

Abstract
Liver cancer is a common malignant disease, with high incidence and mortality rates. The study on the proteomics of liver cancer has attracted particular attention. The quantitative study method of proteomics depends predominantly on two-dimensional (2D) gel electrophoresis. In the present study we reported a rapid and accurate proteomics quantitative study method of high repeatability that includes the use of stable isotope labeling for the extraction of proteins and peptides via enzymolysis to achieve new type 2D capillary liquid chromatography-mass spectrometry separation using the separation mode of cation-exchange chromatography in conjunction with reversed-phase chromatography. LTQ OrbiTrap mass spectrometry detection was also performed. A total of 188 differential proteins were analyzed, including 122 upregulating [deuterium/hydrogen ratio (D/H) >1.5)] and 66 downregulating proteins (D/H<0.67). These proteins may play an important role in the occurrence, drug resistance, metastasis and recurrence of cancer or other pathological processes. Such a proteomics technology may provide biological data as well as a new methodological basis for liver cancer research.
Keywords: stable isotope labeling, liver cancer, quantitative proteomics, mass spectrum
Introduction
Liver cancer is the fifth most common malignant tumor worldwide and ranks third in terms of mortality, with ∼500,000 new cases annually. The incidence of liver cancer is high in develo ping countries, which account for 81% of all liver cancer cases, with liver cancer patients in China accounting for 54% of the total. Liver cancer is the second most common cause of cancer-related mortality (1,2) and is characterized by late diagnosis, poor prognosis, metastatic tendency and insensitivity to chemotherapy or radiotherapy. A previous study indicated that the occurrence of liver cancer is a slow process of gradual changes that develop mainly as a consequence of chronic hepatitis and hepatic fibrosis. Those pathological processes include alterations in gene and protein expression (3).
With the advent of the post-genomic era, the study focus of bioscience has shifted from genomics to proteomics. The research areas of proteomics mainly cover three aspects: differential protein expressions during disease generation and progress, polypeptide or protein identification, and their post-translational modification and interactions between proteins (4). Quantitative proteomics have been a study focus over the last few years and they involve the quantitative description of protein expression levels and their alterations according to variations in time and space or under various physiological or pathological conditions. This has become an important novel approach to studying proteomics. Quantitative proteomics may promote proteomics research, provide a new method of identifying new potential tumor labeling (5), provide a new methodological basis for the research on novel chemotherapeutic drugs and facilitate improvement of comprehensive cancer treatment.
At present, two-dimensional (2D) gel electrophoresis is mainly employed in the research protocol of biomass spectrometry-based quantitative proteomics. 2D gel electrophoresis separates proteins according to their two physicochemical properties, isoelectric point (IEP) and varying molecular weight, in order to isolate the proteins of complex biological samples on a 2D plane. Through silver staining or fluorescent dye imaging, the 2D map is scanned by the computer and PDQuest (Bio-Rad Hercules, CA, USA) or Image Master (Imatest LLC, Boulder, CO, USA) software is used to analyze results and identify differential proteins. Subsequently, the proteins are extracted from the gel for enzymolysis and biomass spectrometry detection (6,7). 2D gel electrophoresis is based on the traditional separation approach of biomass spectrometry proteomics and it is not able to isolate proteins that are overly acidic or alkaline or proteins and membrane proteins of very high or low molecular weight. This method has certain disadvantages, such as poor repeatability, low accuracy, difficulty in automation and failure to meet high-flux analysis requirements.
In this study, human liver cancer cells and normal liver cells were isolated, cultured and used as control models. Stable isotope labeling was introduced chemically and LTQ ObiTrap biomass spectrometry detection was performed. Differential proteins were screened for biological identification. This study may enrich the available biological data on liver cancer and provide a methodological basis for further elucidation of generation, drug resistance, metastasis and recurrence following resection or other pathological process.
