

Abstract
A dual stimuli responsive nanogel-polyelectrolyte complex based on electrostatic coating has been developed. The nanoassembly is designed to elicit two disparate responses (viz. surface property change and guest encapsulation stability) from two different stimuli (viz. pH and redox variations). The components of the nanogel and the polyelectrolyte have been conveniently achieved from a simple homopolymer, poly(pentafluorophenylacrylate).
Supramolecular assemblies that respond to the presence of multiple stimuli have attracted tremendous interest due to their potential in being more selective.1 The anticipated selectivity is due to the requirement that more than one stimulus has to be concurrently present to elicit the appropriate response. Popular among the stimuli-responsive elements involve change in the surface features of a host assembly or change in the host properties of the assembly.2–8 The former causes stimulus-responsive changes to the interfacial interactions of the assembly with its surroundings, while the latter often results in release of the sequestered guest molecules in response to the stimuli. Among the stimuli investigated, pH and redox sensitive assemblies have attracted particular attention.9–33 In prior systems, the focus has been to develop an assembly, where the combined effect of two different stimuli is much better than either of the stimuli alone.34–37 We have been interested in developing assemblies where two different features of an assembly are sequentially impacted by the presence of the dual stimuli. We focused specifically on variations in pH and redox conditions as the stimuli impacting change in surface properties and guest molecule encapsulation capabilities as the responses respectively.
To achieve a supramolecular assembly with these stimuli responsive characteristics, we have developed a polyelectrolyte-nanogel complex (Scheme 1). In this complex, the polyelectrolyte has charge-conversional features – i.e. the charge in the polyelectrolyte will change from negative to positive charge at low pH. The nanogel is capable of sequestering hydrophobic guest molecules that are released in response to change in the redox environment. The nanogel will have a positively charged surface so as to complement the polyelectrolyte during the complex formation. The hypothesis here is that a change in the polyelectrolyte charge, in response to lowered pH, will compromise this electrostatic complementarity. The dissociation event will cause a change in the surface properties of the nanoassembly. We also conceived that the electrostatically bound polyelectrolyte will enhance the encapsulation stability of the hydrophobic guest molecules inside the nanogel. Dissociating the polyelectrolyte from the nanogel and then subjecting it to a reducing condition will cause the guest molecules to be released from the nanogel. These expectations are illustrated in Scheme 1.
Scheme 1.
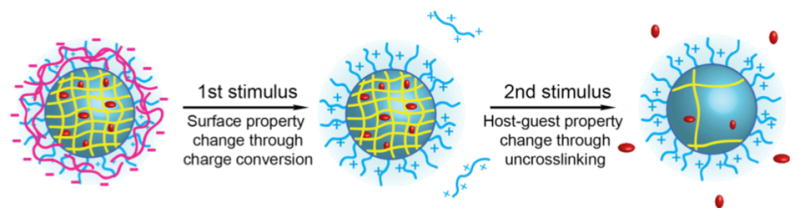
Representation of dual stimuli - dual responsive features of the reported polyelectrolyte-
The structures of the cationic polymeric nanogel, its precursor, the complementary anionic polyelectrolyte, and the products of the pH-induced reaction (the non-complementary cationic polyelectrolyte and the anionic small molecule) are all shown in Scheme 2. The nanogel is synthesized through the formation of an amphiphilic random copolymer nanoassembly, which is crosslinked through an in situ reduction reaction.19 The pyridyldisulfide units provide the hydrophobic component of the amphiphilic polymer and afford the handle to execute the crosslinking reaction to generate the disulfide crosslinked nanogels. The quaternary ammonium moiety provides the cationic charge to the nanogel, while the N-isopropyl acrylamide unit plays the role of charge-neutral, hydrophilic units that can be used to modulate the cationic charge density in the nanogel. The anionic moiety in the polyelectrolyte is based on the monoamide formed from tetrahydrophthalic acid. The oligoethyleneglycol units present in the polyelectrolyte is used to tune its charge density.
Scheme 2.
