

Abstract
Background
SMC migration and proliferation critically influence the clinical course of vascular disease. We tested the effect of the novel small leucine-rich repeat protein podocan on SMC migration and proliferation using a podocan deficient mouse in combination with a model of arterial injury and aortic explant SMC culture. In addition, we examined the effect of overexpression of the human form of podocan on human SMC and tested for podocan expression in human atherosclerosis. In all these conditions we evaluated concomitantly the Wnt-TCF-pathway.
Methods and Results
Podocan was strongly and selectively expressed in arteries of WT mice after injury. Podocan−/− mice showed increased arterial lesion formation as compared to WT littermates in response to injury (P<0.05). Also, SMC proliferation was increased in arteries of podocan −/− mice compared to WT (P<0.05). In vitro, migration and proliferation were increased in podocan−/− SMC and were normalized by transfection with the WT podocan gene (P<0.05). In addition, upregulation of the Wnt-TCF-pathway was found in SMC of podocan−/− mice both in vitro and in vivo. On the other hand, podocan overexpression in human SMC significantly reduced SMC migration and proliferation inhibiting the Wnt-TCF-pathway. Podocan and a Wnt-TCF-pathway marker were differently expressed in human coronary restenotic versus primary lesions.
Conclusions
Podocan appears to be a potent negative regulator of the migration and proliferation of both murine and human SMC. The lack of podocan results in excessive arterial repair and prolonged SMC proliferation, which likely is mediated by the Wnt-TCF-pathway.
Keywords: Extracellular Matrix, Smooth Muscle Cells, Proliferation, Arteries
INTRODUCTION
Extracellular matrix (ECM) molecules are highly effective and selective modulators of important cell functions such as migration and proliferation 1-4. The small leucine-rich repeat proteins (SLRP) found in the ECM are also potent regulators of cell phenotype 5. This growing family of SLRP’s is comprised of 5 classes defined by the number of leucine-rich repeats, the N-terminal composition, and the number of exons. Members of class I, biglycan and decorin, are among the best studied ECM molecules in fibrosis and cancer 5-10. We cloned podocan, a novel member of the SLRP family, which differed in all three classifying categories and, as a result, established a new (fifth) class of this protein family. We identified podocan by representational difference analysis of cDNA in HIV-1 transgenic and non-transgenic podocytes 11. Podocan mRNA and protein expression increases in sclerotic glomerular lesions of HIV-associated nephropathy (HIVAN) but is also present albeit at lower levels in normal heart, kidney and in smooth muscle cells (SMC) in vivo and in vitro 12. Human and murine podocan share a greater than 91% homology 11. Recently, podocan has also been shown by other investigators to be present in human aortic tissue 13.
Given the inhibitory effect of decorin on SMC proliferation and the capability of biglycan to enhance SMC proliferation, we hypothesized that podocan could also modulate SMC migration and proliferation 9, 14-17. Human atheroma has a varying content of fibrotic tissue depending on the prevailing driving factors of lesion formation such as hyperlipidemia, smoking, diabetes or mechanical injury post PCI 18, 19. The close regulation of SMC migration and proliferation within the intimal space is critical in maintaining a delicate balance between insufficient and excessive plaque repair. When SMC proliferation is too suppressed, the ensuing weakening of the fibrous cap can result in plaque vulnerability underlying acute coronary syndrome and when SMC proliferation is excessive, intimal hyperplasia can follow such as in restenosis post PCI 20, 21. Several important SMC growth-regulatory pathways and molecules have been shown to modulate arterial lesion formation – among them PDGF and TGF-beta 3, 22. Recently, an important developmental pathway - the Wnt-TCF-pathway - has been implicated in the regulation of SMC proliferation in vitro 23, 24 and also in vivo 25. Wnt activation via its cell surface receptors leads to an increase in non-phosphorylated beta-catenin (stable form) and a reduction in phosphorylated beta-catenin (form marked for degradation). Their ratio is used as a marker of Wnt-activation. Subsequent nuclear translocation of beta-catenin – a hallmark of complete Wnt-TCF-pathway activation - controls the transcription of multiple target genes affecting cell proliferation, migration and survival 23, 24.
To test the effect of podocan on SMC proliferation and arterial response to injury in vivo, we generated mice deficient in podocan and performed a femoral arterial denudating injury as previously described 26, 27. We also generated primary aortic SMC explant cultures with podocan−/− and WT genotypes to examine the effects of podocan deficiency on SMC migration and proliferation in vitro. In addition, we overexpressed the human form of podocan to assess the effect of increased amounts of podocan on human SMC. To further determine the relevance of podocan for human arterial lesion formation we also tested for podocan expression in different forms of human atherosclerosis. In all these conditions we concomitantly examined the Wnt-TCF-pathway.
METHODS
Generation of Podocan Deficient Mice
A podocan-targeting vector was constructed by inserting a neomycin cassette, which led to the deletion of exons III through VIII, abolishing podocan expression (see also Supplement section). After ES cell transfection, selection of positive ES cells and blastocyst injection, the resulting chimeric males were crossed with C57/BL6 female mice. Heterozygous offspring were bred to homozygosity. Genotyping was achieved by using podocan-specific primers in PCR. Mice were housed at the Center for Laboratory Animal Sciences at The Mount Sinai Medical Center, New York. Mice received standard rodent chow (Nutrition International) and tap water ad libitum. Procedures and animal care were approved by the Institutional Animal Care and Use Committee, and were in accordance with the “Guide for the Care and Use of Laboratory Animals’ (National Research Council. Washington, D.C.: National Academy Press 1996).
Endothelial Denudation Injury of Mouse Femoral Artery
Mice were anesthetized with intra-peritoneal pentobarbital sodium (40 mg/kg) (Nembutal®, Abbott Laboratories). Removal of the endothelium of the common femoral artery using a surgical microscope was achieved by 3 passages of a 0.25 mm angioplasty guide wire (Advanced Cardiovascular Systems) in 51 podocan−/− and WT mice. The protocol, as well as the degree of injury applied to the vessel wall has been standardized, validated, and described in detail in previous studies 26, 27.
Tissue Preparation, Histology and Immunostaining
Animals were sacrificed 1, 2, 4, and 6 weeks after arterial injury and perfusion-fixed with 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS) at 100 mm Hg for 10 minutes and their hindlimbs excised en bloc. Animals at the 4 and 6 week time points were injected with BRDU (Sigma-Aldrich) 24 hours prior to sacrifice. Specimens were fixed overnight in 4% PFA in PBS and decalcified in 10% formic acid. Two 2-mm thick cross sections were cut from each hindlimb at the level of the femoral injury and processed for paraffin embedding. Sequential sections (4μm thick) were stained with Masson’s Trichrome and hematoxylin-eosin. Immunohistochemistry was performed with polyclonal rabbit antibodies against murine and human podocan (generated in our lab, 1:45 and 1:25, respectively), von Willebrand Factor (Dako; 1:1000), smooth muscle alpha-actin (Sigma; 1:300), non-phospho beta-catenin (Cell Signaling; 1:150), anti-BRDU antibody (Accurate; 1:400) and Ki-67 (R&D Systems; 1:150). Slides were quenched with 3% H2O2, blocked with 1% BSA in PBS and incubated with the primary antibodies at 37°C for 2 hours. After washing in PBS, bound primary antibody was detected using an appropriate biotinylated secondary antibody for 15 minutes at 37°C. Sections were washed in PBS, reacted with horseradish peroxidase-conjugated streptavidin, developed with 3,3′-diaminobenzidine and counterstained with hematoxylin. Negative controls were prepared by substitution of primary antibody with the respective control IgG. Double labeling was performed using FITC- and Texas Red-conjugated secondary antibodies (Jackson Immuno Laboratories) with DAPI counterstaining.
