

Abstract
Bacterial biofilms are becoming a significant societal problem: biofilms form dental plaque, coat ships causing biofouling, and cling onto medical instruments and implants. Understanding how these surface-bound communities are formed is crucial for the development of suitable strategies for their dispersal. At the heart of a switch that commits Bacilli and related species to form biofilms is a transcriptional regulator called SinR and its multiple antagonists. In this addendum, we discuss an alternative model to account for how one of the antagonists is regulated by controlled proteolysis.
Keywords: Bacillus, SinR, adaptive responses, biofilm, protein interactions, transcriptional repression
In response to environmental challenges, such as lack of nutrients, exposure to chemical threats or increased cell density, microorganisms are required to rapidly change gene expression profiles to facilitate “lifestyle switches” that increase the collective odds of survival. For the Gram positive model organism, Bacillus subtilis, these adaptive changes include the over-production of flagella and motility genes (controlled by the σD regulon), the formation of a spore (controlled by the Spo0A regulon), or the attachment to a solid surface in a complex community of cells called a biofilm. The understanding of the processes involved in biofilm formation is an increasingly important topic because of the role of biofilms in microbial infections and contamination of medical instruments. Unlike motility or sporulation, in which hundreds of genes are under the direct control of the regulatory element, the decision to participate in a biofilm in B. subtilis is primarily the result of the actions of the transcriptional repressor SinR on just 2 operons, epsA-O and tapA-sipW-tasA, which encode the genes responsible for the production of the exopolysaccharide and protein component of the extracellular matrix, respectively.1-4 A second major protein component of the biofilm matrix BslA was recently identified5,6 and appears to confer hydrophobicity to the biofilm surface, although in this case the expression of bslA is under control of the DegS-DegU 2-component system. The control of SinR in the cell is achieved through the actions of 2 dedicated anti-repressors, SinI and SlrA, which bind to SinR and perturb its tetrameric arrangement, resulting in the loss of the ability of SinR to bind to its DNA operator sequences.7 A further protein, SlrR, also influences SinR activity through the formation of a heteromeric complex, although in this case SinR is a modulator of SlrR activity, transforming it into a repressor of operons involved in autolysis and motility.8,9 The fact that slrR is itself under transcriptional control by SinR (being located adjacent to the epsA-O operon) establishes a number of feedback loops that may serve to amplify small environmental changes or stochastic differences between cells in a population, leading to heterogeneity or the formation of a bistable state. The structural, kinetic and thermodynamic basis of these interactions was the focus of a recent investigation that aims to understand the functioning of this “epigenetic switch” on a systems level.10 One aspect of this genetic circuitry that is still unclear is the precise roles that the proteins SlrR and SlrA play in modulating the activity of SinR.
Role of SlrA in the Epigenetic Switch
Pull-down experiments using a tagged version of SlrA established that it is able to interact with both SinR and SlrR.11 These 2 protein interaction activities would appear to be in conflict because SlrA would both promote and repress biofilm formation simultaneously by interacting with and inhibiting the functions of SinR and SlrR, respectively. Using a combination of surface plasmon resonance and isothermal titration calorimetry, a high affinity interaction between SlrA and SinR but not between SlrA and SlrR10 was found, necessitating a reassessment of the role of SlrA as a dedicated antagonist of SinR. While this reassessment would simplify the role of SlrA in the circuitry of the switch, the questions would arise as to why 2 dedicated antagonists are required and how the activities or functions of these 2 antagonists differ. One possible answer to these questions may lie in the genetic contexts of the 2 proteins: SlrA is under the transcriptional control of the TetR family repressor YwcC,12 whereas SinI is expressed from a locus immediately upstream of SinR, which has a complex transcriptional profile that includes a significant amount of bicistronic sinI-sinR mRNA. This may exert its effects in 2 ways: first, by allowing further environmental signals to enter the system through the YwcC repressor; second, because the expression of SinI and SinR are temporally and spatially coupled, SinI would be better positioned to interact with SinR prior to its engagement with DNA, whereas SlrA may be in the better position to compete in the cytoplasmic pool with SlrR for SinR binding (Fig. 1). Support for this hypothesis comes from the fact SlrA mutants are only mildly repressed at the biofilm loci in comparison to SinI mutants,11 despite the similar overall affinities of the SlrA-SinR and SinI-SinR complexes (10 nM and 2 nM, respectively).10 Furthermore, it has been noted by 2 independent studies that it is not possible to displace directly the SinR tetramer from DNA10,13 in vitro, indicating that temporal, non-equilibrium effects may play an important role in repression.