Materials and methods
Materials
Human liver cancer and normal liver tissues were obtained by surgery and provided by the Hepatopancreatobiliary Surgery Department of the Affiliated Hospital of Guilin Medical University, with the approval of the local Ethics Committee. The cocktail of protease inhibitors was purchased from Roche Diagnostics (Mannhaim, Germany). CH2O, CD2O, pancreatin and cyano sodium borohydride were purchased from Sigma (St. Louis, MO, USA). The Finnigan liquid chromatograph, cell culture chamber and LTQ ObiTrap mass spectrometer were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). All the antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Separation and culture of human liver cancer and normal liver cells
Human liver cancer and normal liver tissues were retrieved from 40 adult male patients who underwent liver cancer resection and 30 adult male patients who underwent partial liver resection. The study was approved by the Ethics Committee of the Affiliated Hospital of Guilin Medical University. Cell isolation was conducted as previously described (8). The viability of isolated hepatocytes was determined by trypan blue exclusion. Cell suspensions with viabilities >85% were plated and cultured for subsequent experiments. After the cells were homogenized by pipetting lightly until uniform, they were transferred in a culture bottle with a bottom area of 25 cm2 at a density of 1×106 cells/ml and placed into the hatch chamber under 5% CO2 and 95% O2 for culture. The culture medium comprised 15% heat-inactivated fetal calf serum (Gibco, Carlsbad, CA, USA), 84% Dulbecco’s modified Eagle’s medium (Sigma) and 1% penicillin and streptomycin.
Sample preparation
The cells were collected when they reached ∼80% confluence. A total of 1×107 cells were added to 3 ml of cell lysis solution [200 mM NaCl, 50 mM Tris-HCl (pH 8.0), 65 mM dithiothreitol (DTT)] with 1 mM of the protease inhibitor phenylmethylsulfonyl fluoride (PMSF) and 60 μl of the protease inhibitor cocktail. Ultrasonication was performed under the following conditions: 3 sec/cycle with 3-sec intervals between the cycles and the cells were cooled on ice for 60 min after 180 cycles. A protein precipitation solution was then added, consisting of acetone:ethanol:acetic acid (50:50:1) and precipitation was allowed to occur for 24 h at −20°C. Centrifugation at 25,000 × g was performed for 30 min at 4°C and the supernatant was discarded. The precipitate was washed with acetone and ethanol precooled at 4°C. Centrifugation at 25,000 × g was again performed for 15 min at 4°C and the supernatant was discarded. The samples were freeze-dried and dissolved again in 8 M urea and 100 mM NH4HCO3. The protein concentration was determined with Coomassie Brilliant Blue (Sigma). A total of 100 μg protein was added to 10 mM DTT in a water bath at 60°C for 60 min. Indole-3 acetic acid (IAA, 20 mM) was then added for a light-tight reaction of 30 min. NH4HCO3 (100 mM) was used to dilute the urea to a concentration of 1 M. Trypsin (1:25) was then added in a water bath at 37°C for 24 h. The samples were desalted, eluted and freeze-dried.
Stable isotope dimethyl labeling
Stable isotope dimethyl labeling was performed according to the Boersema method (9) with the appropriate improvements. The freeze-dried samples were redissolved in 100 μl of 100 mM tetraethylammonium bromide (TEAB, pH 8.0). CH2O [4 μl 4% (v/v)] and CD2O [4 μl 4% (v/v)] were used for marking and redissolution of 25 μg TEAB for liver and normal cancer cells, respectively, in enzymolysis peptides. The samples were lightly oscillated for even blending. A total of 4 μl 0.6 M NaBH3CN was added for the labeling reaction at room temperature for 60 min (Fig. 1). Sixty minutes after the labeling reaction, 16 μl of 1% ammonia water was added and the samples were oscillated lightly for even blending to terminate the reaction. Subsequently, 10% (v/v) trifluoroacetic acid (TFA) was added to further terminate the labeling reaction and acidize the solution. The two were then blended for desalination, elution and freezing-drying, followed by redissolution in 30 μl 0.1% TFA for LTQ OrbiTrap mass spectrometric detection.
Figure 1.

Labeling reaction of stable isotope dimethyl labeling
Preparation of monolithic column
Two types of monolithic columns were prepared, according to the method previously described (10,11). The polymerization solution was prepared via ultrasonic blending of 80 μl ethylene glycol methacrylate phosphate (EGMP), 60 mg methylenebisacrylamide, 270 μl dimethyl sulfoxide, 200 μl dodecanol, 50 μl dimethylformamide and 2 mg azobisisobutyronitrile (AIBN) for 15 min. The prepared polymerization solution entered the capillary of inner diameter (ID) 150 μm via siphoning. Both ends of the capillary were sealed with silicon rubber, followed by immersion in a water bath at 60°C for 12 h. Methanol was used to remove unreacted monomer and porogen to complete the preparation of a strong cationic exchange capillary monolithic column. The preparation process of the inverted capillary monolithic column was similar to that of the strong cationic exchange capillary monolithic column. The only difference was the composition of the polymerization solution [100 μl low melting point agarose (LMA), 100 μl 3,4-ethylenedioxy-N-methylamphetamine, 170 μl normal propanol, 130 μl 1,4-butanediol, 20 μl water and 2 mg AIBN prepared with ultrasonic blending], which entered a capillary of ID 75 μm via siphoning. Furthermore, the outlet of the prepared inverted capillary monolithic column was bent into an electric spray needle of ID 5 μm with a butane torch and filled with C18 nanoparticles.
