

Abstract
The sequence diversity of individual human genomes has been extensively analyzed for variations and phenotypic implications for mRNA, miRNA, and long non-coding RNA genes. TRNA (tRNA) also exhibits large sequence diversity in the human genome, but tRNA gene sequence variation and potential functional implications in individual human genomes have not been investigated. Here we capitalize on the sequencing data from the 1000-genomes project to examine the diversity of tRNA genes in the human population. Previous analysis of the reference human genome indicated an unexpected large number of diverse tRNA genes beyond the necessity of translation, suggesting that some tRNA transcripts may perform non-canonical functions. We found 24 new tRNA sequences in > 1% and 76 new tRNA sequences in > 0.2% of all individuals, indicating that tRNA genes are also subject to evolutionary changes in the human population. Unexpectedly, two abundant new tRNA genes contain base-pair mismatches in the anticodon stem. We experimentally determined that these two new tRNAs have altered structures in vitro; however, one new tRNA is not aminoacylated but extremely stable in HeLa cells, suggesting that this new tRNA can be used for non-canonical function. Our results show that at the scale of human population, tRNA genes are more diverse than conventionally understood, and some new tRNAs may perform non-canonical, extra-translational functions that may be linked to human health and disease.
Keywords: tRNA, isodecoder, SNP, 1000 genomes project
Introduction
The availability of whole genome sequencing at low cost enabled sequencing of many organisms, and many individuals of the same organism. The recently completed 1000-genomes project provides DNA sequencing data for more than 1000 human individuals from many diverse ethnic groups.1,2 The 1000-genomes data have been extensively analyzed for variations in protein coding regions, DNA regulatory elements, or long non-coding RNA genes (e.g., refs. 3–9). These results have provided significant insights into human genetic variations.
An often neglected class of genes in large scale genome analysis is sequence variation of tRNA genes. tRNAs read mRNA codons in protein synthesis and bridge the nucleic acid and the amino acid worlds. Analysis by tRNAscan-SE indicates that the hg19 reference human genome contains 613 tRNA genes.10 Among these, 446 genes have a tRNAscan score of ≥ 50: these tRNAs can fold into the canonical cloverleaf structure with zero or at most one mismatch in the stem; these tRNAs are divided into 280 different sequences (Table 1). One hundred and sixty-seven genes have a tRNAscan score of < 50: more than one mismatch is present in the stems and these tRNAs are divided into 160 different sequences (Table 2). A mature tRNA contains 74–93 nucleotides; therefore, the entire repertoire of human tRNA genes is made of only ~60 000 base pairs, representing just 1.9 × 10−5 or 0.0019% of all genome sequences. Since individual human genomes are ~99.5% identical, we expect to see ~300 new tRNA single nucleotide polymorphisms (SNP), assuming random mutation frequencies and distributions. On the other hand, tRNA sequences are highly restricted through secondary and tertiary structural as well as functional constraints. Given that the reference human genome already contains an unexpectedly large number of diverse tRNA sequences,10,11 we may find very few new tRNA sequences in the population analysis. In any case, finding additional diversity in the human population may enhance the potential usefulness of tRNA in a myriad of ways such as fine-tuning translation or performing extra-translational functions where tRNA molecules may be used to regulate a wide range of cellular processes.12-16
Table 1. tRNA isodecoder genes in the hg19 reference human genome with scores ≥ 50 (from ref. 10).
| Amino acid | Anticodon | # Genes | # Isodecoders | # Reads | # New variant reads | # New sequences (> 1% population) |
|---|---|---|---|---|---|---|
| Ala | AGC | 26 | 19 | 73 488 | 10 | 2 |
| CGC | 5 | 5 | 3645 | |||
| TGC | 9 | 8 | 11 887 | |||
| Arg | ACG | 7 | 2 | 26 614 | 30 | 1 (1) |
| CCG | 4 | 2 | 2981 | |||
| TCG | 6 | 6 | 11 101 | |||
| CCT | 5 | 5 | 10 903 | |||
| TCT | 6 | 5 | 50 969 | 10 | 2 | |
| Asn | ATT | 1 | 1 | 2048 | ||
| GTT | 23 | 15 | 76 577 | 18 | 3 | |
| Asp | GTC | 15 | 5 | 34 214 | ||
| Cys | GCA | 30 | 24 | 207 063 | 79 | 6 (1) |
| Gln | CTG | 13 | 7 | 53 467 | ||
| TTG | 6 | 4 | 11 616 | |||
| Glu | CTC | 8 | 2 | 40 167 | ||
| TTC | 8 | 5 | 18 824 | 6 | 1 | |
| Gly | GCC | 14 | 5 | 43 046 | 3 | 1 |
| CCC | 7 | 5 | 13 301 | |||
| TCC | 9 | 4 | 67 427 | 14 | 3 | |
| His | GTG | 10 | 2 | 16 427 | ||
| Ile | AAT | 14 | 9 | 62 572 | 21 | 1 (1) |
| GAT | 3 | 1 | 823 | |||
| TAT | 5 | 3 | 88 809 | 21 | 2 (1) | |
| Leu | AAG | 9 | 4 | 28 073 | ||
| CAG | 9 | 2 | 86 177 | |||
| TAG | 3 | 3 | 9130 | |||
| CAA | 7 | 6 | 441 612 | 1181 | 4 (3) | |
| TAA | 4 | 4 | 10 577 | |||
| Lys | CTT | 16 | 11 | 41 580 | 13 | 2 |
| TTT | 14 | 10 | 28 393 | |||
| Met-i | CAT | 9 | 2 | 12 798 | ||
| Met-e | CAT | 10 | 7 | 29 580 | ||
| Phe | GAA | 12 | 7 | 39 527 | 97 | 3 (2) |
| Pro | AGG | 9 | 2 | 5250 | ||
| CGG | 4 | 2 | 2378 | |||
| TGG | 7 | 3 | 4,372 | |||
| SeC | TCA | 2 | 2 | 5737 | ||
| Ser | AGA | 10 | 5 | 24 101 | ||
| CGA | 4 | 4 | 12 249 | |||
| TGA | 4 | 4 | 11 047 | |||
| GCT | 8 | 6 | 75 198 | |||
| Sup | TTA | 1 | 1 | 1288 | ||
| Thr | AGT | 9 | 6 | 16 476 | ||
| CGT | 5 | 5 | 5354 | |||
| TGT | 6 | 6 | 17 215 | |||
| Trp | CCA | 7 | 5 | 14 745 | ||
| Tyr | ATA | 1 | 1 | 7672 | 7 | 2 |
| GTA | 12 | 8 | 236 582 | 262 | 4 (2) | |
| Val | AAC | 10 | 6 | 13 976 | ||
| CAC | 15 | 10 | 24 976 | |||
| TAC | 5 | 4 | 8809 | |||
| Total | 446 | 280 | 2 141 895 | 37 (11) |
Table 2. tRNA isodecoder genes in the hg19 reference human genome with scores < 50 (from ref. 10).
