

Summary
Phenotypic variation of microbial populations is a well-known phenomenon and may have significant impact on the success of industrial bioprocesses. Flow cytometry (FC) and the large repertoire of fluorescent dyes bring the high-throughput analysis of multiple parameters in single bacterial cells into reach. In this study, we evaluated a set of different fluorescent dyes for suitability in FC single cell analysis of the biotechnological platform organism Corynebacterium glutamicum. Already simple scattering properties of C. glutamicum cells in the flow cytometer were shown to provide valuable information on the growth activity of analysed cells. Furthermore, we used DAPI staining for a FC-based determination of the DNA content of C. glutamicum cells grown on standard minimal or complex media. Characteristic DNA patterns were observed mirroring the typical uncoupled DNA synthesis in the logarithmic (log) growth phase and are in agreement with a symmetric type of cell division of C. glutamicum. Application of the fluorescent dyes Syto 9, propidium iodide, and DiOC2(3) allowed the identification of subpopulations with reduced viability and membrane potential within early log and stationary phase populations. The presented data highlight the potential of FC-based analyses for online monitoring of C. glutamicum bioprocesses and provide a first reference for future applications and protocols.
Introduction
It is a well-known fact that even isogenic microbial populations often exhibit significant variability at the single cell level, which consequently may impact the productivity and the economic success of a whole bioprocess (Lidstrom and Konopka, 2010). Conventional approaches for the analytical monitoring of biotechnological processes are, however, still dominated by bulk measurements providing average data for cellular fitness, productivity or physical parameters. Consequently, there is a lack in receiving statistically valid information about the complex phenotypic structure within populations of industrial microorganisms (Nebe-von-Caron et al., 2000; Müller and Nebe-von-Caron, 2010; da Silva et al., 2012).
In this context, flow cytometry (FC) represents a high resolution technique to assess phenotypic patterns of microbial populations and paves the way for novel high-throughput screening approaches in microbial strain development (Dietrich et al., 2010; Tracy et al., 2010). Based on the principle of hydrodynamic focusing, cells in a flow cytometer are transported in a fluid stream in a pearl chain-like arrangement. At the laser intercept cells passing the laser beam are analysed on the basis of their scattering (forward scatter, FSC, and side scatter, SSC) and fluorescence properties (dyes, natural fluorescence or recombinant proteins). Previous studies revealed that even simple parameters as the cell size, granularity or DNA content give already valuable insights into the metabolic status and the fitness of the whole population (Erlebach et al., 2000; Hewitt and Nebe-von-Caron, 2004; Müller, 2007). Membrane potential (MP) and integrity are further parameters, which can be analysed by the use of specific fluorescent dyes and do provide important information regarding the cellular stress response and viability parameters, which were shown in several studies to significantly correlate with productivity and economic efficiency of the bioprocess (Hewitt et al., 1999; David et al., 2011). When equipped with a cell sorting capability for separation of single bacterial cells, FC analyses can be complemented with further downstream investigations, such as microscopy, cultivation in liquid or on agar plates, or even Omics-based technologies (Bernas et al., 2006; Wiacek et al., 2006; Jehmlich et al., 2010).
The small cell size (1/1000 of the volume of a mammalian cell) and huge diversity with respect to cell wall composition and metabolism challenges FC approaches in the field of microbiology and affects measurement quality (Shapiro and Nebe-von-Caron, 2004; Müller and Nebe-von-Caron, 2010). Nowadays, a variety of fluorescent dyes is available to assess different phenotypic parameters, such as growth, MP or integrity. The majority of current protocols was, however, established and verified for only a small number of model species (Shapiro and Nebe-von-Caron, 2004). To end up with detailed and meaningful insights into the phenotypic structure of a species of interest, suitability of fluorescent dyes and staining protocols have to be adapted and optimized for every single bacterial species to be studied (Vives-Rego et al., 2000; Müller and Nebe-von-Caron, 2010).
Besides species like Escherichia coli or Saccharomyces cerevisiae, the Gram-positive soil bacterium Corynebacterium glutamicum represents one of the most important platform organisms in industrial biotechnology (Eggeling and Bott, 2005; Burkovski, 2008). Currently, C. glutamicum is used for the industrial production of more than 3 million tons of amino acids per year, mainly l-glutamate and l-lysine. Only few studies have yet applied single cell approaches for strain development or for the analysis of phenotypic variation of C. glutamicum populations (Frunzke et al., 2008; Binder et al., 2012; Grünberger et al., 2012; Mustafi et al., 2012). A study of our lab could show that even under standard cultivation conditions wild type populations of the strain C. glutamicum ATCC 13032 exhibit significant variation with respect to the activity of the large prophage CGP3 (Frunzke et al., 2008). FC was first used in two very recent studies for the development of biosensor-based high-throughput screening approaches for the isolation of metabolite producing mutants (Binder et al., 2012; Mustafi et al., 2012). In these studies the intracellular amino acid levels were monitored in single cells by using transcriptional regulators as natural sensor devices to turn on expression of a fluorescent protein in response to the internal amino acid concentration.
In this work, we used FC and cell sorting to analyse population dynamics of C. glutamicum with respect to cell size, granularity and DNA content at single cell resolution. Fluorescent dyes and staining protocols were established to assess membrane integrity and potential. Altogether, these data represent a first comprehensive insight into the phenotypic distribution of C. glutamicum populations and, thus, provide an important reference for future applications of FC as powerful tool for process monitoring and high-throughput screening for this important model organism.
