

Summary
Invadopodia are protrusive structures used by tumor cells for degradation of the extracellular matrix to promote invasion [1]. Invadopodia formation and function are regulated by cytoskeletal remodeling pathways and the oncogenic kinase Src. The guanine nucleotide exchange factor Vav1, which is an activator of Rho family GTPases, is ectopically expressed in many pancreatic cancers, where it promotes tumor cell survival and migration [2, 3]. We have now determined that Vav1 is also a potent regulator of matrix degradation by pancreatic tumor cells, as depletion of Vav1 by siRNA-mediated knockdown inhibits the formation of invadopodia. This requires the exchange function of Vav1 toward the GTPase Cdc42, which is required for invadopodia assembly [4, 5]. In addition, we have determined that Src-mediated phosphorylation and activation of Vav1 is both required for, and, unexpectedly, sufficient for, invadopodia formation. Expression of Vav1 Y174F, which mimics its activated state, is a potent inducer of invadopodia formation through Cdc42, even in the absence of Src activation and phosphorylation of other Src substrates, such as cortactin. Thus, these data identify a novel mechanism by which Vav1 can enhance the tumorigenicity and invasive potential of cancer cells. These data suggest that Vav1 promotes the matrix-degrading processes underlying tumor cell migration, and further, under conditions of ectopic Vav1 expression, that Vav1 is a central regulator and major driver of invasive matrix remodeling by pancreatic tumor cells.
Results and Discussion
Vav1 expression promotes degradation of the extracellular matrix
Ectopic expression of Vav1 in pancreatic cancers leads to increased tumor cell survival, enhanced cell migration, and a poor prognosis [2, 3]. Accordingly, RNAi-mediated depletion of Vav1 in DanG pancreatic adenocarcinoma cells inhibited transwell invasion (Figure 1A). In addition to upregulating migratory signaling pathways, tumor cells degrade and remodel the extracellular matrix (ECM) to allow for escape from the primary tumor and metastasis. The actin cytoskeletal changes required for migration and invasion are regulated by Rho GTPases, including Rac1, RhoA, and Cdc42, the activation of which are controlled by guanine nucleotide exchange factors (GEFs) [6, 7]. As Vav1 is a GEF and activator of Rho GTPases, and is ectopically expressed in tumor cells, we hypothesized that Vav1 could promote the invasive process of matrix degradation (Supplemental Figure S1A) [7]. To test this, DanG cells were depleted of Vav1 using siRNA, then plated on fluorescent gelatin-coated coverslips for seven hours. Control transfected cells showed robust matrix degradation (Figure 1B). However, Vav1 depletion, confirmed by western blot analysis (Figure 1C), reduced both the number of cells competent to degrade matrix (Figure 1D) and the area of degradation per cell (Figure 1E). Similar results were observed in three other Vav1-expressing pancreatic tumor cell lines (CFPAC, Panc04.03, and HPAF-II; Supplemental Figure S1B-E). Thus, in addition to regulating tumor cell survival and migration, Vav1 also promotes degradation of the ECM by pancreatic cancer cells.
Figure 1. Vav1 expression promotes matrix degradation by pancreatic tumor cells.
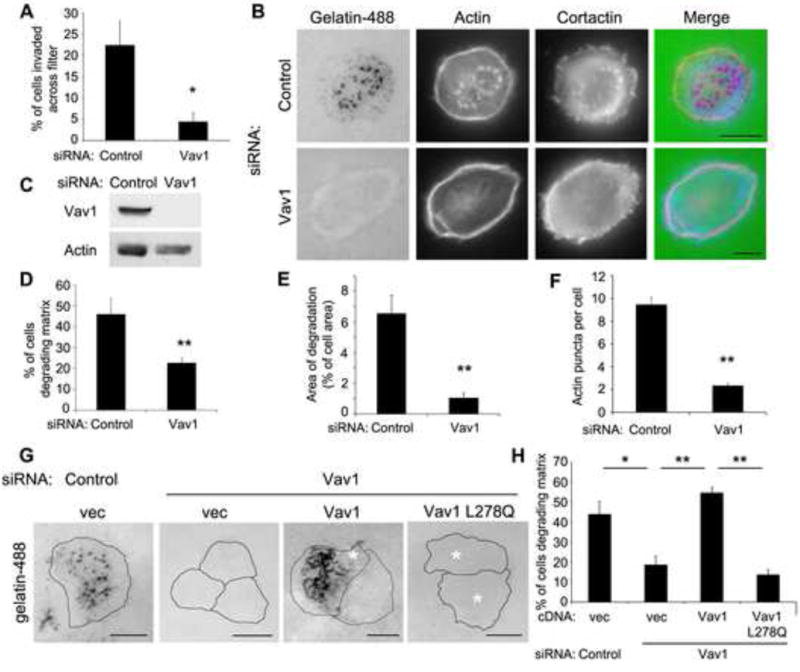
(A) Vav1 is required for invasive migration of pancreatic tumor cells. DanG cells were transfected with siRNA against Vav1 or a control siRNA and plated in a transwell invasion assay. The percentage of cells invaded across the filter after 72h was measured. (B) Depletion of Vav1 using siRNA inhibits degradation of a gelatin matrix. DanG cells were transfected with siRNA against Vav1 or a control siRNA and then plated on fluorescent gelatin for 7 hours. Cells were stained with TRITC-Phalloidin and cortactin to detect the actin cytoskeleton and invadopodial puncta. While control cells degraded substantial amounts of matrix, the cells treated to reduce Vav1 levels did not. (C) Parallel samples from (B) were lysed and immunoblotted to confirm protein knockdown. (D) The percentage of cells degrading the gelatin matrix was scored. At least 100 cells were scored per condition. (E) The area of degradation was quantified in at least 20 cells per condition. (F) The number of actin puncta per cell was scored (n>100 cells per condition). (G) The GEF activity of Vav1 is required for matrix degradation. DanG cells were depleted of Vav1 using siRNA, and then transfected with WT Vav1 or GEF-inactive Vav1 (L278Q). Cells were plated on fluorescent gelatin for 7 hours, stained for ectopic Vav1 expression (not shown), and then scored for matrix degradation. Re-expression of WT Vav1, but not GEF-inactive Vav1, restored matrix degradation. Asterisks indicate transfected cells. (H) The percentage of cells degrading the gelatin matrix was scored (n>100 cells per condition). All graphed data indicate the mean +/- SEM of at least three independent experiments. * indicates p<0.05, ** indicates p<0.01. Scale bars = 10 μm. See also Supplemental Figure S1.
