

Abstract
Multi-drug resistance (MDR) to chemotherapy is the major challenge in the treatment of cancer. MDR can develop by numerous mechanisms including decreased drug uptake, increased drug efflux and the failure to undergo drug-induced apoptosis. Evasion of drug-induced apoptosis through modulation of ion transporters is the main focus of this paper and we demonstrate how pro-apoptotic ion channels are downregulated, while anti-apoptotic ion transporters are upregulated in MDR. We also discuss whether upregulation of ion transport proteins that are important for proliferation contribute to MDR. Finally, we discuss the possibility that the development of MDR involves sequential and localized upregulation of ion channels involved in proliferation and migration and a concomitant global and persistent downregulation of ion channels involved in apoptosis.
Keywords: cancer, drug resistance, tumour proliferation, apoptosis, ion channels in cancer
1. Introduction
Multi-drug resistance (MDR) to chemotherapy is a major challenge in the treatment of cancer and is one cause of cancer chemotherapy failure. The cellular resistance of tumour cells to chemotherapeutic agents can be an innate property (termed intrinsic resistance) or can be acquired during chemotherapy (termed extrinsic resistance). Intrinsic resistance is often associated with cell differentiation or with genetic changes that occur during the initiation of tumour formation. Extrinsic resistance arises through the expansion of rare genetic variants in a tumour cell population owing to the proliferation of drug-resistant cells with selective advantages. As suggested by the name, MDR refers to resistance to numerous drugs that have different chemical structures and distinct mechanisms of action. Several molecular mechanisms have been proposed to explain MDR, including tumour cell-specific mechanisms such as decreased drug accumulation in the cell, sequestration of the drug in intracellular vesicles, activation of DNA repair pathways that counteract the effects of the drugs and evasion of apoptosis or cell cycle arrest [1–3]. Extracellular mechanisms have also been proposed, such as involvement of the stromal cell compartment in drug uptake and activation of alternative escape pathways. In addition, genes that control cell death and survival signalling, including the genes encoding Bcl-2 and p53, can acquire mutations that lead to drug resistance through modulation or impairment of apoptosis. Moreover, activation of alternative signalling pathways that modulate cell migration, proliferation and apoptosis may be involved in development of drug-resistance pathways [4,5].
Decreased intracellular drug accumulation can result from a decrease in drug influx via drug solute carriers (SLC) [6] or from an increase in drug efflux via ATP-binding cassette (ABC) drug efflux pumps such as the P-glycoprotein (MDR1), multi-drug-resistance-associated protein (MRP) and mitoxantrone-resistance protein (MXR) [7]. These pumps are targeted by several anti-cancer drugs. The use of fluorescent calcein, which is an ABC transporter substrate, makes it possible to identify drugs that compete with calcein for the ABC transporter. Using similar methods, many chemotherapeutic drugs have been shown to be substrates/inhibitors of MDR1, MRP and MXR (figure 1). Some drugs used in chemotherapy have been specifically selected for use because they are not substrates for the ABC drug efflux pumps [8]. For example, this is true for the platinum drugs, which are used for treatment of solid tumours in more than 50% of all cancer patients. For these platinum-based drugs, drug resistance is often caused by decreased drug accumulation via the copper transporter 1 (CRT1), increased drug efflux via copper-transporting ATPases (ATP7A, ATP7B), increased detoxification of the drug by thiol-containing molecules within the cell [9–11] or, as described below, by evasion of programmed cell death (apoptosis).
Figure 1.
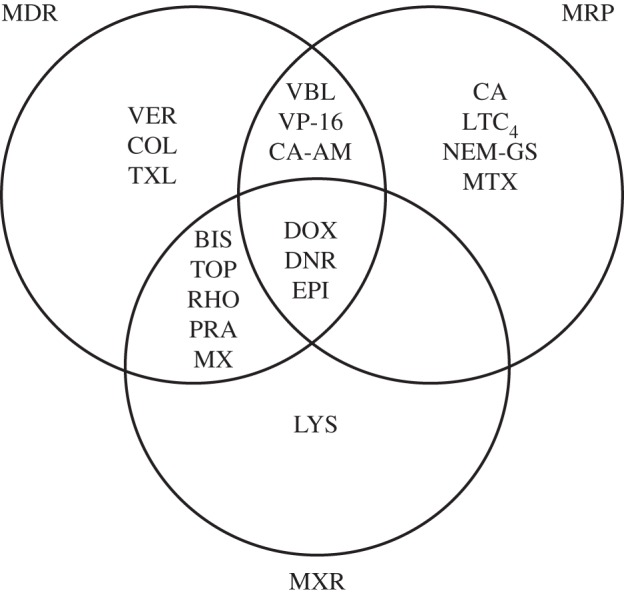
Substrate overlaps between the transporters P-glycoprotein/MDR1, multi-drug-resistance-associated protein (MRP) and mitoxantrone-resistance protein (MXR). The substrate and inhibitor profiles for the transporters were obtained from micrographs that showed the steady-state accumulation of fluorescent drugs (60 min incubation at 37°C); adapted from [7]. BIS, bisantrene; CA, calcein; CA-AM, calcein-AM ester; COL, colchicine; DNR, daunorubicin; DOX, doxorubicin; EPI, epirubicin; LTC4, leukotriene C4; LYS, LysoTracker; MTX, methotrexate; MX, mitoxantrone; NEM-GS, N-ethyl maleimide glutathione; PRA, prazosin; RHO, rhodamine 123; TXL, taxol; TOP, topotecan; VBL, vinblastine; VER, verapamil; VP-16, etoposide.
In recent years, it has become increasingly clear that downregulation of ion channels and transporters is an important mechanism in the development of drug resistance via impairment of programmed cell death [12]. In this review, we discuss how drug resistance can develop through the modulation of membrane-bound ion transporters, limitation of cell shrinkage and, thus, impairment of apoptosis. We discuss the ion transporters that are involved in this process and try to clarify the mechanisms by which downregulation of channels can make the cancer cells apoptosis-resistant. It is noted that several reports have shown that ion channel overexpression can also be associated with apoptosis resistance. These examples will also be discussed below. Because modulation of ion channels is also involved in changes in cell migration and cell proliferation, we also briefly mention examples in which upregulation of ion channels contributes to tumour cell drug resistance. Finally, we point out that the cell-surface accessibility of ion channels suggests that they have strong potential as diagnostic and therapeutic targets in tumour treatment.