Results and discussion
Size distribution of C. glutamicum wild type cells
Cells passing the laser beam in the flow chamber of a cytometer can be characterized on the basis of their scattering properties. It is a general assumption that the light scattering axial to the laser beam, so-called FSC, can be correlated with the size of a cell, whereas light scattering perpendicular to the laser beam (SSC) is indicative for the internal complexity or granularity of a cell (Müller and Nebe-von-Caron, 2010).
In the case of C. glutamicum ATCC 13032, we observed a characteristic FSC distribution during growth in CGXII minimal medium containing 4% glucose as carbon source (Fig. 1). Cells of the early logarithmic (log) or stationary phase exhibited the smallest FSC signal. This signal significantly increased during the mid-log phase and reached a maximum approximately 5.5 h after inoculation (Fig. 2A). Microscopic analysis confirmed these findings demonstrating a difference in cell size of log phase cells (5.5 h: 2.58 μm ± 0.31) and stationary cells (24 h: 1.66 μm ± 0.31) (Fig. 2B). In Fig. 2C the FSC versus SSC distribution is shown as contour plots highlighting the formation of a second subpopulation in log phase populations. In order to verify whether the high-FSC/SSC subpopulation in fact represents cells in a proliferating state we used DAPI (4′,6-diamidino-2′-phenylindole) staining to correlate scattering properties with the DNA content of the respective populations. The fluorescent dye DAPI is routinely used for DNA quantification of bacterial and eukaryotic cells and binds to A/T-rich regions of DNA resulting in a bright and stable fluorescence signal. Due to its low emission maximum (463 nm) DAPI can also nicely be combined with other stains. As expected, cells with a low-FSC signal (P1) also showed a lower DAPI fluorescence than high-FSC cells (P2) (Fig. 3).
Figure 1.
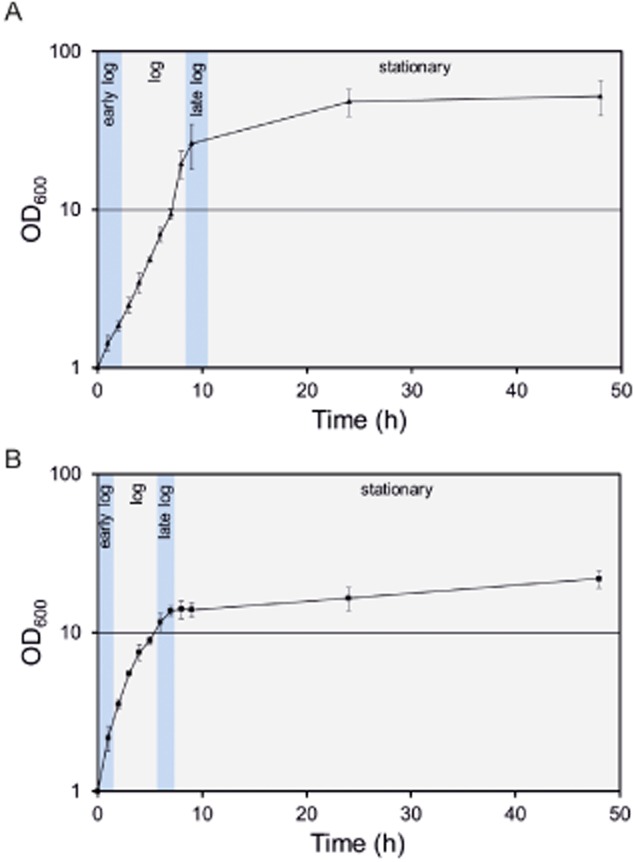
Growth of C. glutamicum ATCC 13032 on (A) CGXII minimal medium with 4% glucose and (B) BHI complex medium in a 50 ml shaking flask culture at 30°C and 120 r.p.m.
Figure 2.

Size distribution of C. glutamicum cells during growth on CGXII minimal medium.A. Mean in forward scatter (FSC). Bars represent the mean ± SD of three independent biological replicates. The asterisks depict significant differences between different time points (*P ≤ 0.05, **P ≤ 0.01).B. Microscopic analysis of C. glutamicum cells of the exponential (5.5 h after inoculation) and stationary growth phase (24 h).C. Contour plots of C. glutamicum cells (FSC versus SSC).
Figure 3.
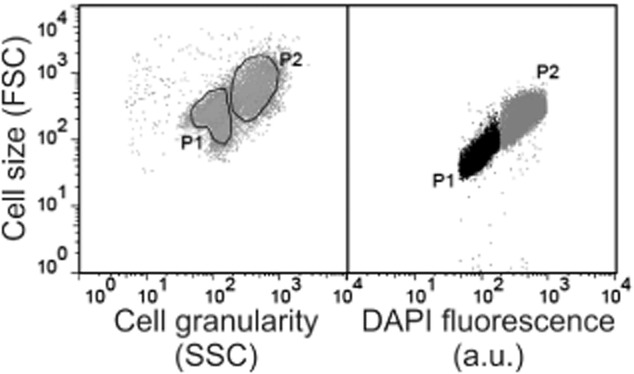
Correlation between cell size (FSC signal) and DAPI fluorescence. Gated populations (P1 and P2) are shown as dot plots: Cell size (FSC) against granularity (SSC) and FSC against DAPI fluorescence.
When interpreting cell size distributions, the unique and multilayered cell wall structure of C. glutamicum has to be considered (Eggeling et al., 2008). A characteristic feature of the group of mycolata, including the genera Corynebacterium, Mycobacterium and Nocardia, is the formation of so-called V-shaped cells caused by a snapping cell division (see arrows in Fig. 2B). The cell wall of the mycolata is surrounded by a lipid-rich envelope, regarded as outer membrane and permeability barrier. After cell division, the daughter cells remain together and form a characteristic V-shape (Thanky et al., 2007; Letek et al., 2008). These cell cluster and aggregates most likely also contribute to the observed increase in FSC and DAPI signals in log phase populations where V-cells mainly occur (Fig. 2).
