

Abstract
Purpose
Lynch syndrome is an autosomal dominant syndrome of familial malignancies resulting from germ-line mutations in DNA mismatch repair (MMR) genes. Our goal was to take a pathway-based approach to investigate the influence of polymorphisms in cell-cycle related genes on age of onset for Lynch syndrome using a tree-model.
Experimental Design
We evaluated polymorphisms in a panel of cell-cycle related genes (AURKA, CDKN2A, TP53, E2F2, CCND1, TP73, MDM2, IGF1 and CDKN2B) in 220 MMR gene mutation carriers from 129 families. We applied a novel statistical approach, tree-modeling (Classification and Regression Tree), to the analysis of data on Lynch syndrome patients to identify individuals with a higher probability of developing colorectal cancer at an early age and explore the gene-gene interactions between polymorphisms in cell-cycle genes.
Results
We found that the subgroup with CDKN2A C580T wild-type genotype, IGF1 CA-repeats ≥19, E2F2 variant genotype, AURKA wild-type genotype, and CCND1 variant genotype had the youngest age of onset, with a 45-year median onset age. While the subgroup with CDKN2A C580T wild-type genotype, IGF1 CA-repeats ≥19, E2F2 wild-type genotype and AURKA variant genotype had the latest median age of onset, which was 70 years. Furthermore, we found evidence of a possible gene-gene interaction between E2F2 and AURKA genes related to CRC age of onset.
Conclusions
Polymorphisms in these cell-cycle related genes work together to modify the age at onset of CRC in patients with Lynch syndrome. These studies provide an important part of the foundation for development of a model for stratifying age of onset risk among those with Lynch syndrome.
Keywords: Tree model, cell cycle pathway, Polymorphisms, Lynch syndrome, Age of onset
Introduction
Lynch syndrome is an autosomal dominant disorder caused by germline mutations in mismatch repair (MMR) genes with MSH2 and MLH1 being the most frequently mutated [1-2]. It is also commonly known as hereditary nonpolyposis colorectal cancer, which may be a misnomer because cancer predisposition is not restricted to the colon. The term Lynch syndrome has been specifically referred to as individuals diagnosed based on an inherited germline mutation in a DNA MMR gene and has been used as such herein. Patients with this syndrome are at increased risk for developing a variety of different cancers with colorectal cancer (CRC) and endometrial cancer being the most common. Extracolonic neoplasms such as small-bowel, ureter, renal pelvis, stomach, ovary, and hepatobiliary cancer are seen less frequently [3-4]. The age of CRC onset varies considerably in Lynch Syndrome and is thought to be due to a combination of other low penetrance genetic factors and environmental exposures. Therefore, it is of great important to examine low-penetrance genes as potential modifiers of risk for cancer onset at an early age in individuals with Lynch syndrome.
In previous studies we reported that polymorphisms in CCND1, TP53, IGF1, and AURKA influenced age-associated risk for CRC in Lynch syndrome [5-8). Reeves et al confirmed our study that the IGF1 polymorphism is an important modifier of disease onset in Lynch syndrome [9]. Talseth et al reported that the CCND1 polymorphism was associated with a significant difference in age of disease onset in patients harboring MSH2 mutations, which was not observed in MLH1 mutation carriers [10]. However they did not find the AURKA polymorphism was associated with age of onset of CRC in Lynch syndrome. In addition, Talseth et al did not find an association between the TP53 polymorphism and age at diagnosis of CRC in Lynch syndrome [11]. In light of the many controversial results published about the influence of some of the candidate SNPs on age associated risk for CRC in patients with Lynch syndrome. We took a systematic multigenic pathway-based approach.
Focusing on specific pathways such as the cell cycle pathways which assess the combined effects of a panel of polymorphisms in genes that interact with each other, may allow us to see the influence of SNPs that are not seen when only main effects are evaluated. These SNPs may only be significant in the context of a specific genetic background. This approach which assesses the combined effects of a panel of polymorphisms interacting in the same pathway may amplify the effects of individual polymorphisms and enhance predictive power. Efficient cell-cycle regulation is important for accurate and efficient replication and repair of the genome. Cell-cycle genes play an important role in response to DNA damage by interacting with the DNA repair pathways, coordinately causing cells to undergo cell-cycle arrest while DNA is being repaired [12]. We have selected 9 candidate genes because they are known to interact with each other in the cell cycle pathways. We selected 10 polymorphisms from the nine cell-cycle genes based on their allele frequencies, functional significance, or published association studies. In addition to selecting the controversial SNPs described above (CCND1 [5, 10], AURKA [8, 10], TP53 [6, 11], and IGF1 [7, 9), we selected SNPs that have been reported to influence risk for other cancers (TP73 [13], MDM2 [14], CDKN2A [15], and CDKN2B [15]). The E2F2 SNP was selected because it showed an association with age-associated risk for Lynch syndrome in our preliminary studies. This panel of polymorphisms was assessed using classification and regression tree analysis (CART), a tree-based methodology, to identify groups with a higher probability of developing CRC at an early age and explore the gene-gene interactions between polymorphisms of cell-cycle genes. The CART method uses a binary recursive partitioning to identify subgroups of subjects at higher risk and its results are presented as a decision tree, which is easier to interpret than other statistical methods [16-19]. CART analysis is often able to uncover complex interactions between predictors that may be difficult or impossible to uncover using traditional multivariate techniques [20]. Furthermore, tree-based modeling is adept in uncovering predictors that may be largely operative within specific patient subgroups but may have minimal effect or none in other patient subgroups [21]. To our knowledge, this is the first systematic pathways-based study on the role of polymorphisms of cell-cycle related genes on age associated risk for Lynch syndrome.