Computer Assisted Morphometry
Investigators blinded to the study design performed the histomorphometric evaluation. A computer-assisted planimetry system was used (Image Pro Plus). Neointima formation was assessed by H&E and Masson’s trichrome staining. SMC density and proliferation (Ki-67 and BRDU labeling) were quantified as alpha-actin-positive cells per area and as percentage Ki-67/BRDU-positive cells from total cells with nuclear counterstaining. No significant inter- or intra-observer variations were noted.
Culture of Murine and Human SMC and Podocan Transfection
Aortic SMCs were prepared by the explant method from podocan−/− mice or WT littermates. Briefly, the aortas were freed of any connective tissue and adherent peri-vascular fat, the endothelial cell layer was removed, and the arteries were cut into approximately 3mm rectangular pieces. The pieces were placed in DMEM (Gibco) supplemented with 20%FBS, 100 U/ml penicillin, 100g/ml streptomycin and 0.25μg/ml amphotericin B in a humidified atmosphere of 5% CO2 and 95% air at 37°C. The SMC generated exhibited a typical “hill and valley” growth pattern and morphological examination and smooth muscle alpha-actin staining confirmed the cell type. Human primary aortic SMC were obtained commercially (Promo Cell) and seeded in 10 ml culture flasks. Medium was replaced every other day. All SMCs were serially passaged before reaching confluence, and all experiments were performed on SMCs from passages 2 to 4. Cells were washed three times with HBSS and rendered quiescent in serum free DMEM for 24 hours prior to experiments. Podocan transfection experiments were performed according to standard protocols. In brief, the expression vectors encoding the full-length mouse/human podocan protein (pCDNA3.1-m/hPodocan) and control vector (pCDNA3.1) were transfected into SMC using Fugene 6.0 (Roche). The cells were harvested at 48 h post transfection for evaluation of SMC proliferation and migration. Protein analyses confirmed the expression of podocan in podocan −/− cells transfected by pCDNA3.1-mPodocan and in human SMC transfected by pCDNA3.1-hPodocan.
Cell Proliferation Assay
To assess the proliferation of SMCs cells were trypsinized, washed 2 times with PBS and added to gelatin-coated 96 well plates at a density of 5×103 cells/well in DMEM containing either 10% FBS or recombinant PDGF (R&D Systems). After culture for 72 hours, cell number was assessed using the MTS assay (Promega). For human SMC treated with either podocan expressing vector or empty vector and for untreated cells a colorimetric BRDU-incorporation assay (Roche) was used.
Cell Migration Assay
The migration of SMCs was examined using a colorimetric cell migration assay (Chemicon) based on the Boyden chamber principle using inserts with a pore size of 8 μm. SMCs were trypsinized, washed 2 times with PBS, resuspended in 1% FBS in DMEM, and added to the top wells (2.5×104 cells/300 μL). DMEM with 10% FBS or recombinant mouse PDGF (R&D Systems) was added to the bottom chamber. After 6 hours at 37°C, non-migrating cells were scraped from the upper surface of the filter. Cells on the bottom surface were incubated with Cell Stain Solution, then subsequently extracted and detected by spectrophotometry (absorbance at 560 nm).
Wnt-TCF-Pathway Evaluation
For evaluation of Wnt-TCF-pathway related protein expression, SMC lysates were prepared for protein electrophoresis and Western blotting using RIPA lysis buffer (Santa Cruz Biotech.) and PARISTM Kit (Ambion). Imaging and analysis were performed by FluorchemTM 8800 system and AlphaEasy FC software (Alpha Innotech). Specific antibodies against phosphorylated and non-phosphorylated beta-catenin (Cell Signaling) were used for Western-blotting. To determine Wnt-TCF pathway activation by measuring beta-catenin/Tcf/Lef-1 transcriptional activity directly, we used a Luciferase-based transcriptional reporter assay. TOPflash/FOPflash plasmids (Upstate) were transfected into cultured SMCs. Cells were cotransfected with pRL-SV40 (Promega) as internal control. 48 h later, reporter luciferase activity was measured by dual luciferase reporter assay (Promega) and normalized to Renilla luciferase activity. TCF reporter luciferase activity was represented by the ratio of TOPflash and FOPflash luciferase activity. All in vitro experiments were performed in triplicates and repeated a minimum of three times. See also the Supplement section for a more detailled description of this assay.
Origin and Analysis of Human Arterial Specimens
Paraffin blocks of formalin-fixed atherosclerotic carotid plaque tissue were obtained from carotid endarterectomy specimens (n=7). Use of excess anonymous surgical pathology tissue was approved by the institutional review board. Percutaneous directional atherectomy was performed in patients presenting with stable angina attributed to the presence of stenotic primary atherosclerotic lesions or restenotic lesions after previous balloon angioplasty or atherectomy (2.2 to 20 months after the initial interventional procedure). Tissue samples were obtained by atherectomy from a total of 18 coronary target lesions, including 7 restenotic and 11 primary lesions (angiographic stenosis degree >75%) as shown in Table 1. The origin of these atherectomy samples was the left anterior descending artery in 12 cases, the right coronary artery in 5 cases, and the circumflex coronary artery in 1 case. Restenosis was defined according to previously reported clinical and angiographic criteria 28, 29. Informed consent for the analysis of tissue samples was obtained from all patients prior to revascularization. Immediately after percutaneous atherectomy, all specimens were fixed in 4% paraformaldehyde in PBS. Subsequently, specimens were processed for paraffin embedding. Sequential sections (4μm thick) were cut and stained with Masson’s Trichrome and hematoxylin-eosin. Immunohistochemistry was performed with polyclonal rabbit antibodies against human podocan (generated in our lab, 1:45), smooth muscle alpha-actin (Sigma; 1:300), and non-phospho beta-catenin (Cell Signaling; 1:150). Hematoxylin- and Masson’s Trichrome-stained sections allowed for the counting of cells in the intima; adjacent medial areas of the vessels were not analyzed. Assessment of cell density as well as expression of podocan and non-phospho beta-catenin was performed using a computer-assisted morphometry system as described above. Nuclei were counted per area and used to calculate the cell density per mm2, podocan expression was measured as percentage of intimal area covered by podocan staining and expression of nonphospho beta-catenin was measured as percentage of intimal cells with nuclear non-phospho beta-catenin labeling. Ten randomly selected intimal areas, each encompassing 0.04 mm2, were assessed per tissue sample as previously described 30.
Table 1.
Coronary Atherectomy Samples: Patient and Lesion Characteristics
| Lesion | Age | Gender | Stenosis % Pre/Post |
Lesion | Cells/mm2 | Time Interval (Months) |
|
|---|---|---|---|---|---|---|---|
| Primary coronary lesions | |||||||
| 1 | 42 | M | 90 | 40 | LAD | 298 | |
| 2 | 59 | M | 90 | 0 | LAD | 30 | |
| 3 | 50 | M | 95 | 30 | RCA | 21 | |
| 4 | 48 | M | 99 | 20 | LAD | 73 | |
| 5 | 70 | M | 90 | 30 | LAD | 189 | |
| 6 | 58 | F | 85 | 10 | RCA | 94 | |
| 7 | 48 | M | 90 | 30 | LAD | 23 | |
| 8 | 44 | M | 80 | 40 | LAD | 233 | |
| 9 | 60 | M | 99 | 20 | RCA | 218 | |
| 10 | 61 | M | 90 | 10 | RCA | 460 | |
| 11 | 63 | M | 80 | 20 | LAD | 291 | |
| Restenotic coronary lesions | |||||||
| 12 | 77 | M | 99 | 25 | LAD | 491 | 2.6 (PTCA) |
| 13 | 68 | M | 80 | 20 | LAD | 1167 | 3.7 (PTCA) |
| 14 | 67 | M | 90 | 20 | LAD | 583 | 3.8 (PTCA) |
| 15 | 69 | F | 90 | 20 | RCx | 627 | 2.2 (PTCA) |
| 16 | 60 | M | 80 | 10 | LAD | 819 | 3.2 (Ath) |
| 17 | 61 | M | 80 | 25 | RCA | 404 | 20.0 (PTCA) |
| 18 | 53 | M | 80 | 30 | LAD | 338 | 11.0 (PTCA) |
Statistical Analysis
SPSS/PC+ software was used for data analysis. Data are shown as mean±SEM (in vivo data) and as mean±SD (in vitro data). Two-way ANOVA testing was used to evaluate neointima area, reendothelialization, SMC-density, and expression of Ki-67/BRDU with podocan−/− and WT genotype. After testing for normal distribution and equality of variances with Levene’s F-test, the independent sample t-test was used to compare intimal SMC density (cells per mm2), podocan expression (percentage of intimal area covered by podocan staining) and expression of the Wnt-TCF pathway marker non-phospho beta-catenin in SMC (percentage of intimal SMCs labeled positive) in primary versus restenotic coronary lesions. Absorption at OD588 (migration assay) and OD490 (proliferation assay) were also compared using the independent sample t-test. Probability values were two-tailed and corrected for ties. P values <0.05 were considered significant.