Heterogeneity with respect to cell size distribution was also described in previous microscopy-based studies of C. glutamicum (Gayen and Venkatesh, 2008; Grünberger et al., 2012). In the study of Gayen and Venkatesh the authors reported a difference in the dividing properties of small and large cells of this species. Cells with a surface area greater than 1.6 μm2 were categorized as maximally dividing cells; smaller cells exhibited significantly reduced proliferation capacity. A correlation between cell size and growth phase was also observed in a picolitre bioreactor microfluidic device were cells of C. glutamicum showed a size of ≥ 1.3 μm during exponential growth and a size of < 1.3 μm under carbon source limiting conditions (Gayen and Venkatesh, 2008; Grünberger et al., 2012). In general, reduction in cell size (in FSC signal, respectively) under carbon source limiting or stressful conditions has been described for various species (Erlebach et al., 2000; Hewitt and Nebe-von-Caron, 2004; Müller and Lösche, 2004; Díaz et al., 2010). For example, a significant decline in FSC values was reported for S. cerevisiae alcoholic fermentations, which was in fact associated with a reduction of actively proliferating yeast cells (Díaz et al., 2010). Furthermore, bacteria typically show a clear correlation between cell size and chromosome equivalents in the proliferation phase (Müller et al., 2010) – a fact which is also supported by the present study for C. glutamicum (Fig. 3). Thus, the measurement of the FSC and SSC distributions of cells in a bioreactor already provides valuable and easy accessible information on the overall fitness and metabolic activity of the bacterial culture.
DNA pattern of C. glutamicum
In several recent studies the analysis of the DNA pattern of microbial populations has been proven as excellent tool for the determination of proliferation and cell cycle activities of microorganisms (Müller, 2007). In this study, we used DAPI staining to record the DNA pattern of C. glutamicum grown on CGXII minimal medium or BHI (brain heart infusion) complex medium in shake flasks. Samples were taken as indicated, DAPI stained and analysed in a flow cytometer equipped with a 355 nm solid state laser (see Experimental procedures). The strain C. glutamicum ATCC 13032 is known to contain one circular chromosome (Kalinowski et al., 2003). Since an absolute quantification of chromosome copy numbers requires more elaborated measurements, the first peak in a DAPI histogram is typically regarded as benchmark population Cn, where C stands for chromosome equivalent and n for the number thereof. Figure 4B shows the characteristic medium-dependent pattern that was observed for C. glutamicum cells grown on minimal or complex medium. Cells cultured in CGXII minimal medium typically showed two distinct peaks (Cn and C2n) in DAPI fluorescence at the early log phase, 1 h after inoculation into fresh medium. In the mid log phase, where C. glutamicum typically exhibits maximal growth rates, uncoupled DNA synthesis was observed as indicated by the shift of the population towards > C2n chromosome equivalents. In the late log phase a backshift occurred and cells arrested within distinct CXn peaks in the stationary phase. On complex medium a similar tendency was observed, however, in comparison to minimal medium the distribution of DAPI peaks shifted towards higher CXn equivalents suggesting the occurrence of cells containing elevated numbers of chromosomes (Fig. 4B). This finding is in line with even better growth conditions on BHI medium resulting in higher growth rates of C. glutamicum (μ ∼ 0.6−h) compared with CGXII minimal medium (μ ∼ 0.4−h). In the stationary phase, again, the formation of distinct CXn peaks was detected with a maximum of chromosome equivalents of about C16n (versus max. C4n in CGXII).
Figure 4.

DNA patterns of C. glutamicum cells grown on minimal or complex medium.A. Schematic overview on the uncoupled cell cycle of bacteria (adapted from Müller et al., 2010). Within the B-phase cells increase their biomass before they are able to enter the C-phase. The duration of the B-phase depends on the environmental conditions. In the C-phase the DNA is replicated and chromosomes are segregated to the cell poles. The duration of the C-phase is species-dependent. When growth conditions are limiting, some species may remain in a so-called pre-D. The D-phase represents the phase of actual cell division.B. DNA patterns of C. glutamicum cells of the early logarithmic (log), log and stationary phase. Cells were cultured in CGXII minimal medium with 4% glucose or in BHI complex medium. After sampling, cells were fixed with 10% sodium azide (in PBS) and stained with DAPI (see Experimental procedures).C. Detailed view on DAPI histogram of stationary phase C. glutamicum cells (pre-D phase) grown on BHI complex medium. Indicated peaks correspond to different numbers of chromosome equivalents (Cn).D. Time-course measurements of C. glutamicum cells during growth on CGXII minimal medium with 4% glucose.
The formation of distinct peaks within the late log and stationary phase can be explained due to the fact that bacteria finish replication (C-phase), but do not continue throughout cell division (D-phase) (Fig. 4A). Thus, our data reveal that C. glutamicum arrests in a pre-division (pre-D) phase when growth conditions become limiting. The occurrence of a pre-D phase under substrate limiting conditions has already been reported for several species, such as Rhodococcus erythropolis or Acinetobacter species, and was furthermore shown to readily pass over into cell division phase when growth conditions improve (Goodfellow et al., 1998; Müller et al., 2002). This is in agreement with the immediate entrance into D-phase when C. glutamicum stationary cultures were inoculated into fresh medium (see Fig. 4D, 1 h versus 48 h). Remarkably, previous studies revealed that occurrence of the pre-D phase is in fact a species-specific characteristic (Müller, 2007). The analysis of DNA patterns furthermore supports a symmetrical cell division of C. glutamicum, typical for rod-shaped species. We identified distinct peaks in the histogram corresponding to Cn (119), C2n (237), C4n (416), C8n (763), C16n (1454) etc. (Fig. 4) (Müller, 2007). However, we also observed minor peaks of odd numbers of chromosome equivalents, which is in fact indicative for a small fraction of asymmetrically dividing cells.