Patients and Methods
Subjects
We studied 220 MLH1 or MSH2 mutation carriers from 129 families from the M. D. Anderson Hereditary Colorectal Cancer Registry (n=210) and the Mayo Clinic Familial Cancer Program (FCP) (n=10). The patients in the M.D. Anderson registry came from families suspected to have Lynch syndrome or were very young (<45 years of age) at CRC diagnosis or were referred to the University of Texas M.D. Anderson Cancer Center because of a known mutation. Those enrolled in the Mayo Clinic FCP were referred for clinical medical genetic evaluation due to personal or family history suggestive of a hereditary disorder (no specific criteria required) and were subsequently enrolled in the FCP. Of the 220 confirmed MMR mutation carriers, 125 (56.8%) had colorectal cancer and the remaining 95 (43.2%) were unaffected MMR mutation carriers. The median age of individuals with and without colorectal cancer was 42 and 45 years, respectively. The proportion of women (55.5%) was greater than that of men (44.5%) but more men were affected with colorectal cancer (64.3%) than women (50.8%). Of the 129 families, 37 families (29%) had more than one mutation positive family member enrolled in the study. Written informed consent was obtained from all subjects from both registries. The Institutional Review Boards of the University of Texas M.D. Anderson Cancer Center and of the Mayo Clinic Rochester both approved the study, in accordance with the U.S. Department of Health and Human Services. Age of onset of CRC was defined as the patient’s age at diagnosis. For the unaffected carriers, the age recorded was the age at last follow-up. The mutation status of all participants was determined in a Clinical Laboratory Improvement Act (CLIA) certified laboratory. The patients’ demographic data are summarized in Table 1.
Table 1.
Demographic and genetic characteristics of the study population
| Colorectal Cancer |
Without Colorectal Cancer |
Total | |
|---|---|---|---|
| Gender | |||
| Male | 63 | 35 | 98 |
| Female | 62 | 60 | 122 |
| Race/Ethnicity | |||
| Caucasian | 102 | 83 | 185 |
| African American | 15 | 6 | 21 |
| Hispanic | 7 | 6 | 13 |
| Asian | 1 | 0 | 1 |
| Age | |||
| Median | 42 | 45 | 43 |
| Range | 22−84 | 18−84 | 18−84 |
| MMR mutation type* | |||
| MSH2 | 69 | 58 | 127 |
| MLHl | 57 | 37 | 94 |
| Mutation type | |||
| Missense † | 35 | 22 | 57 |
| Deletion/insertion/ truncating |
90 | 73 | 163 |
One patient with hoih MSH2 and MLHI mutated.
These missense mutations were confirmed to be pathogenic mutations by a Clinical Laboratory Improvement Act – certified laboratory or from the International Collaborative Group-HNPCC InSiGHT database or from the published literature (36, 37).
DNA Extraction and Genotyping
Whole blood was collected from each patient. DNA was isolated using an AUTOPURE LS (Gentra Systems, Inc., Minneapolis, MN) according to the manufacturer’s instructions. Genotyping of single nucleotide polymorphisms (SNPs) for CDKN2A, CDKN2B, TP53, CCND1, AURKA, MDM2, E2F2, and TP53 and the CA-repeat polymorphism for IGF1 was performed as previously described [7, 8]. Primers (Sigma/Genosys, The Woodlands, TX) for polymerase chain reaction (PCR) and pyrosequencing are listed in Table 2. For each polymorphism, 10% of the samples were randomly selected and genotyping repeated with 100 % concordance. The genotypes were read independently by two different persons. The genes, nucleotide substitutions, amino acid changes, minor allele frequency, dbSNP ID, and location of the 10 polymorphisms evaluated are summarized in Table 2.
Table 2.