2. Evasion of drug-induced apoptosis
A hallmark of apoptosis is cell shrinkage, which is also termed ‘apoptotic volume decrease’ (AVD); hence, disordered or altered cell volume regulation is associated with apoptosis (reviewed in [13]). AVD results from a loss of KCl via K+ and Cl− channels and a concomitant loss of water [14–19], and it has turned out that downregulation of K+ channels [20] and Cl− channels [19,21,22] courses resistance in cancer cells towards apoptosis. Cell shrinkage is usually followed by regulatory volume increase (RVI) [23,24] which counteracts AVD and thereby apoptosis [25,26]. The most important transport systems involved in RVI that have potential anti-apoptotic effects are the Na+, K+, 2Cl− co-transporter NKCC1, the Na/K ATPase, cation channels and the Na+/H+ exchanger NHE1 [13,24] (figure 2, left-hand side). It has been demonstrated in several cell types that hypertonic cell shrinkage results in apoptosis (reviewed in [13]). For example, in NIH3T3 cells, caspase 3 activity increases fivefold following a twofold increase in extracellular osmolarity [27]. Bortner et al. [28] recently demonstrated that repetitive hypertonic exposure of lymphocytes resulted in a cell line with improved RVI and an attendant resistance towards shrinkage-induced apoptosis. In accordance with these observations, Chinese hamster ovary cells do not exhibit RVI because of lack of NHE1, and these cells are more prone to apoptosis compared with cells expressing NHE1 [25]. The activation of apoptosis following cell shrinkage may involve activation of p38/p53 signalling [27], CD95 death receptor trafficking to the plasma membrane [29], and inhibition of growth factor-mediated signalling [30]. The cooperation and coordination of signalling networks as a phenotypic hallmark of MDR is discussed in greater detail by Chen & Sikic [31].
Figure 2.

Anti- and pro-apoptotic plasma membrane-bound ion transporters involved in MDR. The anti-apoptotic transporters include the plasma membrane Ca2+ ATPase (PMCA), hypertonicity-induced cation channels (HICCs), the Na+/H+ exchanger (NHE1), the Na+/K+-ATPase, the Na+-dependent taurine transporter (TauT) and the 1Na+, 1K+, 2Cl− cotransporter (NKCC1). The pro-apototic transporters include the membrane-bound Ca2+ channel (Orai1) and various transient receptor potential channels (Trps) and K+ and Cl− channels.
3. Prevention of cell shrinkage protects against apoptosis
Experiments in Ehrlich ascites tumour cells (EATC) have demonstrated that the addition of a Ca2+ ionophore to cells in culture elicits a substantial net loss of KCl with concomitant cell shrinkage; this is followed by RVI, so that the cells regain their original volume within 10–15 min [32]. On the other hand, inducing a net loss of KCl in EATC with the chemotherapy drug cisplatin induces AVD, which has three stages. As seen in figure 3, these stages are designated AVD1, AVDT and AVD2 and are characterized by a net loss of KCl, by a compensatory net uptake of NaCl and then by a net loss of KCl, respectively. AVDT represents an unsuccessful RVI response in which the continuous loss of K+ reflects impaired function of the Na, K-ATPase [33]. Organic osmolytes are lost throughout the entire AVD process [19]. Figure 4a shows that multi-drug-resistant EATC (MDR EATC) obtained by treating EATC with daunorubicin for more than 70 passages [34] show no AVD1 response after the addition of cisplatin. While wild-type EATC (Wt EATC) enter apoptosis after addition of cisplatin, as reflected by a fourfold increase in caspase 3 activity within 14 h of the addition, MDR EATC show no significant increase in caspase 3 activity within the 14 h time frame (figure 4b). After 18 h of cisplatin exposure, both Wt and MDR EATC cells show eightfold and threefold increases in caspase 3 activities, respectively (figure 4b). Hence, the lack of AVD1 in MDR EATC correlates with prevention of apoptosis.
Figure 3.

Time-dependent changes in cellular water content and ion content in Wt EATC following exposure to 5 µM cisplatin. (a) The water content (millilitre per gram cell dry weight) was normalized to values obtained prior to cisplatin exposure. (b) Cl− content (micromole per gram cell dry weight) was obtained by Ag+ titration. (c,d) K+ and Na+ content was determined using emission flame photometry. The values are reported as means with the standard error of the mean. Asterisks (*) and plus symbols (++) indicate values that were significantly different from the initial control value. Adapted from [19].
Figure 4.
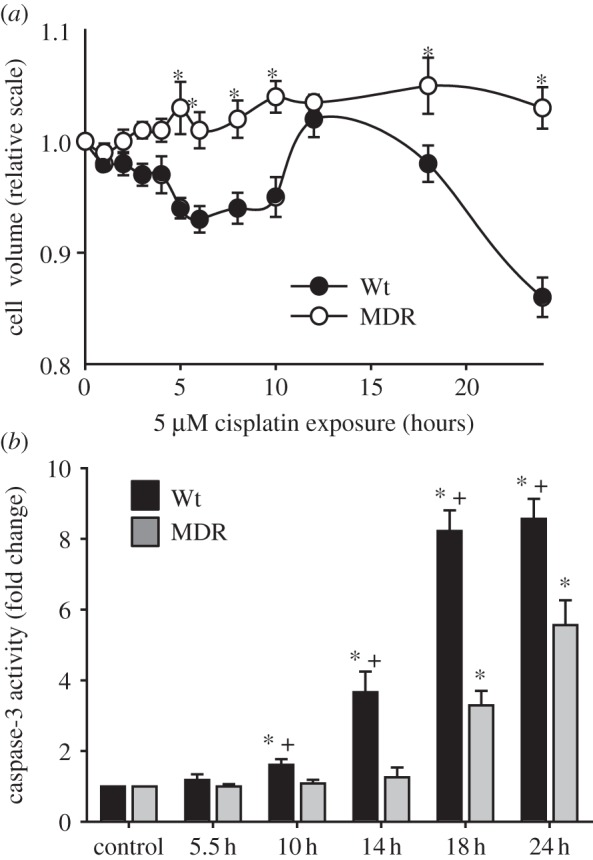
Changes in cell volume and caspase 3 activity in wild-type (Wt) and multi-drug resistant (MDR), EATC. (a) Cell volume was estimated by electronic cell sizing using the Coulter counter technique. (b) Caspase 3 activity was determined using a calorimetric assay to detect production of p-nitroanilide by cleavage of the substrate acetyl-Asp-Glu-Val-Asp p-nitroanilide. The values are reported as means with the standard error of the mean. In (a), asterisk (*) indicates a significant difference between Wt and MDR EATC cells. In (b), asterisk (*) indicates a significant difference compared with control, and plus symbol (+) indicates a significant difference between Wt and MDR EATC cells. Adapted from [19].
4. The role of ion channels in resistance to drug-induced apoptosis
Figure 2 (right-hand side) shows the pro-apoptotic ion channels. Notably, there is downregulation of these channels in MDR. These channels include the K+ and Cl− channels, which are responsible for AVD, as well as Ca2+ channels, which are involved in Ca2+ influx and hence modulation of Ca2+-sensitive steps during apoptosis.
(a). Cl− channels
Reduction in volume-regulated anion current (VRAC) has been related to MDR in several cell lines [19,21,22,35]. However, VRAC activity is in HT-29 cells irrespective of MDR1 expression [36], and overexpression of MDR1 is accompanied by increases in VRAC current in the multi-drug-resistant cell line H69AR [37]. Gollapudi et al. [35] demonstrated that the Cl− conductance was reduced in multi-drug-resistant HL60/AR cells compared with the HL60 parent cells, and that in vitro treatment of drug-sensitive HL60 cells with a Cl− channel blocker resulted in increased resistance to daunorubicin. Likewise, Okada and co-workers [21] demonstrated that VRAC is absent in the multi-drug-resistant human epidermoid cancer cell line KCP-4 and that treatment with a histone deacetylase inhibitor causes partial restoration of VRAC activity and, concomitantly, cisplatin sensitivity. The effects in KCP-4 were blocked by simultaneous treatment of the cells with a VRAC channel blocker [21].