Tumor cells degrade the ECM through invadopodia, invasive protrusions which are sites for targeted secretion of metalloproteases [8]. Invadopodia consist of an actin core and require the activity of Cdc42 for the actin nucleation and polymerization necessary for their formation. We hypothesized that Vav1 was required for either the formation or the maturation of invadopodia. To test this, DanG cells were depleted of Vav1 by siRNA, plated on fluorescent gelatin, and then stained for actin or cortactin. Control cells formed numerous puncta, which stained positive for both actin and cortactin, and often coincided with sites of matrix degradation, indicating the presence of functional invadopodia. However, depletion of Vav1 dramatically reduced the number of actin puncta (Figure 1F). These data suggest that, when Vav1 is ectopically expressed in pancreatic tumor cells, it promotes ECM degradation through the formation of invadopodia.
Vav1 regulates invadopodia formation and matrix degradation through Cdc42
While Vav1 is an exchange factor for Rho family GTPases, it does have GEF-independent adapter functions [9, 10]. Thus, we tested if matrix degradation required Vav1 GEF activity. Re-expression of WT Vav1 completely rescued matrix degradation in the Vav1 knockdown cells (Figure 1G, H). However, GEF-inactive Vav1 L278Q was unable to restore matrix degradation, demonstrating that the GEF activity of Vav1 is required and suggesting that Vav1 regulates ECM degradation through its action as an exchange factor for Rho GTPases.
Vav1 is primarily a GEF for Rac1, though it also has exchange activity toward Cdc42 and RhoA [11, 12]. Therefore, we sought to determine which Rho GTPase mediated matrix degradation downstream of Vav1 in pancreatic cancer cells. Expression of a constitutively active form of Rac1, Cdc42, or RhoA should overcome the requirement for a GEF and restore matrix degradation in cells depleted of Vav1. Vav1-depleted DanG cells were transfected with empty vector or active Rac1 (Q61L), RhoA (Q63L), or Cdc42 (Q61L). Only expression of active Cdc42, but not Rac1 or RhoA, restored matrix degradation in Vav1-knockdown cells, both in the percentage of cells degrading matrix and in the area of degradation per cell, suggesting that Cdc42 activation is downstream of Vav1 (Figure 2A-C). Even though Vav1 regulates Rac1 activity, matrix degradation did not require Rac1, as knockdown of Rac1 did not affect the ability of Cdc42 to rescue matrix degradation in Vav1-depleted cells (Supplemental Figure S2A). Further, Vav1 mediates invadopodia formation through Cdc42, as expression of constitutively active Cdc42 also restored the presence of actin puncta in Vav1 knockdown cells (Figure 2D and Supplemental Figure S2B). Consistent with this observation, expression of dominant negative Cdc42 (T17N) or siRNA-mediated knockdown of Cdc42 inhibited matrix degradation (Figure 2E, Supplemental Figure S2C) [5]. These data suggest that Vav1-mediated Cdc42 activation promotes invadopodia function and matrix degradation.
Figure 2. Vav1 regulates matrix degradation through Cdc42.
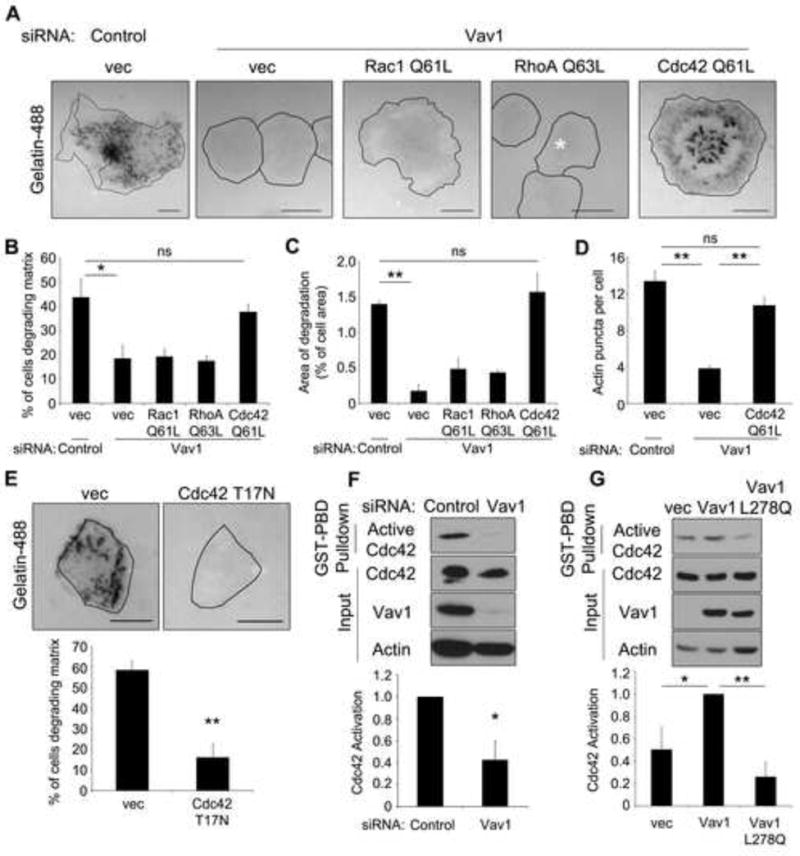