Polymorphisms evaluated in this study and their primers for genotyping
| GENE | Polymorphism | Type of change | Location | MAF* | dbSNP ID | PCR & Pyrosequencing Primers |
|---|---|---|---|---|---|---|
| CDKN2A | C to G | NA | C540G, 3’UTR | 0.14 | rsll515 | F Bio 5’7GTGCCACACATCTTTGACCTCAG3’ |
| R 5’TACGAAAGCGGGGTGGGT3’ | ||||||
| S 5’GACTGATGATCTAAGTTTCC3’ | ||||||
| C to T | NA | C580T, 3’UTR | 0.12 | rs3088440 | Same F & R as above | |
| S 5’TGTGGCGGGGGCAGT3’ | ||||||
| CDKN2B | C to A | NA | Intron 1 | 0.10 | rs2069426 | F Bio 5’7CCTCTGCACTGGGTGAAAACTT3’ |
| R 5’ATCATGACCTGCCAGAGAGAGC3’ | ||||||
| S 5’GAGAGCAGAGTGGTCAG3’ | ||||||
| TP53 | G to C | Arg to Pro | Codon 72 | 0.25 | rs1042522 | F Bio 5’7AGACCCAGGTCCAGATGAAGC3’ |
| R 5’CCGGTGTAGGAGCTGCTGG3’ | ||||||
| S 5’GGTGCAGGGGCCACG3’ | ||||||
| CCNDl | G to A | Pro to Pro, alternate | Codon 242 | 0.40 | rs603965 | F Bio 5’7TCCTACTACCGCCTGACACGC3’ |
| splicing | R 5’GCACTAGGTGTCTCCCCCTGTAA3’ | |||||
| S 5’GCACATCACCCTCACTTA3’ | ||||||
| AURKA | T to A | Phe to lie | Codon 31 | 0.21 | rs2273535 | F 5’CCATTCTAGGCTACAGCTCCA3’ |
| R Bio 5’7 ATTCTGAACCGGCTTGTGAC3’ | ||||||
| S 5’TCTCGTGACTCAGCAA3’ | ||||||
| MDM2 | T to G | NA | Promoter SNP309 | 0.38 | rs2279744 | F 5’GGGGTGGTTCGGAGGTCT3’ |
| R Bio 5’7GTGACCCGACAGGCACCT3’ | ||||||
| S 5’GGGCTGCGGGGCCGCT3’ | ||||||
| E2F2 | G to T | Gln to His | Codon 226 | 0.45 | rs2075995 | F Bio 5’7AGGAGCTGATGAACACGGAG3’ |
| R 5’ACTTGTCCTCAGTCAGGTGCTTGA3’ | ||||||
| S 5’AGCAGCTCTGGATGAG3’ | ||||||
| TP73 | G to A | NA | 5’UTR Ex2+4 | 0.21 | rs2273953 | F 5’AGTTCCCAGGGTGCTCAGGT3’ |
| R Bio 5’7GGTGGACTGGGCCATCTTC3’ | ||||||
| S 5’CCTTCCTTCCTGCAGA3’ | ||||||
| IGFl | CA repeat | NA | 5’UTR | † | F 5’6 -GCTAGCCAGCTGGTGTTATT−3’ | |
| R 5’ACCACTCTGGGAGAAGGGTA−3’ |
MAP = minor allele frequency.
No dbSNP ID for the IGFl polymorphism because it is composed of CA repeats, not single nucleotide polymorphism.
F = Forward, R = Reverse, S = Sequencing Primer, Bio = Biotinylated
Statistical Methods
We used the χ2 goodness-of-fit test to test Hardy-Weinberg (HW) equilibrium. Time to onset was computed from date of birth to clinically detectable cancer. Subjects who had not yet developed cancer were censored at the time of last follow-up. CART analysis was used to construct survival trees to identify subgroups of patients with differing median times to onset and to evaluate gene-gene interactions. We grew a tree and then pruned the tree back using a pruning rule based on the log-rank statistic, since the response is a time-to-event outcome [20]. The significance of each split was evaluated using the log-rank statistic for each node split. If the log-rank statistic at the split and subsequent splits is not significant, then the branch was pruned (significance level of P ≤ 0.05). We applied the RPART function written in the S-PLUS software (Version 7.0, Insightful Corporation, Seattle, Washington, USA) to construct the tree [22]. The Kaplan Meier method was used to calculate and compare the median age of onset between the subgroups that CART identified and the log-rank statistic was used to evaluate significant (two-sided alpha = 0.05) differences in age of onset. Hazard Ratios (HRs) for genotypes of interest were estimated by fitting a Cox proportional hazards model with a robust variance correction, adjusting for intrafamilial correlations in time to onset for cancer. We tested the null hypotheses of multiplicative gene-gene interaction by including main effect variables and their product terms in the Cox regression model. Analyses were performed using S-PLUS software.