Altogether, these data highlight the value of DNA pattern analysis as rapid online-compatible tool for process monitoring and provide a first reference for C. glutamicum in standard minimal and complex media. Analysis of DNA patterns for the estimation of growth and activity states may also serve as a convenient alternative in turbid (e.g. CaCO3-buffered) production media which hamper the measurement of biomass as optical density or cell dry weight.
FC-based measurement of the membrane potential
The MP plays an important role in the physiology of bacteria as it is intimately involved in the generation of ATP and has been implicated in bacterial autolysis, glucose transport, chemotaxis or bacterial survival at low pH. Therefore, the MP is a further adequate parameter to evaluate the cellular activity and metabolic state of a microbial population. Here, we used the carbocyanine dye 3,3′-diethyloxacarbocyanine iodide (DiOC2(3)) that exhibits a green fluorescence in all bacteria and shifts towards red emission due to self-association of dye molecules in dependency of the MP (high MP → increased red fluorescence) (Novo et al., 1999; 2000).
In a first set of experiments we verified suitability of DiOC2(3) for measurements in C. glutamicum. As a control, C. glutamicum cells were incubated with the uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP), an inhibitor of oxidative phosphorylation, to a final concentration of 50 μM. CCCP-treated cells showed a considerable decrease in the red/green ratio of DiOC2(3) fluorescence indicative for a collapsed MP (Fig. 5A). For further validation we used the potassium ionophore valinomycin, which facilitates transport of potassium ions across the cell membrane. As a matter of fact, bacteria are depolarized when the external potassium concentration exceeds the cytoplasmic concentration (Novo et al., 2000). As expected, C. glutamicum log phase cells treated with 5 μM valinomycin showed a decrease in the red/green ratio of DiOC2(3) fluorescence in response to increasing external potassium concentrations. The MP collapsed rapidly in presence of potassium concentrations between 1 and 10 mM. A constant ratio was maintained for potassium concentrations > 15 mM (Fig. 5B).
Figure 5.
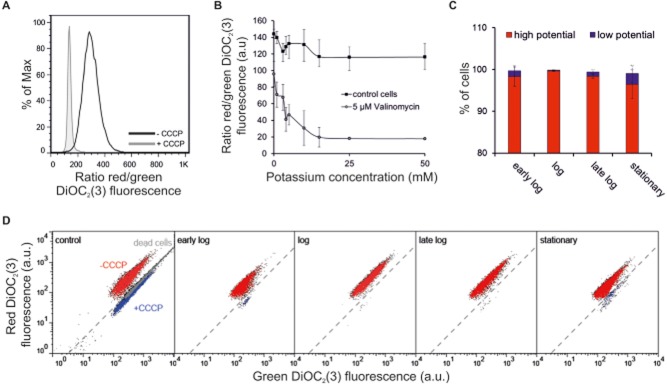
Determination of the MP of C. glutamicum cells.A. Control experiment ± 50 μM CCCP.B. Control experiments ± 5 μM valinomycin and different external potassium concentrations.C. Percentage of cells with a high or low MP. High and low MP were defined by the gating of the ± CCCP control shown in D. Bars represent the mean ± SD of three independent experiments.D. Representative dot plots of C. glutamicum cells of different growth phases (green DiOC2(3) fluorescence against the red DiOC2(3) fluorescence).
Next, we analysed DiOC2(3) stained C. glutamicum cells grown in CGXII medium at different growth phases. Especially in early log and stationary phase populations we observed the formation of a second ‘low potential’ population, which resides at the position of CCCP-treated or dead cells (Fig. 5C and D). In the mid log phase, where cells grow at maximum rate, all cells showed vital states and almost no ‘low potential’ cells were detected. However, it has to be considered that a direct conclusion on cellular viability cannot be derived from FC measurements using fluorescent dyes as all dyes itself affect viability of treated cells (Table 1) (Nebe-von-Caron et al., 2000). We therefore rather emphasize MP measurements either for direct comparisons of different strains of the same species under well-defined conditions or as indirect tool to assess cellular viability during cultivation of a particular species.
Table 1.
Survival rate of log and stationary phase C. glutamicum cells after treatment with different fluorescence dyes
| Survival rates (%)a |
||||||||
|---|---|---|---|---|---|---|---|---|
| Unstained control |
SYTO®9 |
DiOC2(3) |
Propidium iodide |
|||||
| Incubation time (h) | BHI | CGXII | BHI | CGXII | BHI | CGXII | BHI | CGXII |
| Log phase | 96 ± 2 | 95 ± 4 | 80 ± 13 | 71 ± 34 | < 1 | < 1 | < 1 | < 1 |
| Stat phase | 63 ± 3 | 49 ± 4 | 66 ± 15 | 59 ± 29 | < 1 | < 1 | < 1 | < 1 |
Data represent the percentage of visible colonies of sorted single cells of three independent experiments ± SD. Stained cells were considered as ‘dead’ when the survival rate was below 1%.
