

Abstract
Background
Polymorphisms at cleavage sites (CS) can influence Gag and Pol proteins processing by the viral protease (PR), restore viral fitness and influence the virological outcome of specific antiretroviral drugs. However, data of HIV-1 variant-associated CS variability is scarce.
Methods
In this descriptive research, we examine the effect of HIV-1 variants on CS conservation using all 9,028 gag and 3,906 pol HIV-1 sequences deposited in GenBank, focusing on the 110 residues (10 per site) involved at 11 CS: P17/P24, P24/P2, P2/P7, P7/P1, P1/P6gag, NC/TFP, TFP/P6pol, P6pol/PR, PR/RTp51, RTp51/RTp66 and RTp66/IN. CS consensus amino acid sequences across HIV-1 groups (M, O, N, P), group M 9 subtypes and 51 circulating recombinant forms (CRF) were inferred from our alignments and compared to the HIV-1 consensus-of-consensuses sequence provided by GenBank.
Results
In all HIV-1 variants, the most conserved CS were PR/RTp51, RTp51/RTp66, P24/P2 and RTp66/IN and the least P2/P7 and P6pol/PR. Conservation was significantly lower in subtypes vs. recombinants in P2/P7 and TFP/P6pol and higher in P17/P24. We found a significantly higher conservation rate among Group M vs. non-M Groups HIV-1. The late processing sites at Gag (P7/P1) and GagPol precursors (PR/RTp51) presented a significantly higher conservation vs. the first CS (P2/P7) in the 4 HIV-1 groups. Here we show 52 highly conserved residues across HIV-1 variants in 11 CS and the amino acid consensus sequence in each HIV-1 group and HIV-1 group M variant for each 11 CS.
Conclusions
This is the first study to describe the CS conservation level across all HIV-1 variants and 11 sites in one of the largest available sequence HIV-1 dataset. These results could help other researchers for the future design of both novel antiretroviral agents acting as maturation inhibitors as well as for vaccine targeting CS.
Introduction
The human immunodeficiency virus type 1 (HIV-1) Gag proteins are essential for the virus, as they have a structural and functional role in the viral cycle. They coordinate viral trafficking, membrane binding, assembly, cofactor packaging, budding, and viral modulation. Gag proteins are generated through viral maturation, essential in the viral life cycle by enabling the generation of mature infectious viral particles through the proteolytic process in specific cleavage sites (CS) of Gag precursor (Pr55gag) and GagPol precursors (Pr160GagPol) proteins by the viral protease (PR) [1], [2]. Gag precursor is cleaved within the virion in three main structural Gag proteins: matrix (P17 or MA), capsid (P24 or CA) and nucleocapsid (P7 or NC), flanked by two spacer segments (P1 and P2) with regulatory functions [3]. Gag P6, a sixth protein of Gag precursor, plays an essential role in the release of the virus from infected cell membranes [3]. During translation of the Gag precursor an occasional ribosomal frameshift leads to the production of a GagPol precursor protein, the abundance of which is approximately 5% that of Gag precursor [4]. GagPol precursor contains the main structural proteins matrix P17, P24, P7, a transframe protein (TFP), P6pol and the three viral replication enzymes, PR, reverse transcriptase (RT) and integrase (IN) [3]. PR is activated concomitant with viral budding. As PR is only active as a dimer, it is thought that autoprocessing is initiated by dimerization of two PR domains that are embedded in the GagPol precursor [5]. Maturation triggers a second assembly event that generates a condensed conical capsid core, which organizes the viral RNA genome and viral proteins to facilitate viral replication in the next round of infection [6].
Processing of both HIV-1 Gag and GagPol polyproteins by the viral PR is highly specific, temporally regulated, and essential for the production of infectious HIV-1 particles. The differential rate of processing at each of the 11 proteolytic reactions by cleavage exists [6] and is determined by the context surrounding processing sites of the CS [7]. However, the precise mechanisms governing the rates of the cleavage events are still not fully understood [7].
The physical consequence of Gag cleavage is a morphological rearrangement of the non-infectious immature particle to a mature infectious particle. For this reason, amino acid substitutions on Gag proteins, included in CS, could influence processing [2], [8], morphogenesis, budding [9], the virus replicative capacity or viral fitness [3], [10] and the virological outcome of specific regimens, particularly to protease inhibitors (PI) [5], [11]–[20]. In fact, several Gag substrate mutations, included in CS, can confer PI resistance in the absence and/or presence of PR mutations [17]–[20]. The fundamental role of proteolytic maturation in the generation of infectious particles makes inhibition of this process an attractive target for therapeutic intervention. Thus, a new class of potential antiretroviral drugs targeting individual Gag CS has entered development [21].
Whether or not the processing regulation is different across HIV-1 variants remains unclear. It is well known that HIV-1 shows a high genetic diversity due to its high replication rate, the error-prone RT and the recombination events between HIV-1 variants occurring during the viral replication after co-infection and/or superinfection events [22]–[24]. A large number of HIV-1 variants have been described based on viral sequences homology and HIV-1 has been divided into four groups: M (main), O (outlier), N (non-M, non-O) and P [23]. HIV-1 Group M is subdivided into 9 subtypes (A–D, F–H, J, K), at least 58 circulating recombinant forms (CRF) (http://www.hiv.lanl.gov/content/sequence/HIV/CRFs/CRFs.html) -designated by a number and the genetic subtypes present in their genome- and multiple unique recombinant forms (URF), widely spread throughout the world and with different recombination breakpoints from those found in CRFs. At least 20% of the 34 million infected humans have an inter-subtype URF or CRF [25] and new inter-subtype recombinants have increasing prevalence and complexity in the pandemic, including in some European countries [26]. Genetic variability in PR and CS provide the potential to modulate PR activity and susceptibility to PI [20]. For instance, CS polymorphisms in certain HIV-1 group M variants can influence the virological outcome of a first-line LPV/r single drug regimen [19].