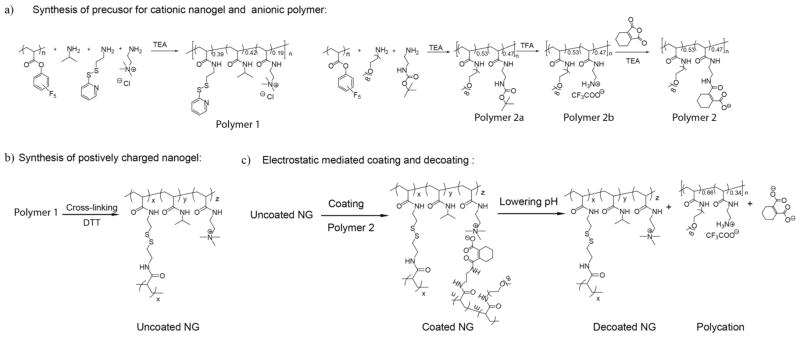
Preparation of a) precursor for cationic nanogel and anionic polymer; b) nanogel; c) structures of nanogel-polyanion complexes and the disassociated products induced by lowering the pH of the aqueous solution.
Both the nanogel polymer precursor and the anionic polyelectrolyte were prepared through a simple substitution reaction of poly(pentafluorophenyl acrylate) (PPFPA) with appropriately functionalized primary amines (Scheme 2). To synthesize the cationic nanogel, the activated acrylate ester PPFPA was treated with isopropylamine (0.3 equiv.), pyridyldisulfide containing 2-aminoethanethiol (0.5 equiv.), and 2-aminoethyl-trimethylammonium chloride (0.2 equiv.). The targeted copolymer 1 was obtained in 85% yield. The percentage of the co-monomers in the polymer was assessed to be 0.42:0.39:0.19 using NMR, which shows that the ratio corresponds to the feed ratio of the monomers in the substitution reaction. The substitution reaction was also analyzed using the C=O stretching bands in IR,38 where we found that the reaction to be complete (Figure 1). The polymer was then converted to the corresponding disulfide crosslinked nanogel by treating the polymer with dithiothreitol (DTT) based on the previously reported procedure.39,40 Similarly, the polyelectrolyte was synthesized by first treating PPFPA with mono-amino-oligoethyleneglycol and N-Boc-ethylenediamine in 0.7:0.3 ratio to afford polymer 2a. The ratio of the co-monomer in the polymer 2a was found to be 0.53:0.47 using NMR. As with the syntheses of the polymer nanogel precursor, quantitative substitution of the pentafluorophenyl moiety in this case was also ascertained using IR (Figure 1). The Boc-protecting group was removed by treating the polymer with trifluoroacetic acid (TFA). The resulting primary amine polymer 2b was treated with 3,4,5,6-tetrahydrophthalic anhydride to convert this cationic polymer to the carboxylic acid containing anionic polymer 2. Note that this substitution reaction, which has an associated charge conversion feature, can be conveniently reversed by lowering the pH of the aqueous solution (Figure S5). This reversal indeed forms the basis for the pH-induced charge conversion here.
Figure 1.

FTIR characterization of polymer precursor reactions for the preparation of the cationic nanogel and anionic coating polymer.
Next, we resorted to prepare the nanogel-polyanion complex was through electrostatic interactions. Briefly, to a polymer 2 solution with a concentration of 10 mg/mL at pH 9.0, a calculated amount of positively charged nanogel with a concentration of 1 mg/mL was added dropwise and the mixture was stirred for 30 min. Then, free polyanion was removed from the mixture by ultrafiltration to afford pure nanogel-polyanion complex.