(A) Constitutively active Cdc42, but not constitutively active Rac1 or RhoA, rescues matrix degradation in Vav1 knockdown cells. DanG cells were depleted of Vav1 by siRNA, and then transfected with myc-tagged Rac1 (Q61L), RhoA (Q63L), or Cdc42 (Q61L). Cells were plated on a fluorescent gelatin matrix for 7 hours, and then fixed and stained for the myc epitope tag (not shown). (B) The percentage of cells degrading the gelatin matrix was scored (n> 100 cells per condition) and (C) the area of degradation was quantified (n> 20 cells per condition). (D) Cells transfected as described above were stained for actin and the myc epitope tag (not shown), and the number of actin puncta per cell was scored (n>40 cells per condition). (E) Dominant negative Cdc42 inhibits matrix degradation in Vav1-expressing cells. DanG cells were transfected with empty vector or Cdc42 T17N and matrix degradation was assessed as described. Cdc42 T17N expression was verified by immunofluorescence for Cdc42 (not shown). The percentage of cells degrading matrix was scored in at least 50 cells per condition. (F) Cdc42 activation is impaired in the absence of Vav1. DanG cells were depleted of Vav1 by siRNA and subjected to a GST-PBD pulldown for active Cdc42. (G) Ectopic Vav1 expression enhances Cdc42 activation. PANC1 cells were transfected with empty vector, WT Vav1, or GEF-inactive Vav1 (L278Q) and were analyzed by GST-PBD pulldown for active Cdc42. For both (F) and (G), levels of active Cdc42 were normalized to total Cdc42. All graphed data indicate the mean +/- SEM of at least three independent experiments. * indicates p<0.05, ** indicates p<0.01, and ns indicates no statistically significant difference. Scale bars = 10 μm. See also Supplemental Figure S2.
Further, RNAi-mediated depletion of Vav1 in DanG cells reduced Cdc42 activation by 60% in pancreatic cancer cells as measured by a biochemical pulldown for active Cdc42 (Figure 2F). In addition, overexpression of WT Vav1, but not GEF inactive Vav1 L278Q, caused a twofold increase in Cdc42 activation in PANC1 pancreatic cancer cells (which do not express endogenous Vav1, Figure 2G). Taken together, these data indicate that Vav1 regulates matrix degradation in pancreatic cancer cells through activation of Cdc42.
Vav1 mediates the effects of Src on matrix degradation
The tyrosine kinase Src is aberrantly activated in many cancers and is a potent inducer of invadopodia formation and matrix degradation [13-15]. Src family kinases phosphorylate multiple substrates involved in invadopodial structure and dynamics, including N-WASP, cortactin, the scaffold Tks5, and the PI phosphatase synaptojanin 2 [15-20]. Vav1 is a substrate for Src family kinases in hematopoietic cells, where phosphorylation of critical residues (Tyr142, Tyr160, and Tyr174) induces a conformational change to allow activation of Vav1 [21-24]. Mutation of Vav1 to mimic its activated phosphorylated state increases Rac1 activation and migration in breast cancer cells [25]. Therefore, we hypothesized that Src signals through Vav1 in pancreatic tumor cells to modulate invadopodia formation and matrix degradation. Vav1 is phosphorylated downstream of Src in DanG cells, as EGF-stimulated tyrosine phosphorylation of Vav1 was inhibited by the Src inhibitors PP2 or dasatinib (Figure 3A, Supplemental Figure S3A). Consistently, Vav1 tyrosine phosphorylation was increased twofold by expression of active Src Y530F, but not inactive Src Y419F (Supplemental Figure S3B). Together, these data suggest that Src family kinases promote Vav1 tyrosine phosphorylation, and therefore Vav1 activation.
Figure 3. Src-mediated activation of Vav1 is a central regulator of matrix degradation by pancreatic tumor cells.