Establishment of 2D nano LC-MS/MS and sample analysis
The 2D nano HPLC-MS/MS was performed with a Finnigan Surveyor liquid chromatography pump and an LTQ ObiTrap mass spectrometer with a flow rate of ∼120 nl/min via split-flow. The flowing phase was solution A (0.1% formic acid solution), solution B was 0.1% formic acid in acetonitrile solution and solution C was 1,000 mM NH4Ac (pH 2.7) and 0.1% formic acid solution. Mass spectra were detected using an LTQ OrbiTrap spectrometer in cationic mode by applying a voltage of 1.8 kV. The mass spectrometric (MS) scanning was used to achieve full scan (range, 400–1,800 m/z) followed by MS/MS scan of 6 peaks of full scan. The basic process was as follows: First, the Finnigan automatic loading pump was loaded with 25 μl of redissolved labeling sample at the speed of 10 μl/min onto the phosphate integral material enriching column (150 μm × 7 cm ID). NH4Ac brine solution was loaded at 10 different concentrations resulting from the blending of the flowing phases A and B at the flow rate of 200 nl/min in order to classify and elute the peptides enriched on the strong cation-exchange monolithic column to C18 (75 μm × 10 cm ID) invert isolation column. The 10 concentrations of the NH4Ac brine solution were 50, 100, 150, 200, 250, 300, 350, 400, 500 and 1,000 mM NH4Ac. The elution time for each saline concentration was 10 min. Rebalancing was performed for 10 min with 0.1% of formic acid solution. After each balancing, 2D nano HPLC-MS/MS had a gradient elution of 155 min with 5 min for 0–5% flowing phase B, 120 min for 5 to 35% flowing phase B, 10 min for 35 to 80% flowing phase B, 10 min for maintaining 80% flowing phase B, 2 min of 80 to 0% flowing phase B and 8 min for 100% phase A balance.
Protein identification and quantification
The original data were consolidated via DTA Super Charge (v2.0a7; http://www.xentrik.net/software/mass_spectrometry_-_dta_supercharge.html) in Mascot generic format and the Mascot search engine (http://www.matrix-science.com) was used to search for consolidated data at International Protein index (IPI) human protein database (http://www.ebi.ac.uk), with carbamidomethylation + 57,0215 selected as the fixed modification and oxidation of methionine (Met) + 15,9949, light-marked dimethylation + 28,0313 (C- and N-terminal) and heavy-marked dimethylation + 32,0564 (C- and N-terminal) set as the variable modifications. The peptide mass tolerance was set at 20 ppm and MS/MS tolerance was set at 0.8 Da. The trypsin enzymolysis maximum leakage cut-off value was set as 2 and the important threshold value was set as 0.01 to ensure a false discovery rate of <1%. The protein quantification was obtained via dimethylation-based MSQuant (http://msquant.sourceforge.net) and a protein identified by at least three peptides was considered as credible to investigate the standard deviation of the identified protein. The protein was selected with peptide MSQuant ≥25 (Grade 1, P≤0.05) as the ratio and proportion of peptides were obtained from the calculation of extracted ion chromatograms of the peptide of the hydrogen and weight scale from the isotopic peak, the proportion of proteins was the average value of all the peptide proportions with standardization by StatQuant software (https://trac.nbic.nl/statquant) (12–14).