RESULTS
Expression of Podocan in Injured Mouse Femoral Artery
In non-injured femoral arteries of WT animals podocan expression could not be detected by immunostaining (Fig.1 a and d). In contrast, podocan was found consistently in arteries of WT mice after injury. Podocan deposition was seen surrounding medial and neointimal SMCs (Fig.1 b and e). Injured arteries of podocan−/− mice were completely devoid of podocan, as expected, confirming the specificity of the podocan antibody (Fig.1 c and f). The complete time course analysis of podocan expression in WT arteries using antibodies for podocan and alpha-actin showed barely detectable podocan staining at 1 week in alpha-actin positive media (Fig.1 g and h). At 2 weeks post injury a strong, albeit patchy, podocan expression emerged in the media alongside with strong alpha-actin expression in the media (Fig.1 i and j). At 4 weeks most neointimal cells expressed alpha-actin and were surrounded by podocan staining largely of the ECM (Fig.1 k and l).
Figure 1.

Effect of injury and podocan genotype on arterial podocan expression. (a and d): Podocan is not detected in non-injured wild type femoral arteries, x200, x1000; scale bar=50 μm. (b and e): After injury brown labeling (arrows) for podocan is clearly seen in media (me) and neointima (ni) of wild type arteries at 4 weeks; x200, x1000; scale bar=50 μm. (c and f): After injury an artery with podocan−/− genotype exhibits a large neointima (ni) and shows, as expected, no podocan labeling serving as negative control for podocan immunostaining; x200, x1000; scale bar=50 μm. Time course of podocan expression post injury in wild type artery: 1 week (g-h): At 1 week after injury podocan is hardly detectable in the media (me) while alpha-actin is expressed strongly. x200; scale bar=50 μm. 2 weeks (i-j): Much stronger, albeit patchy, medial (me) podocan expression appears (arrows). Of note, podocan is also seen in early neointima (ni), x200; scale bar=50 μm. 4 weeks (k-l): Podocan expression (arrows) remains strong in neointima and media in close spatial association with alpha-actin signals, x200; scale bar=50 μm.
Effect of Podocan on Arterial Response to Injury
We examined the effect of podocan genotype on arterial response to injury in WT (n=27) and podocan−/− mice (n=28). At 1 and 2 weeks, no significant difference in neointima size was found between the groups (1week: 2.0±0.9 vs. 1.8±0.8 ×10−3mm2, P=NS; 2weeks: 3.8±1.0 vs. 2.9±0.9 ×10−3mm2, P=NS) (Fig.2a-f and m). At 4 weeks, however, the neointima area was significantly greater with podocan−/− genotype compared to WT (11.6±1.8 vs. 4.4±1.3 ×10−3mm2, P<0.05) (Fig.2c, f, m). The neointima to media ratio was also increased with podocan−/− genotype at 4 weeks (3.04±0.44 vs. 1.14±0.15; P<0.01). SMC-density did not show a significant difference between both groups early post injury (1week: 2078±978 vs. 1958±934 ×103 cells/mm2, P=NS; 2weeks: 8822±2078 vs. 7823±1934 ×103 cells/mm2, P=NS) (Fig.2g-l and n). At 4 weeks, however, SMC-density of neointima was significantly increased with podocan−/− genotype (9989±2778 vs. 5813±2012 ×103 cells/mm2, P<0.05) (Fig.2n). At 1 week, 4.4±1.0% of cells expressed the proliferation marker Ki-67 with podocan−/− and 4.1±0.8% with WT genotype (P=NS) (Fig.3). At 2 weeks, Ki-67 expression decreased in both groups (2.3±1.1% vs. 2.2±0.9%; P=NS). However, with podocan−/− genotype Ki-67 expression increased again at 4 weeks (7.3±1.9% vs. 2.4±1.0%; P<0.05) (Fig.3). Reendothelialization did not differ between the groups (1week: 27±2% vs. 29±4%, P=NS; 2weeks: 57±5% vs. 54±4%, P=NS; 4weeks: 79±4% vs. 84±4%, P=NS). Of note, neointima area (12.8±1.7 vs. 4.6±1.4 ×10−3mm2, P<0.05) and expression of the proliferation marker Ki-67 (6.0±1.3% vs. 0.0±0.0%, P<0.05) remained significantly increased with podocan−/− genotype even 6 weeks after injury (Figure 4a-f and j). Measuring proliferation by BRDU-incorporation confirmed the increased SMC proliferation found with podocan−/− genotype (18±3% vs. 2±2%, P<0.05) (Fig.4g to k).
Figure 2.

Time course of arterial response to injury with wild type and podocan−/− genotype. Combined Masson Elastin staining: WT genotype (a-c), podocan−/− genotype (d-f): 1 week (a and d): Cells adhere along the arterial surface on the luminal side of the media (me) and an adventitial cellular infiltrate forms in both groups in a similar fashion; x200; scale bar=50 μm. 2 weeks (b and e): A comparably sized typical early neointimal lesion (ni) with densely packed cells has formed in both groups; x200; scale bar=50 μm. 4 weeks (c and f): A moderately sized arterial lesion (ni) has formed with WT genotype; in contrast, with podocan−/− genotype the neointima shows a strongly increased size; x200; scale bar=50 μm. Bar graph (m): Comparison of neointima formation with WT and podocan−/− genotype: neointima area in ×10−2 mm2 (independent sample t-test). Smooth Muscle Alpha-Actin: WT genotype (g-i), podocan−/−genotype (j-l): 1 week (g and j): Alpha-actin expression (brown labeling) is predominantly seen in the media (me) in both groups, no neointima has formed yet; x200. 2 weeks (h and k): Nascent neointimal (ni) alpha-actin expression and a trend towards higher SMC numbers with the podocan−/− genotype can be seen; x200; scale bar=50 μm. 4 weeks (i and l): A steep increase in the numbers of alpha-actin positive cells can be observed in neointima (ni) with podocan−/−genotype; x200; scale bar=50 μm. Bar graph (n): Comparison of neointimal SMC density with WT and podocan−/− genotype: cell density in ×103 mm2 (independent sample t-test).
Figure 3.

Arterial proliferation with wild type and podocan−/− genotype. Ki-67 (FITC) and smooth muscle alpha-actin (Texas Red) double labeling: WT genotype (a-f): podocan−/−genotype (g-l): 1 week (a and g): Early after injury Ki-67 positive (green) and alpha-actin positive (red) SMC (arrow) are seen in the media (me) in both groups; x200; scale bar=50 μm. (d and j represent matching IgG-isotype control stainings). 2 weeks (b and h): At this time only few Ki-67 signals (arrows) are seen in both groups consistent with a gradual decline in proliferation after the first week; x200; scale bar=50 μm. (e and k negative controls). 4 weeks (c and i): An unusually late rise in proliferation of SMC (red alpha-actin labeling) is detected by nuclear Ki-67 (green) labeling (arrows) with podocan−/− genotype; x200; scale bar=50 μm. (f and l negative controls). Bar graph (m): Comparison of Ki-67 expression with WT and podocan−/− genotype after arterial injury: expression in % cells (independent sample t-test).