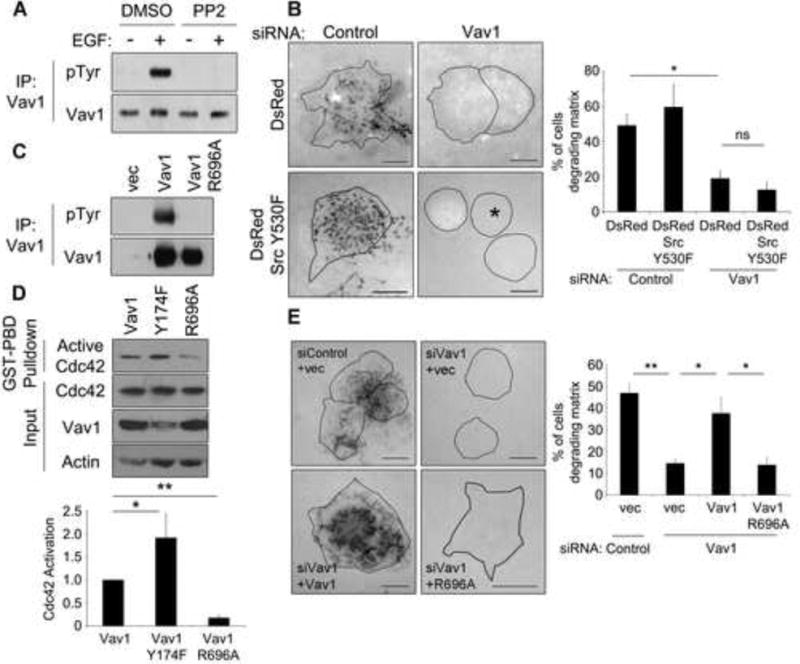
(A) Vav1 is phosphorylated downstream of Src family kinases in pancreatic tumor cells. DanG cells were treated with PP2 (25 μM) for 4 hours, then stimulated with EGF (100 ng/ml) for 5 min. Lysates were immunoprecipitated for Vav1 and immunoblotted for phospho-tyrosine. PP2 treatment abolished Vav1 tyrosine phosphorylation. (B) Vav1 is required for Src-mediated matrix degradation. DanG cells depleted of Vav1 were transfected with DsRed vector or DsRed-tagged active Src (Y530F) and plated on fluorescent gelatin for 7 hours. The asterisk indicates a transfected cell. The percentage of cells degrading matrix was scored in >50 cells per condition. Even in the presence of active Src, cells depleted of Vav1 do not degrade the gelatin matrix. (C) The mutation R696A blocks Vav1 phosphorylation. PANC1 cells were transfected with control vector, WT Vav1, or Vav1 R696A, and stimulated with EGF and processed as described in (A). Tyrosine phosphorylation of Vav1 was severely inhibited by the R696A mutation. (D) Vav1 phosphorylation and activation regulate Cdc42. PANC1 cells were transfected with WT Vav1, active Vav1 (Y174F), or Vav1 R696A and subjected to a GST-PBD pulldown. Active Cdc42 was normalized to total Cdc42, and compared to cells expressing WT Vav1. (E) Vav1 activation is required for matrix degradation. DanG cells were depleted of Vav1 by siRNA, then transfected to express either WT Vav1 or Vav1 R696A and plated on fluorescent gelatin for 7 hours. The percentage of cells capable of degrading the matrix was scored in >50 cells per condition. WT Vav1 rescued matrix degradation, but not Vav1 R696A. All graphed data represent the mean +/- SEM of three independent experiments. * indicates p<0.05, ** indicates p<0.01, and ns indicates no statistically significant difference. Scale bars = 10 μm. See also Supplemental Figure S3.
To test if Vav1 is required for Src-mediated matrix remodeling, DanG cells were transfected with constitutively active Src (Y530F), and then depleted of Vav1 by siRNA. Remarkably, matrix degradation in the Src-expressing cells was almost completely ablated in the absence of Vav1 (Figure 3B), suggesting that Vav1 functions downstream of Src to support matrix remodeling. As DanG cells have high levels of active Src and matrix degradation, additional expression of active Src did not increase matrix remodeling. Significantly, in Panc04.03 cells, which have lower levels of endogenous matrix degradation, expression of active Src Y530F caused a 3-fold increase in matrix degradation, which was completely blocked by knockdown of Vav1 (Supplemental Figure S3C). Together, these data indicate a requirement for Vav1 in Src-mediated matrix degradation.
We next tested if Src-mediated phosphorylation and activation of Vav1 was required for matrix degradation by preventing Vav1 phosphorylation. Paradoxically, structural studies have determined the Y174F mutation induces the open, “active” conformation, even in the absence of a phosphate, inducing constitutive activation [26, 27]. Therefore, an indirect, inactive Vav1 form was used by mutating the SH2 domain of Vav1 (R696A), which impairs its ability to interact with tyrosine-phosphorylated substrates required for Vav1 recruitment and activation by Src [28]. Consistent with a defect in recruitment and activation, Vav1 R696A is not tyrosine-phosphorylated in EGF-stimulated cells (Figure 3C). Further, Vav1 phosphorylation and activation correspond with an ability to activate Cdc42. PANC1 cells transfected to express WT Vav1, active Vav1 Y174F, or phospho-deficient Vav1 R696A showed marked differences in Cdc42 activation. Cells expressing phospho-defective, inactive R696A Vav1 exhibited much less activation than cells expressing WT Vav1 (Figure 3D), whereas active Vav1 Y174F induced a twofold increase in Cdc42 activation compared to WT Vav1. These data suggest that Vav1 phosphorylation and activation downstream of Src family kinases regulates Cdc42 activation in pancreatic cancer cells.
We then tested if phosphorylation of Vav1 controls matrix degradation. Notably, the defect in matrix degradation induced by Vav1 knockdown was completely rescued by re-expression of WT Vav1, but not the R696A mutant (Figure 3E), suggesting that Vav1 recruitment and phosphorylation are required for Cdc42-mediated degradation of the ECM.
Vav1-Cdc42 activation is sufficient for matrix degradation downstream of Src
From these findings, we hypothesized that Vav1 phosphorylation alone could be sufficient to induce invadopodia formation and matrix degradation downstream of Src. Thus, expression of an active Vav1 could potentially drive matrix degradation, even when Src activity is ablated by a Src inhibitor. The Src inhibitor PP2 blocked matrix degradation in control cells (Figure 4A-C). While expression of the active Vav1 Y174F markedly enhanced matrix degradation in control cells, most remarkable was that PP2-treated cells exhibited the same massive increase in degradation. Similar results were observed in the presence of the Src inhibitors SU6656 and dasatinib, and in CFPAC cells (Supplemental Figure S4A-C). Similarly, PP2 treatment inhibited the formation of invadopodia, and this defect was rescued by expression of Vav1 Y174F (Figure 4D-E). Thus, even in the presence of a Src inhibitor, activation of Vav1 induces degradation of the matrix, suggesting that Vav1 activation is sufficient for invadopodial matrix degradation downstream of Src.
Figure 4. Vav1 activation is sufficient for matrix degradation downstream of Src.