Western blot analysis
Cells were collected and added to the lysis solution (50 mM Tris-HCl, 137 mM sodium chloride, 10% glycerin, 100 mM sodium vanadate, 1 mM PMSF, 10 mg/ml aprotinin, 10 mg/ml leupeptin, 1% NP-40, 5 mM cocktail; pH 7.4) to extract proteins. The protein concentration was determined with the bicinchoninic acid method and proteins were dyed with bromophenol blue. The same amount of protein was added to each well and isolated with 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis. The proteins were transferred to a polyvinylidene fluoride membrane with the semi-dry method and sealed with 5% skimmed milk powder. After washing with Tris-buffered saline with Tween-20 (TBST), primary antibody was added for incubation for 1 h, followed by the addition of secondary antibody for incubation for 1 h. Chemiluminescence was used for X-ray film exposure. The stripe was scanned for grey-scale analysis.
Results and Discussion
Liver cancer is one of the most common malignancies worldwide, causing ∼10,000 deaths annually. The identification of the early molecular events of tumorigenesis may facilitate and improve diagnostic efficiency and lead to the development of more effective treatment strategies. In this study, a 2D nano LC-MS/MS-based quantitative proteomics analysis of the human liver cancer cells and normal liver cells was conducted using stable isotope labeling technology to provide a new method for the research on quantitative proteomics.
Currently, the main research method of biomass spectrometric quantitative proteomics is 2D gel electrophoresis. In a study conducted by Xu et al (15), 2D gel electrophoresis was used to analyze the glycoproteomics of Chang liver cells and MHCC97-H cells in order to select 63 differential proteins, including 7 glycoproteins with significant upregulation. Zhang et al (16) used 2D gel electrophoresis to analyze the proteomics of G1 phase hepatitis B-relevant liver cancer and normal liver tissue in order to select 15 differential proteins and proved the significance of the downregulating protein proteasome activator subunit 1 in the early diagnosis of liver cancer. In addition, a study by Suo et al (17) combined 2D gel electrophoresis with mass spectrum analysis to investigate the proteomics of the HepG2 liver cell strain with sorafenib therapy to identify 19 differential proteins, including 6 upregulating and 13 downregulating proteins. We identified 188 differential proteins in human liver cancer cells, including 122 upregulating and 66 downregulating proteins, via stable isotope labeling technology combined with LTQ OrbiTrap mass spectrometric detection. These differential proteins may play important roles in the occurrence, drug resistance, metastasis and recurrence of the liver cancer or other pathological processes.
In the present study the 14-3-3 proteins and TCP1 were screened. The 14-3-3 proteins specifically bind to serine-phosphorylated proteins and interact with Raf-1, PI-3K, ASK-1, PKC or other protein kinases, regulating signalling pathways (18–21). Previous studies demonstrated that the expression of 14-3-3 proteins β, γ, δ and θ is high in lung cancer tissue and are associated with malignant potential (22). The expression of 14-3-3 proteins β and η are high in nerve astrocytoma and are associated with malignant potential (23). Furthermore, 14-3-3 protein β has been found to promote the proliferation of the K2 rat liver cancer cell line (24). Through mass spectrum detection, we demonstrated that the expression of 14-3-3 proteins α, β, δ, ε, ζ, η and θ in human liver cancer cells was higher compared to that in normal liver cells (Table I). Among these, the expression of 14-3-3 proteins ζ and δ was the most pronounced (Figs. 2 and 3), suggesting that the ζ and δ subtypes of the 14-3-3 protein may be involved in the development of human liver cancer. However, we noted that the expression level of 14-3-3 protein γ in human liver cancer cells was not significantly different from that in normal liver cells, which was different from the results reported by Lee et al (25). Western blot analysis was then used to assess 14-3-3 proteins ζ and δ and our results were identical to those obtained from the mass spectrum detection (Fig. 4).
Table I.
Screened differential proteins.
| IPI | Peptides | D/H | Protein name |
|---|---|---|---|
| IPI00018146 | 3 | 1.535149217 | 14-3-3 protein θ |
| IPI00013122 | 3 | 1.722638965 | Hsp90 co-chaperone Cdc37 |
| IPI00018465 | 9 | 2.432924271 | T-complex protein 1 subunit η |
| IPI00217223 | 3 | 2.724430084 | Multifunctional protein ADE2 |
| IPI00216318 | 7 | 2.994079351 | 14-3-3 protein β/α |
| IPI00009342 | 4 | 3.14406848 | Ras GTPase-activating-like protein |
| IPI00000816 | 15 | 3.976726055 | 14-3-3 protein ε |
| IPI00021263 | 4 | 16.9955101 | 14-3-3 protein ζ/δ |
| IPI00013890 | 4 | 17.93141747 | 14-3-3 protein σ |
| IPI00215911 | 4 | 0.304679751 | DNA-(apurinic or apyrimidinic site) lyase |
| IPI00007750 | 5 | 0.323970616 | Tubulin α-1 chain |
| IPI00003362 | 16 | 0.371136308 | Hypothetical protein |
| IPI00008274 | 4 | 0.428145468 | Adenylyl cyclase-associated protein 1 |
| IPI00302925 | 9 | 0.461544245 | T-complex protein 1 subunit θ |
| IPI00465430 | 7 | 0.510590911 | 70-kDa protein |
IPI, international protein index; D/H, deuterium/hydrogen ratio.