Application of Syto 9 and propidium iodide – ‘live/dead’ staining
A classical approach for the determination of membrane integrity is the combination of the fluorescent dyes propidium iodide (PI) and Syto 9, known as ‘live/dead staining’ or BacLight (Hammes et al., 2008). Both dyes bind specifically to DNA and RNA by intercalation between bases. Whereas Syto 9 (green fluorescence) is membrane permeable and stains all cells, PI only penetrates cells with damaged membranes and masks in these cells the Syto 9 signal. For verification of the staining protocol we performed a calibration using defined mixtures of live and dead (isopropanol-killed) cells stained with PI and Syto 9. FC analysis of these mixtures revealed significant agreement of measured and assumed cell fractions (Fig. 5A). Application of this dye combination on C. glutamicum cells of different growth phases resulted in the identification of a PI positive subpopulation in the early log phase, which significantly declines in the log phase and reappears in the stationary phase (Fig. 6B and C). This increase in PI positive cells correlated well with the distribution of a low MP population (Fig. 5). However, absolute values differ between the applied staining approaches and again highlight that these approaches are not suitable for the exact counting of live and dead cells in a population, but rather show a characteristic pattern in a particular growth phase.
Figure 6.

Live/dead staining of C. glutamicum cells using the fluorescent dyes PI and Syto 9. C. glutamicum ATCC 13032 was grown on CGXII minimal medium with 4% glucose, sampled as indicated and stained with PI (red) and Syto 9 (green) as described in Experimental procedures.A. Calibration: The assumed fraction of dead (isopropanol-killed) cells was plotted against the measured fraction of dead cells.B. Percentage of Syto 9 and Syto 9/PI positive (live) and PI positive (dead) cells in dependency of cultivation time. Bars represent the mean ± SD of three independent experiments.C. Dot plots (PI versus Syto 9) of the control experiment (calibration), early log, log, and stationary phase C. glutamicum cells (live: green; dead: red).D. Microscopic analysis (fluorescence channel) of log phase C. glutamicum cells (5.5 h after inoculation) stained with Syto 9 and PI. The majority of cells showed a green fluorescence (I), a small amount of cells stained PI positive (II). Often V-shaped sister cells were observed where one cell stained Syto 9 positive whereas the sister cell stained PI positive (III/IV).
Remarkably, we also observed a shift of log phase cells towards a higher PI and Syto 9 signal (Fig. 6C). This behaviour was also described for several other species and is supposed to be due to loosen cell wall structures during periods of fast cell size growth and high division rates. However, the actual reason why the cell walls of some dividing cells might become permeable for PI remains hitherto unknown (Lybarger and Maddock, 2001; Shi et al., 2004). Interestingly, in microscopic analysis we often observed (besides green or red cells, Fig. 6D I/II) V-shape cells where one cell stained green and the other one red (Fig. 6D III/IV). This phenomenon was observed for more than 50% of V-shape cells in the exponential phase and for about 33% in the stationary phase. This finding has also been described for Corynebacterium diphtheria (Ott et al., 2010), but the reason for this staining pattern is so far unknown. However, it is very unlikely that red cells of V-shapes, occurring during fast cell division in log phase populations, represent ‘dead’ C. glutamicum cells. It can rather be suggested that this phenomenon is caused by the specific corynebacterial cell wall composition and cell division resulting in one PI impermeable and one permeable sister cell.
Conclusion
In the present study we have applied FC as rapid and convenient tool to evaluate growth behaviour and viability of C. glutamicum at different growth stages. The reported FC approaches combine the advantage of rapid, online-compatible measurements with high-throughput analysis at single cell resolution. The large repertoire of available stains provides access to a variety of cellular parameters and can be combined in multiparameter FC analysis. Recent efforts of several groups aim on the development of downstream analytics, such as transcriptomics or proteomics approaches, which can be applied for the further analysis of FC sorted subpopulations to obtain insights into phenotypic variation at the molecular level (Bernas et al., 2006; Wiacek et al., 2006; Jehmlich et al., 2010). Recently, our group also reported the development of a genetically encoded single cell biosensor for the intracellular detection of l-methionine and branched-chain amino acids in C. glutamicum (Mustafi et al., 2012). Application of this sensor in the l-valine-producing C. glutamicum strain ΔaceE already suggested significant variation with respect to amino acid production at the single cell level. In this context, the application of multiparameter cytometry, making use of genetical biosensors and/or fluorescent dyes, will provide more insights into phenotypic variation of industrial production strains and the underlying mechanisms thereof.
Our study, as well as previous ones with other model species, highlights the importance of a thorough verification of available fluorescent dyes and the adaptation of the respective staining protocols for each particular species of interest. Overcoming this obstacle, muliparameter cytometry provides an efficient online tool to rapidly monitor growth states and metabolic activity of microbial populations in bioprocesses.
Experimental procedures
Bacterial strains, media and growth conditions
In the following experiments C. glutamicum ATCC 13032 was used as wild type strain (Kalinowski et al., 2003). A first pre-culture of C. glutamicum was inoculated with colonies from a fresh BHI agar plate (brain heart infusion, Difco™ BHI, BD, Heidelberg, Germany) and grown in 5 ml BHI liquid medium for 8 h at a temperature of 30°C and a shaking rate of 170 r.p.m. Afterwards, cells were washed with 0.9% (w/v) NaCl and were transferred to 50 ml CGXII minimal medium with 4% (w/v) glucose (Keilhauer et al., 1993). Cells were cultured overnight at 30°C with a shaking rate of 120 r.p.m. For the main culture, CGXII medium was inoculated to an OD600 of 1 and C. glutamicum was grown at 30°C for 48 h in 500 ml shaking flasks in 50 ml medium. The growth of C. glutamicum in CGXII medium with 4% glucose or BHI complex medium is shown in Fig. 1.
Staining of bacteria
Samples of C. glutamicum were harvested from 50 ml cultures at different time points as indicated. OD600 was measured and the samples were diluted in phosphate-buffered saline (PBS) (129 mM NaCl, 2.5 mM KCl, 7 mM Na2HPO4 × 2H2O, 1.3 mM KH2PO4, pH 7.0) or FACSFlow™ sheat fluid buffer (BD, Heidelberg, Germany). Afterwards, cells were stained with the different fluorescence dyes as described below. Table 2 provides an overview on used dye concentrations and incubation times.