Despite the high biological relevance of CS during HIV-1 maturation and the importance of the knowledge of CS conservation for the design of both novel antiretroviral agents acting as maturation inhibitors as well as for vaccine targeting CS in future, scarce data of HIV-1 variant-associated CS variability is available. Previous reports only analyzed a limited number of HIV-1 variants and site sequences [3], [27], [28]. Thus, the goal of our descriptive analysis was to analyze, for the first time, the conservation rate at amino acid level of each individual protease CS located within Gag or Pol for all HIV-1 groups, Group M subtypes and recombinants circulating in the HIV/AIDS pandemic. For this purpose we used a large dataset of HIV-1 sequences routinely deposited at Los Alamos National Center for Biotechnology Information or GenBank. We also defined the consensus sequences at each CS in all HIV-1 variant, identifying the highly conserved amino acids residues in each CS.
Methods
Sequence Data
All the available HIV-1 gag/pol sequences were retrieved from GenBank, (http://www.ncbi.nlm.nih.gov/). The 12,934 gag/pol sequences comprised 2,844 nucleotides, located from 790 to 2,292 in gag and from 2,253 to 5,096 in pol encoding the proteins shown in Table 1 . These sequences belonged to 4 groups (M, O, N, P), 9 Group M subtypes (A: sub-subtypes A1 and A2, B, C, D, F: sub-subtypes F1 and F2, G, H, J and K), 51 of the 58 CRF currently described, and with available sequences at GenBank and URF ( Figure 1 ). For the subsequent analysis, we grouped in 12 recombinant families the closely related CRF sharing the same parental subtypes and very similar recombination patterns ( Figure 2 ), as previously recommended [23]. All gag/pol nucleotides sequences were retrieved in FASTA format, including the subtype B HXB2 reference sequence. The MEGA (Molecular Evolutionary Genetics Analysis. Arizona States University, Tempe) program version 5.05 (http://www.megasoftware.net/) [29] was used to perform the nucleotides alignments and to translate them into amino acids.
Table 1. Gag and Pol HIV-1 proteins numbered in HXB2 genome.
| Gene | Protein | ||||
| HIV-1 proteins | Length (nucleotide) | Position (nucleotide) | Length (amino acid) | Position (amino acid) | |
| Gag | P17 | 396 | 790–1185 | 132 | 263–394 |
| P24 | 693 | 1186–1878 | 231 | 395–625 | |
| P2 | 42 | 1879–1920 | 14 | 626–639 | |
| P7 | 165 | 1921–2085 | 55 | 640–694 | |
| P1 | 48 | 2086–2133 | 16 | 695–710 | |
| P6 | 156 | 2134–2289 | 52 | 711–762 | |
| Total | 1500 | 790–2289 | 500 | 263–762 | |
| Pol | PR | 297 | 2253–2549 | 99 | 751–849 |
| RTp51 | 1320 | 2550–3869 | 440 | 850–1289 | |
| RTp66 | 360 | 3870–4229 | 120 | 1290–1409 | |
| IN | 864 | 4230–5093 | 288 | 1410–1697 | |
| Total | 2841 | 2253–5093 | 947 | 751–1697 | |
Nucleotides and amino acids numbered according to HXB2 subtype B reference strain (GenBank accession number K03455). P17, matrix; P24, capsid; P2, spacer peptide 1; P7, nucleocapside; P1, space peptide 2; PR, protease; RT, retrotranscriptase; IN, integrase; TFP, transframe protein. Retrieved from http://www.hiv.lanl.gov/.
Figure 1. Distribution of HIV-1 Group M subtypes and CRF families.

A total of 12,848 HIV-1 Group M sequences were retrieved from GenBank: 8,985 gag (A) and 3,863 pol (B) sequences.
Figure 2. Gag and Pol HIV-1 recombinants sequences grouped by families. Availability of consensus sequences at GenBank.

CRF sequences were grouped in 12 recombinant families; no, number; CRF, circulating recombinant forms http://www.hiv.lanl.gov/content/sequence/HIV/CRFs/CRFs.html; URF, unique recombinant forms. Other variants with consensus sequences from GenBank were: A1, A2, B, C, D, F1, G, H and K subtypes for gag and: A1, A2, B, C, D, F1, F2, G and H subtypes for pol. http://www.hiv.lanl.gov/content/sequence/NEWALIGN/align.html.
Identification of gag and pol Coding Regions and CS Sequences Defined at GenBank
After performing the alignments, we determined the residues and their location in Gag and Pol proteins ( Table 1 ), identifying their nucleotides and amino acids and numbering them according to HXB2 subtype B reference strain (GenBank accession number K03455). We then identified the residues and the location of 11 cleavage sites (CS) within Gag and GagPol precursors: P17/P24, P24/P2, P2/P7, P7/P1, P1/P6gag, P7/TFP, TFP/P6pol, P6pol/PR, PR/RTp51, RTp51/RTp66 and RTp66/IN according to HXB2 sequence.
Inferred Consensus Sequences
The consensus sequence is considered the sequence carrying the most frequent residues, either nucleotides or amino acids, at each position in a multiple sequence alignment. We collected all Gag and Pol consensus sequences available in GenBank (http://www.hiv.lanl.gov/content/sequence/NEWALIGN/align.html). The HIV-1 Group M variants with inferred consensus sequences in GenBank are indicated in Figure 2 , and were calculated as explained in http://www.hiv.lanl.gov/content/sequence/NEWALIGN/align.html#consensus. Using our amino acid alignment, composed of 12,934 sequences, we determined new consensus sequences for each HIV-1 group and each Group M subtype, CRF and URF in the 11 CS ( Figures 3 and 4 ). Then, we manually compared our inferred variant-associated consensus sequences at each CS with the ones provided by GenBank when available, showing the discrepancies.
Figure 3. HIV-1 variants showing differences in CS1–CS5 amino acid vs. consensus-of-consensuses sequence from GenBank.