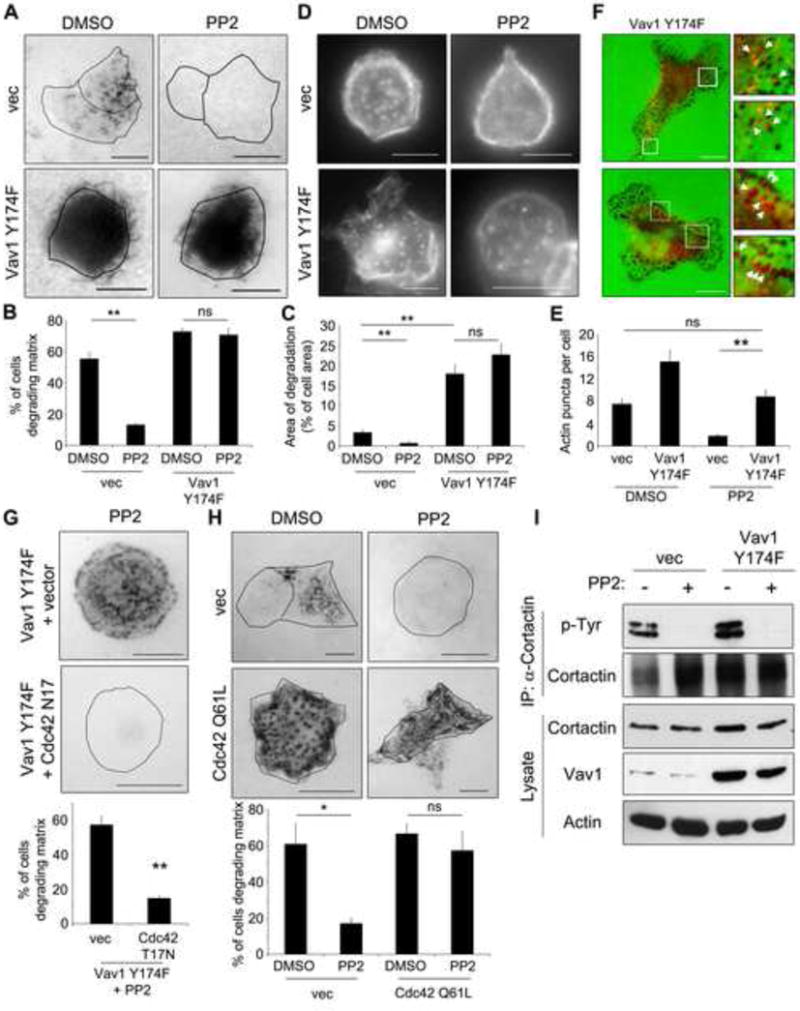
(A) DanG cells were transfected with control vector or active Vav1 Y174F, then treated with the Src inhibitor PP2 (10 μM) or DMSO vehicle control while plated on a fluorescent gelatin matrix for 7 hours. (B) The percentage of cells degrading the matrix was scored (n>100 cells per condition). (C) The area of degradation was quantified in n>20 cells per condition. Note that Vav1 Y174F potently induced matrix degradation, even in the presence of PP2. (D) Vav1 Y174F rescues the formation of invadopodia. DanG cells were transfected as described in (A), and plated on fluorescent gelatin overnight in the presence of BB94 (1 μM). The BB94 was washed out and cells were incubated for 4 hours +/- PP2 prior to staining for actin to mark invadopodia, and Vav1 to detect transfected cells (not shown). (E) The number of actin puncta per cell was scored in at least 20 cells per condition. (F) Vav1 Y174F can be detected at sites of matrix degradation. DanG cells were transfected with Vav1 Y174F, and plated on fluorescent gelatin (green) for 7 hours. Cells were fixed and stained for Vav1 (red). The boxed regions are magnified at right. Arrows indicate Vav1 Y174F puncta that colocalize with sites of matrix degradation. (G) DanG cells virally transduced to stably express Vav1 Y174F were transfected with empty vector or dominant negative Cdc42 (T17N, myc-tagged), then plated on fluorescent gelatin in the presence of 10 μM PP2. Overexpressed Vav1 and Cdc42 T17N were identified by immunofluorescence for Vav1 and myc, respectively (not shown). Cdc42 T17N inhibits matrix degradation induced by Vav1 Y174F. (H) DanG cells were transfected with myc-tagged active Cdc42 (Q61L) or empty vector, and plated on fluorescent gelatin +/- PP2 (10 μM) for 7 hours. Cdc42 Q61L expression was verified by anti-myc immunofluorescence (not shown). Cdc42 Q61L was sufficient to rescue matrix degradation in the presence of PP2. For E and F, the percentage of cells degrading matrix was scored in at least 50 cells per experiment. (I) The Src substrate cortactin is not phosphorylated upon PP2 treatment. DanG cells stably expressing GFP vector or Vav1 Y174F were treated with PP2 (10 μM) or DMSO control, then immunoprecipitated for cortactin and blotted for phospho-tyrosine. Note that cortactin is not phosphorylated in the PP2-treated cells, even in the cells expressing Vav1 Y174F. All graphed data represent the mean +/- SEM of three independent experiments. * indicates p<0.05, ** indicates p<0.01, and ns indicates no statistically significant difference. Scale bar = 10 μm. See also Supplemental Figure S4.
Consistent with these findings, active Vav1 Y174F showed increased localization to sites of matrix degradation and to actin puncta marking invadopodia compared to cells expressing WT Vav1. Importantly, the Src inhibitory drug PP2 did not reduce this colocalization (Figure 4F and Supplemental Figure S4D). We speculate that Vav1 localization is transient or occurs only at early stages of invadopodia formation. These stages may then be enhanced by the constitutive activation of Vav1 using the Y174F mutation, thereby facilitating Vav1 detection at these sites. The localization of Vav1 Y174F to sites of matrix degradation supports the premise that Vav1 is a component of invadopodial assembly, and further, our data suggest that Vav1 activation is dominant to upstream Src-mediated signaling pathways.