| Amino acid | Anticodon | # Genes | # Isodecoders | # Reads | # New variant reads | # New sequences (> 1% population) |
|---|---|---|---|---|---|---|
| Ala | AGC | 4 | 4 | 4697 | ||
| CGC | ||||||
| TGC | 2 | 2 | 6377 | 19 | 2 | |
| Arg | ACG | |||||
| CCG | ||||||
| TCG | ||||||
| CCT | 5 | 4 | 7252 | |||
| TCT | ||||||
| Asn | ATT | |||||
| GTT | 12 | 8 | 17 969 | 4 | 1 | |
| Asp | GTC | 5 | 5 | 6647 | ||
| Cys | ACA | 1 | 1 | 206 118 | 2496 | 4 (4) |
| GCA | 1 | 1 | 1,573 | |||
| Gln | CTG | 13 | 7 | 47 285 | 55 | 6 (1) |
| TTG | 6 | 4 | 314 169 | 20 | 5 | |
| Glu | CTC | 16 | 16 | 22 886 | 3 | 1 |
| TTC | 11 | 11 | 20 322 | 6 | 1 | |
| Gly | GCC | 1 | 1 | 1463 | ||
| CCC | 2 | 2 | 6464 | |||
| TCC | 2 | 2 | 18 335 | 17 | 1 (1) | |
| His | GTG | 1 | 1 | 1122 | ||
| Ile | AAT | 3 | 3 | 5018 | ||
| GAT | ||||||
| TAT | ||||||
| Leu | AAG | 4 | 4 | 12 789 | 3 | 1 |
| CAG | 1 | 1 | 3992 | 3 | 1 | |
| TAG | 1 | 1 | 2763 | |||
| CAA | 1 | 1 | 1958 | |||
| TAA | 7 | 7 | 424 907 | 340 | 4 (2) | |
| Lys | CTT | 5 | 5 | 6833 | ||
| TTT | 7 | 7 | 9,184 | |||
| Met-i | CAT | 1 | 1 | 1830 | ||
| Met-e | CAT | |||||
| Phe | GAA | 5 | 5 | 7052 | ||
| Pro | AGG | 4 | 4 | 24 926 | 4 | 1 |
| GGG | 1 | 1 | 1470 | |||
| TGG | 3 | 3 | 8978 | 26 | 1 (1) | |
| SeC | TCA | 1 | 1 | 1460 | ||
| Ser | AGA | 2 | 2 | 4207 | ||
| CGA | ||||||
| TGA | 3 | 3 | 199 533 | 16 | 2 | |
| ACT | 1 | 1 | 654 | |||
| Sup | TTA | 2 | 2 | 70 512 | 254 | 4 (3) |
| CTA | 1 | 1 | 38 092 | 353 | 2 (1) | |
| Thr | AGT | 1 | 1 | 2314 | ||
| CGT | 1 | 1 | 1360 | |||
| TGT | ||||||
| Trp | CCA | 2 | 2 | 10 549 | 7 (1) | 1 |
| Tyr | ATA | |||||
| GTA | 4 | 4 | 10 881 | |||
| Val | AAC | 1 | 1 | 1345 | ||
| CAC | 5 | 4 | 11 349 | 3 | 1 | |
| TAC | 1 | 1 | 121 980 | |||
| Total | 167 | 160 | 1 668 615 | 3,629 | 39 (13) |
Distinct tRNAs with the same anticodon but with sequence variations in the body—termed tRNA isodecoders11—can serve biologically non-redundant roles. Selective tRNA isodecoder expression analyzed by Chip-seq using RNA pol III antibodies and by direct RNA-seq has been shown to depend on cell type and cell state.17 The silkworm Bombix Mori displays differential tRNA gene expression for tRNAAla(AGC) isodecoders.18,19 A unique tRNA isodecoder is exclusively transcribed in silk glands, whereas another is transcribed ubiquitously in all cell types. An E. coli tRNATrp isodecoder contains a sequence change in the D-stem, yet, this isodecoder completely switches its decoding from UGG to UGA codon.20 A systematic study of human tRNA isodecoders as UAG suppressors showed a 20-fold difference in stop codon suppression efficiency in a human cell line, even though all isodecoders shared similar aminoacylation capacity and stability in vivo.21 A human tRNA isodecoder modulates the expression level of a tRNA synthetase gene through direct interaction with the 3′UTR of its mRNA, leading to the formation of alternate polyadenylation sites.15 Other roles for tRNAs have been suggested,22 including a recent demonstration that tRNAs interact with a wide range of human proteins in the cell, these proteins are previously not known to interact with any RNA.16
Here we capitalize on the data provided by the 1000-genomes project to examine the diversity of tRNA genes in the human population. Starting from the known tRNA genes in the hg19 reference genome, we first identify SNPs in all tRNA genes that contain new sequence variations in the tRNA body. We then examine the distribution of these new SNPs among tRNA species and map the new sequence change hotspots on the tRNA structure. We then focus on the tRNA genes with tRNAScan score of ≥ 50 and break down how these new genes are distributed across the ancestries of people participating in the 1000-genomes project. We link human ancestries to the occurrence of specific new genes and vice versa. Surprisingly, two abundant new isodecoders contain sequence changes in the anticodon stem that disrupt Watson-Crick base pairing. We experimentally compare the structural differences between three pairs of isodecoder transcripts—with and without the disrupted stem—and found that two new isodecoders with mismatch in the anticodon stem indeed fold differently compared with the standard tRNA. We also show that one new isodecoder with anticodon stem mismatch has the same half-life in cells as its counterpart, but is charged at much lower levels compared with the standard tRNA in vivo. Our results suggest that some new tRNA isodecoders may perform non-canonical functions in distinct human populations.
Results
Bioinformatics pipeline
The huge amount of data stemming from the sequencing of more than 1000 people warrants a careful plan for bioinformatics surveys (Fig. S1). Two databases were used to carry out our analysis: the genomic tRNA database10 (Fig. S1A), in which all tRNAs from the hg19 version of the human genome have been tabulated (Fig. S1B), and the 1000 genomes project23 (Fig. S1C) containing genome-wide, deep-sequenced DNA fragments for n = 1617 individuals (Fig. S1D). The 1000 genomes project was launched to provide a comprehensive resource on human genetic variations. DNA from a pool of cells from a given individual was fragmented to small pieces and then sequenced at a depth of ~4x coverage. This coverage was not enough to reconstruct each individual’s genomes, but sufficient to find most genetic variants that have frequencies of at least 1% in the studied populations. About two weeks of computations on 20 quad-cores CPU cluster (~1100 CPU-days) were required to perform ~93 000 of a three-step job (average of 17 min/job) (Fig. S1E): (1) download deep-sequencing reads data file pertaining to an individual from the 1000-genomes project website; (2) map the data on tRNA genes; and (3) save the mappings aside and wipe out the reads data file (as their cumulative size would be too large to store on our file server).