Table 2.
Filter set-ups, concentrations, incubation times and function of used fluorescence dyes
| Fluorescence dye | Excitation/emission (nm) | Long-pass filter (nm) | Band-pass filter (nm) | Concentration (μM) | Incubation time (min) | Function |
|---|---|---|---|---|---|---|
| DAPIa | 355/463 | – | 450/65 | 0.25 | 60 | DNA dye |
| DiOC2(3) | 30 | 30 | Membrane potential | |||
| Green fluorescence | 488/497 | 502 | 530/30 | |||
| Red fluorescence | 488/610 | 565 | 610/20 | |||
| PI | 538/617 | 595 | 610/20 | 20 | 15 | Nucleic acid dye/membrane integrity |
| Syto9 | 485/498 | 502 | 530/30 | 3.3 | 15 | Nucleic acid dye/cell count |
Analyses were performed with a MoFlo cytometer (see Experimental procedures).
DNA patterns
Bacteria were harvested by centrifugation (4.000 g, 5 min, 4°C) and washed in phosphate buffer (0.4 M Na2HPO4/NaH2PO4, 150 mM NaCl, pH 7.2). Subsequently, cells (diluted to an OD600 of 1) were fixed with phosphate buffer containing 10% sodium azide and stored at 4°C until cytometric measurement for at least 1 h and no longer than 1 week. Two ml of the cell suspension (diluted to OD600 = 0.035) were treated with 1 ml solution A (0.1 M citric acid and 4.1 mM Tween 20) for 10 min at room temperature (RT). Afterwards, cells were stained with 0.25 μM DAPI (Sigma-Aldrich, Taufkirchen, Germany) in 0.4 M sodium phosphate solution (Na2HPO4) pH 7.0 for 60 min at RT in the dark using a modification of a standard procedure (Meistrich et al., 1978).
Membrane potential
C. glutamicum cells were diluted to an OD600 of 0.05 and stained with 30 μM DiOC2(3) [(stock solution: 3 mM in dimethylsulphoxide (DMSO)] (Sigma-Aldrich) for 30 min at RT. We chose this long incubation time, since the DiOC2(3) signal is shifting rapidly during the first 30 min towards higher fluorescence intensities. This would require a to the split second measurement to achieve reproducible results. An experiment where cells were stained for only 1 min showed a similar pattern/as cells stained for 30 min (data not shown). For CCCP (carbonyl cyanide m-chlorophenylhydrazone) control experiments, cells were incubated with 50 μM CCCP (stock solution 5 mM in DMSO) (Sigma-Aldrich). In a further proof of principle experiment, cells were treated with 5 μM of the potassium ionophore valinomycin (stock solution 5 mM in DMSO) (Sigma-Aldrich) and stained in phosphate buffer (pH 7) containing different concentrations of KCl (0–300 mM) and appropriate amounts of NaCl (300-x, x = KCl concentration) (Novo et al., 2000).
Live-dead staining
Cells with an OD600 of 0.05 were incubated with 20 μM propidium iodide (PI stock solution: 20 mM in DMSO) (Molecular Probes, Leiden, Netherlands) and 3.3 μM Syto 9 (stock solution: 3.34 mM in DMSO) (Molecular Probes, Leiden, Netherlands) for 15 min at RT. For validation of the protocol, intact cells and cells with injured membranes, (treated with 70% isopropyl alcohol for 30 min) were mixed in different ratios. Afterwards, cells were stained as described above.
Flow cytometry
Flow cytometry analyses and cell sorting experiments were performed with a FACSAria II flow cytometer (Becton Dickinson, Heidelberg, Germany) using a blue solid state laser (Sapphire™ 488-20) with an excitation wavelength of 488 nm and a power of 13 mW. Cytometer set-up and performance tracking was performed with Cytometer Setup 0026; Tracking Beads [bright (3 μm), mid (3 μm), and dim (2 μm) beads] labelled with a mixture of fluorochromes (Becton Dickinson).
DNA analyses were performed on a MoFlo (DakoCytomation, FortCollins, CO) equipped with a water-cooled argon-ion laser (Innova 70C from Coherent, Santa Clara, California, USA). Excitation of 400 mW at 488 nm was used to analyse the FSC and SSC as trigger signal at the first observation point. DAPI was excited by 150 mW with a 355 nm solid state laser XCyte (JDS Uniphase) at the second observation point. The orthogonal signal was first reflected by a beamsplitter and then recorded after reflection by a 555 nm long-pass dichroic mirror, passage by a 505 nm short-pass dichroic mirror and a BP 488/10. DAPI fluorescence was monitored using a 450/65 band pass filter. Amplification was carried out at logarithmic scales. Fluorescent beads (0.5 μm bright blue Fluoresbrite carboxylate microspheres, 360/407, Polyscience, Warrington, Pennsylvania, USA) were used to align the MoFlo. Also, an internal DAPI stained bacterial cell standard was introduced for tuning the device up to a CV value not higher than 6%. Table 2 summarizes filter set-ups used for the analysis of the different fluorescence dyes. Ten thousand cells per sample (40 000 cells for DAPI measurements) were recorded using FACSDiva software 6.0 (FACSAria II) or Summit v4.3 software (MoFlo). Data were analysed with the FlowJo FC analysis software 7.6.5 (Tree Star, Ashland, USA).