As shown in figure 5a,b, VRAC, as well as the volume-sensitive leak pathway for organic osmolytes, is reduced in MDR EATC compared with Wt EATC. Addition of NS3728, which is an effective VRAC inhibitor [38], reduces the apoptotic response to cisplatin in a dose-dependent manner (figure 5c) in Wt and MDR EATC and at 17 µM NS3728 Wt EATC is as cisplatin resistant as the MDR EATC. This indicates that impaired VRAC activity in MDR EATC correlates with the impaired AVD response and with cisplatin resistance. Similarly, Min et al. [22] demonstrated that impaired VRAC activity contributes to cisplatin resistance in human lung adenocarcinoma (A549/CDDP) cells. Apoptosis is accompanied by DNA fragmentation and it has been shown that T cells lacking the type I transmembrane phosphatase CD45 have a reduced capacity to activate Cl− channels and show less DNA fragmentation following induction of apoptosis via mitochondria perturbing agents [39]. It is suggested that loss of Cl− increases DNA fragmentation. This is in agreement with the observation that inhibition of Cl− channels blocks UV-C induced DNA degradation in human Jurkat cells [40]. However, data for the Jurkat cells indicate that the effect of Cl− reduction is limited to intrinsic activation of apoptosis [40].
Figure 5.
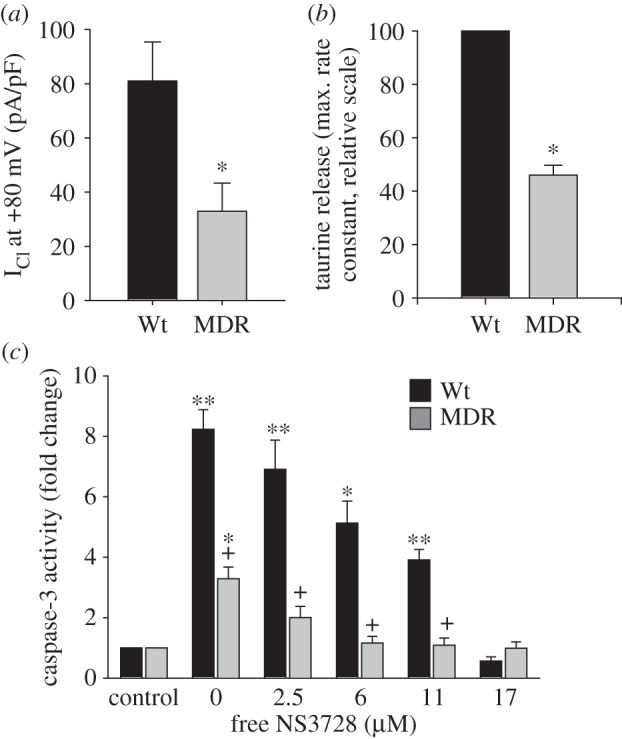
Downregulation of the volume-regulated Cl− current/taurine release pathway in multi-drug resistant (MDR) Ehrlich ascites cells (EATC) and elimination of cisplatin-induced apoptosis following addition of the Cl− channel blocker NS3728. (a) The volume-activated Cl− current was measured using a whole-cell patch-clamp technique following hypotonic exposure (reduction of the extracellular medium to two-third of the isotonic value). (b) Volume-activated release of the organic osmolyte taurine was estimated as the maximal obtainable rate constant following hypotonic exposure. The MDR value is relative to the value in Wt cells. (c) Caspase 3 activity was measured using a calorimetric assay to detect production of p-nitroanilide by cleavage of the substrate acetyl-Asp-Glu-Val-Asp p-nitroanilide. NS3728 was added to block the Cl− current, and the free concentration of NS3728 was determined using Centrifree YM-30 micropartition devices and 14C-labelled NS3728. In (a,b), asterisk (*) indicates significant differences compared with Wt EATC. In (c), asterisk (*) indicates a significant difference compared with control cells without cisplatin, and plus symbol (+) indicates a significant difference between Wt and MDR EATC cells. Adapted from [19].
(b). K+ channels
Potassium channel activity, and hence K+ loss, play an essential role in the initiation of apoptosis owing to (i) decay of the membrane potential and the associated Ca2+ influx; (ii) AVD; and (iii) activation of various enzymes involved in the apoptotic process [12,13,41,42]. Addition of clofilium, which is a TASK2 K+ channel blocker [24], prevents AVD and abrogates cisplatin-induced caspase 3 activity in Wt EATC [19]. Similarly, targeting the big conductance K+ channel with the inhibitor tetraethyl ammonium attenuates cisplatin-induced apoptosis in type I spiral ligament fibrocytes [43] and mouse neocortical neurons [44]. The TASK3 gene (Kcnk9) is overexpressed in several types of human carcinomas which has been associated with resistance towards apoptosis [45]. This is in contrast to what is seen in different glioma cell lines where application of the TASK3 channel opener isoflurane significantly reduces cell survival and the TASK channel blockers bupivacaine and spermine completely reverses this effect [46]. Downregulation of K+ channels as a resistance mechanism is observed in many malignant cancer cells—for example, the expression of Kv1.5 is suppressed in several cancer cell lines [47]. Furthermore, Han et al. [48] demonstrated that upregulation of Kv1.5 increases the K+ current and concomitantly the sensitivity to multiple chemotherapeutic drugs in gastric cancer cells (SGC7901), whereas downregulation of the channel enhances the drug-resistant phenotype. An additional example of downregulation of K+ channels in MDR cells is that the gene expression index for small (SK1/KCNN1) and intermediate (IK/KCNN4) conductance calcium-activated potassium channels is lower in MDR EATC (table 1). KCNN4 was recently associated with proliferation and invasion in colorectal cancer [49]. Moreover, the expression index for the K+ channel modulatory factor 1 (KCMF1) is reduced in MDR EATC (table 1). KCMF1 is broadly overexpressed in human cancer tissues, such as pancreatic carcinomas [50]. However, there are conflicting data as to whether KCMF1 has a pro-oncogenic [50,51] or a more tumour-suppressive [52] function, and its role in apoptosis needs to be investigated further.
Table 1.
Downregulation in the expression of K+ channels in the MDR phenotype. The expression index was determined using the Affymetrix GeneChip Mouse Genome 430 2.0 microarray and the GeneChip Expression Analysis system. (T Litman & EK Hoffmann 2009, unpublished data.)
| gene name | Wt EATC | MDR EATC |
|---|---|---|
| gene expression index | ||
| kcnn1 | 131 ± 2 | 108 ± 2 |
| kcnn4 | 1451 ± 12 | 1246 ± 14 |
| kcmf1 | 1450 ± 10 | 968 ± 15 |
(c). Ca2+ channels
MDR can be achieved via downregulation of proteins involved in Ca2+ homeostasis, so targeting Ca2+ transporters in order to enhance the pro-apoptotic potential of malignant cells may be a useful strategy in the treatment of cancer. The calcium dependence of apoptosis is well described and seems to involve elevation of the intracellular Ca2+ concentration and decreases in the Ca2+ concentration in the endoplasmic reticulum (ER) for review [53,54]. To become resistant cancer cells could either reduce Ca2+ influx by downregulation of Ca2+ permeable channels and/or adapt to chronic-reduced ER Ca2+ [53]. The main plasma membrane-bound Ca2+ transporters that may be involved in the development of MDR include store-operated channels (SOC), transient receptor potential channels (Trps), voltage-gated Ca2+ channels and plasma membrane Ca2+ ATPases (PMCAs), which are briefly discussed below. The ER Ca2+-ATPase (Serca) and the inositol phosphate- (IP3-) sensitive receptor are not discussed in this review.