The nanogel-polyelectrolyte complex was characterized using dynamic light scattering (DLS) and zeta potential measurements. The size of nanogel itself was found to be 30 nm as measured by dynamic light scattering (DLS), as shown in Figure 2a. After being coated by polymer 2, the size increased to 55 nm indicating the formation of nanogel-polyanion complex. Since the surface of the nanogel contains positively charged quaternary ammonium moieties, the nanogel will be expected to be cationic and thus exhibit a positive zeta potential. Indeed the zeta potential of the nanogel was found to be about +30 mV (Figure 2b). If the nanogel was efficiently coated with the negatively charged polymer, then the surface charge of the nanogel-polyelectrolyte complex should be anionic. Indeed, the zeta potential of this complex was found to be about −30 mV.
Figure 2.

Changes on (a) size and (b) zeta potential of cationic nanogel after coating and decoating
Our design hypothesis is that the pH-induced conversion of the anionic coating polymer to a cationic polymer will cause the polymer to dissociate from the nanogel due to electrostatic repulsion. To test this possibility, we observed the size and the zeta potential of the nanogel upon lowering the pH of the solution. As shown in Figure 2a, the size of the nanogel changed back to ~30 nm upon decreasing the pH of the solution to 4.4 for 30 min. This size corresponds to the uncoated nanogel suggesting that the polyelectrolyte has dissociated from the nanogel. This was further supported by the zeta potential measurements. As shown in Figure 2b, lowering pH of the solution causes the zeta potential to positively shift to +18 mV. The fact that the charge did not fully recover to that of the cationic nanogel itself (+30 mV) might suggest that the dissociation is not complete. This possibility cannot be unambiguously ruled out. However, it is also possible that the smaller zeta potential (+5 mV) of the dissociated polymer contributes to make the overall zeta potential in the nanogel-polyelectrolyte mixture lower. We tested this possibility by mixing the low pH treated anionic polymer 2 and the cationic nanogel, and measured the zeta potential (Figure S7). The fact that increasing concentrations of the polymer can reduce the overall zeta potential suggests that the latter possibility of the dissociated polymer contributing to the observed lower zeta potential indeed exists. Additionally, the disassociation of nanogel complex was investigated under various pHs by following the zeta potential of the complex (Figure S8). The zeta potential measurement suggests the disassociation kinetics is increases with decreasing pH..
Next, we were interested in investigating the possibility of tuning the size of these assemblies by simply varying the ratio of the nanogel to the polyelectrolyte. It is conceivable that when excess polyelectrolyte is used to form the complex, the polyelectrolyte will form more or less a monolayer coating on the surface of the nanogel without much aggregation. However, when the ratio of polyelectrolyte to the nanogel is smaller, nanogel aggregation should be possible. The complexes, studied above, were prepared using a 1:5 ratio of the nanogel to polyelectrolyte, where the complex size was 55 nm compared to the nanogel size of 30 nm. When this ratio was changed to 1:10, the size of the complex was only slightly increased or essentially unchanged from the original nanogel size (Figure 3). Note that this size increase is much smaller than that observed with the 1:5 mixture. To insure that the complexation has indeed occurred in this case, we also investigated the zeta potential of the nanogel-polyanion complex. The zeta potential of this complex has indeed changed to −30 mV (Figure S6). We then investigated the effect of changing the ratio to 2:1. As anticipated, the size of the nanogel increased to ~100 nm (Figure 3). Interestingly here too, the zeta potential of the nanogel was found to be −30 mV. These results are taken to suggest that 0.5 equivalent (in terms of weight %) is already sufficient to fully neutralize the positive charges on the surface of the nanogels and render the overall surface charge of the complex to be negative. However, it seems that this charge neutralization can not be achieved without causing the nanogels to aggregate during the complexation event. All these nanogel complexes exhibit pH-induced surface property change (Figure S6).
Figure 3.

Tunable size of nanogel/polyanion complex observed by DLS (top) and TEM (bottom).