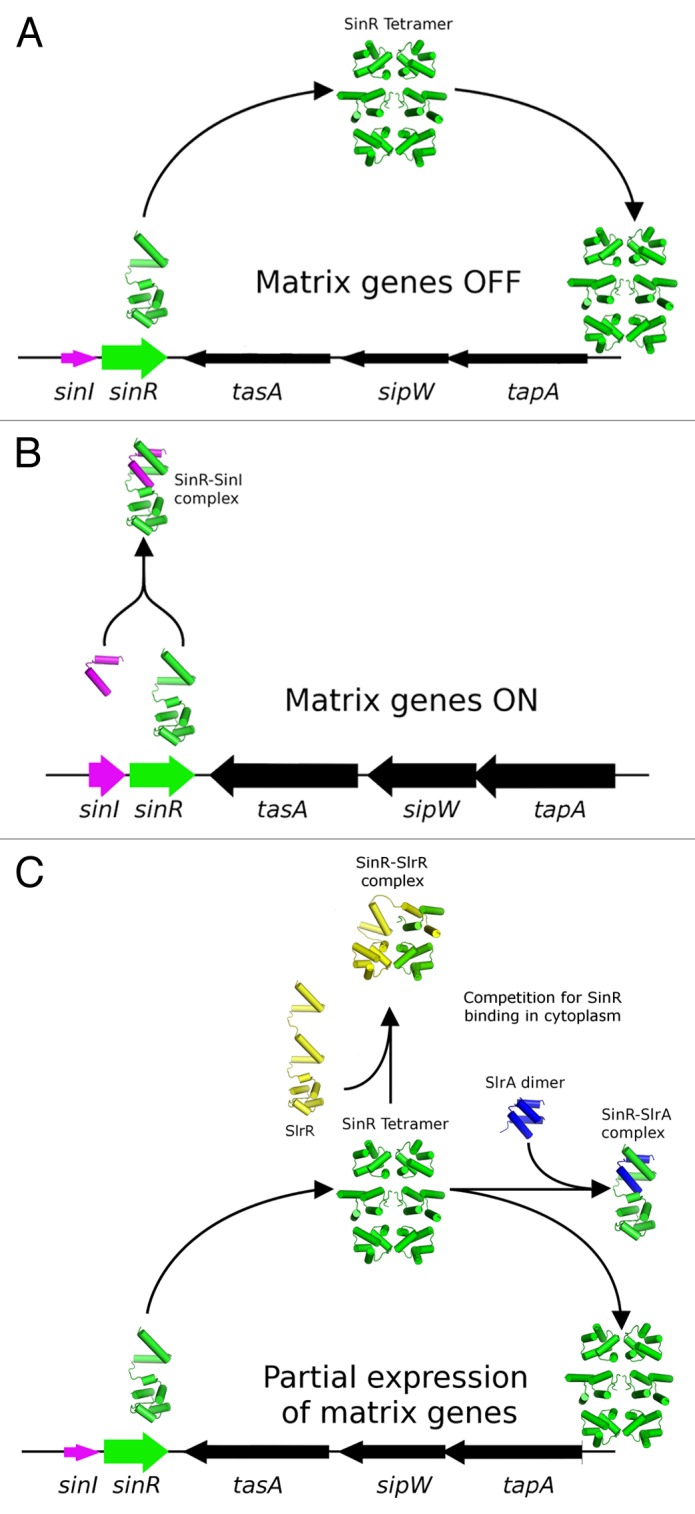
Figure 1. Schematic diagram illustrating the different roles of the antagonists SinI and SlrA in modulating the activity of SInR. (A) SinR, when expressed in the absence of antagonists, must first form the SinR tetramer before binding and repressing the activity of the nearby tapA-sipW-tasA operon. (B) SinI is located immediately adjacent to SinR in a bicistronic sinI-sinR operon, and as such is well positioned to immediately form a 1:1 complex with SinR, thereby blocking tetramerization, and the ability to repress the matrix operons. (C) While SlrA is able to bind SinR with a similar affinity, its expression from a distant chromosomal location means that it would have to compete with other agents in the cytoplasmic pool for the binding of the SlrR tetramer, and as such is less efficient at antagonising the DNA binding activity of SlrR. Levels of expression of the genes in the schematic are indicated by the thickness of the arrows that represent each gene.