Figure 2.

Identification of the peptide sequence of protein 14-3-3 ζ/δ.
Figure 3.

Quantitative analysis of protein 14-3-3 ζ/δ.
Figure 4.
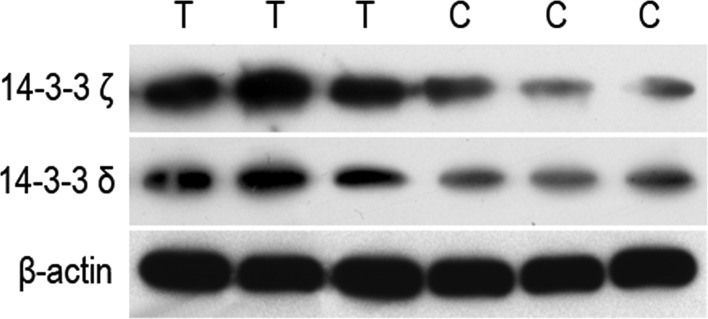
Western blot analysis of 14-3-3 protein ζ and δ expression levels. T, liver cancer cell; C, normal liver cell.
TCP1 is a molecular chaperone protein and its subtypes are involved in numerous pathways, including the assembly and folding of various intracellular proteins. Coghlin et al (26) reported a high expression of TCP1β and TCP1ε in colon cancer and suggested that TCP1β may be associated with the clinical outcome of colon cancer patients via the use of 2D gel electrophoresis based on biomass spectrum. Iijima et al (27) demonstrated that TCP1α is able to prompt cell proliferation. We identified the differences in the expression of TCP1η and TCP1θ between human liver cancer cells and normal liver cells and we obtained identical results via western blot analysis (Fig. 5). This suggested that TCP1η and TCP1θ may also participate in the progression of human liver cancer. In this study, we screened for differential 14-3-3 and TCP1 protein families and also observed that the expression of P70, hypothetical protein or other proteins in human liver cancer cells were different from those in normal liver cells. The differential proteins, including 14-3-3 and TCP1 protein families, may be a potential target in the treatment of liver cancer and a tumor labeling index associated with liver cancer, playing a critical role in the occurrence, drug resistance, metastasis and recurrence of liver cancer or other pathological processes.
Figure 5.
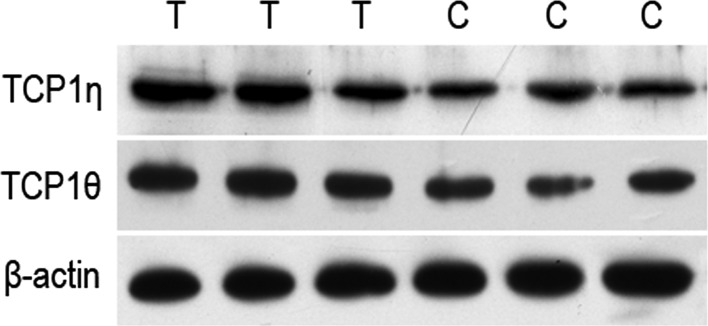
Western blot analysis of TCP1η and TCP1θ protein expression levels. T, liver cancer cell; C, normal liver cell.
In summary, we observed that stable isotope labeling may overcome the limitations of 2D gel electrophoresis and it has such advantages as high repeatability, stability, sensitivity and precise quantification. This chemical reaction is highly stable with high labeling efficiency. High-flux LTQ OrbiTrap mass spectrometer is able to effectively measure the molecular weight difference of 4 Da generated after labeling reaction. This labeling method may label two or three samples simultaneously and is suitable for various biological samples. The required sample quantity ranges from a microgram to a milligram, which is a wide mass range. The type of MSQuant software applicable to dimethylation may be used to automatically process data in batch conveniently and easily. This study has provided extensive biological data for the research on liver cancer, as well as a methodological basis for further elucidation of the generation, drug resistance, metastasis and recurrence following resection or other pathological process.