Changes are only indicated when they appeared in a specific position in at least 50% of the GenBank downloaded sequences in order to compare them with the GenBank consensus-of-consensuses sequence. Asterisks indicate the HIV-1 variants shown in Figure 2 with non available consensus sequence in GenBank. Black represents highly conserved amino acid residues and present in more than 99% of the 9,028 Gag and 3,906 GagPol HIV-1 sequences with respect to the consensus-of-consensuses sequence. When two residues within the analyzed sequences from each HIV-1 variant showed a conservation of 50% the two code letters were written in the upper case. When 3 or more residues appear in the same position and none presented a conservation of more than 50%, they were shown in lower case letters, which represented higher to lower conservation.
Figure 4. HIV-1 variants showing differences in CS6-CS11 amino acid vs. consensus-of-consensuses sequence from GenBank.

See legend of Figure 3 .
We also retrieved the consensus-of-consensuses sequence provided by GenBank in order to generate an alignment of gag and pol individual consensus sequences that were used to analyze the conservation rate across sites and HIV-1 variants ( Figure 5 ).
Figure 5. Amino acids CS conservation located in Gag and GagPol precursors in all HIV-1 variants.

The conservation was determined by comparing our inferred consensus sequences with sequences from each HIV-1 variant vs. consensus-of-consensuses sequence retrieved from GenBank. Coloured boxes indicate the CS conservation rate at amino acid level: green (≥90%), orange (51–89%) and red (≤50%). The number in each coloured box shows the rate of conserved amino acid in each CS in all sequences of the corresponding HIV-1 variant. CS, cleavage site; P17, matrix; P24, capsid; P2, spacer peptide 1; P7, nucleocapside; P1, space peptide 2; TFP, transframe protein; PR, protease; RT, retrotranscriptase; IN, integrase; CRF, circulant recombinant form; URF, unique recombinant form.
Amino Acid Conservation Rate at CS Across HIV-1 Variants
All gag and pol sequences from GenBank were grouped according to the HIV-1 variant. We manually compared the degree of amino acid conservation in each CS, determined by the number of coincident amino acids among the 10 residues of each CS, in all downloaded sequences from each given variant with respect to the consensus-of-consensuses sequence provided by GenBank. The exact percentage of conserved amino acid residues for each HIV-1 variant and site with respect to the GenBank consensus-of-consensuses amino acid sequence was calculated counting the number of coincident residues in each of the 10 positions in the site in all sequences ascribed to a given variant divided by the total number of retrieved sequences for each variant and multiplied by the 10 residues of the site, expressing the result in percentages. To clarify results, we established a color code to show the conservation level in each site and HIV-1 variant ( Figure 5 ).
Data Analysis
Changes in rates were assessed using the chi-square analysis. Statistical analyses were performed using Epi Info v6.0 (Centers for Disease Control and Prevention, Atlanta, GA, USA). Significance was set at p<0.05.
Results
Gag/Pol HIV-1 Sequences Used for the Analysis and Variants Distribution
A total of 9,028 Gag and 3,906 Pol HIV-1 sequences were downloaded from GenBank database. They included 43/43 Gag/Pol sequences from 3 HIV-1 Groups (O, N, P) and 8,985/3,863 Gag/Pol sequences from Group M. Figure 1 shows the Group M variants distribution of our retrieved sequence dataset, including a total of 7,913/3,269 Gag/Pol sequences from 9 HIV-1 group M subtypes (A: subsubtypes A1 and A2, B, C, D, F: sub-subtypes F1 and F2, G, H J and K), 1,060/583 Gag/Pol sequences ascribed to 51 CRF and 12/11 Gag/Pol URF sequences.
In order to simplify the analysis, we grouped all the sequences from the 51 CRFs in 12 different CRF families according to a similar recombination pattern ( Figure 2 ). The downloaded sequences for each subtype and CRF family are detailed in Figure 5 . Despite the large difference in the number of 8,985 Gag/3,863 Pol retrieved sequences, the specific distribution of HIV-1 Group M subtypes and CRF families was similar for both genes ( Figure 1 ). Recombinants displayed 11.9% gag and 15.4% pol sequences. Among subtypes, sequences from subtype B were the most represented in both gag/pol (43%/74.3%) coding regions, followed by sequences ascribed to subtype C (27.4%/16.7%), sub-subtype A1 (21.6%/4.3%) and subtype D (5.6%/1.9%). There were no gag sequences from sub-subtype F2 and subtypes J and K available in our dataset. Within the recombinants, family 01 (69.7%/44.4%) was the most represented, followed by families BC (7.2%), AG (5.9%), cpx (5.6%) and BF (4.5%) in gag and by families AG (16.8%), cpx (12.5%), BF (9,4%), and BG (4.3%) in pol, among others. URF sequences represented less than 0.3% of downloaded sequences (12 gag and 11 pol sequences).
HIV-1 Variant-specific gag/pol Consensus Sequences Available at GenBank
Figure 2 shows the specific subtypes and recombinants with consensus sequences in gag and pol available in GenBank, which carries the most frequent residue, either nucleotide or amino acid, at each position in a multiple sequence alignment. Table 2 summarizes the amino acids involved in each of the 11 CS (10 amino acids per site) in the HXB2 isolate as well as the consensus-of-consensuses sequence for each CS, defined by GenBank after the alignment of 28 gag/24 pol individual consensus sequences, corresponding to 8/7 subtypes among 9 in Group M and to 11/10 CRF within the 58 described ( Figure 2 ). The consensus-of-consensuses sequence was taken as reference for the analysis of the conservation at amino acid level across variants in the 110 residues (10 amino acids in each of the 11 CS), as described in Methods.