Induction of apoptosis in Bcl-2-overexpressing cells requires sustained Ca2+ influx via activated channels (SOCs), and downregulation of these channels seems to be a key component of the protective action of Bcl-2 against apoptosis in hormone-insensitive cancer cells [55]. Moreover, the apoptosis resistance of neuroendocrine (NE) differentiated prostate cancer cells seems to suggest that NE differentiation of prostate cancer epithelial cells involves reduction in the replenishment of the ER Ca2+ store, decreased expression of SERCA and substantial downregulation of SOCs [56]. SOCs are activated through a mechanism in which depletion of intracellular calcium stores leads to aggregation of STIM1, i.e. the Ca2+ sensor in ER, and Orai1, the membrane-bound Ca2+ channel protein. Reduced expression of Orai1, and, consequently, reduced SOC activity, prevents Ca2+ overload in response to pro-apoptotic stimuli and thus establishes the MDR phenotype in prostate cancer cells [57]. On the other hand, Faouzi et al. [58] suggest that Orai3 promotes apoptosis resistance in breast cancer cells. Several of the TRP channels have been discussed in relation to the regulation of Ca2+ influx during apoptosis and development of MDR, e.g. TRPC1, TRPV2 and TRPV6 [12,53]. The eventual role of the voltage-gated Ca2+ channels in MDR is complicated thus Cav3.2 seems to be involved in apoptotic resistance in a prostate cancer cell line [12], whereas Cav3.1, which possess comparable biophysical properties to Cav3.2, promotes apoptosis in breast cancer cells [59].
5. Improvement of regulatory volume increase protects against apoptosis
Cell shrinkage is normally accompanied by an RVI response that reflects net uptake of Na+, K+ and Cl− via the Na+/H+ exchanger, NKCC1, and via non-selective cation channels followed by exchange of cellular Na+ for extracellular K+ via the Na+/K+-ATPase [24]. As seen in figure 3, AVDT represents an inadequate RVI response, i.e. the Na+/K+-ATPase is insufficient and the EATC cells continue to lose K+. The effect of inhibition of the Na+/K+-ATPase on apoptosis was reviewed previously [60]. Na+-dependent transporters for organic osmolytes contribute to the RVI response, while overexpression of the taurine transporter TauT protects kidney cells against cisplatin-induced apoptosis [61], TauT knockdown increases cisplatin-induced apoptosis in Ehrlich Lettré cells [62]. In agreement with this, Warskulat and co-workers demonstrated that mice lacking a functional TauT (TauT–/–) lack cellular taurine and become more prone to apoptosis, as seen in retinal degeneration [63,64].
(a). Role of NKCC1, HICCs, NHE1 and PMCA
The literature concerning the role of NKCC1 and hypertonicity-induced cation channels (HICCs) in MDR is quite limited. In glioblastoma cancer cells, inhibition of NKCC1 with bumetanide augments temozolomid-induced AVD and apoptosis [65]. This raises the possibility that a combination of chemotherapeutic drugs with NKCC1 inhibitors might increase the efficiency of the chemotherapeutic treatment. In HeLa cells, HICCs rescue cells from staurosporine-elicited apoptosis [26].
In a number of cancer types, inhibition or knockdown of the Na+/H+ exchanger NHE1 has been shown to enhance chemotherapeutic cell death. In HeLa cells, which are a human cervical cancer-derived cell line, inhibition of RVI during hypertonic stress through application of NHE and anion exchanger blockers prolongs cell shrinkage and augments caspase-3 activation [25]. In agreement with this, hypertonic conditions induce apoptosis in NHE1-deficient PS120 fibroblasts, whereas transfection of HeLa cells with NHE1 restores RVI and prevents apoptosis [25]. In breast cancer cells, NHE1 is an essential player in paclitaxel-induced apoptosis; importantly, simultaneous inhibition of NHE1 results in synergistic potentiation of low-dose paclitaxel pro-apoptosis effects [66]. More recently, it was demonstrated that inhibition or knockdown of NHE1 sensitizes deltaNErbB2-expressing cells to cisplatin-induced apoptosis [67]. Overexpression of BCR-ABL and P-glycoprotein (Pgp) is a known mechanism underlying imatinib resistance, and NHE1 is an important target that has been implicated in the reversal of imatinib resistance in resistant leukaemia (K562) cell lines and in BCR-ABL-positive patient cells [68]. Notably, the role of NHE1 in drug resistance is not limited to its participation in RVI, since it is also involved in acidification of the extracellular nano-environment [69] and hence decreases the passive uptake of weakly basic chemotherapeutic drugs, e.g. doxorubicin, mitoxantrone, vincristine and vinblastine [70].
The plasma membrane Ca2+ ATPases (PMCAs) are low-capacity, high-affinity systems that export Ca2+ from the cytosol to the extracellular environment. There are four isoforms of PMCA: while PMCA1 and 4 are expressed ubiquitously, PMCA2 and 3 show more specific expression patterns [71]. Overexpression of PMCA seems to play a role in breast cancer progression by conferring resistance to apoptosis, and breast cancer patients with increased PMCA2 expression have a poor prognosis [72]. Baggott and co-workers [73] demonstrated that PMCA2-mediated inhibition of the calcineurin/NFAT signalling pathway is implicated in PMCA2-dependent apoptosis resistance in breast cancer cells.
6. Can upregulation of ion transport proteins that are important for proliferation contribute to multi-drug resistance?
Abnormal expression and/or activity of K+, Na+, Ca2+, Cl− channels and TRP channels is involved in the growth and proliferation of cancer cells [74–76]. Thus, developing specific channel blockers represents a promising strategy for cancer treatment. It is not known whether upregulation of certain ion channels plays a role in MDR. TMEM16A (ANO1) is a Ca2+-activated Cl− channel that is overexpressed in several carcinomas where it controls cell proliferation, migration and metastasis [76–79]. Targeting ANO1 has been proposed as a possible treatment for malignant tumours [80]. TMEM16F (ANO6) was also shown recently to be a Ca2+-activated Cl− channel with delayed Ca2+ activation [81,82]; in addition, it has been associated with phospholipid scrambling and apoptosis [83]. Our group investigated whether ANO1 and ANO6 were upregulated during MDR development. Using QPCR with ARP as a reference gene, we found that ANO1 and ANO6 are strongly upregulated in MDR EATC compared with Wt EATC (ANO1 to ARP ratio: 0.00016 ± 0.00005 (n = 3) in Wt and 0.0021 (n = 2) in MDR; ANO6 to ARP ratio: 0.00002 ± 0.00001 (n = 3) in Wt and 0.0017 (n = 2) in MDR).