Acknowledgments
The authors would like to thank Professor Ye (Dalian Institute of Chemical Physics, Chinese Academy of Sciences) for providing valuable assistance and critical comments on this manuscript. This study was supported by the National High Technology Research and Development Program (863 Program).
References
- 1.Shariff MI, Cox IJ, Gomaa AI, Khan SA, Gedroyc W, Taylor-Robinson SD. Hepatocellular carcinoma: current trends in worldwide epidemiology, risk factors, diagnosis and therapeutics. Expert Rev Gastroenterol Hepatol. 2009;3:353–367. doi: 10.1586/egh.09.35. [DOI] [PubMed] [Google Scholar]
- 2.He J, Gu D, Wu X, Reynolds K, Duan X, Yao C, Wang J, Chen CS, Chen J, Wildman RP, Klag MJ, Whelton PK. Major causes of death among men and women in China. N Engl J Med. 2005;353:1124–1134. doi: 10.1056/NEJMsa050467. [DOI] [PubMed] [Google Scholar]
- 3.Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339–346. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- 4.Pandey A, Mann M. Proteomics to study genes and genomes. Nature. 2000;405:837–846. doi: 10.1038/35015709. [DOI] [PubMed] [Google Scholar]
- 5.Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208–1236. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]
- 6.Abdallah C, Dumas-Gaudot E, Renaut J, Sergeant K. Gel-based and gel-free quantitative proteomics approaches at a glance. Int J Plant Genomics. 2012;2012:494572. doi: 10.1155/2012/494572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fisher A, Sekera E, Payne J, Craig P. Simulation of two dimensional electrophoresis and tandem mass spectrometry for teaching proteomics. Biochem Mol Biol Educ. 2012;40:393–399. doi: 10.1002/bmb.20651. [DOI] [PubMed] [Google Scholar]
- 8.Tang B, Zhang Y, Liang R, Yuan P, Du J, Wang H, Wang L. Activation of the δ-opioid receptor inhibits serum deprivation-induced apoptosis of human liver cells via the activation of PKC and the mitochondrial pathway. Int J Mol Med. 2011;28:1077–1085. doi: 10.3892/ijmm.2011.784. [DOI] [PubMed] [Google Scholar]
- 9.Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJ. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc. 2009;4:484–494. doi: 10.1038/nprot.2009.21. [DOI] [PubMed] [Google Scholar]
- 10.Wang F, Dong J, Jiang X, Ye M, Zou H. Capillary trap column with strong cation-exchange monolith for automated shotgun proteome analysis. Anal Chem. 2007;79:6599–6606. doi: 10.1021/ac070736f. [DOI] [PubMed] [Google Scholar]
- 11.Wang F, Dong J, Ye M, Jiang X, Wu R, Zou H. Online multidimensional separation with biphasic monolithic capillary column for shotgun proteome analysis. J Proteome Res. 2008;7:306–310. doi: 10.1021/pr700562b. [DOI] [PubMed] [Google Scholar]
- 12.Mortensen P, Gouw JW, Olsen JV, Ong SE, Rigbolt KT, Bunkenborg J, Cox J, Foster LJ, Heck AJ, Blagoev B, Andersen JS, Mann M. MSQuant, an open source platform for mass spectrometry-based quantitative proteomics. J Proteome Res. 2010;9:393–403. doi: 10.1021/pr900721e. [DOI] [PubMed] [Google Scholar]
- 13.Wang F, Chen R, Zhu J, Sun D, Song C, Wu Y, Ye M, Wang L, Zou H. A fully automated system with online sample loading, isotope dimethyl labeling and multidimensional separation for high-throughput quantitative proteome analysis. Anal Chem. 2010;82:3007–3015. doi: 10.1021/ac100075y. [DOI] [PubMed] [Google Scholar]