Resetting the Switch by SlrR Degradation, an Alternative Model
It has been established that SlrR is subject to degradation in vivo, appearing to degrade proteolytically with a half-life of approximately 100 min,9 leading to the suggestion that SlrR undergoes autocleavage in a manner similar to the LexA family of repressors. LexA family members catalyze their autocleavage at a conserved 4-residue consensus motif “VAAG,” which matches almost perfectly to a “VQAG” sequence found within SlrR. While it is beyond doubt that SlrR is subject to proteolytic degradation, it would seem highly unlikely that SlrR contains autoendopeptidase activity. Though a LexA-like consensus motif is present in the SlrR sequence, a catalytic domain would also be required for this activity, which is certainly not present in SlrR. Moreover, the LexA-like motif is located within the first of 2 consecutive helical hook regions, the first of which appears to have diverged more significantly than the second, with the closest homologs being SinI and SlrA at 33% and 50% identities, respectively. In LexA, the cleavage site is in a flexible and accessible loop, whereas the motif in SlrR (based on its likely homology to SinR) will form an ordered α-helical structure, a site that is unlikely to be a protease substrate.
Here we present an alternate model for how SlrR is targeted for degradation. Central to this model is the fact that SlrR uniquely contains 2 helical hooks, which are capable of participating in both heterodimeric protein–protein interactions and oligomeric self-interactions. Assuming that both helical hook domains are active and, in accordance with the situation for SinR, SinI and SlrA, the hetero-oligomeric interactions are of higher affinity than the self-interactions, SlrR possesses the potential to form both “open” and “closed” complexes (Fig. 2). The open complexes would contain the potential to form aggregates in a concentration dependent manner (Fig. 2), which would then be targeted for degradation by Clp-type proteases that are specific to mis-folded or aggregated proteins.14 This model agrees with the concentration dependent aggregation behavior of the purified protein in vitro,10 and also the increased stability of SlrR in a clpC-deficient background in vivo.9 Furthermore, mutation of the putative “VQAG” consensus motif to “VQVV” increases significantly the stability of SlrR. It is most likely that this substitution, being within the helical hooks, affects the ability of the protein to form these oligomeric interactions. The experimental validation of these ideas as well as an investigation of the DNA binding activities of the SinR-SlrR complex will be the subject of further work.