It was demonstrated previously that the Cl− channel CLC3 is upregulated in drug-resistant prostate cancer cells [84]. However, Cl− channels are, as described above, generally considered to be pro-apoptotic and downregulated in MDR. These seemingly conflicting findings mandate further investigation. Indeed, cell proliferation and apoptosis both require activation of K+ and Cl− channels. It is possible that activation of ion channels at localized/restricted areas of the plasma membrane (Ca2+-activated and agonist controlled) or sequentially activated channels (cell cycle dependent) are mainly involved in proliferation/migration, whereas a more global and persistent activation of channels (e.g. volume sensitive, voltage sensitive) is involved in apoptosis. Consistent with this hypothesis, we found that VRAC is downregulated in MDR EATC, which prevents apoptosis, whereas ANO1, which is normally associated with cell proliferation, is upregulated. VRAC is an example of an anion channel that has roles both in apoptosis and in cell cycle progression (VRAC activity decreases as cells go from G0 to G1) (figure 6). Hence, cells in G0 can either progress into apoptosis if VRAC levels increase (see §4) or continue into G1 and proliferate if VRAC levels decrease. In the latter case, reduced VRAC activity in MDR EATC (figure 5a) would both facilitate cell cycle progression and prevent apoptosis.
Figure 6.
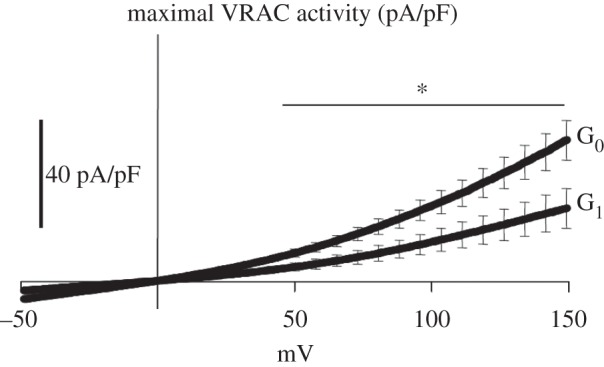
Cell cycle-dependent changes in maximal volume-regulated anion channel (VRAC) activity in ELA cells. The VRAC current was measured using a whole-cell patch-clamp technique as the Cl− current in G0 and G1 phase ELA cells following exposure to hypotonic extracellular solution (190 mOsm) and at nominally zero [Ca2+]i (no added Ca2+, 10 mM EGTA in the pipette solution). The data shown are the I/V relationships based on the mean current density obtained from six to nine cells at each cell cycle phase; error bars indicate the standard error of the mean. Asterisk (*) indicates that the current densities in G0 are significantly different from those in G1 (p < 0.05). Adapted from [38].
7. Conclusion
MDR is one of the most serious challenges when treating cancer using chemotherapy drugs. Many mechanisms are involved in MDR development, and the involvement of changes in the expression and function of ion channels and transport systems has only become clear in recent years. During the development of MDR, several pro-apoptotic ion channels are downregulated, while anti-apoptotic ion transporters are upregulated; these changes act to protect the cancer cells from cell death. However, there are also examples in which ion channels that are important for cell proliferation and migration are upregulated during the development of resistance. We do not yet have a clear picture of the differences between ion channels involved in apoptosis and ion channels involved in proliferation and migration. If ion channels are to be targeted by cancer therapies, then it is essential to know which channels are predominantly downregulated in MDR cells to prevent apoptosis and which predominantly promote growth and proliferation and thus are likely to be upregulated. One possibility is that the development of MDR involves sequential and localized upregulation of ion channels involved in proliferation and migration and a concomitant global and persistent downregulation of ion channels involved in apoptosis. To develop specific activators for the pro-apoptotic channels and specific blockers for the channels that are involved in tumour growth, migration and invasion, it is essential to distinguish between these types of channels and the mechanisms underlying their activation. Further research in this area is needed.
Funding statement
This work was supported by The Danish Council for Independent Research (Natural Sciences and Medical Sciences), FP7 Curie Initial Training Network “IonTraC” (Ion Transport Proteins in Control of Cancer Cell Behaviour), and by the Novo Nordisk Foundation.
References
- 1.Krishna R, Mayer LD. 2000. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur. J. Pharm. Sci. 11, 265–283. ( 10.1016/S0928-0987(00)00114-7) [DOI] [PubMed] [Google Scholar]
- 2.Lothstein L, Israel M, Sweatman TW. 2001. Anthracycline drug targeting: cytoplasmic versus nuclear--a fork in the road. Drug Resist. Update 4, 169–177. ( 10.1054/drup.2001.0201) [DOI] [PubMed] [Google Scholar]
- 3.Stavrovskaya AA. 2000. Cellular mechanisms of multidrug resistance of tumor cells. Biochemistry (Mosc). 65, 95–106. [PubMed] [Google Scholar]
- 4.El MG, Le TC, Batty GN, Faivre S, Raymond E. 2009. Markers involved in resistance to cytotoxics and targeted therapeutics in pancreatic cancer. Cancer Treat Rev. 35, 167–174. ( 10.1016/j.ctrv.2008.10.002) [DOI] [PubMed] [Google Scholar]
- 5.Tamburrino A, Piro G, Carbone C, Tortora G, Melisi D. 2013. Mechanisms of resistance to chemotherapeutic and anti-angiogenic drugs as novel targets for pancreatic cancer therapy. Front Pharmacol. 4, 56 ( 10.3389/fphar.2013.00056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giacomini KM, et al. 2010. Membrane transporters in drug development. Nat. Rev. Drug Discov. 9, 215–236. ( 10.1038/nrd3028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Litman T, Druley TE, Stein WD, Bates SE. 2001. From MDR to MXR: new understanding of multidrug resistance systems, their properties and clinical significance. Cell Mol. Life Sci. 58, 931–959. ( 10.1007/PL00000912) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breier A, Gibalova L, Seres M, Barancik M, Sulova Z. 2013. New insight into p-glycoprotein as a drug target. Anticancer Agents Med. Chem. 13, 159–170. ( 10.2174/187152013804487380) [DOI] [PubMed] [Google Scholar]
- 9.Dmitriev OY. 2011. Mechanism of tumor resistance to cisplatin mediated by the copper transporter ATP7B. Biochem. Cell Biol. 89, 138–147. ( 10.1139/O10-150) [DOI] [PubMed] [Google Scholar]