- 14.Lemeer S, Jopling C, Gouw J, Mohammed S, Heck AJ, Slijper M, den Hertog J. Comparative phosphoproteomics of zebrafish Fyn/Yes morpholino knockdown embryos. Mol Cell Proteomics. 2008;7:2176–2187. doi: 10.1074/mcp.M800081-MCP200. [DOI] [PubMed] [Google Scholar]
- 15.Xu Z, Zhou X, Lu H, Wu N, Zhao H, Zhang L, Zhang W, Liang YL, Wang L, Liu Y, Yang P, Zha X. Comparative glycoproteomics based on lectins affinity capture of N-linked glycoproteins from human Chang liver cells and MHCC97-H cells. Proteomics. 2007;7:2358–2370. doi: 10.1002/pmic.200600041. [DOI] [PubMed] [Google Scholar]
- 16.Zhang D, Lim SG, Koay ES. Proteomic identification of down-regulation of oncoprotein DJ-1 and proteasome activator subunit 1 in hepatitis B virus-infected well-differentiated hepatocellular carcinoma. Int J Oncol. 2007;31:577–584. [PubMed] [Google Scholar]
- 17.Suo A, Zhang M, Yao Y, Zhang L, Huang C, Nan K, Zhang W. Proteome analysis of the effects of sorafenib on human hepatocellular carcinoma cell line HepG2. Med Oncol. 2012;29:1827–1836. doi: 10.1007/s12032-011-0013-y. [DOI] [PubMed] [Google Scholar]
- 18.Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 19.Chong ZZ, Maiese K. Erythropoietin involves the phosphatidylinositol 3-kinase pathway, 14-3-3 protein and FOXO3a nuclear trafficking to preserve endothelial cell integrity. Br J Pharmacol. 2007;150:839–850. doi: 10.1038/sj.bjp.0707161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cockrell LM, Puckett MC, Goldman EH, Khuri FR, Fu H. Dual engagement of 14-3-3 proteins controls signal relay from ASK2 to the ASK1 signalosome. Oncogene. 2010;29:822–830. doi: 10.1038/onc.2009.382. [DOI] [PubMed] [Google Scholar]
- 21.Gurusamy N, Watanabe K, Ma M, Zhang S, Muslin AJ, Kodama M, Aizawa Y. Inactivation of 14-3-3 protein exacerbates cardiac hypertrophy and fibrosis through enhanced expression of protein kinase C beta 2 in experimental diabetes. Biol Pharm Bull. 2005;28:957–962. doi: 10.1248/bpb.28.957. [DOI] [PubMed] [Google Scholar]
- 22.Qi W, Liu X, Qiao D, Martinez JD. Isoform-specific expression of 14-3-3 proteins in human lung cancer tissues. Int J Cancer. 2005;113:359–363. doi: 10.1002/ijc.20492. [DOI] [PubMed] [Google Scholar]
- 23.Yang X, Cao W, Lin H, Zhang W, Lin W, Cao L, Zhen H, Huo J, Zhang X. Isoform-specific expression of 14-3-3 proteins in human astrocytoma. J Neurol Sci. 2009;276:54–59. doi: 10.1016/j.jns.2008.08.040. [DOI] [PubMed] [Google Scholar]
- 24.Sugiyama A, Miyagi Y, Komiya Y, Kurabe N, Kitanaka C, Kato N, Nagashima Y, Kuchino Y, Tashiro F. Forced expression of antisense 14-3-3beta RNA suppresses tumor cell growth in vitro and in vivo. Carcinogenesis. 2003;24:1549–1559. doi: 10.1093/carcin/bgg113. [DOI] [PubMed] [Google Scholar]
- 25.Lee IN, Chen CH, Sheu JC, Lee HS, Huang GT, Yu CY, Lu FJ, Chow LP. Identification of human hepatocellular carcinoma-related biomarkers by two-dimensional difference gel electrophoresis and mass spectrometry. J Proteome Res. 2005;4:2062–2069. doi: 10.1021/pr0502018. [DOI] [PubMed] [Google Scholar]
- 26.Coghlin C, Carpenter B, Dundas SR, Lawrie LC, Telfer C, Murray GI. Characterization and over-expression of chaperonin t-complex proteins in colorectal cancer. J Pathol. 2006;210:351–357. doi: 10.1002/path.2056. [DOI] [PubMed] [Google Scholar]
- 27.Iijima M, Shimizu H, Tanaka Y, Urushihara H. A Dictyostelium discoideum homologue to Tcp-1 is essential for growth and development. Gene. 1998;213:101–106. doi: 10.1016/s0378-1119(98)00190-5. [DOI] [PubMed] [Google Scholar]