Results
Subject Characteristics
Table 1 summarizes the demographic and genetic characteristics of the study population. The median age of onset (Table 1) was 43 years (range, 18 to 84 years). There was no significant difference between the age of onset of the subjects with MLH1 mutations and those with MSH2 mutations as assessed by the log-rank test (P = 0.35). Similarly, we did not observe a difference in age of onset between subjects with missense mutations and deletion/insertion/truncating mutations when the data were analyzed by the same procedures (P = 0.52). Genotype frequencies of all 10 SNPs were found to be consistent with Hardy-Weinberg equilibrium (2 = 0.01-3.02; P > 0.08).
CART Analysis
CART was performed using genotypes of the 10 polymorphisms. Figure 1 depicts the final resulting tree. There was an initial split on CDKN2A C580T genotype. The latest age of onset of CRC subgroup (group 1) included patients with CDKN2A C580T wild-type genotype (WW), IGF1 CA-repeats ≥19, E2F2 WW, and AURKA variant genotype (WM/MM). These patients had a 70-year median onset age. The subgroup (group 7) with CDKN2A C580T WW, IGF1 CA-repeats ≥19, E2F2 WM/MM, AURKA WW, and CCND1 WM/MM had the youngest median age of onset (45 years). Interestingly, when E2F2 was WW, patients with WW for AURKA developed CRC earlier (median age 48 years) than patients with AURKA WM/MM (median age 70 years). When E2F2 was WM/MM, the reverse was true. Patients with AURKA WW developed CRC later (median age 51 years) than patients with WM/WM (median age 46 years). This observation suggests a gene-gene interaction between E2F2 and AURKA.
Fig. 1.

Time to onset tree for age of onset of CRC. Inside each node is the number of affected subjects /the total number of subjects. Med = median age of onset. The hazard ratios (HRs) are calculated for all terminal nodes such that terminal node 1 is the reference group. WW = wild type, WM= Heterozygote, MM= Homozygous polymorphism. The terminal nodes are shown in color, the nodes giving rise to gene-gene interaction are shown in bold.
In Figure 2, we used the Kaplan-Meier method to plot time-to-onset curves for the seven CART subgroups that were presented in Figure 1. The color of these curves corresponds to the color of subgroups in the final tree. The log-rank test (P = 0.007) demonstrated a statistically significant difference among the time-to-onset curves of these subgroups. Furthermore we used the Cox proportional hazards model to estimate HRs for all the groups jointly and using the subgroup with the latest age of CRC onset (group 1) as the referent (Fig. 1). Because it is possible that there is a correlation of time to cancer onset in individuals from the same family due to genetic or familial factors, we applied a robust variance correction in the Cox regression analysis to adjust for the differences (23). For groups 2-7, we found HRs of 1.66 (95% CI, 0.47-5.86), 2.57 (95% CI, 0.81-8.10), 3.73 (95% CI, 1.15-12.09), 4.20 (95% CI, 1.34-13.17), 5.23 (95% CI, 1.66-16.54), and 4.14 (95% CI, 1.36-12.60), respectively (Fig. 1). In the tree, among the patients at the IGF1 CA ≥ 19 repeat node (n=126), and using E2F2 WW with AURKA WM/MM as the referent genotype, we found a HR of 5.08 (95% CI, 1.17-22.12, P = 0.033) for patients carrying the E2F2 WW with AURKA WW genotype, which showed that the SNP of AURKA was protective in this context. In contrast, among the patients at the IGF1 CA≥19 repeat node using E2F2 WM/MM with AURKA WM/MM as the referent genotype, we found a HR of 0.57 (95% CI, 0.35-0.95, P = 0.030) for patients carrying the E2F2 WM/MM and AURKA WW genotype, which showed that the AURKA SNP is a risk factor in this group. This suggested a gene-gene interaction between E2F2 and AURKA as the influence of the AURKA SNP on risk varies depending on the E2F2 genotypes.
Fig. 2.
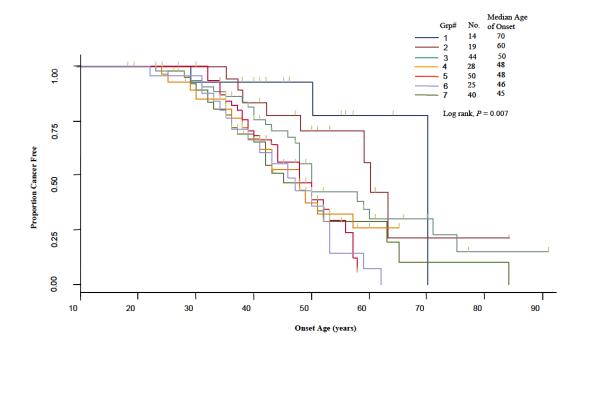
Time-to-onset curves for comparing terminal nodes from the time-to-onset tree.