Mapped deep sequencing reads
Deep-sequencing reads were mapped on the known tRNA genes in the reference human genome and subsequently assigned a score (Fig. S2). The score of an aligned read is simply the number of matches minus the number of mismatches. Hence, if the same read can be aligned at two or more genomic loci, the locus with the highest score retains the read, and other mappings are discarded (Fig. S2A). As tRNA isodecoder genes often share significant sequence overlap, mappings of equal scores are all kept, regardless of the number of targeted genes. About 70% of individuals contain at least 500 mapped reads on their tRNA genes (Fig. S2B). The number of mapped reads depends on the extent of the genomic footprint of tRNA genes (estimated to be ~60 000 on 3.2 billion nucleotides or 0.0019%) and also on the deep-sequencing depth or coverage. A grand total of 3 810 510 reads found a hit on tRNA genes, with input read lengths between 26–174 nucleotides (Fig. S2C). The resulting mapped alignment lengths (Fig. S2D) showed an expected exponential decrease with spikes corresponding to input lengths. In an effort to understand how degenerate tRNA fragments can be with respect to the whole human genome, we broke down all tRNA genes into 22 nucleotides fragments and mapped them back on RNA transcripts and repetitive elements. Three genomic loci showed up with significant overlap with some tRNA genes (Fig. S3)—sequencing reads mapping on these loci will not be possible to determine their true genomic origin with certainty.
To uncover new SNPs in tRNA, hence new tRNA isodecoder genes, one ideally starts by assembling a person’s chromosomes from DNA sequencing data. The program tRNAscan-SE24,25 can then be used to identify all tRNA genes. This method has been previously applied to identify tRNA genes in the reference human genome.25 The sequencing reads from the 1000-genomes project are too short and sequencing coverage not deep enough to enable assembly of individual chromosomes. The application of tRNAscan-SE would not work in our case to identify new tRNA SNPs. Therefore, we matched any potential new tRNA SNP to the existing pool of known tRNA genes in the reference human genome (Fig. 1).
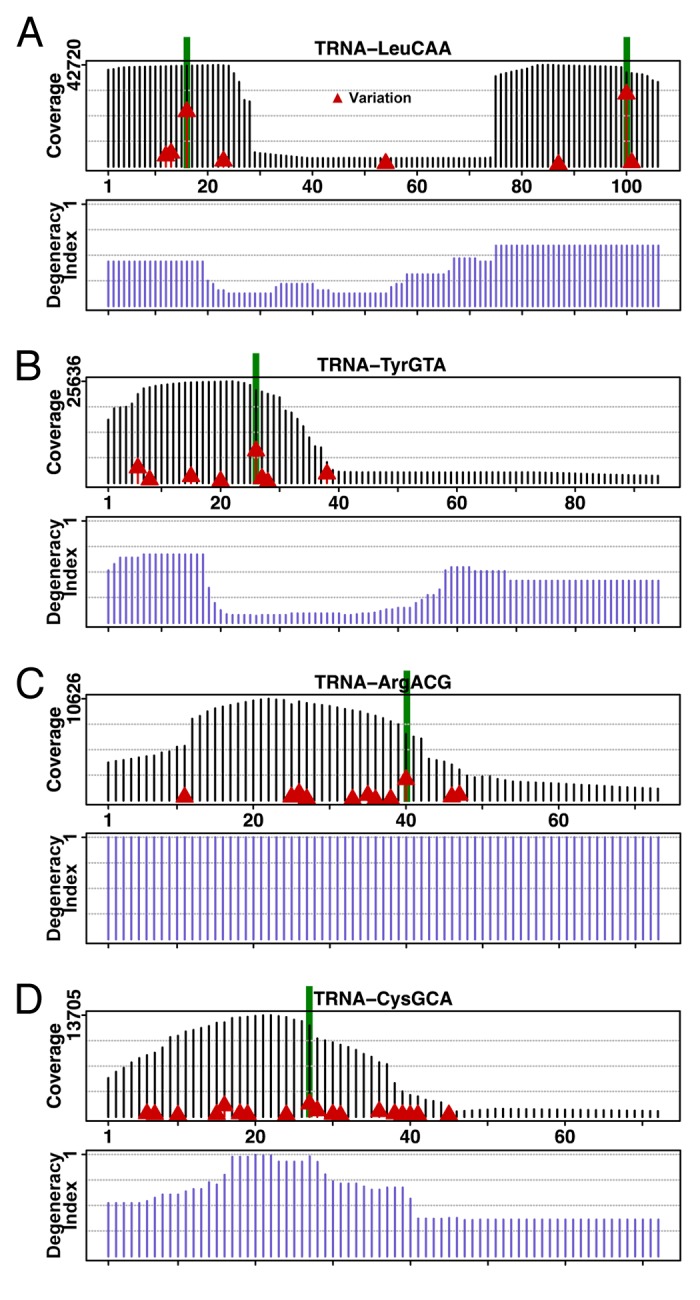
Figure 1. Coverage and sequence mismatch frequencies on selected tRNA genes. Deep sequencing data of DNA of individuals participating in the 1000-genomes project mapped on tRNA genes shows coverage along the gene and highlights sequence mismatches for each gene’s positions. All baseline sequences are from the corresponding tRNA gene sequences in the hg19 reference genome.10 The coverage is determined by piling up all deep sequencing reads that map to the gene, and shown as vertical black bars. Sequence mismatch frequencies per position are marked by red triangles set at heights proportional to the frequencies: higher red bars indicate higher mismatch frequencies. Five highest mismatched columns are highlighted in green. Selected tRNAs are: (A) tRNALeu(CAA). The well-shaped coverage is due to the fact that only a few tRNALeu genes have introns at positions corresponding to the bottom of the well. (B) tRNATyr(GUA). Greater coverage at the 5′ region of the tRNA gene gives an indication of the degeneracy of that region among the isodecoders. (C) tRNAArg(ACG). (D) tRNACys(GCA).
The coverage along a given tRNA gene is generally not uniform because most tRNA isodecoder genes share identical sub-sequences in certain regions. To get a quantitative view on how “degenerate” are these sub-sequences in a family of tRNA isodecoders, we devised a “degeneracy index” plot (Fig. 1). This plot shows what fraction of tRNA isodecoders share the same 22-mer starting at a given gene position. For instance, the degeneracy index for the 5′ end of tRNALeu(CAA) is ~0.5, meaning that half of tRNALeu(CAA) isodecoders have the same 5′ sub-sequence (Fig. 1A). The sequence around the anticodon stem-loop is more unique to each isodecoder family as the degeneracy index drops then rises again toward the 3′ end. We found that the deep-sequencing coverage qualitatively agrees with the degeneracy index.