- 10.Howell SB, Safaei R, Larson CA, Sailor MJ. 2010. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Mol. Pharmacol. 77, 887–894. ( 10.1124/mol.109.063172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalayda GV, Wagner CH, Buss I, Reedijk J, Jaehde U. 2008. Altered localisation of the copper efflux transporters ATP7A and ATP7B associated with cisplatin resistance in human ovarian carcinoma cells. BMC Cancer 8, 175 ( 10.1186/1471-2407-8-175) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehen'kyi V, Shapovalov G, Skryma R, Prevarskaya N. 2011. Ion channnels and transporters in cancer. 5. Ion channels in control of cancer and cell apoptosis 11. Am. J. Physiol. Cell Physiol. 301, C1281–C1289. ( 10.1152/ajpcell.00249.2011) [DOI] [PubMed] [Google Scholar]
- 13.Lang F, Hoffmann EK. 2012. Role of ion transport in control of apoptotic cell death. Compr. Physiol. 2, 2037–2061. [DOI] [PubMed] [Google Scholar]
- 14.Bortner CD, Cidlowski JA. 1998. A necessary role for cell shrinkage in apoptosis. Biochem. Pharmacol. 56, 1549–1559. ( 10.1016/S0006-2952(98)00225-1) [DOI] [PubMed] [Google Scholar]
- 15.Lang F, Foller M, Lang K, Lang P, Ritter M, Vereninov A, Szabo I, Huber SM, Gulbins E. 2007. Cell volume regulatory ion channels in cell proliferation and cell death. Methods Enzymol. 428, 209–225. ( 10.1016/S0074-7696(08)62498-5) [DOI] [PubMed] [Google Scholar]
- 16.Okada Y, Maeno E. 2001. Apoptosis, cell volume regulation and volume-regulatory chloride channels. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 130, 377–383. ( 10.1016/S1095-6433(01)00424-X) [DOI] [PubMed] [Google Scholar]
- 17.Okada Y, Maeno E, Shimizu T, Dezaki K, Wang J, Morishima S. 2001. Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD). J. Physiol. 532, 3–16. ( 10.1111/j.1469-7793.2001.0003g.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okada Y. 2004. Ion channels and transporters involved in cell volume regulation and sensor mechanisms. Cell Biochem. Biophys. 41, 233–258. ( 10.1385/CBB:41:2:233) [DOI] [PubMed] [Google Scholar]
- 19.Poulsen KA, Andersen EC, Hansen CF, Klausen TK, Hougaard C, Lambert IH, Hoffmann EK. 2010. Deregulation of apoptotic volume decrease and ionic movements in multidrug-resistant tumor cells: role of chloride channels. Am. J. Physiol. Cell Physiol. 298, C14–C25. ( 10.1152/ajpcell.00654.2008) [DOI] [PubMed] [Google Scholar]
- 20.Prevarskaya N, Skryma R, Shuba Y. 2010. Ion channels and the hallmarks of cancer 7. Trends Mol. Med. 16, 107–121. ( 10.1016/j.molmed.2010.01.005) [DOI] [PubMed] [Google Scholar]
- 21.Lee EL, Shimizu T, Ise T, Numata T, Kohno K, Okada Y. 2007. Impaired activity of volume-sensitive Cl– channel is involved in cisplatin resistance of cancer cells. J. Cell Physiol. 211, 513–521. ( 10.1002/jcp.20961) [DOI] [PubMed] [Google Scholar]
- 22.Min XJ, Li H, Hou SC, He W, Liu J, Hu B, Wang J. 2011. Dysfunction of volume-sensitive chloride channels contributes to cisplatin resistance in human lung adenocarcinoma cells 1. Exp. Biol. Med. (Maywood). 236, 483–491. ( 10.1258/ebm.2011.010297) [DOI] [PubMed] [Google Scholar]
- 23.Hoffmann EK, Dunham PB. 1995. Membrane mechanisms and intracellular signalling in cell volume regulation. Int. Rev. Cytol. 161, 173–262. [DOI] [PubMed] [Google Scholar]
- 24.Hoffmann EK, Lambert IH, Pedersen SF. 2009. Physiology of cell volume regulation in vertebrates. Physiol. Rev. 89, 193–277. ( 10.1152/physrev.00037.2007) [DOI] [PubMed] [Google Scholar]
- 25.Maeno E, Takahashi N, Okada Y. 2006. Dysfunction of regulatory volume increase is a key component of apoptosis. FEBS Lett. 580, 6513–6517. ( 10.1016/j.febslet.2006.10.074) [DOI] [PubMed] [Google Scholar]
- 26.Numata T, Sato K, Okada Y, Wehner F. 2008. Hypertonicity-induced cation channels rescue cells from staurosporine-elicited apoptosis. Apoptosis 13, 895–903. ( 10.1007/s10495-008-0220-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friis MB, Friborg C, Schneider L, Nielsen MB, Lambert IH, Christensen ST, Hoffmann EK. 2005. Cell shrinkage as a signal to apoptosis in NIH3T3 fibroblasts. J. Physiol. 567.2, 427–443. ( 10.1113/jphysiol.2005.087130) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bortner CD, Scoltock AB, Sifre MI, Cidlowski JA. 2012. Osmotic stress resistance imparts acquired anti-apoptotic mechanisms in lymphocytes 1. J. Biol. Chem. 287, 6284–6295. ( 10.1074/jbc.M111.293001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reinehr R, Haussinger D. 2007. Hyperosmotic activation of the CD95 system. Methods Enzymol. 428, 145–160. ( 10.1016/S0076-6879(07)28008-5) [DOI] [PubMed] [Google Scholar]
- 30.Nielsen MB, Christensen ST, Hoffmann EK. 2008. Effects of osmotic stress on the activity of MAPKs and PDGFR-beta-mediated signal transduction in NIH-3T3 fibroblasts. Am. J. Physiol. Cell Physiol. 294, C1046–C1055. ( 10.1152/ajpcell.00134.2007) [DOI] [PubMed] [Google Scholar]
- 31.Chen KG, Sikic BI. 2012. Molecular pathways: regulation and therapeutic implications of multidrug resistance. Clin. Cancer Res. 18, 1863–1869. ( 10.1158/1078-0432.CCR-11-1590) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoffmann EK, Lambert IH, Simonsen LO. 1986. Separate, Ca2+-activated K+ and Cl– transport pathways in Ehrlich ascites tumor cells. J. Membr. Biol. 91, 227–244. ( 10.1007/BF01868816) [DOI] [PubMed] [Google Scholar]
- 33.Panayiotidis MI, Franco R, Bortner CD, Cidlowski JA. 2010. Ouabain-induced perturbations in intracellular ionic homeostasis regulate death receptor-mediated apoptosis. Apoptosis 15, 834–849. ( 10.1007/s10495-010-0494-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Litman T, Pedersen SF, Kramhoft B, Skovsgaard T, Hoffmann EK. 1998. pH regulation in sensitive and multidrug resistant Ehrlich ascites tumor cells. Cell Physiol. Biochem. 8, 138–150. ( 10.1159/000016277) [DOI] [PubMed] [Google Scholar]
- 35.Gollapudi S, McDonald T, Gardner P, Kang N, Gupta S. 1992. Abnormal chloride conductance in multidrug resistant HL60/AR cells. Cancer Lett. 66, 83–89. ( 10.1016/0304-3835(92)90284-3) [DOI] [PubMed] [Google Scholar]