We subsequently tested departure from multiplicative model of interaction between these genes, E2F2 and AURKA, using the Cox regression model. This model showed a significant interaction (P-interaction = 0.003) among patients at the IGF1 CA≥19 repeat node. Kaplan-Meier curves for E2F2 and AURKA among those patients are shown in figure 3 (log-rank, P = 0.021). This shows a much earlier time to onset for CRC among individuals with variant genotypes for both E2F2 and AURKA. When the 126 patients at the IGF1 CA≥ 19 repeat node were analyzed for the main effect of the E2F2 genotype, the HR for E2F2 WM/MM was 0.79 (95% CI=0.43-1.45) using E2F2 WW as the reference. For the main effect of AURKA, the HR for the AURKA WM/MM genotype was 0.25 (95% CI=0.07-0.87) using AURKA WW as the reference. The HR for the interaction effect for the presence of both E2F2 WM/MM and AURKA WM/MM genotypes was 6.87 (95% CI=1.72-27.32). The test for departure from multiplicative model of interaction between these two genes in all patients was not statistically significant (P-interaction = 0.132). Our data suggested a gene-gene interaction between E2F2 and AURKA only in the context of the genetic background of CDKN2A WW, IGF1 CA repeat≥19.
Fig. 3.
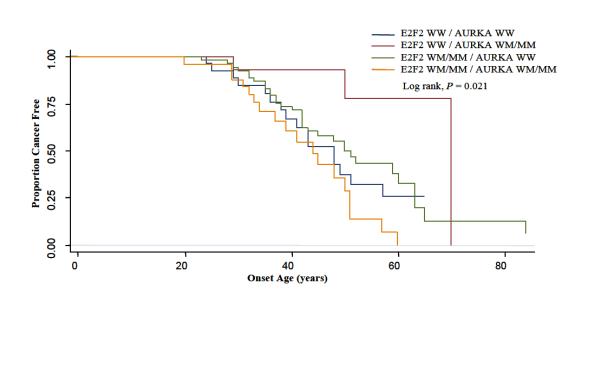
Time-to-onset curves for patients at the IGF1 CA≥19 repeat node by genotypes for E2F2 and AURKA. WW = wild type, WM= Heterozygote, MM= Homozygous polymorphism.
In addition, in the subgroup with E2F2 WM/MM and AURKA WW, patients with CCND1 WM/MM (group 7) had a greater risk for development of CRC at a young age than those with CCND1 wild-type genotype (group 2) (HR=2.27, 95%CI= 1.01-5.09 using group 2 as the referent).
Discussion
In this study, we have taken a pathway-based approach to elucidate genetic risk modifiers influencing age of onset of CRC in Lynch syndrome patients. We used CART, which is a novel tree-based statistical approach, to identify groups with a higher probability of developing CRC at an early age and explore the gene-gene interactions between polymorphisms of cell-cycle genes. We identified a subgroup with high probability of cancer occurrence at younger ages of onset, a 45-year median onset age, and a subgroup with the latest age of onset, a 70-year median onset age for CRC. The overall median onset age of CRC was 50. We would expect to see some subgroups having median ages below the overall median age. The two groups with the latest age of onset (70 and 60 years) were the two groups with the lowest number of individuals. We would expect that they would have the lowest numbers of individuals since there are not very many patients with onset ages after the age of 60 or 70. Furthermore, CART analysis identified a gene-gene interaction between E2F2 and AURKA genes related to Lynch syndrome.
CART analysis identified CDKN2A C580T and IGF1 CA-repeat as the initial splits, indicating that the polymorphisms in these genes are the most informative for separating patients into those Lynch syndrome patients who are more likely to develop CRC early vs. those who are more likely to develop CRC at a later age. Germline mutations in the CDKN2A gene are known to predispose individuals to pancreatic cancer and melanoma [24]. The SNP of CDKN2A C580T was reportedly associated with tumor aggressiveness for melanoma [15]. High circulating levels of IGF1 are associated with increased risk for several common cancers, including colorectal, breast, prostate, and lung cancer [25]. The IGF1 promoter cytosine-adenine (CA) dinucleotide–repeat length polymorphism is thought to influence levels of IGF1, with shorter repeat lengths being associated with higher levels of IGF1 expression. We previously reported that a shorter CA-repeats was associated with an earlier age at onset of CRC in Lynch syndrome [7]. In the present study, CART analysis selected subgroups of those with CA-repeat length <19 and ≥19.
The results of CART analysis suggest a novel gene-gene interaction between E2F2 and AURKA as the influence of the AURKA SNP on risk varies depending on the E2F2 genotypes. In the final pruned tree, individuals who are E2F2-WW and AURKA-WM/MM have a later median age at onset compared to individuals who are E2F2 WM/MM and AURKA-WM/MM (70 years versus 46 years, P = 0.007). We included this interaction in a Cox proportional hazard model and observed that the HRs for the interaction effect for the presence of both E2F2 WM/MM and AURKA WM/MM genotypes was 6.87 (95% CI=1.72-27.32) (P-interaction = 0.003), indicating association to an earlier age of onset. In contrast, at this node of the tree the main effect for the presence of AURKA WM/MM genotypes was protective which was consistent with our previous findings in Caucasians [8], and also with the recent study reporting that the polymorphism was associated with a significantly reduced risk for lung cancer in Caucasians [26], although Talseth et al did not find the AURKA polymorphism was associated with age of onset of CRC in Lynch syndrome [10]. The E2F2 SNP showed a protective main effect too, but it was not statistically significant. Our findings suggested that the SNPs of the two genes have a more-than-multiplicative joint effect on the age-associated risk for CRC in the patients of this subgroup. The findings also suggested that genetic variants and their influence on risk can differ in the context of different genetic backgrounds, and highlighting the potential power of CART to uncover complex gene-gene interactions between predictors of risk within specific patient subgroups.