Mapped reads on tRNA genes can give hints as to where the hotspots of variation on particular tRNAs are. Indeed, a census of the most varied tRNA positions points to a few tRNA genes where the matched sequences do not agree with the tRNA genes in the hg19 reference genome (Fig. 1). Sequencing error rates as high as those seem unlikely, and thus, these positions are candidates for new tRNA SNPs. A more thorough examination of tRNA position or column coverage shows that about 65% of the total tRNA gene positions have at least 1000-fold coverage (Fig. S4A), with more than half of tRNA gene positions (55%) exhibiting a variation rate less than or equal to 0.5% (Fig. S4B). We found no correlation between the coverage in a tRNA gene position and its variation rate (Fig. S4C) nor its sequence entropy (Fig. S4D). Finally, most tRNA gene positions have low sequence entropies (Fig. S4E), and entropy increases as a function of a column’s variation rate (Fig. S4F). Figure S4G shows the variation rates vs. column coverage for the 5′ 30 nucleotides of tRNA genes with scores ≥ 50, and another plot for the 3′ 40 nucleotides with scores < 50. There is no correlation between deep sequencing coverage and the variation rates for these specific areas.
Mapped reads per individual
The distribution of ancestry codes from the sampled individuals shows a rough uniform coverage per ancestry code (Fig. S5A). If we instead look at the number of individuals for whom we have deep-sequencing information for a particular tRNA isodecoder, only 20 isodecoders of the possible 280 (with tRNAscan-SE score ≥ 50) are present in at least half of the population (Fig. S5B). Furthermore, only about 30% of the individuals have more than 1000 tRNA gene positions with > 10x coverage among the total of 21 787 unique tRNA sequence positions (Fig. S5C). Most individuals contain no new positions, and the vast majority of people carry less than 10 new SNPs (Fig. S5D). Even though these numbers seem low, they are consistent with the small footprint of tRNA genes on the whole human genome. It also validates the hg19 genome as a good “reference” genome as few new tRNA isodecoders are found.
New isodecoder gene occurrences
Because we focus on tRNA gene diversity, we need to define what we call a “new” isodecoder gene. For a given individual, if more than 2/3 of a tRNA position with ≥ 10x coverage disagree with the reference genome sequence but consistently display the same nucleotide change, we flag that tRNA position as changed. Isodecoder sequences are almost all identical to one another so it is difficult to say exactly which isodecoder gene in the reference genome (Tables 1 and 2) is altered in a given individual. At the least, the very presence of an altered position in one or several tRNA isodecoders hints at the presence of a new tRNA isodecoder instance in the individual’s genome. Unfortunately, the limited sequencing depth does not allow us to flag those tRNA isodecoder genes that are absent in an individual with respect to the reference hg19 genome.
We separated the reference tRNA genes into two groups, one with tRNAscan-SE score ≥ 50 (Table 1) and the other with the score < 50 (Table 2) to facilitate analysis. The high scoring tRNAs are generally expected to fold into the canonical cloverleaf structure needed to function in translation. Among the high scoring tRNAs, the most abundant new tRNA isodecoder genes are tRNALeu(CAA) (53%) followed by tRNATyr(GUA) (22%) (Fig. 2A). For tRNALeu(CAA), the majority of sequence variations are found in the D stem-loop, and others in the acceptor stem. The most frequent sequence change is U16 to C, which is not expected to affect tRNA structure and function in any significant way.26 Other sequence changes switch the C12 = G23 base pair to a U = G wobble, G6 = U67 wobble to a G = C, and G13 = U22 to an A = U. All these base pair changes are isosteric or near isosteric.27 For tRNATyr(GUA), the most frequent change replaces the G26 = A44 purine pair with an A = A pair. In the tertiary structure of tRNA, the G = A mismatch is in imino-form rather than sheared, so that the imino A = A base pair would stay near isosteric27 but slightly weaken the overall structure upon the loss of an out-of-plane hydrogen bond provided by G.28 The other sequence change of G6 = C67 would introduce an A6 = C67 base pair, also near isosteric, but may require the A to be protonated.29

Figure 2. Distribution of new tRNA genes. All reference genome tRNA genes are separated into two groups according to their tRNAscan-SE scores to facilitate analysis. (A–C) Scores ≥50 listed in Table 1; (D–E) Scores <50 listed in Table 2. (A) High-scoring tRNA group. Distribution of new isodecoder instances per isoacceptor family. The most frequently occurring new isodecoder codes for leucine (53%), followed by tyrosine (22%). Only those tRNAs with more than five new instances are shown. Unique new isodecoder instances found in >1% of the population is also shown. Each instance is tagged with a Roman number. The nucleotide positions are adjusted according to the standard tRNA nomenclature. The new genes indicated by a star were tested experimentally for their folding and in vivo charging and stability. (Inset) Distribution of new isodecoder instances among individuals. Over half (54%) of all individuals carry at least one new isodecoder. Most individuals (81%) carry only one or none new isodecoder, while few individuals have as many as 11 new tRNA isodecoders. (B) Sequence change for the 11 new genes that are present in > 1% population mapped onto the canonical tRNA structure. Roman numerals correspond to those of the previous panel. (C) Box-and-whisker plot showing the distribution of new isodecoder instances per ancestry codes. Thick horizontal bars indicate the median, while the bottom and top of the boxes the first and third quartiles of the distributions, respectively. (D) Low-scoring tRNA group. Distribution of new isodecoder instances per isoacceptor family. The most frequently occurring new isodecoder codes for cysteine (57%), followed by a suppressor tRNA (17%). Only those tRNAs with more than five new instances are shown. Unique new isodecoder instances found in > 1% of the population is also shown. Each instance is tagged with a Roman number. The nucleotide positions are adjusted according to the standard tRNA nomenclature. (E) Sequence change for the 13 new genes that are present in >1% population mapped onto the canonical tRNA structure.
A total of 11 new isodecoders are found in at least 1% of the population (Fig. 2A). Six of the 11 sequence changes are in the D stem-loop (Fig. 2B). Analysis of tRNA function highlights the fact that the D-stem/loop contains fewest tRNA determinant and antideterminant elements for aminoacylation and in ribosome decoding compared with the acceptor stem, anticodon, and T stem-loops.30 Two of the 11 sequence changes are in the acceptor stem and three of the 11 in the anticodon stem. The T stem-loop is completely devoid of any new isodecoders. We did not thoroughly analyze sequence changes occurring in the variable loop because those positions are difficult to align due to a greater number of insertions and deletions.
We further assessed the 11 new tRNA isodecoder sequences with all isodecoders in the reference genome (Fig. S6). For six new isodecoders, the assessment is unambiguous because no isodecoder in the reference genome contains the specific altered nucleotide (Fig. S6A–F). In five other cases, other isodecoder sequences from the same isoacceptor family in the reference genome contain the same nucleotide identity as the new isodecoder. In these cases, more thorough analysis shows that the entire sequencing reads containing the altered nucleotide in our new isodecoders matches only to specific isodecoders in the reference genome (Fig. S6G–K). For example, the new tRNALeu(CAA) isodecoder contains a C at position 16 and CAGA in the anticodon stem located 13–16 nucleotides downstream of C16 (Fig. S6G). Although several known genes in the tRNALeu(CAG) isoacceptor family also contain C16, these tRNA genes have UGCG instead in the anticodon stem. Therefore, the new isodecoder with C16/CAGA only matches the U16/CAGA tRNALeu(CAA) in the reference genome.