Table 2. Gag and Pol HIV-1 CS numbered in HXB2 genome.
| HIV-1 | Gene | Protein | |||||
| No. | Name | Consensus-of-consensusesequence from GenBank | HXB2 sequence | Length (nucleotide) | Position (nucleotide) | Length (amino acid) | Position (amino acid) |
| #1 | P17/P24 | VSQNY/PIVQN | VSQNY/PIVQN | 30 | 1171–1200 | 10 | 390–399 |
| #2 | P24/P2 | KARVL/AEAMS | KARVL/AEAMS | 30 | 1864–1893 | 10 | 621–630 |
| #3 | P2/P7 | TT-IM/MQRGN | SATIM/MQRGN | 30 | 1906–1935 | 10 | 635–644 |
| #4 | P7/P1 | ERQAN/FLGKI | ERQAN/FLGKI | 30 | 2071–2100 | 10 | 690–699 |
| #5 | P1/P6 gag | RPGNF/LQSRP | RPGNF/LQSRP | 30 | 2119–2148 | 10 | 706–715 |
| #6 | P7/TFP | ERQAN/FFREN | ERQAN/FFRED | 30 | 2071–2100 | 10 | 690–699 |
| #7 | TFP/P6 pol | ENLAF/QQGEA | EDLAF/LQGKA | 30 | 2094–2123 | 10 | 698–707 |
| #8 | P6 pol /PR | VSLSF/PQITL | VSFNF/PQVTL | 30 | 2238–2267 | 10 | 746–755 |
| #9 | PR/RTp51 | CTLNF/PISPI | CTLNF/PISPI | 30 | 2535–2564 | 10 | 845–854 |
| #10 | RTp51/RTp66 | GAETF/YVDGA | GAETF/YVDGA | 30 | 3855–3884 | 10 | 1285–1294 |
| #11 | RTp66/IN | IRKVL/FLDGI | IRKVL/FLDGI | 30 | 4215–4244 | 10 | 1405–1414 |
Nucleotides and amino acids numbered according to HXB2 subtype B reference strain (GenBank accession number K03455). Pr160GagPol includes CS #1 to 11 and Pr55Gag includes CS #1 to 5. Underlined amino acids show the changes in the HXB2 sequence vs. the consensus-of-consensuses sequence from GenBank. P17, matrix; P24, capsid; P2, spacer peptide 1; P7, nucleocapside; P1, space peptide 2; PR, protease; RT, retrotranscriptase; IN, integrase; TFP, transframe protein; No., CS position in Gag and GagPol precursors; CS, cleavage site. Retrieved from http://www.hiv.lanl.gov/.
New Inferred Consensus Sequence in HIV-1 Groups, Subtypes and Recombinant vs. that Provided by GenBank
Since gag and pol consensus sequences were not defined by GenBank in all HIV-1 subtypes and CRF, we deduced our personal consensus sequence for all HIV-1 variants using our generated alignment of 9,028 Gag and 3,906 Pol HIV-1 sequences. We determined that the rate of amino acid residues among the retrieved sequences coincided with the consensus-of-consensuses in the corresponding site. For the first time, we inferred the consensus sequence in each site for the different HIV-1 groups and for all subtypes, sub-subtypes and recombinants within Group M. Figures 3 and 4 show the HIV-1 variants that carry amino acid differences with the corresponding consensus-of-consensuses sequence from GenBank in CS. We identified when our inferred consensus sequence presented the same amino acid residue as consensus-of-consensuses provided by GenBank. All discrepancies found between our inferred variant-specific CS consensus sequences with the consensus-of-consensuses provided by GenBank were also identified (see Table S1).
Identification of Highly Conserved Residues at CS
Interestingly, we identified 52/110 (47.3%) amino acids conserved in more than 99% of the 9,028 Gag and 3,906 Pol HIV-1 sequences with respect to the consensus-of-consensuses sequence and these are marked in black in Figures 3 and 4 . Thus, nearly half of the residues involved in the 11 CS could accept a different degree of variant-dependent variability. Among sites, PR/RTp51 presented the highest number of highly conserved residues (9/10), followed by RTp51/RTp66 and P24/P2 (7/10), RTp66/IN (6/10), P7/P1 and P7/TFP (5/10), P1/P6gag and P6pol/PR (4/10), P17/P24 (3/10) and TFP/P6pol and P2/P7 (1/10).
Observed Differences in CS Conservation Rates Across HIV-1 Variants and Sites
We evaluated the percentage of conserved residues in the retrieved sequences for each HIV-1 variant and site with respect to the GenBank consensus-of-consensuses amino acid sequence, as explained in Methods. We established a color code to indicate the different levels of conservation, and the exact amino acid conservation rate in each CS and variant ( Figure 5 ). Interestingly, despite the structural and functional roles of proteins in the viral cycle, we observed different conservation rates across the sites and HIV-1 variants.
Conservation Among CS
In all HIV-1 variants, including all sequences ascribed to the 4 groups, we defined the conservation rate in each site ( Figure 5 ). The CS with the highest number of conserved residues were CS9 (PR/RTp51, 99%), CS10 (RTp51/RTp66, 98%), CS2 (P24/P2, 98%), CS11 (RTp66/IN, 97%), CS1 (P17/P24, 96%), CS4 (P7/P1, 96%) and CS6 (P7/TFP, 96%). The least conserved CS across HIV-1 groups, Group M subtypes and recombinants were CS3 (P2/P7, 71%), CS8 (P6pol/PR, 80%), CS7 (TFP/P6pol, 81%) and CS5 (P1/P6gag, 89%). CS8 and CS3 showed different lengths across variants (data not shown). We observed a significantly higher conservation at the last processing sites in Gag (CS4, P7/P1) and GagPol (CS9, PR/RTp51) precursors compared to the first processing site (CS3, P2/P7) in the 4 HIV-1 groups ( Figure 6 ), according to the proccessing order previously defined [3], [5], [30].
Figure 6. Conservation of the first and late processing sites at Gag and GagPol precursors.