- 36.Kunzelmann K, Slotki IN, Klein P, Koslowsky T, Ausiello DA, Greger R, Cabantchik ZI. 1994. Effects of P-glycoprotein expression on cyclic AMP and volume-activated ion fluxes and conductances in HT-29 colon adenocarcinoma cells. J. Cell Physiol. 161, 393–406. ( 10.1002/jcp.1041610302) [DOI] [PubMed] [Google Scholar]
- 37.Jirsch J, Deeley RG, Cole SP, Stewart AJ, Fedida D. 1993. Inwardly rectifying K+ channels and volume-regulated anion channels in multidrug-resistant small cell lung cancer cells. Cancer Res. 53, 4156–4160. [PubMed] [Google Scholar]
- 38.Klausen TK, Bergdahl A, Hougaard C, Christophersen P, Pedersen SF, Hoffmann EK. 2007. Cell cycle-dependent activity of the volume- and Ca2+-activated anion currents in Ehrlich lettre ascites cells 7. J. Cell Physiol. 210, 831–842. ( 10.1002/jcp.20918) [DOI] [PubMed] [Google Scholar]
- 39.Dupere-Minier G, Desharnais P, Bernier J. 2010. Involvement of tyrosine phosphatase CD45 in apoptosis. Apoptosis 15, 1–13. ( 10.1007/s10495-009-0413-z) [DOI] [PubMed] [Google Scholar]
- 40.Heimlich G, Cidlowski JA. 2006. Selective role of intracellular chloride in the regulation of the intrinsic but not extrinsic pathway of apoptosis in Jurkat T-cells. J. Biol. Chem. 281, 2232–2241. ( 10.1074/jbc.M507367200) [DOI] [PubMed] [Google Scholar]
- 41.Remillard CV, Yuan JX. 2004. Activation of K+ channels: an essential pathway in programmed cell death. Am. J. Physiol. Lung Cell Mol. Physiol. 286, L49–L67. ( 10.1152/ajplung.00041.2003) [DOI] [PubMed] [Google Scholar]
- 42.Yu SP. 2003. Regulation and critical role of potassium homeostasis in apoptosis. Prog. Neurobiol. 70, 363–386. ( 10.1016/S0301-0082(03)00090-X) [DOI] [PubMed] [Google Scholar]
- 43.Liang F, Schulte BA, Qu C, Hu W, Shen Z. 2005. Inhibition of the calcium- and voltage-dependent big conductance potassium channel ameliorates cisplatin-induced apoptosis in spiral ligament fibrocytes of the cochlea. Neuroscience 135, 263–271. ( 10.1016/j.neuroscience.2005.05.055) [DOI] [PubMed] [Google Scholar]
- 44.Yu SP, et al. 1997. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science 278, 114–117. ( 10.1126/science.278.5335.114) [DOI] [PubMed] [Google Scholar]
- 45.Pei L, Wiser O, Slavin A, Mu D, Powers S, Jan LY, Hoey T. 2003. Oncogenic potential of TASK3 (Kcnk9) depends on K+ channel function 203. Proc. Natl Acad. Sci. USA 100, 7803–7807. ( 10.1073/pnas.1232448100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meuth SG, Herrmann AM, Ip CW, Kanyshkova T, Bittner S, Weishaupt A, Budde T, Wiendl H. 2008. The two-pore domain potassium channel TASK3 functionally impacts glioma cell death 75. J. Neurooncol. 87, 263–270. ( 10.1007/s11060-008-9517-5) [DOI] [PubMed] [Google Scholar]
- 47.Bonnet S, et al. 2007. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth 125. Cancer Cell. 11, 37–51. ( 10.1016/j.ccr.2006.10.020) [DOI] [PubMed] [Google Scholar]
- 48.Han Y, Shi Y, Han Z, Sun L, Fan D. 2007. Detection of potassium currents and regulation of multidrug resistance by potassium channels in human gastric cancer cells 39. Cell Biol. Int. 31, 741–747. ( 10.1016/j.cellbi.2007.01.008) [DOI] [PubMed] [Google Scholar]
- 49.Lai W, Liu L, Zeng Y, Wu H, Xu H, Chen S, Chu Z. 2013. KCNN4 channels participate in the EMT induced by PRL-3 in colorectal cancer. Med. Oncol. 30, 566 ( 10.1007/s12032-013-0566-z) [DOI] [PubMed] [Google Scholar]
- 50.Beilke S, Oswald F, Genze F, Wirth T, Adler G, Wagner M. 2010. The zinc-finger protein KCMF1 is overexpressed during pancreatic cancer development and downregulation of KCMF1 inhibits pancreatic cancer development in mice. Oncogene 29, 4058–4067. ( 10.1038/onc.2010.156) [DOI] [PubMed] [Google Scholar]
- 51.Jang JH. 2004. FIGC, a novel FGF-induced ubiquitin-protein ligase in gastric cancers. FEBS Lett. 578, 21–25. ( 10.1016/j.febslet.2004.10.071) [DOI] [PubMed] [Google Scholar]
- 52.Kreppel M, Aryee DN, Schaefer KL, Amann G, Kofler R, Poremba C, Kovar H. 2006. Suppression of KCMF1 by constitutive high CD99 expression is involved in the migratory ability of Ewing's sarcoma cells. Oncogene 25, 2795–2800. ( 10.1038/sj.onc.1209300) [DOI] [PubMed] [Google Scholar]
- 53.Dubois C, Vanden Abeele F, Prevarskaya N. 2013. Targeting apoptosis by the remodelling of calcium-transporting proteins in cancerogenesis. FEBS J. 280, 5500–5510. ( 10.1111/febs.12246) [DOI] [PubMed] [Google Scholar]
- 54.Orrenius S, Zhivotovsky B, Nicotera P. 2003. Regulation of cell death: the calcium–apoptosis link. Nat. Rev. Mol. Cell Biol. 4, 552–565. ( 10.1038/nrm1150) [DOI] [PubMed] [Google Scholar]
- 55.Vanden Abeele F, Skryma R, Shuba Y, Van CF, Slomianny C, Roudbaraki M, Mauroy B, Wuytack F, Prevarskaya N. 2002. Bcl-2-dependent modulation of Ca2+ homeostasis and store-operated channels in prostate cancer cells 27. Cancer Cell. 1, 169–179. ( 10.1016/S1535-6108(02)00034-X) [DOI] [PubMed] [Google Scholar]
- 56.Vanoverberghe K, et al. 2004. Ca2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells 8. Cell Death Differ. 11, 321–330. ( 10.1038/sj.cdd.4401375) [DOI] [PubMed] [Google Scholar]
- 57.Flourakis M, et al. 2010. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells 2. Cell Death Dis. 1, e75 ( 10.1038/cddis.2010.52) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Faouzi M, Hague F, Potier M, Ahidouch A, Sevestre H, Ouadid-Ahidouch H. 2011. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell Physiol. 226, 542–551. ( 10.1002/jcp.22363) [DOI] [PubMed] [Google Scholar]
- 59.Ohkubo T, Yamazaki J. 2012. T-type voltage-activated calcium channel Cav3.1, but not Cav3.2, is involved in the inhibition of proliferation and apoptosis in MCF-7 human breast cancer cells. Int. J. Oncol. 41, 267–275. [DOI] [PubMed] [Google Scholar]
- 60.Panayiotidis MI, Bortner CD, Cidlowski JA. 2006. On the mechanism of ionic regulation of apoptosis: would the Na+/K+-ATPase please stand up? Acta Physiol. (Oxf). 187, 205–215. ( 10.1111/j.1748-1716.2006.01562.x) [DOI] [PubMed] [Google Scholar]