New isodecoder gene occurrences are not distributed equally among individuals. More than half of all individuals (54%) carry one new occurrence; about one-quarter (27%) have no new occurrence (Fig. 2A inset). Only one person in five (20%) has two or more new occurrences. A breakdown by ancestry shows that individuals from European ancestry have more new isodecoder occurrences; individuals from East Asian ancestry seem to have fewer new isodecoders (Fig. 2C). The reason for this discrepancy is unclear, it may be related to the choice of individual samples in the 1000-genomes sequencing project. Alternatively, it is possible that the Europeans indeed have a slightly higher tRNA genome diversity than East Asians.
The low-scoring tRNAs generally have two or three mismatches in the stems. Among these tRNAs, the most abundant new tRNA isodecoder genes fall in two groups from tRNACys(ACA) (36% for the top of the group) followed by tRNASup(CTA/TTA) (9.6% for the top of the group) (Fig. 2D). The reference genome contains one annotated tRNACys(ACA), one annotated tRNASup(CTA), and two annotated tRNASup(TTA) genes (Table 2). The high abundance of new isodecoder occurrences in these low-copy tRNA genes suggests that low scoring tRNA genes are under low evolutionary pressure for conservation. In two cases, the new sequence variants restore base pair mismatches to Watson-Crick base pairs (Fig. 2D; CysACA from CxU to C-G and SupTTA from AxC to A-U), which could “rescue” a poorly folded tRNA into a canonical structure. In contrast to the high-scoring tRNAs, most new variants among this group contain sequence changes in the acceptor and the T stems (Fig. 2E). These tRNA regions are more subject to canonical tRNA function constraints including aminoacylation and ribosome decoding. The high degree of variation in these regions is consistent with the low scoring tRNAs serving potential non-canonical roles, e.g., in gene expression control through direct binding to specific mRNA.15
New isodecoder genes vs. ancestries
We applied Bayes’ theorem to uncover any relationship between specific nucleotide content in specific tRNA positions with respect to ancestries. Bayes’ theorem computes conditional probabilities. A conditional probability reassesses a probability of an event given the conditional knowledge that another event took place. Consider the probability that a person in our analysis is from the Finnish ancestry, noted as P(FIN). Consider also the probability that a person features a C at position 26 in one of its tRNA isodecoders, noted as P(C@26). These two events may or may not be linked. In a contingency table (Fig. S7), the counts of various ancestry codes are compiled in columns and the nucleotide content at position 26 in rows, we can compare the marginal probability P(FIN) with the conditional probability P(FIN|C@26), the latter being the probability of a person to be of Finnish ancestry given that it contains C26 in a given tRNA. Likewise, we can compare the marginal P(C@26) with the conditional P(C@26|FIN). When a conditional probability is higher than the marginal a priori probability, the knowledge of the condition yields higher probabilities than the a priori ones: hence a gain—the ratio of the conditional to the marginal probabilities. Selected conditional probabilities with gains greater than 1.5 are presented in Figure 3.

Figure 3. Population analysis of the most frequent new tRNA sequences among the high scoring tRNA group. (A) Sequence change mapped onto the canonical tRNA secondary structure for tRNALeu(CAA), the tRNA gene with the most abundant new instances. Square bubbles indicate the nucleotide substitution frequencies. Conserved nucleotides are shown in black, while the anticodon nucleotides in gray. Dotted lines show base paired positions. (B) Probability table conditioned on ancestry for position 16 of tRNALeu(CAA). The SoAs label stands for South Asian, Amer for Americas, WeAf for West Africa and Euro for European. (C) Sequence change mapped onto the canonical tRNA secondary structure for tRNATyr(GUA), the tRNA gene with the second most abundant new instances. (D) Probability table conditioned on ancestry for position 26 of tRNATyr(GUA). The SoAs label stands for South Asian, EaAs for East Asian. (E and F) Sequence changes in the anticodon stem in two other abundant new isodecoders. The sequence changes introduce a purine mismatch in the anticodon stem. (E) Position 40 of tRNAArg(ACG) occurring in 15.5% population. (F) Position 27 of tRNACys(GCA) occurring in 11.3% population.
Some ancestry codes can be deduced with almost certainty when considering the presence of certain nucleotides at given positions (Fig. 3; Fig. S7). For instance, finding C26 in tRNATyr(GUA) almost certainly predicts a Finnish ancestry. Other such conditional probabilities are also present (Fig. S7D). On the other hand, there is only a ~2% probability for all individuals with Finnish ancestry with C26 in tRNATyr(GUA). For tRNATyr(GUA), the probability to find A26 for individuals from East Asia is 50%, whereas the probably to find G26 for individuals from West Asia is 90% (Fig. S7D). For tRNALeu(CAA), South Asians have a C at position 16, while West Africans and Europeans a U half of the time (Fig. 3A and B).
We further focused on the three new isodecoders that alter base pairs in the tRNA anticodon stem, a critical region for tRNA function (Fig. 3C–F). These three isodecoders occur in 11–27% of all individuals. For tRNATyr(GUA), the reference genome tRNA features a G26 = A44 purine pair, the new isodecoder changes this to an A26 = A44 pair (Fig. 3C and D). As discussed previously, the effect of the G26-to-A change on the tRNA structure may be negligible. For tRNAArg(ACG), the reference genome tRNA features a G30 = C40 Watson-Crick base pair, the new isodecoder changes this to G30 = G40 (Fig. 3E). Not only would the G = G mismatch impact the double-helix grooves leading to a greater distortion, but depending on the flanking base pairs, G = G can also oscillate between two mirror-image conformations, going from (syn)H/W(anti) to (anti)W/H(syn) and back.31 For tRNACys(GCA), the reference genome tRNA features a U28 = A42 Watson-Crick base pair, the new isodecoder changes this to G28 = A42 (Fig. 3F). The surrounding structural context may drive this G = A mismatch in a (syn)H/W(anti) base pair, where the adenosine would be protonated.32
Comparative structural mapping of tRNA isodecoders in vitro
We were surprised to find two new tRNA isodecoders containing anticodon stem mismatches that are abundantly present in the human population (Fig. 3E and F). These mismatches may not significantly affect the tRNA structure so that they can also be used in translation. Alternatively, these mismatches could have strong effects on tRNA structure and consequently hinder its function in translation. In the latter case, either the presence of these isodecoders is simply tolerated in cells or these isodecoders perform extra-translational functions that avoid the competition by the ribosome.22 We therefore experimentally tested whether the isodecoders containing anticodon stem mismatches indeed affect the tRNA structure in vitro (Fig. 4) and its stability and aminoacylation in vivo (Fig. 5).