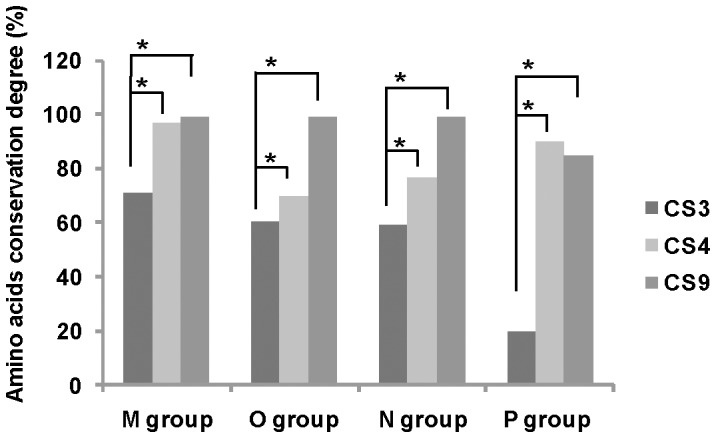
Late processing sites at Gag (CS4, P7/P1) and GagPol (CS9, PR/RTp51) precursors and first CS site (CS3, P2/P7) according to the CS order previously described [3], [5], [30]. *Significant difference, p<0.01.
Conservation among HIV-1 Groups
We observed differences in the CS conservation rate across HIV-1 groups and sites ( Figure 5 ). Interestingly, CS10 (RTp51/RTp66) showed more than 90% of conserved residues regarding consensus-of-consensuses amino acid sequence in the 4 HIV-1 groups. Comparing M and non-M Groups, we observed higher conservation in CS9 (99% and 98%, respectively) and in CS10 (98% and 91%, respectively). However, CS7 (TFP/P6pol) presented the poorest conservation rate across non-M Groups (41%), followed by CS5 (P1/P6gag, 54%), CS3 (P2/P7, 56%), CS8 (P6pol/PR, 60%) and CS1 (P17/P24, 68%). Group O showed the lowest conservation in 6 of the 11 CS (CS1, CS2, CS4, CS6, CS8 and CS11), Group N in CS1 and CS7 and Group P in CS3, CS5, CS9 and CS10 ( Figures 3 and 4 ). Considering the 11 CS, we found a significantly higher conservation rate among Group M vs. non-M Groups HIV-1 variants (91% vs. 71%, p<0.001), probably due to the use of group M consensus for comparison.
Conservation among Group M Subtypes and Recombinants
Seven sites (CS1, CS2, CS4, CS6, CS9, CS10 and CS11) were well conserved within the total HIV-1 Group M subtypes and recombinants, showing more than 90% conservation ( Figure 5 ). Four sites (CS3, CS5, CS7 and CS8) were more variable in a large number of HIV-1 variants. The lowest conservation rate in the 11 CS was found in the following HIV-1 Group M subtypes and recombinants: CS1 (P17/P24) in sub-subtype A2 (89%) and in AG recombinant family (77%); CS2 (P24/P2) in sub-subtype F2 (90%) and recombinant families AD and URF (both 94%); CS3 (P2/P7) in subtypes G (52%) and recombinant family DF (53%); CS4 (P7/P1) in subtype G (84%) and recombinant family DF (87%); CS5 (P1/P6gag) in sub-subtype A1 (76%) and recombinant family AU (73%); CS6 (P7/TFP) in subtype G (84%) and recombinant family BG (88%); CS7 (TFP/P6pol) in subtype B (77%) and recombinant families AB and BC (both 75%); CS8 (P6pol/PR) in sub-subtype A2 and subtype C (both 68%) and recombinant family BG (67%); CS9 (PR/RTp51) sub-subtype A2 (90%) and recombinant family A1 (95%); CS10 (RTp51/RTp66) in clades H (88%) and G (89%) and recombinant family A1 (90%); and CS11 (RTp66/IN) in sub-subtype F1 (90%) and recombinant family BF (91%). Thus, subtype G showed the highest variability in CS3, CS4 and CS6 and subtype B in CS7 compared to other Group M subtypes.
The recombinant families DF, BG, A1 and BF showed the highest variability in CS3 ( Figure 5 ). The CS2, CS6, CS9, CS10 and CS11 were highly conserved (94%–99%) across all CRF families and URF. Overall, recombinants showed the same conservation as subtypes (91%) in the 11 analyzed CS, the BG recombinant family with 87%, subtype G with 86%, sub-subtype A2 with 88% and subtype H with 89%. The HIV-1 variants that presented the lowest CS conservation were significantly lower in subtypes vs. recombinants in CS3 (70% vs.76%, p<0.001) and in CS7 (80% vs. 87%, p<0.001) and higher in CS1 (97% vs. 90%, p<0.001).
Conservation among Sub-subtypes in Specific Sites
Sub-subtype A2 presented significantly lower conservation than sub-subtype A1 in CS1 (89% vs. 98%, p<0.001), in CS3 (70% vs. 85%, p<0.001) and in CS9 (90% vs. 99%, p<0.001), significantly higher conservation in CS5 (80% vs. 76%, p = 0.04) and the conservation was of great significance in CS11 (100% vs. 92%, p = 0.06). Sub-subtype F2 showed superior conservation compared to sub-subtype F1 in CS11 (100% vs.90%, p = 0.04), although the number of available F2 sequences was very low (see Figure 5 ).
Discussion
HIV-1 genomes analysis provides useful biological information in terms of the structure and function of viral proteins [31]. The correct core formation is essential for the production of infectious HIV particles and this is known to be dependent on accurate proteolytic processing of Gag. Thus, mutations that disrupt the cleavage of individual sites or alter the order in which sites are cleaved result in aberrant particles that have significantly reduced infectivity [6]. Although other publications previously reported that certain CS were more conserved than others, they only analyzed a very limited number of HIV-1 variants and site sequences [3], [27], [28]. Thus, to our knowledge, our study is the first to evaluate the conservation rate in 11 CS within Gag and GagPol precursors and to define the consensus sequences in each site using a large sequence dataset including all Group M subtypes and most CRF. Furthermore, it is the first study that includes sequences from Groups N, O and P, identifying completely conserved residues at CS present in all 4 groups. We showed the conservation rate in each HIV-1 variant and CS, finding different conservation rates across sites in the 4 HIV-1 groups and in Group M variants, including a complete panel of recombinants, whose prevalence and complexity is increasing in the pandemic [23]. In fact, the different clade distribution for gag and pol sequences retrieved for GenBank used in the study could be explained by the large number of recombinants circulating in pandemic, with different clades in different viral genome genes.
