synthase kinase-3 beta in adrenal chromaffin cells: enhancement of Na-22+ influx, Ca-452+ influx and catecholamine secretion. Neuropharmacology 61, 1265–1274. ( 10.1016/j.neuropharm.2011.07.029) [DOI] [PubMed] [Google Scholar]
- 93.Brackenbury WJ, Djamgoz MBA. 2007. Nerve growth factor enhances voltage-gated Na+ channel activity and transwell migration in Mat-LyLu rat prostate cancer cell line. J. Cell. Physiol. 210, 602–608. ( 10.1002/jcp.20846) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Diss JKJ, Calissano M, Gascoyne D, Djamgoz MBA, Latchman DS. 2008. Identification and characterization of the promoter region of the Nav1.7 voltage-gated sodium channel gene (SCN9A). Mol. Cell. Neurosci. 37, 537–547. ( 10.1016/j.mcn.2007.12.002) [DOI] [PubMed] [Google Scholar]
- 95.Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, Frenette PS. 2013. Autonomic nerve development contributes to prostate cancer progression. Science 341, 1236361/1–1236361/10 ( 10.1126/science.1236361) [DOI] [PubMed] [Google Scholar]
- 96.Aloe L, Rocco ML, Bianchi P, Manni L. 2012. Nerve growth factor: from the early discoveries to the potential clinical use. J. Transl. Med. 10, 239 ( 10.1186/1479-5876-10-239) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. 2006. VEGF receptor signalling in control of vascular function. Nat. Rev. Mol. Cell Biol. 7, 359–371. ( 10.1038/nrm1911) [DOI] [PubMed] [Google Scholar]
- 98.Eccles SA. 2004. Parallels in invasion and angiogenesis provide pivotal points for therapeutic intervention. Int. J. Dev. Biol. 48, 583–598. ( 10.1387/ijdb.041820se) [DOI] [PubMed] [Google Scholar]
- 99.Pan H, Zhao Q, Zhan Y, Zhao L, Zhang W, Wu Y. 2012. Vascular endothelial growth factor-C promotes the invasion of cervical cancer cells via up-regulating the expression of voltage-gated sodium channel subtype Nav1.6. Tumor 32, 313–319. [Google Scholar]
- 100.Andrikopoulos P, et al. 2011. Angiogenic functions of voltage-gated Na+ channels in human endothelial cells: modulation of vascular endothelial growth factor (VEGF) signalling. J. Biol. Chem. 286, 16 846–16 860. ( 10.1074/jbc.M110.187559) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Andrikopoulos P, Baba A, Matsuda T, Djamgoz MBA, Yaqoob MM, Eccles SA. 2011. Ca2+ influx through reverse mode Na+/Ca2+ exchange is critical for vascular endothelial growth factor-mediated extracellular signal-regulated kinase (ERK) 1/2 activation and angiogenic functions of human endothelial cells. J. Biol. Chem. 286, 37 919–37 931. ( 10.1074/jbc.M111.251777) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sun G-C, Ma Y-Y. 2013. Vascular endothelial growth factor modulates voltage-gated Na+ channel properties and depresses action potential firing in cultured rat hippocampal neurons. Biol. Pharm. Bull. 36, 548–555. ( 10.1248/bpb.b12-00841) [DOI] [PubMed] [Google Scholar]
- 103.Malykhina AP, Lei Q, Erickson CS, Saban MR, Davis CA, Saban R. 2012. VEGF induces sensory and motor peripheral plasticity, alters bladder function, and promotes visceral sensitivity. BMC Physiol. 12, 15 ( 10.1186/1472-6793-12-15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang X, Bao L, Yang L, Wu QF, Li S. 2012. Roles of intracellular fibroblast growth factors in neural development and functions. Sci. China-Life Sci. 55, 1038–1044. ( 10.1007/s11427-012-4412-x) [DOI] [PubMed] [Google Scholar]
- 105.Laezza F, Lampert A, Kozel MA, Gerber BR, Rush AM, Nerbonne JM, Waxman SG, Dib-Hajj SD, Ornitz DM. 2007. FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol. Cell. Neurosci. 42, 90–101. ( 10.1016/j.mcn.2009.05.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang C, Hennessey JA, Kirkton RD, Wang C, Graham V, Puranam RS, Rosenberg PB, Bursac N, Pitt GS. 2011. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ. Res. 109, 775–782. ( 10.1161/CIRCRESAHA.111.247957) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ge J, Wang Y, Liu H, Chen FF, Cui XL, Liu ZH. 2010. Activin A maintains cerebral cortex neuronal survival and increases voltage-gated Na+ neuronal current. Neural Regen. Res. 5, 1464–1469. ( 10.3969/j.issn.1673-5374.2010.19.004) [DOI] [Google Scholar]