Figure 4.

Late arterial proliferation with wild type and podocan−/− genotype. WT genotype (a, d, g): podocan−/− genotype (b, e, h): H&E staining (a-c): Compared with WT (a) the strong increase in neointima (ni) with podocan−/− genotype (b) persists even at 6 weeks. Non-injured artery (c) of podocan−/− mouse serves as negative control for proliferation labeling; x200; scale bar=50 μm. Ki-67 and smooth muscle alpha-actin double labeling (d-f): At 6 weeks no green (FITC) Ki-67 labeling is seen in neointima (ni) and media (me) with WT genotype (d). With podocan−/− genotype, however, green nuclear Ki-67 labeling (arrows) is seen in neointima (ni) even 6 weeks after injury (e). Non-injured podocan−/− contra-lateral artery does not show any ki-67 signals (f). x200; scale bar=50 μm. BRDU-Labeling (g-i and k): In smaller WT neointima (ni) BRDU-labeling is absent at 6 weeks after injury (g). In contrast, multiple nuclei with brown labeling (arrows) indicating BRDU-incorporation are seen in hypercellular neointima with podocan−/− genotype (h); x200; scale bar=50 μm. Brown BRDU labeling (arrows) is also seen in bone marrow (bm) and serves as positive control (i). Brown labeling is absent with isotype-control staining in bone marrow; x400; scale bar=50 μm (k). Bar graph (j): Comparison of Ki-67 and BRDU expression with WT and podocan−/− genotype 6 weeks after arterial injury: expression in % cells (independent sample t-test).
Effect of Podocan on Migration and Proliferation in Mouse and Human SMC
In WT aortic explants, there was no cellular outgrowth at 3 days in all 8 samples (Fig.5a). In contrast, at the edge of podocan−/− aortic explants, SMC outgrowth was visible in 6 out of 8 samples at 3 days indicating early SMC outgrowth (Fig.5b). Subsequently, we compared migration of the cultured SMCs and found that podocan−/− SMCs grown in 10% FBS migrated significantly faster than WT cells (0.73±0.06 vs. 0.55±0.03, P<0.05) (Fig.5c). Podocan−/− SMCs also grew at a significantly greater rate than WT cells when cultured in 10% FBS or in response to recombinant PDGF (10ng/ml) (10% FBS: 0.76±0.03 vs. 0.69±0.03, P<0.05; 10ng/ml PDGF: 1.01±0.03 vs. 0.89±0.03, P<0.05) as measured by the MTS assay (Fig.5d). In an attempt to restore the WT SMC phenotype we transfected podo−/− SMC with podocan expressing vector. Podocan synthesis was induced in podocan−/− SMCs treated with podocan expressing vector as confirmed by Western Blot (data not shown). Proliferation in both 10% FBS (0.35±0.01 vs. 0.40±0.01, P<0.05) and with PDGF stimulation (0.49±0.02 vs. 0.65±0.02, P<0.05) was significantly reduced as compared to empty vector treatment and approached that seen with WT cells (Fig.5e). Moreover, in human SMC, treated with human podocan expressing vector, podocan was highly enriched compared to vector control and untreated SMC as assessed by Western Blot (Fig.5i). Podocan overexpression resulted in a 29% reduction of SMC migration (0.40±0.08 vs. 0.56±0.09, P<0.05)(Fig.5j). Using a BRDU incorporating assay we also found a time-dependent inhibition of SMC proliferation up to 32% (at 24h) with podocan overexpression (0.15±0.01 vs. 0.22±0.01, P<0.05 at 24 hours)(Fig. 5k). All quantitative data in this section represent Units of optical density resulting from spectrophotometric measurements.
Figure 5.

Effect of WT and podocan−/− genotype on murine aortic SMC: Aortic explant culture (a and b): (a) The edge of WT aortic explant shows no SMC outgrowth at day 3; x400; scale bar=50 μm. (b) In contrast, numerous SMCs are seen at the edge of a podocan−/− aortic explant at the same time point; x400; scale bar=50 μm. SMC migration and proliferation (c-e): (c) SMC migration is increased with podocan −/− genotype compared with WT in a colorimetric test based on the Boyden chamber principle (independent sample t-test). (d) Podocan−/− SMCs also grow faster as measured by the MTS assay (independent sample t-test). (e) Podocan−/− SMC transfected with podocan vector slow their growth to WT-level (independent sample t-test). Wnt-TCF pathway activation (f-h): (f) In SMC with podocan−/− genotype the ratio of phosphorylated to non-phosphorylated beta-catenin is reversed as seen by Western blot. (g) Transcriptional activity of Wnt-TCF pathway measured directly by TOPflash/FOPflash assay is also increased with podocan−/− genotype. (h) Podocan−/− SMCs treated with small inhibitory RNAs to beta-catenin show inhibition of growth. Effect of podocan overexpression on human aortic SMC: Western Blot (i): Western Blot confirms overexpression of the human form of podocan. Podocan in control and empty vector treated SMC is below the detection threshold. Migration (j): SMC migration is reduced by 29% with podocan overexpression. Proliferation (k): SMC proliferation is reduced by 32% with podocan overexpression. Wnt-TCF pathway activation (l): SMC overexpressing podocan show an increase in phosphorylated-beta catenin on Western Blot compared to non-treated or empty vector treated SMC indicating Wnt-TCF pathway suppression.
Effect of Podocan on the Wnt-TCF-Pathway in Mouse and Human SMC
We found a reduction in phosphorylated and an increase in non-phosphorylated beta-catenin in podocan−/− SMC compared with WT indicative of Wnt-pathway activation (Fig.5f). To confirm the increase in transcriptional Wnt-activity in podocan−/− SMCs, we performed TOPflash/FOPflash reporter assays. The TOPflash/FOPflash assays showed greater than 2-fold enhancement in nuclear beta-catenin/Tcf/Lef-1 transcriptional activity in podocan−/− SMCs confirming activation of Wnt-signaling (Fig.5g). When we treated podocan−/− SMCs with beta-catenin small inhibitory (si)RNA, we observed a significant suppression of non-phosphorylated beta-catenin compared to control siRNA treatment (data not shown). Of note, beta-catenin RNA silencing resulted in inhibition of SMC proliferation comparable to the inhibition achieved by WT podocan gene transfection into podocan−/− SMC (Fig.5h). Of note, the in vivo expression of non-phosphorylated (stable) beta-catenin was also strongly increased in podocan−/− neointima compared to WT (2weeks: 10±3% vs. 4±2%, P>0.05; 4weeks: 38±8% vs. 8±3%, P<0.05) (Fig.6a-f and l). Importantly podocan−/− SMC in the neointima displayed nuclear non-phospho beta-catenin staining indicative of nuclear beta-catenin translocation, a hallmark of Wnt-activation (Fig. 6g-i). Conversely, enriching the human form of podocan in human SMC by treatment with podocan expressing vector resulted in a significant increase in phosphorylated beta-catenin over non-phosphorylated beta-catenin seen by Western Blot (Fig.5l) consistent with Wnt-TCF pathway suppression.
Figure 6.