We sought to confirm that this Src-independent degradation was in fact mediated through Vav1-induced Cdc42 activation, as shown above (Figure 2). Expression of dominant negative Cdc42 (T17N) or siRNA-mediated knockdown of Cdc42 markedly reduced the degradation induced by Vav1 Y174F in PP2-treated DanG cells (Figure 4G, Supplemental Figure S4E). Taken together, these data indicate that Cdc42 is necessary downstream of active Vav1 to promote matrix degradation downstream of Src.
As an extension of these findings we predicted that Vav1 Y174F should promote Cdc42 activation, even in the presence of the Src inhibitor. While this activation was observed, we also found an unexpected activation of Cdc42 following PP2 treatment in both control cells and cells expressing Vav1 Y174F. Even though Vav1 potently regulates Cdc42 activation (Figure 2) this suggests that Cdc42 activity could also be regulated by other Src-dependent factors, as Vav1 would be inactive in these cells. Further, as Src inhibitors clearly reduce invadopodia formation (Figure 4), this finding suggests that Cdc42 activation, while necessary, may not be sufficient for matrix degradation downstream of Src. As an alternative approach to this question, we tested if constitutively active Cdc42 (Q61L) was sufficient to restore matrix degradation even upon inhibition of Src. As observed previously, PP2 treatment inhibited matrix degradation by DanG cells, and notably, Cdc42 Q61L was sufficient to overcome the loss of Src activity and restore matrix degradation, similar to expression of Vav1 Y174F (Figure 4H). These data suggest that hyper-activation of Cdc42 via mutation or expression of the mutant GEF is sufficient for matrix degradation downstream of Src. While we cannot exclude that Vav1 and Cdc42 may act in parallel pathways, our data do support the model that Src-mediated activation of Vav1 drives Cdc42 activity, which is then sufficient for invadopodia formation and matrix degradation. Cdc42 promotes invadopodia formation by signaling through N-WASP and Arp2/3 to induce actin nucleation and branching [29, 30]. It remains to be determined if Cdc42 promotes matrix degradation solely through the defined N-WASP-Arp2/3 pathway, or if other downstream effectors are also required.
Unexpectedly, this finding suggests that phosphorylation of other Src substrates may not be required for invadopodia formation. Therefore, we wanted to test the phosphorylation of cortactin, a known substrate downstream of Src via Arg/Abl involved in invadopodia formation [31], in Vav1 Y174F-expressing cells treated with a Src inhibitor. If cortactin was not phosphorylated, it would suggest that active Vav1 could induce invadopodia-mediated matrix degradation even in the absence of cortactin phosphorylation. In control cells, PP2 treatment blocked tyrosine phosphorylation of cortactin, confirming that it is phosphorylated downstream of Src (Figure 4I). Importantly, cortactin phosphorylation was unaffected by the Vav1 Y174F mutation. Cortactin phosphorylation was blocked by PP2 treatment even in cells expressing Vav1 Y174F, which form invadopodia and potently degrade the matrix. These data demonstrate that active Vav1 drives invadopodia-mediated matrix degradation, even in the absence of Src activation and phosphorylation of a known substrate. Further, the data suggest that Src phosphorylation of cortactin may be dispensable for matrix degradation in tumor cells with activated Vav1. Perhaps Cdc42-driven N-WASP activation is sufficient, and further enhancement regulated by cortactin phosphorylation is not necessary. Alternatively, a recent report described phosphorylation-independent interactions between cortactin and SH2 domains of its binding partners, suggesting that cortactin phosphorylation may not be required for its effects [32].
These data strongly support a central role for Vav1 in promoting invadopodia and matrix degradation. Pancreatic cancer cell lines that express Vav1 have become dependent upon it, as knockdown of Vav1 inhibits matrix degradation, even in the presence of multiple other Cdc42 GEFs. However, while Vav1 is ectopically expressed in many pancreatic cancers, it is not expressed in all tumor cells (Supplemental Figure S1B), some of which are still able to form invadopodia and degrade matrix. We hypothesize that Vav1-negative tumors and tumor cell lines have upregulated the invadopodial and invasive machinery by other mechanisms, such as a different Cdc42 GEF. Accordingly, we screened a panel of 9 pancreatic cancer cell lines for the expression of 6 different Cdc42 GEFs, and consistent with our prediction, nearly every tumor cell line exhibited a different pattern of GEF expression (Supplemental Figure S4F). These data reflect the heterogeneity of tumors among individuals, and strengthen the case for an individualized approach to cancer treatment. We hypothesize that differential expression of GEFs is important for defining the pathway regulating invadopodia formation in specific cell types. It will be key to determine how/why different GEFs are utilized (either independently or in combination), and how converging signals may activate one or more GEFs selectively.
Activating mutations in Vav1 have not been described in pancreatic cancers. However, pathways that lead to Vav1 phosphorylation and activation are known to be hyperactivated, including EGFR and Src [13]. Therefore, it is highly likely that, in tumors where Vav1 is ectopically expressed, its activation is upregulated due to hyperactivation of regulatory signaling pathways. Thus, activation of any pathway leading to activation of Src family kinases or, ultimately, Vav1, could potently upregulate the invasive machinery. In addition, while some of the Src inhibitors used in this study have additional kinase targets, this only strengthens the importance of active Vav1 and describes it as a more global regulator of matrix degradation downstream of multiple signaling pathways.