The predicted gene-gene interaction between AURKA and E2F2 is very feasible as both genes play important roles in cell-cycle regulation. This could be due to a direct or indirect physical interaction. AURKA is a serine/threonine kinase, and is best known for its role in proper mitotic entry and maintenance of the G2-M checkpoint. However, like the E2F family members, it also plays an important role in the G1-S checkpoint [27]. AURKA activity has been shown to modulate TP53 stability. TP53 is important in several functions including regulation of the G1-S checkpoint where it positively regulates p21 in response to DNA damage. p21 then binds to cyclin D1/CDK4 or cyclin D1/CDK6 complexes to inhibit their activity and induce cell-cycle arrest [27-28]. Targeted inactivation of E2F1, E2F2, and E2F3 results in elevated p21 protein levels and cell-cycle arrest at G1-S, and G2-M, suggesting a strict requirement for these E2Fs in the control of normal cellular proliferation [29]. Loss of TP53 restores the ability of these cells to progress through both G1-S and G2-M. Whether a gene-gene interaction exists between AURKA and E2F2 as a result of their shared involvement in the control of the TP53-p21 axis, the G1-S checkpoint and/or the G2-M checkpoint will be the focus of future studies.
CART analysis identified subgroup 7 with CDKN2A C580T WW, IGF1 CA-repeats ≥ 19, E2F2 WM/MM genotype, AURKA WW, and CCND1 WM/MM as having the youngest median age of onset (45 years). CCND1 plays an important role in the G1-to-S phase transition of the cell-cycle. We previously reported that the CCND1 G-to-A polymorphism at codon 242 was associated with an earlier age of onset of CRC in MMR mutation carriers [5]. Talseth et al reported that the CCND1 polymorphism was associated with a significant difference in the age of disease onset on patients harboring MSH2 mutations, which was not observed in MLH1 mutation carriers [10]. However, Kruger et al did not find any significant association between the CCND1 polymorphism and age of onset in German population [30]. Bala et al did not observe correlation between a particular allele (A versus G) and age at onset in a Finnish population. However they observed that the presence of the variant transcript b in blood/normal mucosa was associated with a significantly lower age at onset of colon cancer as compared with individuals with transcript a only (35 versus 46 years; P = 0.02) which suggest that the relative abundance of a and b transcripts may modify the age at onset of colon cancer in Lynch syndrome [31]. Previous studies had suggested that the variant allele A is a major source of transcript b [32-33]. The inconsistency in these findings could reflect lifestyle differences among populations or effects of other modifying genetic factors.
A potential limitation of our study is that it is clinic-based rather than population-based, and may not be representative of MMR gene mutation carriers in the general population. However, because the subjects were recruited without any knowledge of their SNP genotypes, no selection bias has occurred with regards to the exposure variables of our analysis. Since some patients were members of the same family, we used robust variance estimators clustering on family membership to account for familial correlation in the risk factors. Larger and independent replication studies are needed to validate our findings.
The strength of our study is that we use a pathways-based multigenetic approach (the cell cycle pathways). The conventional single-gene-based approach to study the role of genetic polymorphisms in carcinogenesis has been unable to yield consistent data across different studies. Included among the reasons for this lack of replication as pointed out by Horne et al [34] is failure to evaluate the effect of multiple pathophysiologically related genes in combination. Given the low risk that is usually conferred by an individual polymorphism, it is unlikely that any one single genetic polymorphism would have a dramatic effect on cancer risk. Recently, Wang et al. reported on the use of CART to evaluate high order gene-gene interactions in lung cancer, providing evidence of the promising potential of a pathways-based approach [35].
In conclusion, our findings support the view that polymorphisms can have different influences on time to onset for CRC in different genetic backgrounds. They also indicate that cell-cycle genes interact with one another in cell-cycle regulation, and that the SNPs in these genes can influence these finely tuned mechanisms of cell-cycle regulation. The tree-based method has great potential for identifying gene-gene interactions. The findings of the study will contribute to our long-term goal to develop a risk model for Lynch syndrome. Future studies should be directed towards confirming these findings in a larger sample population and will require collaborative efforts by multiple investigators to obtain large sample sizes.