- 61.Han X, Yue J, Chesney RW. 2009. Functional TauT protects against acute kidney injury. J. Am. Soc. Nephrol. 20, 1323–1332. ( 10.1681/ASN.2008050465) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tastesen HS, Holm JB, Moller J, Poulsen KA, Moller C, Sturup S, Hoffmann EK, Lambert IH. 2010. Pinpointing differences in cisplatin-induced apoptosis in adherent and non-adherent cancer cells. Cell Physiol. Biochem. 26, 809–820. ( 10.1159/000323990) [DOI] [PubMed] [Google Scholar]
- 63.Heller-Stilb B, van RC, Rascher K, Hartwig HG, Huth A, Seeliger MW, Warskulat U, Häussinger D. 2002. Disruption of the taurine transporter gene (taut) leads to retinal degeneration in mice. FASEB J. 16, 231–233. [DOI] [PubMed] [Google Scholar]
- 64.Warskulat U, Borsch E, Reinehr R, Heller-Stilb B, Roth C, Witt M, Häussinger D. 2007. Taurine deficiency and apoptosis: findings from the taurine transporter knockout mouse. Arch. Biochem. Biophys. 462, 202–209. ( 10.1016/j.abb.2007.03.022) [DOI] [PubMed] [Google Scholar]
- 65.Algharabil J, et al. 2012. Inhibition of Na+-K+-2Cl– cotransporter isoform 1 accelerates temozolomide-mediated apoptosis in glioblastoma cancer cells. Cell Physiol. Biochem. 30, 33–48. ( 10.1159/000339047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reshkin SJ, Bellizzi A, Cardone RA, Tommasino M, Casavola V, Paradiso A. 2003. Paclitaxel induces apoptosis via protein kinase A- and p38 mitogen-activated protein-dependent inhibition of the Na+/H+ exchanger (NHE) NHE isoform 1 in human breast cancer cells. Clin. Cancer Res. 9, 2366–2373. [PubMed] [Google Scholar]
- 67.Lauritzen G, Jensen MB, Boedtkjer E, Dybboe R, Aalkjaer C, Nylandsted J, Pedersen SF. 2010. NBCn1 and NHE1 expression and activity in DeltaNErbB2 receptor-expressing MCF-7 breast cancer cells: contributions to pHi regulation and chemotherapy resistance. Exp. Cell Res. 316, 2538–2553. ( 10.1016/j.yexcr.2010.06.005) [DOI] [PubMed] [Google Scholar]
- 68.Jin W, et al. 2011. Reversal of Imatinib resistance in BCR-ABL-positive leukemia after inhibition of the Na+/H+ exchanger. Cancer Lett. 308, 81–90. ( 10.1016/j.canlet.2011.04.016) [DOI] [PubMed] [Google Scholar]
- 69.Stock C, Mueller M, Kraehling H, Mally S, Noel J, Eder C, Schwab A. 2007. pH nanoenvironment at the surface of single melanoma cells. Cell Physiol. Biochem. 20, 679–686. ( 10.1159/000107550) [DOI] [PubMed] [Google Scholar]
- 70.Tredan O, Galmarini CM, Patel K, Tannock IF. 2007. Drug resistance and the solid tumor microenvironment. J. Natl Cancer Inst. 99, 1441–1454. ( 10.1093/jnci/djm135) [DOI] [PubMed] [Google Scholar]
- 71.Strehler EE, Filoteo AG, Penniston JT, Caride AJ. 2007. Plasma-membrane Ca2+ pumps: structural diversity as the basis for functional versatility. Biochem. Soc. Trans. 35, 919–922. ( 10.1042/BST0350919) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.VanHouten J, Sullivan C, Bazinet C, Ryoo T, Camp R, Rimm DL, Chung G, Wysolmerski J. 2010. PMCA2 regulates apoptosis during mammary gland involution and predicts outcome in breast cancer. Proc. Natl Acad. Sci. USA 107, 11 405–11 410. ( 10.1073/pnas.0911186107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baggott RR, et al. 2012. Disruption of the interaction between PMCA2 and calcineurin triggers apoptosis and enhances paclitaxel-induced cytotoxicity in breast cancer cells. Carcinogenesis 33, 2362–2368. ( 10.1093/carcin/bgs282) [DOI] [PubMed] [Google Scholar]
- 74.Li M, Xiong ZG. 2011. Ion channels as targets for cancer therapy. Int. J. Physiol. Pathophysiol. Pharmacol. 3, 156–166. [PMC free article] [PubMed] [Google Scholar]
- 75.Felipe A, Vicente R, Villalonga N, Roura-Ferrer M, Martinez-Marmol R, Sole L, Ferreres JC, Condom E. 2006. Potassium channels: new targets in cancer therapy 67. Cancer Detect Prev. 30, 375–385. ( 10.1016/j.cdp.2006.06.002) [DOI] [PubMed] [Google Scholar]
- 76.Duvvuri U, et al. 2012. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression 7. Cancer Res. 72, 3270–3281. ( 10.1158/0008-5472.CAN-12-0475-T) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kashyap MK, et al. 2009. Genomewide mRNA profiling of esophageal squamous cell carcinoma for identification of cancer biomarkers. Cancer Biol. Ther. 8, 36–46. ( 10.4161/cbt.8.1.7090) [DOI] [PubMed] [Google Scholar]
- 78.Liu W, Lu M, Liu B, Huang Y, Wang K. 2012. Inhibition of Ca2+-activated Cl– channel ANO1/TMEM16A expression suppresses tumor growth and invasiveness in human prostate carcinoma. Cancer Lett. 326, 41–51. ( 10.1016/j.canlet.2012.07.015) [DOI] [PubMed] [Google Scholar]
- 79.Stanich JE, Gibbons SJ, Eisenman ST, Bardsley MR, Rock JR, Harfe BD, Ordog T, Farrugia G. 2011. Ano1 as a regulator of proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 301, G1044–G1051. ( 10.1152/ajpgi.00196.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arcangeli A, Crociani O, Lastraioli E, Masi A, Pillozzi S, Becchetti A. 2009. Targeting ion channels in cancer: a novel frontier in antineoplastic therapy. Curr. Med. Chem. 16, 66–93. ( 10.2174/092986709787002835) [DOI] [PubMed] [Google Scholar]
- 81.Grubb S, Poulsen KA, Juul CA, Kyed T, Klausen TK, Larsen EH, Hoffmann EK. 2013. TMEM16F (Anoctamin 6), an anion channel of delayed Ca2+ activation. J. Gen. Physiol. 141, 585–600. ( 10.1085/jgp.201210861) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shimizu T, Iehara T, Sato K, Fujii T, Sakai H, Okada Y. 2013. TMEM16F is a component of a Ca2+-activated Cl– channel but not a volume-sensitive outwardly rectifying Cl– channel. Am. J. Physiol. Cell Physiol. 304, C748–C759. ( 10.1152/ajpcell.00228.2012) [DOI] [PubMed] [Google Scholar]
- 83.Suzuki J, Umeda M, Sims PJ, Nagata S. 2010. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838. ( 10.1038/nature09583) [DOI] [PubMed] [Google Scholar]
- 84.Lemonnier L, Shuba Y, Crepin A, Roudbaraki M, Slomianny C, Mauroy B, Nilius B, Prevarskaya N, Skryma R. 2004. Bcl-2-dependent modulation of swelling-activated Cl– current and ClC-3 expression in human prostate cancer epithelial cells. Cancer Res. 64, 4841–4848. ( 10.1158/0008-5472.CAN-03-3223) [DOI] [PubMed] [Google Scholar]