New Findings on CS Variability Across HIV-1 Variants
Only a limited number of studies have previously evaluated the natural variation within gag and CS [3], [5], [12], [28], [32], [33]. However, these have mainly focused on subtypes B and/or C and they have analyzed a smaller dataset or a limited number of CS in most cases. Furthermore, the majority of the studies used HXB2 subtype B as reference sequence for conservation analysis [5], [32], [33], pNL-4-3subtype B [12] or the Group M most recent common ancestor sequence [3]. Only one used the consensus-of-consensuses sequence provided by GenBank as a reference for comparisons [28]. None inferred a consensus sequences for each analyzed HIV-1 variant and site. Other studies included either recombinants or non-M Group sequences. Despite the wide variety in the number of sequences that we downloaded from GenBank for Group M (8,985/3,863 gag/pol sequences) with respect to the rest of groups (43/43 gag/pol sequences) or certain subtypes (H, J, K), sub-subtypes (F2) or CRF, available data permitted the establishment of a comparison among conservation rates at CS and we were able to define specific-variant differences at each CS consensus sequence for each HIV-1 group, subtype, CRF and URF (see Figure 5 ). Our data reflects that the degree of conservation differs between individual amino acid positions at CS and provides significant discrepancies across specific HIV-1 variants and CS, thus improving the available GenBank data for specific-HIV-1 variants consensus sequences.
By using a large dataset of 12,934 sequences from all HIV-1 variants, our study revealed that the CS3, CS5, CS7 and CS8 were the least conserved processing sites across all HIV-1 variants. This finding is in agreement with previous publication using a smaller dataset with 32 subtype C, 34 subtype B and 18 other subtypes sequences [3]. Additional studies are necessary to understand the higher variability in these CS with important roles in the viral cycle. In more detail, CS3 is the first processing site in Gag and GagPol precursors and it is critical for RNA dimer maturation [34]; CS7 is involved in the activation of the viral PR and in the timing and specificity of GagPol cleavage [35]; CS5 is responsible for protein P6gag synthesis which is required for the mature and infectious virion release [36]; CS8 is essential for PR autoprocessing and, it could probably be involved in the correct required PR dimerization [37].
Structural Constrains to CS Variation
Complex interactions of the substrate amino acids within the active site of the viral PR are required for efficient Gag and GagPol cleavage by the PR. HIV-1 PR is only functional in dimeric form and a single monomer is embedded within each precursor. Two individual monomers in different GagPol chains must, therefore, come together to form an embedded dimeric PR, which ultimately cleaves itself into a mature form [37]. HIV-1 maturation requires the recognition by PR of the asymmetric, three-dimensional conformation of the Gag substrate, rather than a particular peptide sequence [38] and, afterwards, PR mediates the cleavage of the HIV-1 structural Gag and GagPol polyproteins by interacting with specific CS [6], [39]. Each substrate has a unique structure that differs in amino acid composition [3]. It is thought that these small differences in substrate structure impact affinity for the viral PR and contribute to the highly regulated and ordered stepwise process of maturation in which the individual cleavages occur at different times and rates [3], [30]. Additional determinants beyond amino acid sequences and local secondary structures of CS are involved in Gag and GagPol processing [7]. As Gag is conserved, there are constraints on the viability of viral strains with multiple mutations due to the fact that combined mutations are likely to destabilize multiprotein structural interactions that are critical for viral function [40]. Thus, amino acid sequence conservation indicates that the specific amino acids are required to maintain basic structure and function, although other authors have suggested an important role of RNA structure in HIV-1 conservation [33], [41]. It is known that physicochemical and structural properties of certain HIV-1 proteins with functional roles in the viral cycle as gp41 can be strongly conserved despite substantial sequence diversity, apparently indicating a delicate balance between evolutionary pressures and the conservation of protein structure [42]. The protein structure, specifically α-helix domains, has been associated with conservation in HIV-1 [33] and is a stable structural element in proteins [43].
Our study reveals which can be the most important CS amino acid sequence for maintaining viral processing by PR and the level of tolerance to amino acid change in each HIV-1 variant. Moreover, the significantly higher conservation observed comparing the late vs. the first CS in Gag and GagPol precursors (flanking the PR) would suggest a higher requirement of structural constrains in the last steps of viral processing. Although the aim of our study is purely descriptive, we strongly believe that it can serve as a working tool for research into the better understanding of the CS structure required for a correct cleavage efficacy across HIV-1 variants and for the design of maturation inhibitors and vaccines targeting CS. Understanding HIV-1 gag and pol co-evolution [44], [45] and the influence of naturally occurring specific-variant polymorphisms at PR [46] in the cleavage process is also crucial for a better interpretation of the biological significance of amino acid changes in CS in the context of a specific HIV-1 variant. Lastly, whether or not the variant specific-residues located in each CS modulate the replicative capacity of the corresponding variant, as was observed for specific natural polymorphisms in the PR in some non-B variants [47], requires further investigation.
It has been suggested that sequences around the CS in Gag are equally conserved as functional motives and sequences targeted by RT inhibitors and are more conserved than non-functional motives [28]. These authors suggested that the amino acid sequences overlapping the CS are immunogenic and, consequently, a vaccine targeting CS could be used for the majority of the world population [28]. Thus, our data on CS conservation across HIV-1 variants could provide useful data to design potential targets for an effective vaccine development against HIV effective for all groups, subtypes and recombinants. Moreover, since mutations within CS have been associated with PI exposure and maturation inhibitor resistance [5], [32], our results could potentially provide a better understanding of the role of gag in antiretroviral resistance and in the development of future maturation inhibitors [4].
Conclusion
This descriptive study firstly determines the CS conservation degree across most HIV-1 variants and sites in a large dataset composed of 12,934 sequences, inferring the consensus sequences at amino acid level in 11 CS in all Group M subtypes and most CRF and URF, as well as in Groups O, N and P. Our results provide new findings that can help for a better understanding of viral evolution, Gag and GagPol precursors’ processing and gag structure-function relationships, among others. Our descriptive research could help other researchers in the design of both novel antiretroviral agents acting as maturation inhibitors and for vaccine targeting CS. The biological significance of HIV-1 variant-associated variability found in each processing site in our study needs further future investigation.