Acknowledgements
We thank Haidee Chancoco and Domitila Patenia for their laboratory technical assistance.
Grant support: National Cancer Institute Grant CA 70759 (Frazier, ML), National Institutes of Health Cancer Center Support Grant CA 16672 (Mendelsohn, J), the Janis Davis Gordon Memorial Postdoctoral Fellowship, Division of Cancer Prevention, The University of Texas M. D. Anderson Cancer Center (Chen, J).
Footnotes
Conflict of interest statement
None declared
References
- 1.Peltomaki P, Vasen H. Mutations associated with HNPCC predisposition -- Update of ICG-HNPCC/INSiGHT mutation database. Dis Markers. 2004;20:269–276. doi: 10.1155/2004/305058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. Jama. 2006;296:1507–1517. doi: 10.1001/jama.296.12.1507. [DOI] [PubMed] [Google Scholar]
- 3.Lynch HT, Boland CR, Gong G, et al. Phenotypic and genotypic heterogeneity in the Lynch syndrome: diagnostic, surveillance and management implications. Eur J Hum Genet. 2006;14:390–402. doi: 10.1038/sj.ejhg.5201584. [DOI] [PubMed] [Google Scholar]
- 4.Aarnio M, Sankila R, Pukkala E, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81:214–218. doi: 10.1002/(sici)1097-0215(19990412)81:2<214::aid-ijc8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 5.Kong S, Amos CI, Luthra R, Lynch PM, Levin B, Frazier ML. Effects of cyclin D1 polymorphism on age of onset of hereditary nonpolyposis colorectal cancer. Cancer Res. 2000;60:249–252. [PubMed] [Google Scholar]
- 6.Jones JS, Chi X, Gu X, Lynch PM, Amos CI, Frazier ML. p53 polymorphism and age of onset of hereditary nonpolyposis colorectal cancer in a Caucasian population. Clin Cancer Res. 2004;10:5845–5849. doi: 10.1158/1078-0432.CCR-03-0590. [DOI] [PubMed] [Google Scholar]
- 7.Zecevic M, Amos CI, Gu X, et al. IGF1 gene polymorphism and risk for hereditary nonpolyposis colorectal cancer. J Natl Cancer Inst. 2006;98:139–143. doi: 10.1093/jnci/djj016. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Sen S, Amos CI, et al. Association between Aurora-A kinase polymorphisms and age of onset of hereditary nonpolyposis colorectal cancer in a Caucasian population. Mol Carcinog. 2007;46:249–256. doi: 10.1002/mc.20283. [DOI] [PubMed] [Google Scholar]
- 9.Reeves SG, Rich D, Meldrum CJ. IGF1 is a modifier of disease risk in hereditary non-polyposis colorectal cancer. Int J Cancer. 2008;123:1339–1343. doi: 10.1002/ijc.23668. [DOI] [PubMed] [Google Scholar]
- 10.Talseth BA, Ashton KA, Meldrum C, et al. Aurora-A and Cyclin D1 polymorphisms and the age of onset of colorectal cancer in hereditary nonpolyposis colorectal cancer. Int J Cancer. 2008;122:1273–1277. doi: 10.1002/ijc.23177. [DOI] [PubMed] [Google Scholar]
- 11.Talseth BA, Meldrum C, Suchy J, Kurzawski G, Lubinski J, Scott RJ. Age of diagnosis of colorectal cancer in HNPCC patients is more complex than that predicted by R72P polymorphism in TP53. Int J Cancer. 2006;118:2479–2484. doi: 10.1002/ijc.21661. [DOI] [PubMed] [Google Scholar]
- 12.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 13.Li G, Wang LE, Chamberlain RM, Amos CI, Spitz MR, Wei Q. p73 G4C14-to-A4T14 polymorphism and risk of lung cancer. Cancer Res. 2004;64:6863–6866. doi: 10.1158/0008-5472.CAN-04-1804. [DOI] [PubMed] [Google Scholar]
- 14.Ruijs MW, Schmidt MK, Nevanlinna H, et al. The single-nucleotide polymorphism 309 in the MDM2 gene contributes to the Li-Fraumeni syndrome and related phenotypes. Eur J Hum Genet. 2007;15:110–114. doi: 10.1038/sj.ejhg.5201715. [DOI] [PubMed] [Google Scholar]
- 15.Sauroja I, Smeds J, Vlaykova T, et al. Analysis of G(1)/S checkpoint regulators in metastatic melanoma. Genes Chromosomes Cancer. 2000;28:404–414. doi: 10.1002/1098-2264(200008)28:4<404::aid-gcc6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 16.Breiman L, Friedman JH, Olshen RA, Stone CJ. Classification and Regression Trees. Wadsworth; Belmont: 1984. [Google Scholar]
- 17.Zhang H, Singer B. Recursive Partitioning in the Health Sciences. Springer; New York: 1999. [Google Scholar]