Finally, we were interested in analyzing the effect of the complex formation on the guest encapsulation. The pH-sensitive features, shown above, demonstrates an approach to alter the surface properties (surface charge) of a nanoassembly in response to the pH stimulus. We were also interested in investigating the effect of the polyelectrolyte coating on the redox-sensitive nature of the nanogel. Note that the nanogel is based on disulfide crosslinks; therefore, the encapsulated guest molecules can be released in response to the presence of thiol-based reducing agents such as glutathione (GSH). Accordingly, we investigated the effect of molecular release using Nile red as the guest molecule. First, since we were interested in showing that the pH and the redox stimuli can independently effect two different properties of the nanogel-polyelectrolyte complex, we investigated the effect of pH on the encapsulation stability of the nanogel. As shown in Figure 4, note that no guest release was observed with the nanogel-polyelectrolyte complex at pH 7.4 (the condition in which polyelectrolyte coating is retained) and pH 4.4 (the condition at which the polyelectrolyte coating is rapidly removed due to charge conversion in the polyelectrolyte).
Figure 4.

Temporal release of Nile red from coated, uncoated and control nanogels in the presence of 1 mM GSH.
Next, it is gratifying to note that the guest release from the nanogel-polyelectrolyte complex is slower than that from the uncoated nanogel itself at pH 7.4. This suggests that the guest encapsulation is further stabilized by the polyelectrolyte coating. When the pH was lowered to 4.4, the pH at which the polyanion would be removed from the complex due to the charge conversion, the release from the nanogel-polyelectrolyte complex was slower than the above two conditions. This observation was indeed surprising. However, it is important to note that GSH itself exhibits substantially different redox activity at lower pH. Therefore, we investigated the GSH-induced release of Nile red at pH 4.4 with the bare nanogel. Indeed, the release profile was very similar to that of the nanogel-polyelectrolyte complex at this pH. These results suggest that the polyelectrolyte is indeed removed from the nanogel to affect guest encapsulation was observed as fast as that from uncoated nanogel. To insure that the redox-sensitive activity can be recovered after subjecting the nanogel complex to a pH change, we investigated the guest encapsulation stability of the nanogel-polyelectrolyte complex at pH 7.4. Here the complex was first subjected to the pH induced dissociation at pH 4.4 for 10 min; the pH of the solution then was raised to pH 7.4. The guest molecule was indeed rapidly released in the presence of GSH at this pH. The guest release profile was once again similar to that of the uncoated nanogel that was subjected to this pH cycling. Put together, these results suggest that the nanogel can stably encapsulate the guest molecules at different pH, but differentially releases the guest molecules due to polyelectrolyte complexation under the influence of the redox stimulus.
In summary, we have shown that: (i) a positively charged nanogel and a negatively charged polyelectrolyte can be conveniently coupled to make a nanogel-polyelectrolyte complex; (ii) since the polyelectrolyte undergoes a charge conversion at low pH, the nanogel-polyelectrolyte complex is dissociated under the influence of the pH stimulus; (iii) simple stoichiometric variations between the nanogel and the polyelectrolyte can be utilized to predictably vary the size of the complex; (iv) while the pH stimulus has a profound impact on the surface features of the nanogel-polyelectrolyte complex, it has no effect on the guest encapsulation; (v) on the contrary, redox stimulus does not affect the surface properties of the complex, but does influence the guest encapsulation in the nanoassembly; (vi) the polyelectrolyte complexation on the nanogel surface affords slightly higher encapsulation stability, compared to bare nanogel. In summary, we have shown that the electrostatics-driven complex between polymeric nanogels and polyelectrolytes can be utilized to design a nanoassembly that responds to two different stimuli, eliciting two diverse responses. This design platform could find use in a variety of applications, including drug delivery and sensing. For example, considering the sequential variations of lower pH in the extracellular environments of tumors and higher GSH concentrations inside the cells, one could envision utilizing the design principles developed here for drug delivery. This will form part of the ongoing efforts in our own laboratory.
Supplementary Material
Acknowledgments
Funding Sources
Any funds used to support the research of the manuscript should be placed here (per journal style).
We thank the National Science Foundation (CHE-1307118) for support. We thank the NIGMS of the NIH for a CBI fellowship to JZ (T32 GM08515). We also thank MRSEC at UMass Amherst for infrastructure support.