Supporting Information
HIV-1 variants showing differences with the CS consensus-of-consensuses sequence inferred by GenBank.
(DOC)
Acknowledgments
The authors wish to thank Carolina Fernández McPhee for her assistance in checking the English of the final version of the manuscript and to Israel Pagán for his helpful comments during the manuscript editing.
Funding Statement
This study was supported by Ministry of Health and Social Policy through the grant of an “Independent clinical project” (EC11-131) and by research project FIS awarded by Instituto de Salud Carlos III (PI12/00240). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Swanstrom R, Wills J (1997) Retroviral gene expression. II. Synthesis, processing, and assembly of viral proteins. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. New York: Cold Spring Harbor Laboratory. pp. 263–334.
- 2. Tessmer U, Kräusslich HG (1998) Cleavage of HIV-1 proteinase from the N-terminally adjacent p6* protein is essential for efficient Gag polyprotein processing and viral infectivity. J Virol 72: 3459–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. de Oliveira T, Engelbrecht S, Janse van Rensburg E, Gordon M, Bishop K, et al. (2003) Variability at HIV-1 subtype C protease cleavage sites: an indication of viral fitness? J Virol 77: 9422–9430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Waheed AA, Freed EO (2012) HIV type 1 Gag as a target for antiviral therapy. AIDS Res Hum Retroviruses 28: 54–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fun A, Wensing AM, Verheyen J, Nijhuis M (2012) Human immunodeficiency virus Gag and protease: partners in resistance. Retrovirology 9: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Adamson CS (2012) Protease-mediated maturation of HIV: inhibitors of protease and the maturation process. Mol Biol Int 2012: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee SK, Potempa M, Kolli M, Özen A, Schiffer CA, et al. (2012) Context surrounding processing sites is crucial in determining cleavage rate of a subset of processing sites in HIV-1 Gag and Gag-Pro-Pol polyprotein precursors by viral protease. J Biol Chem 287: 13279–13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goodenow MM, Bloom G, Rose SL, Pomeroy SM, O’Brien PO, et al. (2002) Naturally occurring amino acid polymorphisms in HIV-1 Gag p7(NC) and the C-cleavage site impact Gag-Pol processing by HIV-1 protease. Virology 292: 137–149. [DOI] [PubMed] [Google Scholar]
- 9. Holguín A, Alvarez A, Soriano V (2006) Variability in the P6gag domains of HIV-1 involved in viral budding. AIDS 20: 624–627. [DOI] [PubMed] [Google Scholar]
- 10. Myint L, Matsuda M, Matsuda Z, Yokomaku Y, Chiba T, et al. (2004) Gag non-cleavage site mutations contribute to full recovery of viral fitness in protease inhibitor-resistant HIV-1. Antimicrob Agents Chemother 48: 444–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Doyon L, Payant C, Brakier-Gingras L, Lamarre D (1998) Novel Gag-Pol frameshift site in HIV-1 variants resistant to protease inhibitors. J Virol 72: 6146–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bally F, Martinez R, Peters S, Sudre P, Telenti A (2000) Polymorphism of HIV type 1 gag p7/p1 and p1/p6 cleavage sites: clinical significance and implications for resistance to protease inhibitors. AIDS Res Hum Retroviruses 16: 1209–1213. [DOI] [PubMed] [Google Scholar]
- 13. Maguire MF, Guinea R, Griffin P, Macmanus S, Elston RC, et al. (2002) Changes in HIV-1 Gag at positions L449 and P453 are linked to I50V protease mutants in vivo and cause reduction of sensitivity to amprenavir and improved viral fitness in vitro. J Virol 76: 7398–7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dam E, Quercia R, Glass B, Descamps D, Launay O, et al. (2009) Gag mutations strongly contribute to HIV-1 resistance to protease inhibitors in highly drug-experienced patients besides compensating for fitness loss. PLoS Pathog 5: e1000345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Banke S, Lillemark MR, Gerstoft J, Obel N, Jørgensen LB (2009) Positive selection pressure introduces secondary mutations at Gag cleavage sites in HIV-1 harboring major protease resistance mutations. J Virol 83: 8916–8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nijhuis M, van Maarseveen N, Schipper P (2005) Novel HIV-1 gag based protease drug resistance mechanism caused by an increased processing of the NC/p1 cleavage site. Antiv Ther 10: S117. [Google Scholar]
- 17. Nijhuis M, van Maarseveen NM, Lastere S, Schipper P, Coakley E, et al. (2007) A novel substrate-based HIV-1 protease inhibitor drug resistance mechanism. PLoS Med 4: e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Verheyen J, Knops E, Kupfer B, Hamouda O, Somogyi S, et al. (2008) Prevalence of C-terminal gag cleavage site mutations in HIV from therapy-naïve patients. Journal of Infection 58: 61–67. [DOI] [PubMed] [Google Scholar]
- 19. Ghosn J, Delaugerre C, Flandre P, Galimand J, Cohen-Codar I, et al. (2011) Polymorphism in Gag gene cleavage sites of HIV-1 non-B subtype and virological outcome of a first-line lopinavir/ritonavir single drug regimen. PLoS One 6: e24798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barrie KA, Perez EE, Lamers SL, Farmerie WG, Dunn BM, et al. (1996) Natural variation in HIV-1 protease, Gag p7 and p6, and protease cleavage sites within gag/pol polyproteins: amino acid substitutions in the absence of protease inhibitors in mothers and children infected by human immunodeficiency virus type 1. Virology 219: 407–416. [DOI] [PubMed] [Google Scholar]
- 21. Adamson CS, Freed EO (2008) Recent progress in antiretrovirals–lessons from resistance. Drug Discov Today 13: 424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang M, Foley B, Schultz AK, Macke JP, Bulla I, et al. (2011) The role of recombination in the emergence of a complex and dynamic HIV epidemic. Retrovirology 7: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhuang J, Jetzt AE, Sun G, Yu H, Klarmann G, et al. (2002) Human immunodeficiency virus type 1 recombination: rate, fidelity, and putative hot spots. J Virol 76 11273–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gao Y, Abreha M, Nelson KN, Baird H, Dudley DM, et al. (2011) Enrichment of intersubtype HIV-1 recombinants in a dual infection system using HIV-1 strain-specific siRNAs. Retrovirology 8: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peeters M (2000) Recombinant HIV sequences: Their role in the global epidemic. In: Kuiken C, Foley B, Hahn B, Korber B, McCutchan F, Marx P, editors. Theorical Biology and Biophysis Group. Los Alamos NM: National Laboratory. pp. I-39-1-54.