Survival rate
For determination of the cellular survival rate after different staining procedures single cells were sorted with the four-way-purity precision mode onto BHI or CGXII agar plates (256 single cells per agar plate). Plates were incubated at a temperature of 30°C for 48 h. Afterwards, the percentage of visible colonies of sorted single cells was determined and was regarded as survival rate (Table 1).
Statistical analysis
Data are expressed as mean ± standard deviation (SD) of three independent biological replicates. Statistical significance was calculated via t-test; P-values ≤ 0.05 were considered to be statistically significant.
Acknowledgments
This work was supported by a Young Investigator grant of the Helmholtz Association (Grant VH-NG-716 to J. F.). Part of this work was integrated in the internal research and development programme of the UFZ and the CITE programme (Chemicals In The Environment).
Funding Information
This work was supported by a Young Investigator grant of the Helmholtz Association (Grant VH-NG-716 to J. F.).
Conflict of interest
None declared.
References
- Bernas T, Gregori G, Asem EK, Robinson JP. Integrating cytomics and proteomics. Mol Cell Proteomics. 2006;5:2–13. doi: 10.1074/mcp.R500014-MCP200. [DOI] [PubMed] [Google Scholar]
- Binder S, Schendzielorz G, Stäbler N, Krumbach K, Hoffmann K, Bott M, Eggeling L. A high-throughput approach to identify genomic variants of bacterial metabolite producers at the single-cell level. Genome Biol. 2012;13:R40. doi: 10.1186/gb-2012-13-5-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkovski A. Corynbebacteria: Genomics and Molecular Biology. Norfolk, UK: Caister Academic Press; 2008. [Google Scholar]
- David F, Berger A, Hansch R, Rohde M, Franco-Lara E. Single cell analysis applied to antibody fragment production with Bacillus megaterium: development of advanced physiology and bioprocess state estimation tools. Microb Cell Fact. 2011;10:1–15. doi: 10.1186/1475-2859-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz M, Herrero M, García LA, Quirós C. Application of flow cytometry to industrial microbial bioprocesses. Biochem Eng J. 2010;48:385–407. [Google Scholar]
- Dietrich JA, McKee AE, Keasling JD. High-throughput metabolic engineering: advances in small-molecule screening and selection. Annu Rev Biochem. 2010;79:563–590. doi: 10.1146/annurev-biochem-062608-095938. [DOI] [PubMed] [Google Scholar]
- Eggeling L, Bott M. Handbook of Corynebacterium glutamicum. Boca Raton, FL, USA: CRC Press; 2005. [Google Scholar]
- Eggeling L, Besra GS, Alderwick L. Structure and synthesis of the cell wall. In: Bukowski A, editor. Corynebacteria: Genomics and Molecular Biology. Norfolk, UK: Caiter Academic Press; 2008. pp. 267–294. [Google Scholar]
- Erlebach CE, Illmer P, Schinner F. Changes of cell size distribution during the batch culture of Arthrobacter strain PI/1-95. Antonie Van Leeuwenhoek. 2000;77:329–335. doi: 10.1023/a:1002632128921. [DOI] [PubMed] [Google Scholar]
- Frunzke J, Bramkamp M, Schweitzer JE, Bott M. Population heterogeneity in Corynebacterium glutamicum ATCC 13032 caused by prophage CGP3. J Bacteriol. 2008;190:5111–5119. doi: 10.1128/JB.00310-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayen K, Venkatesh KV. Quantification of cell size distribution as applied to the growth of Corynebacterium glutamicum. Microbiol Res. 2008;163:586–593. doi: 10.1016/j.micres.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Goodfellow M, Alderson G, Chun J. Rhodococcal systematics: problems and developments. Antonie Van Leeuwenhoek. 1998;74:3–20. doi: 10.1023/a:1001730725003. [DOI] [PubMed] [Google Scholar]
- Grünberger A, Paczia N, Probst C, Schendzielorz G, Eggeling L, Noack S, et al. A disposable picolitre bioreactor for cultivation and investigation of industrially relevant bacteria on the single cell level. Lab Chip. 2012;12:2060–2068. doi: 10.1039/c2lc40156h. [DOI] [PubMed] [Google Scholar]
- Hammes F, Berney M, Wang Y, Vital M, Koster O, Egli T. Flow-cytometric total bacterial cell counts as a descriptive microbiological parameter for drinking water treatment processes. Water Res. 2008;42:269–277. doi: 10.1016/j.watres.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Hewitt CJ, Nebe-Von Caron G, Nienow AW, McFarlane CM. Use of multi-staining flow cytometry to characterise the physiological state of Escherichia coli W3110 in high cell density fed-batch cultures. Biotechnol Bioeng. 1999;63:705–711. doi: 10.1002/(sici)1097-0290(19990620)63:6<705::aid-bit8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Hewitt JC, Nebe-von-Caron G. The application of multiparameter flow cytometry to monitor individual microbial cell physiological states. Adv Biochem Eng Biotechnol. 2004;89:197–223. doi: 10.1007/b93997. [DOI] [PubMed] [Google Scholar]
- Jehmlich N, Hübschmann T, Gesell Salazar M, Volker U, Benndorf D, Müller S, et al. Advanced tool for characterization of microbial cultures by combining cytomics and proteomics. Appl Microbiol Biotechnol. 2010;88:575–584. doi: 10.1007/s00253-010-2753-6. [DOI] [PubMed] [Google Scholar]
- Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, et al. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J Biotechnol. 2003;104:5–25. doi: 10.1016/s0168-1656(03)00154-8. [DOI] [PubMed] [Google Scholar]