- 18.LeBlanc M, Crowley J. Relative risk trees for censored survival data. Biometrics. 1992;48:411–425. [PubMed] [Google Scholar]
- 19.Zhang H, Bonney G. Use of classification trees for association studies. Genet Epidemiol. 2000;19:323–332. doi: 10.1002/1098-2272(200012)19:4<323::AID-GEPI4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 20.Province MA, Shannon WD, Rao DC. Classification methods for confronting heterogeneity. Adv Genet. 2001;42:273–286. doi: 10.1016/s0065-2660(01)42028-1. [DOI] [PubMed] [Google Scholar]
- 21.Shannon WD, Province MA, Rao DC. Tree-based recursive partitioning methods for subdividing sibpairs into relatively more homogeneous subgroups. Genet Epidemiol. 2001;20:293–306. doi: 10.1002/gepi.1. [DOI] [PubMed] [Google Scholar]
- 22.Therneau T, Atkinson E. An introduction to recursive partitioning using the RPART routine. Mayo Clinic, Section of Statistics; Rochester: 1997. [Google Scholar]
- 23.Lin D, Wei, L. The Robust Inference for the Cox Proportional Hazards Model. J. American Statistical Association. 1989;84:1074–1078. [Google Scholar]
- 24.Whelan AJ, Bartsch D, Goodfellow PJ. Brief report: a familial syndrome of pancreatic cancer and melanoma with a mutation in the CDKN2 tumor-suppressor gene. N Engl J Med. 1995;333:975–977. doi: 10.1056/NEJM199510123331505. [DOI] [PubMed] [Google Scholar]
- 25.Yu H, Rohan T. Role of the insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst. 2000;92:1472–1489. doi: 10.1093/jnci/92.18.1472. [DOI] [PubMed] [Google Scholar]
- 26.Gu J, Gong Y, Huang M, Lu C, Spitz MR, Wu X. Polymorphisms of STK15 (Aurora-A) gene and lung cancer risk in Caucasians. Carcinogenesis. 2007;28:350–355. doi: 10.1093/carcin/bgl149. [DOI] [PubMed] [Google Scholar]
- 27.Katayama H, Sasai K, Kawai H, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36:55–62. doi: 10.1038/ng1279. [DOI] [PubMed] [Google Scholar]
- 28.Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 2003;13:65–70. doi: 10.1016/s0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 29.Sharma N, Timmers C, Trikha P, Saavedra HI, Obery A, Leone G. Control of the p53-p21CIP1 Axis by E2f1, E2f2, and E2f3 is essential for G1/S progression and cellular transformation. J Biol Chem. 2006;281:36124–36131. doi: 10.1074/jbc.M604152200. [DOI] [PubMed] [Google Scholar]
- 30.Kruger S, Engel C, Bier A, et al. Absence of association between cyclin D1 (CCND1) G870A polymorphism and age of onset in hereditary nonpolyposis colorectal cancer. Cancer Lett. 2006;236:191–197. doi: 10.1016/j.canlet.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Bala S, Peltomaki P. CYCLIN D1 as a genetic modifier in hereditary nonpolyposis colorectal cancer. Cancer Res. 2001;61:6042–6045. [PubMed] [Google Scholar]
- 32.Betticher DC, Thatcher N, Altermatt HJ, Hoban P, Ryder WD, Heighway J. Alternate splicing produces a novel cyclin D1 transcript. Oncogene. 1995;11:1005–1011. [PubMed] [Google Scholar]
- 33.Sawa H, Ohshima TA, Ukita H, et al. Alternatively spliced forms of cyclin D1 modulate entry into the cell cycle in an inverse manner. Oncogene. 1998;16:1701–1712. doi: 10.1038/sj.onc.1201691. [DOI] [PubMed] [Google Scholar]
- 34.Horne BD, Anderson JL, Carlquist JF, et al. Generating genetic risk scores from intermediate phenotypes for use in association studies of clinically significant endpoints. Ann Hum Genet. 2005;69:176–186. doi: 10.1046/j.1529-8817.2005.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang W, Spitz MR, Yang H, et al. Genetic variants in cell cycle control pathway confer susceptibility to lung cancer. Clin Cancer Res. 2007;13:5974–5981. doi: 10.1158/1078-0432.CCR-07-0113. [DOI] [PubMed] [Google Scholar]
- 36.Lagerstedt Robinson K, Liu T, Vandrovcova J, et al. Lynch syndrome (hereditary nonpolyposis colorectal cancer) diagnostics. J Natl Cancer Inst. 2007;99:291–299. doi: 10.1093/jnci/djk051. [DOI] [PubMed] [Google Scholar]
- 37.Raevaara TE, Korhonen MK, Lohi H, et al. Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology. 2005;129:537–549. doi: 10.1016/j.gastro.2005.06.005. [DOI] [PubMed] [Google Scholar]