Effect of wild type and podocan−/− genotype on Wnt-pathway after arterial injury: Alpha-actin (Texas-Red) and non-phospho beta-catenin (FITC) double-labeling: WT genotype (a and d), podocan−/− genotype (b-c and e-f, and g-k): Low power magnification (a–f): An antibody specific for the non-phosphorylated form of beta-catenin gives much stronger green (FITC) signals in neointima (ni) with podocan −/− genotype (b and e) compared with WT (a and d); x200; scale bar=50 μm. Matching isotype controls are shown for podocan−/− neointima (c and f). High power magnification (g–k): Comparing single DAPI (g), single non-phospho-beta-catenin labeling (h) and both combined (i) under high power magnification demonstrates clearly the nuclear location of beta-catenin signals in podocan−/− neointima indicating beta-catenin nuclear translocation – a hallmark of true Wnt-pathway activation; x1000; scale bar=50 μm. These cells also stain positive for alpha-actin (j) identifying them as SMC and show co-localization with non-phospho beta-catenin signals (k). Bar graph (l): Comparison of non-phospho beta-catenin expression in neointima with WT and podocan −/− genotype after arterial injury: expression in % cells (independent sample t-test).
Podocan and Wnt-TCF-Pathway in Human Atheroma
In atherectomy samples from patients with primary stable atherosclerosis (n=11) podocan expression was abundant (Fig.7). In restenotic lesions podocan expression was significantly decreased with 8±2% of intimal area compared with 30±4% in primary coronary lesions (P<0.05). In contrast, intimal cell density was significantly increased in restenotic compared to primary coronary lesions with 632±107 versus 195±40 cells per mm2 (P<0.05). Of note, the expression of non-phospho beta-catenin was strongly increased in restenotic lesions compared to primary lesions (22±5% vs. 5±1%, P<0.05). Importantly, nuclear staining of non-phospho beta-catenin indicative of nuclear translocation was observed in restenotic lesions (Fig.8). Immunofluorescence labeling showed a co-localization of non-phospho beta-catenin and smooth-muscle alpha-actin in hyperplastic areas of restenotic lesions (Fig.8). In all coronary lesions we observed an inverse correlation between the extent of podocan deposition and nonphospho beta-catenin expression (r=−0.78, P<0.05) and a strong positive correlation between the expression of non-phospho beta-catenin and intimal cell density (r=0.94, P<0.05). Of note, in both lesion types staining with an isotype control antibody that matches the podocan antibody did not show any staining excluding non-specific labeling or autofluorescence artifact (Fig.7).
Figure 7.

Expression of Podocan in Human Atheroma: Primary Carotid Atheroma (a to d), Primary Coronary Lesion (e-f and i-j, and m-n, q-r, u-v), Restenotic Coronary Lesion (g-h and kl, and o-p, s-t, w-x): Podocan immunostaining (a–d): An antibody specific for the human form of podocan gives strong brown labeling in the intima of carotid atheroma (a and c); x100; scale bar=50 μm. Matching isotype staining on adjacent section shows no labeling (b and d). Combined Masson Elastin (CME) staining (e–g): Comparing the histo-architecture of primary and restenotic coronary lesions shows distinct differences. Spindle shaped SMC are surrounded by large spaces of ECM at a rather low cell density in primary lesions (e). In restenotic tissue abundant numbers of SMC are tightly clustered and surrounded by a comparatively smaller ECM space (g); x50, x100, x200; scale bar=50 μm. Two Versions of Podocan and Smooth Muscle Cell Double Labeling (f-v): Smooth muscle alpha-actin (FITC) and podocan (Texas-Red) double-labeling (f, h): Low power magnification images reveal the inverse relation between the degree of intimal podocan labeling (red) and the density of intimal smooth muscle cells (green) in primary (f) compared with restenotic (h) coronary plaque tissue. Smooth muscle alpha-actin (Texas-Red) and podocan (FITC) double-labeling (j, m, n and r, u, v): With reversed double labeling higher power magnification images confirm that large ECM spaces in primary lesions surrounding the red-labeled SMC are enriched with podocan as shown by extensive green (arrows) labeling (j, m, n and r, u, v). In contrast, green podocan labeling (arrows) in restenotic tissue covers a much smaller area and is restricted to the immediate vicinity of red-labeled SMC (l, o, p and t, w, x); x200, x1000; scale bar=50 μm. Corresponding intimal locations in adjacent serial sections are also shown by light microscopy and Combined Masson Elastin (CME) staining (i, q and k, s); x200, x1000; scale bar=50 μm. Of note, in both lesion types staining with an isotype control antibody that matches the podocan antibody does not show any green signals (z, I-IV). Bar graph (y): Comparison of podocan expression (% area) and SMC density (cells per mm2) in primary and restenotic coronary lesions: (independent sample t-test).
Figure 8.
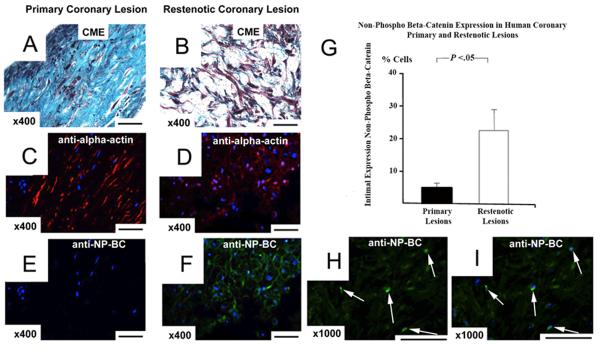
Activation of Wnt-TCF Pathway in Human Atheroma: Primary Coronary Lesion (a, c, e), Restenotic Coronary Lesion (b, d, f, h, i): Alpha-actin (Texas-Red) and non-phospho beta-catenin (FITC) double-labeling: An antibody specific for the non-phosphorylated form of beta-catenin gives strong green (FITC) signals in intimal cells of a restenotic coronary lesion (f), whereas no signals are seen in the intima of a primary lesion (e); x400; scale bar=50 μm. These cells also have typical morphology of SMC (b) and stain positive for alpha-actin (d); x400; scale bar=50 μm. High power magnification (h–i): Comparing single non-phospho-beta-catenin labeling (h) and combined labeling with DAPI (i) demonstrates clearly the nuclear location of beta-catenin signals in restenotic intima indicating beta-catenin nuclear translocation – a hallmark of true Wnt-TCF pathway activation; x1000; scale bar=50 μm. Bar graph (g): Comparison of non-phospho beta-catenin expression in the intima of primary and restenotic coronary lesions: expression in %cells (independent sample t-test).
DISCUSSION
Our results suggest that the novel SLRP podocan is a key regulator of the SMC response after arterial injury. Lack of podocan expression with podocan−/− genotype resulted in late and prolonged SMC proliferation after arterial injury yielding exuberant arterial lesion formation. Arterial response to injury critically involves the migration and proliferation of SMC from the media into the intimal space with subsequent ECM synthesis and remodeling events 31-33. We showed in our study that podocan is selectively enriched in the ECM of arteries post injury in vivo and we demonstrated in vitro, that podocan is capable of inhibiting SMC proliferation and migration. Since the inception of mechanical treatments for human atherosclerosis beginning with balloon angioplasty and later forms of percutaneous coronary interventions such as stenting the control of the SMC response has remained the Achilles heel of this approach. While stents have vastly improved upon the recoil and constrictive remodeling component of restenosis the underlying problem of accelerated intimal SMC growth has remained. A myriad of strategies and an enormous research effort over many years to control the migratory and proliferative response of SMC post PCI have not resulted in a perfect solution for this problem yet. The current approach of delivering stents releasing non-specific agents promoting cell death and/or inhibition of proliferation has evolved in experimental models and multiple clinical trials and has been successful at lowering the need for recurrent vascular interventions 34. However, this success comes at the expense of delaying vascular healing, whose ultimate long-term clinical impact is still being evaluated and debated 34-36. Irrespective of this ongoing debate it remains obvious from a vascular biology point of view that the current agents released by stents to control the SMC proliferative and migratory response are by no means physiologic inhibitors of this process. Rather, they are pretty blunt instruments imparting short- and long-term negative effects on the healing arterial wall such as delayed re-endothelialization, increased inflammation and enhanced thrombogenicity 37-39.