We have recently reported that Vav1 is required for Rac1-mediated formation of lamellipodia and subsequent migration of tumor cells [3]. In addition, Vav1 is a potent regulator of transendothelial migration of leukocytes, and also contributes to CXCL12-induced MT1-MMP expression and invasion by melanoma cells [33, 34]. The current findings further implicate Vav1 in the processes of invasion and migration, namely through the formation of invadopodia and matrix degradation. This process requires Vav1 activation of Cdc42, demonstrating that in pancreatic tumor cells, ectopically expressed Vav1 can signal through multiple pathways. This is consistent with a previous report that Vav1-induced oncogenic transformation requires multiple signaling pathways, including Rac1, Cdc42, and RhoA, as well as NFκB and JNK [35]. Indeed, these data implicate Vav1 in multiple stages of the tumorigenic and metastatic program, and suggest that, upon its aberrant expression, Vav1 is a pivotal signaling node in pancreatic cancers.
Supplementary Material
Highlights.
The Vav1 GEF is ectopically expressed in invasive pancreatic tumor cells.
Vav1 expression significantly promotes matrix degradation by pancreatic tumor cells.
Vav1 promotes invadopodia formation through its GEF activity toward Cdc42.
A Src-mediated activation of Vav1 plays a central role in invadopodia activation.
Acknowledgments
We gratefully acknowledge the members of the McNiven lab for their contributions, in particular, Hong Cao and Eugene Krueger for technical assistance. This work was financially supported by grants from the National Cancer Institute (R01 CA104125 to M.A.M. and Pancreatic Cancer SPORE grant P50 CA102701 to D.D.B.) and the Optical Morphology Core of the Mayo Clinic Center for Cell Signaling in Gastroenterology (NIDDK P30DK084567). G.L.R. was supported by grant T32 CA148073 from the National Institutes of Health.
Abbreviations List
- ECM
Extracellular Matrix
- EGF
Epidermal Growth Factor
- FGD1
FYVE, RhoGEF and PH domain containing 1, faciogenital dysplasia
- GAP
GTPase Activating Protein
- GEF
Guanine nucleotide exchange factor
- MMP
Matrix Metalloprotease
- N-WASP
Neural Wiscott-Aldrich Syndrome Protein
- PAK1
p21-activated kinase 1
- PBD
p21 binding domain
- SH2, SH3
Src homology 2, 3 domains
- Tks5
Tyrosine kinase substrate 5
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Murphy DA, Courtneidge SA. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011;12:413–426. doi: 10.1038/nrm3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fernandez-Zapico ME, Gonzalez-Paz NC, Weiss E, Savoy DN, Molina JR, Fonseca R, Smyrk TC, Chari ST, Urrutia R, Billadeau DD. Ectopic expression of VAV1 reveals an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell. 2005;7:39–49. doi: 10.1016/j.ccr.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 3.Razidlo GL, Wang Y, Chen J, Krueger EW, Billadeau DD, McNiven MA. Dynamin 2 Potentiates Invasive Migration of Pancreatic Tumor Cells through Stabilization of the Rac1 GEF Vav1. Dev Cell. 2013;24:573–585. doi: 10.1016/j.devcel.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakahara H, Otani T, Sasaki T, Miura Y, Takai Y, Kogo M. Involvement of Cdc42 and Rac small G proteins in invadopodia formation of RPMI7951 cells. Genes Cells. 2003;8:1019–1027. doi: 10.1111/j.1365-2443.2003.00695.x. [DOI] [PubMed] [Google Scholar]
- 5.Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, Segall J, Eddy R, Miki H, Takenawa T, et al. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 7.Turner M, Billadeau DD. VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat Rev Immunol. 2002;2:476–486. doi: 10.1038/nri840. [DOI] [PubMed] [Google Scholar]
- 8.Poincloux R, Lizarraga F, Chavrier P. Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J Cell Sci. 2009;122:3015–3024. doi: 10.1242/jcs.034561. [DOI] [PubMed] [Google Scholar]
- 9.Miletic AV, Graham DB, Sakata-Sogawa K, Hiroshima M, Hamann MJ, Cemerski S, Kloeppel T, Billadeau DD, Kanagawa O, Tokunaga M, et al. Vav links the T cell antigen receptor to the actin cytoskeleton and T cell activation independently of intrinsic Guanine nucleotide exchange activity. PLoS One. 2009;4:e6599. doi: 10.1371/journal.pone.0006599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuhne MR, Ku G, Weiss A. A guanine nucleotide exchange factor-independent function of Vav1 in transcriptional activation. J Biol Chem. 2000;275:2185–2190. doi: 10.1074/jbc.275.3.2185. [DOI] [PubMed] [Google Scholar]
- 11.Olson MF, Pasteris NG, Gorski JL, Hall A. Faciogenital dysplasia protein (FGD1) and Vav, two related proteins required for normal embryonic development, are upstream regulators of Rho GTPases. Curr Biol. 1996;6:1628–1633. doi: 10.1016/s0960-9822(02)70786-0. [DOI] [PubMed] [Google Scholar]
- 12.Rapley J, Tybulewicz VL, Rittinger K. Crucial structural role for the PH and C1 domains of the Vav1 exchange factor. EMBO Rep. 2008;9:655–661. doi: 10.1038/embor.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lutz MP, Esser IB, Flossmann-Kast BB, Vogelmann R, Luhrs H, Friess H, Buchler MW, Adler G. Overexpression and activation of the tyrosine kinase Src in human pancreatic carcinoma. Biochem Biophys Res Commun. 1998;243:503–508. doi: 10.1006/bbrc.1997.8043. [DOI] [PubMed] [Google Scholar]
- 14.Guarino M. Src signaling in cancer invasion. J Cell Physiol. 2010;223:14–26. doi: 10.1002/jcp.22011. [DOI] [PubMed] [Google Scholar]