- Keilhauer C, Eggeling L, Sahm H. Isoleucine synthesis in Corynebacterium glutamicum: molecular analysis of the ilvB-ilvN-ilvC operon. J Bacteriol. 1993;175:5595–5603. doi: 10.1128/jb.175.17.5595-5603.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letek M, Fiuza M, Ordonez E, Villadangos AF, Ramos A, Mateos LM, Gil JA. Cell growth and cell division in the rod-shaped actinomycete Corynebacterium glutamicum. Antonie Van Leeuwenhoek. 2008;94:99–109. doi: 10.1007/s10482-008-9224-4. [DOI] [PubMed] [Google Scholar]
- Lidstrom ME, Konopka MC. The role of physiological heterogeneity in microbial population behavior. Nat Chem Biol. 2010;6:705–712. doi: 10.1038/nchembio.436. [DOI] [PubMed] [Google Scholar]
- Lybarger SR, Maddock JR. Polarity in action: asymmetric protein localization in bacteria. J Bacteriol. 2001;183:3261–3267. doi: 10.1128/JB.183.11.3261-3267.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meistrich ML, Gohde W, White RA, Schumann J. Resolution of X and Y spermatids by pulse cytophotometry. Nature. 1978;274:821–823. doi: 10.1038/274821a0. [DOI] [PubMed] [Google Scholar]
- Müller S. Modes of cytometric bacterial DNA pattern: a tool for pursuing growth. Cell Prolif. 2007;40:621–639. doi: 10.1111/j.1365-2184.2007.00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller S, Lösche A. Population profiles of a commercial yeast strain in the course of brewing. J Food Eng. 2004;63:375–381. [Google Scholar]
- Müller S, Nebe-von-Caron G. Functional single-cell analyses: flow cytometry and cell sorting of microbial populations and communities. FEMS Microbiol Rev. 2010;34:554–587. doi: 10.1111/j.1574-6976.2010.00214.x. [DOI] [PubMed] [Google Scholar]
- Müller S, Sträuber H, Lösche A, Babel W. Population analysis of a binary bacterial culture by multi-parametric flow cytometry. J Biotechnol. 2002;97:163–176. doi: 10.1016/s0168-1656(02)00063-9. [DOI] [PubMed] [Google Scholar]
- Müller S, Harms H, Bley T. Origin and analysis of microbial population heterogeneity in bioprocesses. Curr Opin Biotechnol. 2010;21:100–113. doi: 10.1016/j.copbio.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Mustafi N, Grünberger A, Kohlheyer D, Bott M, Frunzke J. The development and application of a single-cell biosensor for the detection of L-methionine and branched-chain amino acids. Metab Eng. 2012;14:449–457. doi: 10.1016/j.ymben.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Nebe-von-Caron G, Stephens PJ, Hewitt CJ, Powell JR, Badley RA. Analysis of bacterial function by multi-colour fluorescence flow cytometry and single cell sorting. J Microbiol Methods. 2000;42:97–114. doi: 10.1016/s0167-7012(00)00181-0. [DOI] [PubMed] [Google Scholar]
- Novo D, Perlmutter NG, Hunt RH, Shapiro HM. Accurate flow cytometric membrane potential measurement in bacteria using diethyloxacarbocyanine and a ratiometric technique. Cytometry. 1999;35:55–63. doi: 10.1002/(sici)1097-0320(19990101)35:1<55::aid-cyto8>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Novo DJ, Perlmutter NG, Hunt RH, Shapiro HM. Multiparameter flow cytometric analysis of antibiotic effects on membrane potential, membrane permeability, and bacterial counts of Staphylococcus aureus and Micrococcus luteus. Antimicrob Agents Chemother. 2000;44:827–834. doi: 10.1128/aac.44.4.827-834.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott L, Holler M, Gerlach RG, Hensel M, Rheinlaender J, Schaffer TE, Burkovski A. Corynebacterium diphtheriae invasion-associated protein (DIP1281) is involved in cell surface organization, adhesion and internalization in epithelial cells. BMC Microbiol. 2010;10:2. doi: 10.1186/1471-2180-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro HM, Nebe-von-Caron G. Multiparameter flow cytometry of bacteria. Methods Mol Biol. 263:33–44. doi: 10.1385/1-59259-773-4:033. [DOI] [PubMed] [Google Scholar]
- Shi L, Günther S, Hübschmann T, Wick LY, Harms H, Müller S. Limits of propidium iodide as a cell viability indicator for environmental bacteria. Cytometry. 2004;71:592–598. doi: 10.1002/cyto.a.20402. 2007. [DOI] [PubMed] [Google Scholar]
- da Silva TL, Roseiro JC, Reis A. Applications and perspectives of multi-parameter flow cytometry to microbial biofuels production processes. Trends Biotechnol. 2012;30:225–232. doi: 10.1016/j.tibtech.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Thanky NR, Young DB, Robertson BD. Unusual features of the cell cycle in mycobacteria: polar-restricted growth and the snapping-model of cell division. Tuberculosis (Edinb) 2007;87:231–236. doi: 10.1016/j.tube.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Tracy BP, Gaida SM, Papoutsakis ET. Flow cytometry for bacteria: enabling metabolic engineering, synthetic biology and the elucidation of complex phenotypes. Curr Opin Biotechnol. 2010;21:85–99. doi: 10.1016/j.copbio.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Vives-Rego J, Lebaron P, Nebe-von Caron G. Current and future applications of flow cytometry in aquatic microbiology. FEMS Microbiol Rev. 2000;24:429–448. doi: 10.1111/j.1574-6976.2000.tb00549.x. [DOI] [PubMed] [Google Scholar]
- Wiacek C, Müller S, Benndorf D. A cytomic approach reveals population heterogeneity of Cupriavidus necator in response to harmful phenol concentrations. Proteomics. 2006;6:5983–5994. doi: 10.1002/pmic.200600244. [DOI] [PubMed] [Google Scholar]