The possible role of podocan as physiologic inhibitor of the SMC migratory and proliferative response is suggested by several observations. Podocan is not expressed constitutively at high levels by vascular SMC at baseline. In the absence of injury podocan expression could not be detected by imunohistochemistry. Only after arterial injury podocan was gradually expressed at increasing and robust levels in the media and neointima between 2 and 4 weeks after injury. Of note, in podocan−/− mice arterial lesion formation was affected specifically between 2 and 4 weeks after injury compared with WT. This is precisely the time when ECM synthesis and podocan deposition typically takes place in the neointima. Occurring after the initial stages of cell adhesion and cell recruitment, this phase of ECM build up and remodeling has been described by several investigators studying different models of arterial injury 33, 40, 41. Consistent with the postulated inhibitory regulatory function of podocan, we found an increase in proliferation in neointima devoid of podocan in animals with podocan−/− genotype as late as 4 weeks after injury. This finding is very unusual because the natural history of arterial wall cell proliferation in most models peaks during the first two weeks and tapers off at later time points 33, 41. In addition to the late increase in proliferation, we also found a late increase in SMC-density at 4 weeks in the podocan−/− neointima. Previous studies reported that by 4 weeks SMC density usually declines due to a decreased rate of SMC proliferation and ongoing ECM synthesis 33, 41. Even as late as 6 weeks after arterial injury robust SMC proliferation was still evident in the neointima of podocan−/− mice.
To extend these in vivo observations and to better define the podocan genotype effect, we explanted SMCs from podocan−/− and WT aortas 42. The podocan genotype determined the rate of migration and proliferation of the cultured SMCs with strongly enhanced migration and proliferation of SMC lacking podocan expression. Transfection of podocan−/− SMC with WT podocan decreased proliferation to WT levels, essentially “normalizing” the podocan−/−phenotype and indicating a specific podocan effect. To test if podocan has a true inhibitory effect also on podocan competent WT cells and to assess the function of the human isoform of podocan we overexpressed human podocan in human SMC. Consistent with our observations in murine SMC, transfection of SMC with the human form of podocan significantly reduced migration and proliferation.
To define the possible mechanism through which podocan exerts its effect on SMC we probed for changes in the Wnt-TCF pathway, which has a central role in controlling proliferation and migration of cells 23, 43, 44. Of note, we detected significant and corresponding alterations in the Wnt-TCF pathway in each of these two different experimental settings. Cultured murine SMCs with podocan−/− genotype showed increased Wnt-TCF pathway activation compared to WT SMC. Conversely, human podocan competent SMC’s overexpressing podocan showed a significantly decreased Wnt-TCF pathway activation. These corresponding changes were verified using two independent and established methods of measuring Wnt-TCF-pathway activation (the relevant methodology is further outlined in the supplemental method section). Further evidence for an important role of the Wnt-TCF pathway in mediating podocan-related effects on SMC also stems from the observation, that beta-catenin siRNA treatment was capable of normalizing increased podocan−/− SMC proliferation in a similar fashion as the restoration of podocan expression with podocan transfection did.
Going back to the in vivo physiology of the arterial response to injury we wanted to test if matching alterations in the Wnt-TCF pathway could also be found in the neointima with podocan−/− and WT genotype. Interestingly, we observed a strong increase in the expression of the non-phosphorylated form of beta-catenin in the neointima with podocan−/− genotype. Podocan−/− neointimal SMC exhibited a nuclear staining pattern suggestive of nuclear translocation of beta-catenin and hence indicative of Wnt-TCF-pathway activation. The peak of this Wnt-TCF-pathway activation in neointimal SMC coincided precisely with the late rise in SMC proliferation in podocan−/− neointima. As final step we set out to determine the relevance of podocan and its effects on the Wnt-TCF pathway for the patho-physiology of intimal hyperplasia in humans.
To this end we examined podocan expression in human coronary restenosis and compared it with podocan expression in primary coronary lesions along with the same marker of Wnt-TCF-pathway activation, whom we found elevated in podocan−/− neointima. Podocan deposition in restenotic coronary lesions was significantly decreased compared with primary lesions whereas the expression of non-phospho beta-catenin was significantly increased in human coronary restenotic tissue yielding a significant inverse correlation. This finding is intriguing since human restenotic tissue is characterized by some of the same features observed in podocan−/− neointima. A steep increase in cell density, increased migration and increased proliferative events 45-48, all features, which are typical of tissues with elevated levels of Wnt-TCF pathway activation as has been extensively described in cancer literature 49. This first report of the inverse relationship between podocan expression and the expression of Wnt-TCF pathway related molecules in human vascular lesions provides further evidence for a possible role of podocan as physiologic inhibitor of the SMC migratory and proliferative response in the arterial response to PCI in patients. Its effects on SMC and its ability to modulate - at least in part - the Wnt-TCF-pathway renders podocan a novel therapeutic target for better controlling arterial repair 50, 51. Further experimental work is needed to delineate the molecular interactions of podocan with Wnt-TCF-pathway related molecules and its effects on other cell types.
Supplementary Material
SMCs critically influence the clinical course of vascular disease. The close regulation of SMC migration and proliferation within the intimal space is critical in maintaining a delicate balance between insufficient and excessive atherosclerotic plaque repair. When SMC proliferation is too suppressed, the ensuing weakening of the fibrous cap can result in plaque vulnerability underlying acute coronary syndrome and when SMC proliferation is excessive, intimal hyperplasia can follow such as in restenosis post PCI. While stents have vastly improved upon the recoil and constrictive remodeling component of restenosis the problem of accelerated intimal SMC growth has remained. The current approach of delivering stents releasing non-specific agents promoting cell death and/or inhibition of proliferation has been successful at lowering the need for recurrent vascular interventions. However, this success comes at the expense of delaying vascular healing, whose ultimate long-term clinical impact is still being evaluated. Short-and long-term negative effects on the healing arterial wall such as delayed reendothelialization, increased inflammation and enhanced thrombogenicity are undisputed. These side effects of DES are being masked by prolonged and aggressive anti-platelet therapy, which is exposing patients – especially the elderly – to increased bleeding risks, complicates clinical decision making through fear of too early treatment cessation, demands rigorous patient compliance and is costly. These issues are not trivial in daily clinical practice.. The possible role of podocan as novel selective inhibitor of the SMC response and the Wnt-TCF pathway opens a door to modulate vascular SMCs in a smarter and more physiologic way.
Acknowledgments
We thank David J. Schneider, MD, for his expert review of the manuscript and we thank Renata Hutter, MA, for expert help in data acquisition and analysis.
Funding Sources: This work was supported in part by grant DK 56492 (to PEK) and by NIH-training grant 5 T32 HL 7824-13 (to RH).