Figure 4. Structural mapping of pairs of tRNA isodecoder transcripts in vitro. Structural mapping of selected tRNA isodecoder pairs was performed by limited nuclease V1 and S1 digestion. Sequences in the context of standard tRNA secondary structure and mapping of tRNAArg(ACG) (A), tRNACys(GCA) (B), and tRNATyr(GUA) (C). Positions of RNA cuts were identified through alkaline hydrolysis (BH lane) and T1 ladder (T1 lane). The sequence change in the new tRNA isodecoder is indicated by red arrows and corresponds to the position of the red dot in gel. tRNA structural regions are indicated on the right side of the gel. (D) tRNA folding analysis by native gel electrophoresis. tRNA transcripts from T7 RNA polymerase reaction was directly loaded on 8% native gel containing 25 mM trisOAc, pH 7.5, 5 mM Mg(OAc)2. The amount of transcription reaction loaded in the left and right panels were 1 and 10 µl, respectively. Consistent with the structural mapping results, tRNATyr(GUA) variants fold in the same manner, whereas tRNAArg(ACG) variants fold very differently.

Figure 5. Stability and charging level of tRNAArg(ACG) isodecoder pair in vivo. (A) tRNA transcripts 32P-labeled at the terminal A76 were transfected into HeLa cells. Total RNA was isolated at designated time points, and equal amount of total RNA was loaded in each lane. tRNAArg(ACG) from the reference genome has C40 and is indicated as C lanes; the new isodecoder has G40 and is indicated as G lanes. (B) Quantitative analysis of the 32P-labeled tRNA bands in panel A. Data are normalized to the 36 h time point where it has the highest level of radioactivity. (C) tRNA transcripts 32P-labeled at the terminal A76 were transfected into HeLa cells. Total RNA under acidic conditions was isolated at designated time points. Equal amount of total RNA was digested with nuclease P1 and run on TLC. Different charging level of tRNA isodecoders at different time points analyzed by TLC. Free and acylated tRNA are indicated on the right side. (D) Quantitative analysis of the p*A and p*A-arg spots in panel A. The y-axis corresponds to the % of p*A-arg spot over p*A+p*A-arg signals.
To test whether the introduction of specific mismatches in the anticodon stem affects tRNA structure, we selected all three isodecoder pairs in Figure 3D–F that are either of standard tRNA configuration (for those in the reference genome) or contain one base pair change in the anticodon stem (new isodecoders). All three tRNA isodecoders from the reference genome are made of standard tRNA configuration as depicted by their predicted secondary structures (Fig. 4). Each of the new isodecoders for tRNAArg(ACG) and tRNACys(GCA) introduces a purine–purine mismatch. The new tRNATyr(GUA) isodecoder changes the G26 = A44 pair to an A26 = A44 pair. The structure of the isodecoder pairs was compared by structural mapping of T7 RNA polymerase transcripts using the single-stranded-specific nuclease S1 and the double-stranded/stacking-specific nuclease V1 (Fig. 4). For tRNAArg(ACG) (Fig. 4A), the S1 digestion signature increases significantly in the new isodecoder (G40), both in the vicinity of G40 and in the D loop. The V1 digestion signature increases in the D stem region and is expanded in the anticodon stem. These results indicate that the C40 to G40 change in the new isodecoder alters the structure of the anticodon and the D stem-loops. For tRNACys(GCA) (Fig. 4B), the S1 digestion signature increases slightly in the new isodecoder (G29), both in the anticodon loop and in the D loop. The V1 digestion signature increases in the D stem and markedly in the anticodon stem-loop region. These results again indicate that the U29 to G29 change in the new isodecoder alters the structure of the anticodon and the D stem-loops. For tRNATyr(GUA) (Fig. 4C), very little difference is observed as anticipated between the reference isodecoder (G26) and the new isodecoder (A26), indicating that this new isodecoder folds into a standard tRNA structure. The structural mapping results support the notion that the two new isodecoders with purine–purine mismatch in the anticodon stem contain a distinct structure compared with a standard tRNA.
We also performed tRNA folding analysis under non-denaturing conditions to validate that the structural difference between the two tRNAArg variants is not derived from the process of denaturation/renaturation (Fig. 4D). Both tRNAArg transcripts in the T7 RNA polymerase transcription mixture were directly loaded on native gels. The folding difference between these two variants was easily visualized.
Comparative analysis of tRNA stability and charging in vivo
To determine whether structural changes affect tRNA function, we further compared the stability and the charging efficiency of the tRNAArg(ACG) isodecoder pair in vivo (Fig. 5). tRNA transcripts were 32P-labeled at the terminal A76 residue using α-32P-ATP and the recombinant CCA-adding enzyme.33 The radiolabeled tRNAs were transfected into HeLa cells for up to 96 h. To measure stability, total RNA was extracted at various time intervals and analyzed using denaturing PAGE (Fig. 5A). For both the reference isodecoder (C40) and the new isodecoder (G40), the relative amount of 32P-labeled tRNA in the total RNA comprises of two phases (Fig. 5B). Within the first 36 h after transfection, the amount increases and reaches a plateau; this result is likely due to the increased uptake of tRNA during this time. From 36–96 h, the amount follows a steady decline by ~50% every 24 h; this result is consistent with the dilution of the radiolabeled tRNA upon additional cell growth. In any case, both isodecoders show similar uptake and decline kinetics, indicating that they are similarly stable in HeLa cells.
We also measured the charging level of both tRNAArg(ACG) isodecoders at 12 and 24 h after transfection (Fig. 5C and D). Here, total RNA from transfected cells was extracted under acidic conditions to maintain charged tRNAs. The charged tRNAs were then treated with nuclease P1, and the free 32P-labeled A76 (p*A) and charged A76 (p*A-arg) were separated by thin-layer chromatography.33 The reference genome isodecoder (C40) is charged at similar levels at both 12 and 24 h, whereas the new isodecoder (G40) is charged at 5–10 times lower levels (Fig. 5D). The absolute charging level of the C40 isodecoder is just a few percent: this result is consistent with the literature showing that transfected tRNA transcripts are generally charged at levels below 10%, presumably due to the lack of modification status, incorrect processing pathway, incorrect localization, or a combination of all three.21,34 For other tRNAs investigated previously, this low percentage charging is still sufficient to demonstrate their usefulness in translation.21 In any case, the extremely low charging levels of the new tRNAArg(ACG) isodecoder indicates that it is not useful for translation, even though it is equally stable as a standard tRNA.