- 26. Yebra G, de Mulder M, Martín L, Rodríguez C, Labarga P, et al. (2012) Most HIV type 1 non-B infections in the Spanish cohort of antiretroviral treatment-naïve HIV-infected patients (CoRIS) are due to recombinant viruses. J Clin Microbiol 50: 407–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liégeois F, Reteno DG, Mouinga-Ondémé A, Sica J, Rouet F (2013) Short communication: high natural polymorphism in the gag gene cleavage sites of non-B HIV type 1 isolates from Gabon. AIDS Res Hum Retroviruses 29: 1179–1182. [DOI] [PubMed] [Google Scholar]
- 28. Luo M, Capina R, Daniuk C, Tuff J, Peters H, et al. (2013) Immunogenicity of sequences around HIV-1 protease cleavage sites: Potential targets and population coverage analysis for a HIV vaccine targeting protease cleavage sites. Vaccine 31: 3000–3008. [DOI] [PubMed] [Google Scholar]
- 29. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pettit SC, Lindquist JN, Kaplan AH, Swanstrom R (2005) Processing sites in the human immunodeficiency virus type 1 (HIV-1) Gag-Pro-Pol precursor are cleaved by the viral protease at different rates. Retrovirology 2: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Doherty RS, De Oliveira T, Seebregts C, Danaviah S, Gordon M, et al. (2005) BioAfrica’s HIV-1 proteomics resource: combining protein data with bioinformatics tools. Retrovirology 2: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Malet I, Roquebert B, Dalban C, Wirden M, Amellal B, et al. (2007) Association of Gag cleavage sites to protease mutations and to virological response in HIV-1 treated patients. J Infect 54: 367–374. [DOI] [PubMed] [Google Scholar]
- 33. Snoeck J, Fellay J, Bartha I, Douek DC, Telenti A (2011) Mapping of positive selection sites in the HIV-1 genome in the context of RNA and protein structural constraints. Retrovirology 8: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shehu-Xhilaga M, Kraeusslich HG, Pettit S, Swanstrom R, Lee JY, et al. (2001) Proteolytic processing of the p2/nucleocapsid cleavage site is critical for human immunodeficiency virus type 1 RNA dimer maturation. J Virol 75: 9156–9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tomasselli AG, Heinrikson RL (1994) Specificity of retroviral proteases: an analysis of viral and nonviral protein substrates. Methods Enzymol 241: 279–301. [DOI] [PubMed] [Google Scholar]
- 36. Müller B, Patschinsky T, Kräusslich HG (2002) The late-domain-containing protein p6 is the predominant phosphoprotein of human immunodeficiency virus type 1 particles. J Virol 76: 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sadiq SK, Noé F, De Fabritiis G (2012) Kinetic characterization of the critical step in HIV-1 protease maturation. Proc Natl Acad Sci U S A 109: 20449–20454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Prabu-Jeyabalan M, Nalivaika E, Schiffer C (2002) Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure 10: 369–381. [DOI] [PubMed] [Google Scholar]
- 39. Kaplan A, Manchester M, Swanstrom R (1994) The activity of the protease of HIV-1 is initiated at the membrane of infected cells before the release of viral proteins and is required for release to occur with maximum efficiency. J Virol 68: 6782–6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dahirel V, Shekhar K, Pereyra F, Miura T, Artyomov M, et al. (2011) Coordinate linkage of HIV evolution reveals regions of immunological vulnerability. Proc Natl Acad Sci U S A 108: 11530–11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van der Kuyl AC, Berkhout B (2012) The biased nucleotide composition of the HIV genome: a constant factor in a highly variable virus. Retrovirology 9: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Steckbeck JD, Craigo JK, Barnes CO, Montelaro RC (2011) Highly conserved structural properties of the C-terminal tail of HIV-1 gp41 protein despite substantial sequence variation among diverse clades: implications for functions in viral replication. J Biol Chem 286: 27156–27166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Richardson J (1981) The anatomy and taxonomy of protein structure. Adv Protein Chem 34: 167–339. [DOI] [PubMed] [Google Scholar]
- 44. Kozísek M, Henke S, Sasková KG, Jacobs GB, Schuch A, et al. (2012) Mutations in HIV-1 gag and pol compensate for the loss of viral fitness caused by a highly mutated protease. Antimicrob Agents Chemother 56: 4320–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rossi AH, Rocco CA, Mangano A, Sen L, Aulicino PC (2013) Sequence variability in p6 gag protein and gag/pol coevolution in human immunodeficiency type 1 subtype F genomes. AIDS Res Hum Retroviruses 29: 1056–1060. [DOI] [PubMed] [Google Scholar]
- 46. Yebra G, de Mulder M, del Romero J, Rodríguez C, Holguín A (2010) HIV-1 non-B subtypes: High transmitted NRTI-resistance in Spain and impaired genotypic resistance interpretation due to variability. Antiviral Research 85: 409–417. [DOI] [PubMed] [Google Scholar]
- 47. Holguín A, Suñe C, Hamy F, Soriano V, Klimkait T (2006) Natural polymorphisms in the protease gene modulate the replicative capacity of non-B HIV-1 variants in the absence of drug pressure. J Clin Virol 36: 264–271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HIV-1 variants showing differences with the CS consensus-of-consensuses sequence inferred by GenBank.
(DOC)