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Daley WP, Peters SB, Larsen M. Extracellular matrix dynamics in development and regenerative medicine. J Cell Sci. 2008;121:255–264. doi: 10.1242/jcs.006064. [DOI] [PubMed] [Google Scholar]
- 2.Geiger B, Bershadsky A, Pankov R, Yamada KM. Transmembrane crosstalk between the extracellular matrix--cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- 3.ten Dijke P, Arthur HM. Extracellular control of tgfbeta signalling in vascular development and disease. Nat Rev Mol Cell Biol. 2007;8:857–869. doi: 10.1038/nrm2262. [DOI] [PubMed] [Google Scholar]
- 4.Raines EW. The extracellular matrix can regulate vascular cell migration, proliferation, and survival: Relationships to vascular disease. Int J Exp Pathol. 2000;81:173–182. doi: 10.1046/j.1365-2613.2000.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iozzo RV. The biology of the small leucine-rich proteoglycans. Functional network of interactive proteins. J Biol Chem. 1999;274:18843–18846. doi: 10.1074/jbc.274.27.18843. [DOI] [PubMed] [Google Scholar]
- 6.Ameye L, Young MF. Mice deficient in small leucine-rich proteoglycans: Novel in vivo models for osteoporosis, osteoarthritis, ehlers-danlos syndrome, muscular dystrophy, and corneal diseases. Glycobiology. 2002;12:107R–116R. doi: 10.1093/glycob/cwf065. [DOI] [PubMed] [Google Scholar]
- 7.Bi Y, Nielsen KL, Kilts TM, Yoon A, M AK, Wimer HF, Greenfield EM, Heegaard AM, Young MF. Biglycan deficiency increases osteoclast differentiation and activity due to defective osteoblasts. Bone. 2006;38:778–786. doi: 10.1016/j.bone.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Kolb M, Margetts PJ, Sime PJ, Gauldie J. Proteoglycans decorin and biglycan differentially modulate tgf-beta-mediated fibrotic responses in the lung. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1327–1334. doi: 10.1152/ajplung.2001.280.6.L1327. [DOI] [PubMed] [Google Scholar]
- 9.Nili N, Cheema AN, Giordano FJ, Barolet AW, Babaei S, Hickey R, Eskandarian MR, Smeets M, Butany J, Pasterkamp G, Strauss BH. Decorin inhibition of pdgf-stimulated vascular smooth muscle cell function: Potential mechanism for inhibition of intimal hyperplasia after balloon angioplasty. Am J Pathol. 2003;163:869–878. doi: 10.1016/S0002-9440(10)63447-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schaefer L, Beck KF, Raslik I, Walpen S, Mihalik D, Micegova M, Macakova K, Schonherr E, Seidler DG, Varga G, Schaefer RM, Kresse H, Pfeilschifter J. Biglycan, a nitric oxide-regulated gene, affects adhesion, growth, and survival of mesangial cells. J Biol Chem. 2003;278:26227–26237. doi: 10.1074/jbc.M210574200. [DOI] [PubMed] [Google Scholar]
- 11.Ross MD, Bruggeman LA, Hanss B, Sunamoto M, Marras D, Klotman ME, Klotman PE. Podocan, a novel small leucine-rich repeat protein expressed in the sclerotic glomerular lesion of experimental hiv-associated nephropathy. J Biol Chem. 2003;278:33248–33255. doi: 10.1074/jbc.M301299200. [DOI] [PubMed] [Google Scholar]
- 12.Shimizu-Hirota R, Sasamura H, Kuroda M, Kobayashi E, Saruta T. Functional characterization of podocan, a member of a new class in the small leucine-rich repeat protein family. FEBS Lett. 2004;563:69–74. doi: 10.1016/S0014-5793(04)00250-9. [DOI] [PubMed] [Google Scholar]
- 13.Didangelos A, Yin X, Mandal K, Baumert M, Jahangiri M, Mayr M. Proteomics characterization of extracellular space components in the human aorta. Mol Cell Proteomics. 2010;9:2048–2062. doi: 10.1074/mcp.M110.001693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kunjathoor VV, Chiu DS, O’Brien KD, LeBoeuf RC. Accumulation of biglycan and perlecan, but not versican, in lesions of murine models of atherosclerosis. Arterioscler Thromb Vasc Biol. 2002;22:462–468. doi: 10.1161/hq0302.105378. [DOI] [PubMed] [Google Scholar]
- 15.O’Brien KD, Olin KL, Alpers CE, Chiu W, Ferguson M, Hudkins K, Wight TN, Chait A. Comparison of apolipoprotein and proteoglycan deposits in human coronary atherosclerotic plaques: Colocalization of biglycan with apolipoproteins. Circulation. 1998;98:519–527. doi: 10.1161/01.cir.98.6.519. [DOI] [PubMed] [Google Scholar]
- 16.Shimizu-Hirota R, Sasamura H, Kuroda M, Kobayashi E, Hayashi M, Saruta T. Extracellular matrix glycoprotein biglycan enhances vascular smooth muscle cell proliferation and migration. Circ Res. 2004;94:1067–1074. doi: 10.1161/01.RES.0000126049.79800.CA. [DOI] [PubMed] [Google Scholar]
- 17.Yamakawa T, Bai HZ, Masuda J, Sawa Y, Shirakura R, Ogata J, Matsuda H. Differential expression of proteoglycans biglycan and decorin during neointima formation after stent implantation in normal and atherosclerotic rabbit aortas. Atherosclerosis. 2000;152:287–297. doi: 10.1016/s0021-9150(99)00475-x. [DOI] [PubMed] [Google Scholar]
- 18.Fuster V, Moreno PR, Fayad ZA, Corti R, Badimon JJ. Atherothrombosis and high-risk plaque: Part i: Evolving concepts. J Am Coll Cardiol. 2005;46:937–954. doi: 10.1016/j.jacc.2005.03.074. [DOI] [PubMed] [Google Scholar]
- 19.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 20.Hultgardh-Nilsson A, Durbeej M. Role of the extracellular matrix and its receptors in smooth muscle cell function: Implications in vascular development and disease. Curr Opin Lipidol. 2007;18:540–545. doi: 10.1097/MOL.0b013e3282ef77e9. [DOI] [PubMed] [Google Scholar]
- 21.Shah PK. Molecular mechanisms of plaque instability. Curr Opin Lipidol. 2007;18:492–499. doi: 10.1097/MOL.0b013e3282efa326. [DOI] [PubMed] [Google Scholar]
- 22.Millette E, Rauch BH, Kenagy RD, Daum G, Clowes AW. Platelet-derived growth factor-bb transactivates the fibroblast growth factor receptor to induce proliferation in human smooth muscle cells. Trends Cardiovasc Med. 2006;16:25–28. doi: 10.1016/j.tcm.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Quasnichka H, Slater SC, Beeching CA, Boehm M, Sala-Newby GB, George SJ. Regulation of smooth muscle cell proliferation by beta-catenin/t-cell factor signaling involves modulation of cyclin d1 and p21 expression. Circ Res. 2006;99:1329–1337. doi: 10.1161/01.RES.0000253533.65446.33. [DOI] [PubMed] [Google Scholar]
- 24.Wang X, Adhikari N, Li Q, Hall JL. Ldl receptor-related protein lrp6 regulates proliferation and survival through the wnt cascade in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2004;287:H2376–2383. doi: 10.1152/ajpheart.01173.2003. [DOI] [PubMed] [Google Scholar]
- 25.Tsaousi A, Williams H, Lyon CA, Taylor V, Swain A, Johnson JL, George SJ. Wnt4/beta-catenin signaling induces vsmc proliferation and is associated with intimal thickening. Circ Res. 2011;108:427–436. doi: 10.1161/CIRCRESAHA.110.233999. [DOI] [PubMed] [Google Scholar]
- 26.Hutter R, Carrick FE, Valdiviezo C, Wolinsky C, Rudge JS, Wiegand SJ, Fuster V, Badimon JJ, Sauter BV. Vascular endothelial growth factor regulates reendothelialization and neointima formation in a mouse model of arterial injury. Circulation. 2004;110:2430–2435. doi: 10.1161/01.CIR.0000145120.37891.8A. [DOI] [PubMed] [Google Scholar]