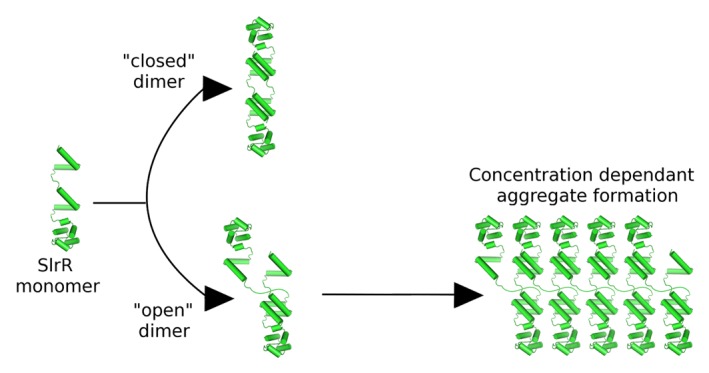
Figure 2. Schematic diagram showing the basis of an alternative model to explain the instability of SlrR, based on the assumption that the 2 interaction domains present within SlrR are able to associate with each other. The result of this is the possibility to form both “closed” and “open” (with unfulfilled interaction potential) complexes, the latter containing the obvious capacity to form large aggregates in a concentration dependent manner. These aggregates would likely be subject to degradation in vivo.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cib/article/25658
References
- 1.Branda SS, González-Pastor JE, Ben-Yehuda S, Losick R, Kolter R. Fruiting body formation by Bacillus subtilis. Proc Natl Acad Sci USA. 2001;98:11621–6. doi: 10.1073/pnas.191384198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kearns DB, Chu F, Branda SS, Kolter R, Losick R. A master regulator for biofilm formation by Bacillus subtilis. Mol Microbiol. 2005;55:739–49. doi: 10.1111/j.1365-2958.2004.04440.x. [DOI] [PubMed] [Google Scholar]
- 3.Chu F, Kearns DB, Branda SS, Kolter R, Losick R. Targets of the master regulator of biofilm formation in Bacillus subtilis. Mol Microbiol. 2006;59:1216–28. doi: 10.1111/j.1365-2958.2005.05019.x. [DOI] [PubMed] [Google Scholar]
- 4.Branda SS, Chu F, Kearns DB, Losick R, Kolter R. A major protein component of the Bacillus subtilis biofilm matrix. Mol Microbiol. 2006;59:1229–38. doi: 10.1111/j.1365-2958.2005.05020.x. [DOI] [PubMed] [Google Scholar]
- 5.Ostrowski A, Mehert A, Prescott A, Kiley TB, Stanley-Wall NR. YuaB functions synergistically with the exopolysaccharide and TasA amyloid fibers to allow biofilm formation by Bacillus subtilis. J Bacteriol. 2011;193:4821–31. doi: 10.1128/JB.00223-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kobayashi K, Iwano M. BslA(YuaB) forms a hydrophobic layer on the surface of Bacillus subtilis biofilms. Mol Microbiol. 2012;85:51–66. doi: 10.1111/j.1365-2958.2012.08094.x. [DOI] [PubMed] [Google Scholar]
- 7.Lewis RJ, Brannigan JA, Offen WA, Smith I, Wilkinson AJ. An evolutionary link between sporulation and prophage induction in the structure of a repressor:anti-repressor complex. J Mol Biol. 1998;283:907–12. doi: 10.1006/jmbi.1998.2163. [DOI] [PubMed] [Google Scholar]
- 8.Chai Y, Norman T, Kolter R, Losick R. An epigenetic switch governing daughter cell separation in Bacillus subtilis. Genes Dev. 2010;24:754–65. doi: 10.1101/gad.1915010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chai Y, Kolter R, Losick R. Reversal of an epigenetic switch governing cell chaining in Bacillus subtilis by protein instability. Mol Microbiol. 2010;78:218–29. doi: 10.1111/j.1365-2958.2010.07335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newman JA, Rodrigues C, Lewis RJ. Molecular basis of the activity of SinR protein, the master regulator of biofilm formation in Bacillus subtilis. J Biol Chem. 2013;288:10766–78. doi: 10.1074/jbc.M113.455592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chai Y, Kolter R, Losick R. Paralogous antirepressors acting on the master regulator for biofilm formation in Bacillus subtilis. Mol Microbiol. 2009;74:876–87. doi: 10.1111/j.1365-2958.2009.06900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi K. SlrR/SlrA controls the initiation of biofilm formation in Bacillus subtilis. Mol Microbiol. 2008;69:1399–410. doi: 10.1111/j.1365-2958.2008.06369.x. [DOI] [PubMed] [Google Scholar]
- 13.Colledge VL, Fogg MJ, Levdikov VM, Leech A, Dodson EJ, Wilkinson AJ. Structure and organisation of SinR, the master regulator of biofilm formation in Bacillus subtilis. J Mol Biol. 2011;411:597–613. doi: 10.1016/j.jmb.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krüger E, Witt E, Ohlmeier S, Hanschke R, Hecker M. The clp proteases of Bacillus subtilis are directly involved in degradation of misfolded proteins. J Bacteriol. 2000;182:3259–65. doi: 10.1128/JB.182.11.3259-3265.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]