Discussion
tRNA isodecoder gene diversity in the human population
The presence of an unexpectedly large tRNA isodecoder diversity in the mammalian genomes has been a paradox since their initial identification.11 In single cell organisms such as bacteria and yeast, many tRNA genes with the same anticodon, i.e., within the same isoacceptor family, have identical sequences. For example, Saccharomyces cerevisiae contains 275 tRNA genes distributed within just 51 different sequences for a gene number/gene sequence ratio of 5.4. The hg19 reference human genome contains 446 tRNA genes (those with tRNAScan-SE score of ≥ 50) distributed within 280 sequences for a gene number/gene sequence ratio of 1.59. At a first glance, large tRNA gene diversity may harm the cell through increased propensity of misacylation by non-cognate tRNA synthetases. Investigations by Chip-seq of RNA polIII subunits and transcription factors indicate that a majority of the tRNA isodecoder genes are transcribed in different cell types.17 Interestingly, different cell types transcribe a small, common set of tRNA genes, but a large, distinct set of tRNA isodecoder genes, suggesting each cell type may have a unique expression pattern of tRNA isodecoder transcripts.35
The human reference genome is composed of pooled DNA from several dozen individuals.36 Hence, the tRNA isodecoder gene collection in the genomic tRNA database10 does not represent simultaneous tRNA genes present in any particular individual. In the absence of assembled genome sequences from the 1000-genomes project, it is very difficult to detect tRNA gene insertions and deletions, so the exact composition of tRNA genes in any particular individual cannot yet be determined. This uncertain knowledge poses an inherent limitation in our approach to determine the composition of all tRNA genes in individuals. Hence, our study only focuses on the identification of new tRNA sequences or SNPs. An initial calculation estimates that if tRNA gene mutations are random, the probability of finding new isodecoders would be ~300, i.e., 16 × 106 (0.5% variations between two individual genomes) * 1.9 × 10−5 (the fraction of all tRNA gene base pairs in a human genome). For the high score tRNA genes, we ended up with 11 new isodecoders present within > 1% of human population. This is one order of magnitude lower than expected as derived from random mutations. This result suggests that the high scoring human tRNA gene sequences are under structural and functional constraints. Structural constraints can be derived from the need to maintain appropriate secondary and tertiary structures; and functional constraints can be derived from the need to efficiently participate in translation, or to perform extra-translational functions.
The sample size of 11 new isodecoders is not large enough to enable marking specific ethnic groups with unique tRNA isodecoder genes. However, if we decrease the threshold of finding new isodecoder sequences to below 1%, the number of new sequence changes increases by many folds, depending on the tRNA species. At this time, we cannot determine whether these additional sequence variations are bone fide tRNA isodecoder genes or derived from sequencing errors. If we assume that some of these are indeed new isodecoder genes, then the total number of tRNA isodecoder genes in the human population would increase significantly. Whether many of these isodecoder genes contain functionally relevant changes or simply are the result of neutral drift remains to be determined. More sequencing data will be needed to investigate the ultimate size and sequence variations of human tRNA isodecoder genes. Nevertheless, decreasing the threshold from 1% to 0.2% population increases the number of new isodecoders derived from high scoring tRNA genes increases from 11 to 37 (Table 1). This kind of diversity would be sufficient to render tRNA isodecoders as markers of human population.
The low scoring tRNA genes show much greater degree of sequence diversity in the reference genome and in the human population (Table 2). From the start, these genes are thought to potentially perform non-canonical functions as was demonstrated for a tRNAAsp gene of this group.15 Among the 167 low scoring tRNA genes, 13 contain new sequence variants in > 1% of population. Expanding this analysis to >0.2% of population identified 39 new sequences. The most significant indicator of these new sequences may perform non-canonical tRNA function is that the sequence variations are located in structurally and functionally more constraint regions in a canonical tRNA (compare Fig. 2B and E).
Potential functions of new tRNA isodecoders
The default functional hypothesis for new tRNA isodecoders is that they fine-tune translation in a cell type or cell state dependent manner. For instance, sequence change in the tRNA body can modulate the efficiency of aminoacylation, modification status or ribosome loading.26,37-39 Using a stop codon suppression assay in HeLa cells, it was shown that tRNA isodecoder sequences from the same isoacceptor family indeed show a wide range of suppression efficiencies.21 Interestingly, the best suppression efficiency was achieved by isodecoder sequences that do not occur naturally.21 This result suggests that not all natural isodecoder sequences are optimized for translational efficiency.
tRNAs are known to participate in a wide range of extra-translational activities. In mammalian cells, charged tRNAs can be used as carriers of chemically activated amino acids for metabolic enzymes or N-terminal amino acid transferases.12,40 Uncharged tRNAs are activators of the protein kinase GCN2 that regulates global translation activity in response to cellular nutrient availability.41 tRNAs can bind to cytochrome C in the cytoplasm to prevent premature apoptosis.14 During oxidative stress, tRNAs are cleaved to produce fragments that are used as small interfering RNAs to regulate the activity of translation factors.42,43 A tRNAAsp isodecoder folds into a non-canonical structure and this tRNA molecule associates with mRNA to regulate alternative 3′ end formation.15 We have shown that tRNA interacts with a wide array of cellular proteins involved in a diverse range of cellular processes.16 For example, direct tRNA interaction with farnesyl-transferase or histone H3K9 methyl-transferase may enable cellular communication between global translation activity with the process of protein modification. In each of these cases, distinct tRNA body sequence likely plays a role to tune the efficiency for these functions.
The most obvious candidates among the 11 new isodecoders to perform extra-translational function are the two isodecoders that introduce base pair mismatch in the anticodon stem. Our results show that these isodecoders from the tRNACys(GCA) and tRNAArg(ACG) isoacceptor families indeed have different structures in the anticodon and D stem-loop regions compared with the standard tRNAs. Further, we show that the G40-tRNAArg(ACG) has the same stability as the standard, C40-tRNAArg(ACG) isodecoder in HeLa cells over several days, and we found no stable tRNA fragments derived from the 3′ portion of this tRNA (Fig. 5). The anticodon stem sequence change in the new tRNACys(GCA) and tRNAArg(ACG) isodecoders should not significantly decrease the overall cellular capacity of tRNACys and tRNAArg in translation. The tRNACys(GCA) isoacceptor family contains 30 genes in the reference genome. Here, the presence of one or even two of the G28 isodecoder genes should hardly make a dent in the translational capacity of this isoacceptor family. The tRNAArg(ACG) isoacceptor family contains 7 genes in the reference genome. Although the G40 isodecoder renders this tRNA incapable to be charged in vivo, this sequence change may occur only in one of the 7 genes, so that the capacity for translation for this isoacceptor family would only change from 7 to 6 genes.
To determine the exact function of these two new isodecoders, one would have to first identify their cellular interacting partners. The extremely low charging level of the G40-tRNAArg(ACG) isodecoder transcript in cells eliminates the competition of the arginyl-tRNA synthetases, so that this isodecoder is now free to interact with other proteins or RNA.