- 15.Boateng LR, Huttenlocher A. Spatiotemporal regulation of Src and its substrates at invadosomes. Eur J Cell Biol. 2012;91:878–888. doi: 10.1016/j.ejcb.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chuang Y, Xu X, Kwiatkowska A, Tsapraillis G, Hwang H, Petritis K, Flynn D, Symons M. Regulation of synaptojanin 2 5′-phosphatase activity by Src. Cell Adh Migr. 2012;6:518–525. doi: 10.4161/cam.22139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bharti S, Inoue H, Bharti K, Hirsch DS, Nie Z, Yoon HY, Artym V, Yamada KM, Mueller SC, Barr VA, et al. Src-dependent phosphorylation of ASAP1 regulates podosomes. Mol Cell Biol. 2007;27:8271–8283. doi: 10.1128/MCB.01781-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park SJ, Suetsugu S, Takenawa T. Interaction of HSP90 to N-WASP leads to activation and protection from proteasome-dependent degradation. EMBO J. 2005;24:1557–1570. doi: 10.1038/sj.emboj.7600586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ayala I, Baldassarre M, Giacchetti G, Caldieri G, Tete S, Luini A, Buccione R. Multiple regulatory inputs converge on cortactin to control invadopodia biogenesis and extracellular matrix degradation. J Cell Sci. 2008;121:369–378. doi: 10.1242/jcs.008037. [DOI] [PubMed] [Google Scholar]
- 20.Stylli SS, Stacey TT, Verhagen AM, Xu SS, Pass I, Courtneidge SA, Lock P. Nck adaptor proteins link Tks5 to invadopodia actin regulation and ECM degradation. J Cell Sci. 2009;122:2727–2740. doi: 10.1242/jcs.046680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han J, Das B, Wei W, Van Aelst L, Mosteller RD, Khosravi-Far R, Westwick JK, Der CJ, Broek D. Lck regulates Vav activation of members of the Rho family of GTPases. Mol Cell Biol. 1997;17:1346–1353. doi: 10.1128/mcb.17.3.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crespo P, Schuebel KE, Ostrom AA, Gutkind JS, Bustelo XR. Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature. 1997;385:169–172. doi: 10.1038/385169a0. [DOI] [PubMed] [Google Scholar]
- 23.Aghazadeh B, Lowry WE, Huang XY, Rosen MK. Structural basis for relief of autoinhibition of the Dbl homology domain of proto-oncogene Vav by tyrosine phosphorylation. Cell. 2000;102:625–633. doi: 10.1016/s0092-8674(00)00085-4. [DOI] [PubMed] [Google Scholar]
- 24.Yu B, Martins IR, Li P, Amarasinghe GK, Umetani J, Fernandez-Zapico ME, Billadeau DD, Machius M, Tomchick DR, Rosen MK. Structural and energetic mechanisms of cooperative autoinhibition and activation of Vav1. Cell. 2010;140:246–256. doi: 10.1016/j.cell.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilsbacher JL, Moores SL, Brugge JS. An active form of Vav1 induces migration of mammary epithelial cells by stimulating secretion of an epidermal growth factor receptor ligand. Cell Commun Signal. 2006;4:5. doi: 10.1186/1478-811X-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li P, Martins IR, Amarasinghe GK, Rosen MK. Internal dynamics control activation and activity of the autoinhibited Vav DH domain. Nat Struct Mol Biol. 2008;15:613–618. doi: 10.1038/nsmb.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez-Lago M, Lee H, Cruz C, Movilla N, Bustelo XR. Tyrosine phosphorylation mediates both activation and downmodulation of the biological activity of Vav. Mol Cell Biol. 2000;20:1678–1691. doi: 10.1128/mcb.20.5.1678-1691.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Margolis B, Hu P, Katzav S, Li W, Oliver JM, Ullrich A, Weiss A, Schlessinger J. Tyrosine phosphorylation of vav proto-oncogene product containing SH2 domain and transcription factor motifs. Nature. 1992;356:71–74. doi: 10.1038/356071a0. [DOI] [PubMed] [Google Scholar]
- 29.Ho HY, Rohatgi R, Lebensohn AM, Le M, Li J, Gygi SP, Kirschner MW. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell. 2004;118:203–216. doi: 10.1016/j.cell.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 30.Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, Kirschner MW. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- 31.Mader CC, Oser M, Magalhaes MA, Bravo-Cordero JJ, Condeelis J, Koleske AJ, Gil-Henn H. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 2011;71:1730–1741. doi: 10.1158/0008-5472.CAN-10-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evans JV, Ammer AG, Jett JE, Bolcato CA, Breaux JC, Martin KH, Culp MV, Gannett PM, Weed SA. Src binds cortactin through an SH2 domain cystine-mediated linkage. J Cell Sci. 2012;125:6185–6197. doi: 10.1242/jcs.121046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bartolome RA, Molina-Ortiz I, Samaniego R, Sanchez-Mateos P, Bustelo XR, Teixido J. Activation of Vav/Rho GTPase signaling by CXCL12 controls membrane-type matrix metalloproteinase-dependent melanoma cell invasion. Cancer Res. 2006;66:248–258. doi: 10.1158/0008-5472.CAN-05-2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ticchioni M, Charvet C, Noraz N, Lamy L, Steinberg M, Bernard A, Deckert M. Signaling through ZAP-70 is required for CXCL12-mediated T-cell transendothelial migration. Blood. 2002;99:3111–3118. doi: 10.1182/blood.v99.9.3111. [DOI] [PubMed] [Google Scholar]
- 35.Palmby TR, Abe K, Karnoub AE, Der CJ. Vav transformation requires activation of multiple GTPases and regulation of gene expression. Mol Cancer Res. 2004;2:702–711. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.