- 108.Kaur K, Zarzoso M, Ponce-Balbuena D, Guerrero-Serna G, Hou LQ, Musa H, Jalife J. 2013. TGF-beta 1, released by myofibroblasts, differentially regulates transcription and function of sodium and potassium channels in adult rat ventricular myocytes. PLoS ONE 8, e55391 ( 10.1371/journal.pone.0055391) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ramos-Mondragon R, Vega AV, Avila G. 2011. Long-term modulation of Na+ and K+ channels by TGF-β1 in neonatal rat cardiac myocytes. Pflügers Arch. 461, 235–247. ( 10.1007/s00424-010-0912-3) [DOI] [PubMed] [Google Scholar]
- 110.Light A, Hammes SR. 2013. Membrane receptor cross talk in steroidogenesis: recent insights and clinical implications. Steroids 78, 633–638. ( 10.1016/j.steroids.2012.12.016) [DOI] [PubMed] [Google Scholar]
- 111.Xu A, Huang P. 2010. Receptor tyrosine kinase coactivation networks in cancer. Cancer Res. 70, 3857–3860. ( 10.1158/0008-5472.CAN-10-0163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moody WJ, Bosma MM. 2005. Ion channel development, spontaneous activity, and activity-dependent development in nerve and muscle cells. Physiol. Rev. 85, 883–941. ( 10.1152/physrev.00017.2004) [DOI] [PubMed] [Google Scholar]
- 113.Dargent B, Couraud F. 1990. Down-regulation of voltage-dependent sodium channels initiated by sodium influx in developing neurons. Proc. Natl Acad. Sci. USA 87, 5907–5911. ( 10.1073/pnas.87.15.5907) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dargent B, Paillart C, Carlier E, Alcaraz G, Martin-Eauclaire MF, Couraud F. 1994. Sodium channel internalization in developing neurons. Neuron 13, 683–690. ( 10.1016/0896-6273(94)90035-3) [DOI] [PubMed] [Google Scholar]
- 115.Klein JP, Tendi EA, Dib-Hajj SD, Fields RD, Waxman SG. 2003. Patterned electrical activity modulates sodium channel expression in sensory neurons. J. Neurosci. Res. 74, 192–198. ( 10.1002/jnr.10768) [DOI] [PubMed] [Google Scholar]
- 116.Shiraishi S, Yokoo H, Yanagita T, Kobayashi H, Minami S, Saitoh T. 2003. Differential effects of bupivacaine enantiomers, ropivacaine and lidocaine on up-regulation of cell surface voltage-dependent sodium channels in adrenal chromaffin cells. Brain Res. 966, 175–184. ( 10.1016/S0006-8993(02)04152-5) [DOI] [PubMed] [Google Scholar]
- 117.Brackenbury WJ, Djamgoz MBA, Isom LL. 2008. An emerging role for voltage-gated Na+ channels in cellular migration: regulation of central nervous system development and potentiation of invasive cancers. Neuroscientist 14, 571–583. ( 10.1177/1073858408320293) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cooper DM, Schell MJ, Thorn P, Irvine RF. 1998. Regulation of adenylyl cyclase by membrane potential. J. Biol. Chem. 273, 27 703–27 707. ( 10.1074/jbc.273.42.27703) [DOI] [PubMed] [Google Scholar]
- 119.Murakami Y, Tanaka J, Koshimura K, Kato Y. 1998. Involvement of tetrodoxin-sensitive sodium channels in rat growth hormone secretion induced by pituitary adenylate cyclase-activating polypeptide (PACAP). Regul. Pept. 73, 119–121. ( 10.1016/S0167-0115(97)01075-6) [DOI] [PubMed] [Google Scholar]
- 120.Brackenbury WJ, Isom LL. 2011. Na channel beta subunits: overachievers of the ion channel family. Front. Pharmacol. 2, 53 ( 10.3389/fphar.2011.00053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chioni AM, Brackenbury WJ, Calhoun JD, Isom LL, Djamgoz MBA. 2009. A novel adhesion molecule in human breast cancer cells: voltage-gated Na+ channel β1 subunit. Int. J. Biochem. Cell Biol. 41, 1216–1227. ( 10.1016/j.biocel.2008.11.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Guillouët M, Rannou F, Giroux-Metges MA, Droguet M, Pennec JP. 2013. Tumor necrosis factor alpha induced hypoexcitability in rat muscle evidenced in a model of ion currents and action potential. Cytokine 64, 165–171. ( 10.1016/j.cyto.2013.07.007) [DOI] [PubMed] [Google Scholar]
- 123.Black JA, Waxman SG. 2013. Noncanonical roles of voltage-gated sodium channels. Neuron 80, 280–291. ( 10.1016/j.neuron.2013.09.012) [DOI] [PubMed] [Google Scholar]