Footnotes
Supporting Information. “This material is available free of charge via the Internet at http://pubs.acs.org.”
References
- 1.Zhuang J, Gordon MR, Ventura J, Li L, Thayumanavan S. Chem Soc Rev. 2013;42:7421–7435. doi: 10.1039/c3cs60094g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wan X, Zhang G, Liu S. Macromol Rapid Commun. 2011;32:1082–1089. doi: 10.1002/marc.201100198. [DOI] [PubMed] [Google Scholar]
- 3.Lee Y, Fukushima S, Bae Y, Hiki S, Ishii T, Kataoka K. J Am Chem Soc. 2007;129:5362–5363. doi: 10.1021/ja071090b. [DOI] [PubMed] [Google Scholar]
- 4.Lee Y, Miyata K, Oba M, Ishii T, Fukushima S, Han M, Koyama H, Nishiyama N, Kataoka K. Angew Chem Int Ed. 2008;120:5241–5244. doi: 10.1002/anie.200800963. [DOI] [PubMed] [Google Scholar]
- 5.Du J, Sun T, Song W, Wu J, Wang J. Angew Chem Int Ed. 2010;49:3621–3626. doi: 10.1002/anie.200907210. [DOI] [PubMed] [Google Scholar]
- 6.Du J, Du X, Mao C, Wang J. J Am Chem Soc. 2011;133:17560–17563. doi: 10.1021/ja207150n. [DOI] [PubMed] [Google Scholar]
- 7.Guo S, Huang Y, Jiang Q, Sun Y, Deng L, Liang Z, Du Q, Xing J, Zhao Y, Wang PC, Dong A, Liang X. ACS Nano. 2010;4:5505–5511. doi: 10.1021/nn101638u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou Z, Shen Y, Tang J, Fan M, Kirk EAV, Murdoch WJ, Radosz M. Adv Funct Mater. 2009;19:3580–3589. [Google Scholar]
- 9.Liu F, Eisenberg A. J Am Chem Soc. 2003;125:15059–15064. doi: 10.1021/ja038142r. [DOI] [PubMed] [Google Scholar]
- 10.Bellomo EG, Wyrsta MD, Pakstis L, Pochan DJ, Deming TJ. Nature Materials. 2004;3:244–248. doi: 10.1038/nmat1093. [DOI] [PubMed] [Google Scholar]
- 11.Doncom KEB, Hansell CF, Theato P, O’Reilly RK. Polym Chem. 2012;3:3007–3015. [Google Scholar]
- 12.Ma N, Li Y, Xu H, Wang Z, Zhang X. J Am Chem Soc. 2010;132:442–443. doi: 10.1021/ja908124g. [DOI] [PubMed] [Google Scholar]
- 13.Duan Q, Cao Y, Li Y, Hu X, Xiao T, Lin C, Pan Y, Wang L. J Am Chem Soc. 2013;135:10542–10549. doi: 10.1021/ja405014r. [DOI] [PubMed] [Google Scholar]
- 14.Dan K, Ghosh S. Angew Chem Int Ed. 2013;125:7441–7446. [Google Scholar]
- 15.Klaikherd A, Nagamani C, Thayumanavan S. J Am Chem Soc. 2009;131:4830–4838. doi: 10.1021/ja809475a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldenbogen B, Brodersen N, Gramatica A, Loew M, Liebscher J, Herrmann A, Egger H, Budde B, Arbuzova A. Langmuir. 2011;27:10820–10829. doi: 10.1021/la201160y. [DOI] [PubMed] [Google Scholar]
- 17.Zelikin AN, Quinn JF, Caruso F. Biomacromolecules. 2006;7:27–30. doi: 10.1021/bm050832v. [DOI] [PubMed] [Google Scholar]
- 18.Lele BS, Leroux JC. Macromolecules. 2002;35:6714–6723. [Google Scholar]
- 19.Zhang Z, Yin L, Tu C, Song Z, Zhang Y, Xu Y, Tong R, Zhou Q, Ren J, Cheng J. ACS Macro Lett. 2013;2:40–44. doi: 10.1021/mz300522n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koo AN, Lee HJ, Kim SE, Chang JH, Park C, Kim C, Park JH, Lee SC. Chem Comm. 2008;48:6570–6572. doi: 10.1039/b815918a. [DOI] [PubMed] [Google Scholar]