- 27.Hutter R, Valdiviezo C, Sauter BV, Savontaus M, Chereshnev I, Carrick FE, Bauriedel G, Luderitz B, Fallon JT, Fuster V, Badimon JJ. Caspase-3 and tissue factor expression in lipid-rich plaque macrophages: Evidence for apoptosis as link between inflammation and atherothrombosis. Circulation. 2004;109:2001–2008. doi: 10.1161/01.CIR.0000125526.91945.AE. [DOI] [PubMed] [Google Scholar]
- 28.Isner JM, Kearney M, Bortman S, Passeri J. Apoptosis in human atherosclerosis and restenosis. Circulation. 1995;91:2703–2711. doi: 10.1161/01.cir.91.11.2703. [DOI] [PubMed] [Google Scholar]
- 29.Serruys PW, Luijten HE, Beatt KJ, Geuskens R, de Feyter PJ, van den Brand M, Reiber JH, ten Katen HJ, van Es GA, Hugenholtz PG. Incidence of restenosis after successful coronary angioplasty: A time-related phenomenon. A quantitative angiographic study in 342 consecutive patients at 1, 2, 3, and 4 months. Circulation. 1988;77:361–371. doi: 10.1161/01.cir.77.2.361. [DOI] [PubMed] [Google Scholar]
- 30.Bauriedel G, Schluckebier S, Hutter R, Welsch U, Kandolf R, Luderitz B, Prescott MF. Apoptosis in restenosis versus stable-angina atherosclerosis: Implications for the pathogenesis of restenosis. Arterioscler Thromb Vasc Biol. 1998;18:1132–1139. doi: 10.1161/01.atv.18.7.1132. [DOI] [PubMed] [Google Scholar]
- 31.Bauriedel G, Windstetter U, DeMaio SJ, Jr., Kandolf R, Hofling B. Migratory activity of human smooth muscle cells cultivated from coronary and peripheral primary and restenotic lesions removed by percutaneous atherectomy. Circulation. 1992;85:554–564. doi: 10.1161/01.cir.85.2.554. [DOI] [PubMed] [Google Scholar]
- 32.Ip JH, Fuster V, Badimon L, Badimon J, Taubman MB, Chesebro JH. Syndromes of accelerated atherosclerosis: Role of vascular injury and smooth muscle cell proliferation. J Am Coll Cardiol. 1990;15:1667–1687. doi: 10.1016/0735-1097(90)92845-s. [DOI] [PubMed] [Google Scholar]
- 33.Jawien A, Bowen-Pope DF, Lindner V, Schwartz SM, Clowes AW. Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J Clin Invest. 1992;89:507–511. doi: 10.1172/JCI115613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drachman DE. Drug-eluting stents in animals and patients: Where do we stand today? Circulation. 2009;120:101–103. doi: 10.1161/CIRCULATIONAHA.109.872473. [DOI] [PubMed] [Google Scholar]
- 35.Finn AV, Vorpahl M, Ladich E, Virmani R. Future directions in stenting. Expert review of cardiovascular therapy. Expert Rev Cardiovasc Ther. 2010;8:1–6. doi: 10.1586/erc.09.144. [DOI] [PubMed] [Google Scholar]
- 36.Virmani R, Wilson GJ, Finn AV. Letter by virmani et al regarding article, “drug-eluting stents in animals and patients: Where do we stand today?”. Circulation. 2010;121:e246. doi: 10.1161/CIR.0b013e3181d870cf. author reply e247. [DOI] [PubMed] [Google Scholar]
- 37.Vorpahl M, Yazdani SK, Nakano M, Ladich E, Kolodgie FD, Finn AV, Virmani R. Pathobiology of stent thrombosis after drug-eluting stent implantation. Curr Pharm Des. 2010;16:4064–4071. doi: 10.2174/138161210794454879. [DOI] [PubMed] [Google Scholar]
- 38.Wilson GJ, Nakazawa G, Schwartz RS, Huibregtse B, Poff B, Herbst TJ, Baim DS, Virmani R. Comparison of inflammatory response after implantation of sirolimus- and paclitaxel-eluting stents in porcine coronary arteries. Circulation. 2009;120:141–149. 141–142. doi: 10.1161/CIRCULATIONAHA.107.730010. [DOI] [PubMed] [Google Scholar]
- 39.Luscher TF, Steffel J, Eberli FR, Joner M, Nakazawa G, Tanner FC, Virmani R. Drug-eluting stent and coronary thrombosis: Biological mechanisms and clinical implications. Circulation. 2007;115:1051–1058. doi: 10.1161/CIRCULATIONAHA.106.675934. [DOI] [PubMed] [Google Scholar]
- 40.Lee RT, Yamamoto C, Feng Y, Potter-Perigo S, Briggs WH, Landschulz KT, Turi TG, Thompson JF, Libby P, Wight TN. Mechanical strain induces specific changes in the synthesis and organization of proteoglycans by vascular smooth muscle cells. J Biol Chem. 2001;276:13847–13851. doi: 10.1074/jbc.M010556200. [DOI] [PubMed] [Google Scholar]
- 41.Reis ED, Roque M, Cordon-Cardo C, Drobnjak M, Fuster V, Badimon JJ. Apoptosis, proliferation, and p27 expression during vessel wall healing: Time course study in a mouse model of transluminal femoral artery injury. J Vasc Surg. 2000;32:1022–1029. doi: 10.1067/mva.2000.109763. [DOI] [PubMed] [Google Scholar]
- 42.Kenagy RD, Hart CE, Stetler-Stevenson WG, Clowes AW. Primate smooth muscle cell migration from aortic explants is mediated by endogenous platelet-derived growth factor and basic fibroblast growth factor acting through matrix metalloproteinases 2 and 9. Circulation. 1997;96:3555–3560. doi: 10.1161/01.cir.96.10.3555. [DOI] [PubMed] [Google Scholar]
- 43.Tsaousi A, Williams H, Lyon CA, Taylor V, Swain A, Johnson JL, George SJ. Wnt4/beta-catenin signaling induces vsmc proliferation and is associated with intimal thickening. Circ Res. 108:427–436. doi: 10.1161/CIRCRESAHA.110.233999. [DOI] [PubMed] [Google Scholar]
- 44.van de Schans VA, Smits JF, Blankesteijn WM. The wnt/frizzled pathway in cardiovascular development and disease: Friend or foe? Eur J Pharmacol. 2008;585:338–345. doi: 10.1016/j.ejphar.2008.02.093. [DOI] [PubMed] [Google Scholar]
- 45.Isner JM, Kearney M, Bauters C, Leclerc G, Nikol S, Pickering JG, Riessen R, Weir L. Use of human tissue specimens obtained by directional atherectomy to study restenosis. Trends Cardiovasc Med. 1994;4:213–221. doi: 10.1016/1050-1738(94)90037-X. [DOI] [PubMed] [Google Scholar]
- 46.Pickering JG, Weir L, Jekanowski J, Kearney MA, Isner JM. Proliferative activity in peripheral and coronary atherosclerotic plaque among patients undergoing percutaneous revascularization. J Clin Invest. 1993;91:1469–1480. doi: 10.1172/JCI116352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simons M, Leclerc G, Safian RD, Isner JM, Weir L, Baim DS. Relation between activated smooth-muscle cells in coronary-artery lesions and restenosis after atherectomy. N Engl J Med. 1993;328:608–613. doi: 10.1056/NEJM199303043280903. [DOI] [PubMed] [Google Scholar]
- 48.Brandl R, Maurer PC, Hofling B, Bauriedel G. Migration behavior of human smooth muscle cells cultivated from restenotic and primary lesions. Angiology. 1995;46:973–980. doi: 10.1177/000331979504601101. [DOI] [PubMed] [Google Scholar]
- 49.Anastas JN, Moon RT. Wnt signalling pathways as therapeutic targets in cancer. Nature reviews. Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 50.Joner M, Finn AV, Farb A, Mont EK, Kolodgie FD, Ladich E, Kutys R, Skorija K, Gold HK, Virmani R. Pathology of drug-eluting stents in humans: Delayed healing and late thrombotic risk. J Am Coll Cardiol. 2006;48:193–202. doi: 10.1016/j.jacc.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 51.Nilsen DW, Melberg T, Larsen AI, Barvik S, Bonarjee V. Late complications following the deployment of drug eluting stents. Int J Cardiol. 2006;109:398–401. doi: 10.1016/j.ijcard.2005.05.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.