- 21.Kim TI, Ou M, Lee M, Kim SW. Biomaterials. 2009;30:658–664. doi: 10.1016/j.biomaterials.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JO, Sahay G, Kabanov AV, Bronich TK. Biomacromolecules. 2010;11:919–926. doi: 10.1021/bm9013364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang S, Zhao Y. J Am Chem Soc. 2010;132:10642–10644. doi: 10.1021/ja103391k. [DOI] [PubMed] [Google Scholar]
- 24.Gillies ER, Frechet JMJ. J Am Chem Soc. 2010;132:10642–10644. [Google Scholar]
- 25.Argentiere S, Blasi L, Morello G, Gigli G. J Phys Chem C. 2011;115:16347–16353. [Google Scholar]
- 26.Jones MC, Ranger M, Leroux JC. Bioconjugate Chem. 2003;14:774–781. doi: 10.1021/bc020041f. [DOI] [PubMed] [Google Scholar]
- 27.Dou H, Jiang M, Peng H, Chen D, Hong Y. Angew Chem Int Ed. 2003;42:1516–1519. doi: 10.1002/anie.200250254. [DOI] [PubMed] [Google Scholar]
- 28.Yan Q, Zhou R, Fu C, Zhang H, Yin Y, Yuan J. Angew Chem Int Ed. 2011;50:4923–4927. doi: 10.1002/anie.201100708. [DOI] [PubMed] [Google Scholar]
- 29.Huynh VT, Binauld S, Souza PLd, Stenzel MH. Chem Mater. 2012;24:3197–3211. [Google Scholar]
- 30.Rodriguez-Hernandez J, Lecommandoux S. J Am Chem Soc. 2005;127:2026–2027. doi: 10.1021/ja043920g. [DOI] [PubMed] [Google Scholar]
- 31.Du J, Tang Y, Lewis AL, Armes SP. J Am Chem Soc. 2005;127:17982–17983. doi: 10.1021/ja056514l. [DOI] [PubMed] [Google Scholar]
- 32.Lu J, Li N, Xu Q, Ge J, Lu J, Xia X. Polymer. 2010;51:1709–1715. [Google Scholar]
- 33.Lee ES, Gao Z, Kim D, Park K, Kwon IC, Bae YH. J Controlled Release. 2008;129:228–236. doi: 10.1016/j.jconrel.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han D, Tong X, Zhao Y. Langmuir. 2012;28:2327–2331. doi: 10.1021/la204930n. [DOI] [PubMed] [Google Scholar]
- 35.Jackson AW, Fulton DA. Macromolecules. 2012;45:2699–2708. [Google Scholar]
- 36.Chen J, Qiu X, Ouyang J, Kong J, Zhong W, Xing MMQ. Biomacromolecules. 2011;12:3601–3611. doi: 10.1021/bm200804j. [DOI] [PubMed] [Google Scholar]
- 37.Ryu J-H, Chacko RT, Jiwpanich S, Bickerton S, Babu RP, Thayumanavan S. J Am Chem Soc. 2010;132:17227–17235. doi: 10.1021/ja1069932. [DOI] [PubMed] [Google Scholar]
- 38.Zhuang J, Jiwpanich S, Deepak VD, Thayumanavan S. ACS Macro Lett. 2012;1:175–179. doi: 10.1021/mz200123f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryu JH, Bickerton S, Zhuang J, Thayumanavan S. Biomacromolecules. 2012;13:1515–1522. doi: 10.1021/bm300201x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryu J-H, Jiwpanich S, Chacko R, Bickerton S, Thayumanavan S. J Am Chem Soc. 2010;132:8246–8247. doi: 10.1021/ja102316a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.