Concluding remarks
We have shown here that the human population has significant tRNA sequence diversity, and some abundant, new tRNA isodecoders do not fold like standard tRNAs. These results raise the possibility that tRNA function, in particular those not directly involved in translation, is also subject to evolutionary change in human population. However, major hurdles remain to accurately determine the function of individual tRNA isodecoders. The first hurdle is the current lack of quantitative methods to accurately determine tRNA isodecoder expression at single nucleotide resolution. Despite the recent advances in RNA sequencing, quantitative tRNA sequencing remains unavailable due to the presence of numerous post-transcriptional modifications that interfere with cDNA synthesis. The second hurdle is the difficulty to manipulate tRNA levels in cells.44 In this regard, recent advances on genomic gene deletion in mammalian cells45 could be useful to assess the function of individual tRNA isodecoders. Once these hurdles are overcome, functional investigations of tRNA isodecoders should reveal why mammalian genomes contain such a large diversity of tRNA genes.
Materials and Methods
Bioinformatics
All deep sequencing reads were mapped on tRNA genes in the hg19 version of the Homo Sapiens genome. The tRNA genes are those proposed in the GtRNAdb database10 with score greater or equal to 50 (Fig. S1A). Introns in tRNA genes are retained, as the deep sequencing data comes from DNA (Fig. S1B). Deep sequencing reads were mapped using an in-house mapper, that allows for at most one mismatch and no gaps (pessimistic mapper). A read aligned to a tRNA gene has a score made up by adding 1 point per match and subtracting 1 point per mismatch. The minimum alignment score allowed is 20: an alignment of length 22 with 1 mismatch (pessimistic specificity). The tRNA isodecoder that features the highest alignment score for a read keeps that read, while other lower-scored matches are discarded. This approach is necessary using the 1000-genomes data that are not assembled into chromosomes. If the genomes of individuals were assembled then we could use the tRNAscan-SE46 program instead.
Deep sequencing reads are provided by the 1000 genomes project1 (Fig. S1C). A grand total of n = 1,617 individuals were analyzed, whose data table has been fetched from the 1000-genomes project FTP site on June 11th, 2012 (Fig. S1D and Table S1). All deep sequencing data pertaining to an individual is gathered together. A genomic position for an individual is declared mutated if that position is covered at least 10x and at least 2/3 of the mapped reads show a common nucleotide distinct from the reference genome at that position. As deep sequencing machines are prone to miscall nucleotide identity at the first read position, mismatches that occur at the first read position are not considered in our study. Because tRNA isodecoders share a great deal of sub-sequences among themselves (Tables 1 and 2), it is impossible to determine exactly which isodecoder sequence is actually being changed in an individual. However, because of the presence of new sequence variations, we can at the least deduce the presence of new isodecoders.
A computer cluster was employed to help plow through the task of aligning all the deep sequencing data from all these individuals (Fig. S1E). Basically, a computer node downloads the deep sequencing data from the FTP site of the 1000 genomes project, align the data on the tRNA genes, then discard the deep sequencing data to facilitate space conservation. Downloading of the data is tricky: only a few concurrent connections are allowed at any time of the FTP server; connection failures would result in a node to sleep a random amount of time then re-attempting a download upon awakening. A total of 20 quad-cores CPUs completed the whole task in about two weeks.
tRNA structural probing
tRNA isodecoder transcripts were synthesized by T7 RNA polymerase using templates generated from PCR with overlapping DNA oligonucleotides. tRNAs were purified on 10% polyacrylamide urea gel and extracted by soaking gel slices overnight at 4 °C in 50 mM KOAc and 200 mM KCl, pH7. After ethanol precipitation, tRNAs were resuspended in RNase-free H2O.
Purified tRNAs (40–80 pmole) were 5′- labeled with [γ-32P]ATP and T4 polynucleotide kinase (USB) after dephosphorylation with calf intestine phosphatase (Roche). The end-labeled and gel-purified RNAs were dissolved in RNase-free H2O. Equivalent amount of labeled tRNA pairs were subjected to limited nuclease cleavage for structural mapping as described previously.21 Before nuclease treatment, tRNAs were renatured at 85 °C for 2 min and folded in folding buffer (50 mM TRIS-HCl, pH 7.4, 10 mM MgCl2, 100 mM KCl, 0.1 μg/μl E. coli tRNA) at 37 °C for 5 min. Double-strand specific nuclease V1 (Ambion) or single-strand specific nuclease S1 (Fermentas) was added at a final concentration of 0.005 U/μl and 1 U/μl, respectively. Cleavage reactions proceeded for 2 min at 37 °C. Equal volume of RNA loading buffer (9 M urea, 50 mM EDTA, XC/BPB) was then added and the samples were split in two and immediately loaded on 8% and 20% polyacrylamide urea gels. Alkaline hydrolysis ladder and RNase T1 hydrolysis ladder are used to indicate the position of RNA cuts. Gels were imaged using phosphorimaging (Biorad).
tRNA stability and charging assays
To profile stability and charging levels of the pair of tRNAArg(ACG) isodecoders, tRNA isodecoder transcripts were labeled with [α-32P]ATP and the recombinant E. coli CCA adding enzyme.47 The reaction condition contains 40 pmole tRNA, 20 mM MgCl2, 50 mM glycine, pH 9.0, 50 mM pyrophosphate, 20 mM CTP, 5.5 pmole [α-32P]ATP, and 0.5 μg CCA adding enzyme, and incubation for 10 min at 37 °C. Gel-purified RNAs were transfected into HeLa cells using Lipofectamine reagent (Invitrogen) according to the manufacturer's instructions. Transfected cell in triplicates were grown in 6-well plates in DMEM at 37 °C under 5% CO2. Total RNAs were extracted at different time points (12 h, 24 h, 36 h, 48 h, 60 h, 72 h, 84 h, 96 h) using acidic phenol/CHCl3 (pH 4.5). After ethanol precipitation, RNAs were resuspended in 10 mM NaOAc/HOAc/1 mM EDTA (pH 4.8). Equal amount of extracted RNAs at different time points were loaded on 10% polyacrylamide urea gel, exposed and imaged using phosphorimager.
To analyze charging level of tRNA isodecoders, the same amount of extracted RNAs were spotted on pre-washed cellulose PEI plate (TLC, Analtech) and migrated with 5% glacial acetic acid and 100 mM NH4Cl. TLC plate was exposed and quantification of radioactive spots was performed using phosphorimager.
tRNA folding under native condition
We performed tRNA folding analysis under non-denaturing conditions to validate that the structural difference between the two tRNAArg variants is not derived from the process of denaturation/renaturation. Twenty µl of the T7 RNA polymerase transcription reaction was mixed with 2 µl 80% glycerol, and either 1 or 10 µl of the whole reaction mixture was directly loaded on 8% native polyacrylamide gels containing 25 mM tris-acetate, pH 7.5 and 5 mM Mg(OAc)2. Gels were run in the same buffer in the cold room for 4h, and stained with ethidium bromide for visualization under UV.
Supplementary Material
Disclosure of Potential Conflicts
No potential conflicts were disclosed.
Acknowledgments
This work was supported by a NIH grant (DP1GM105386 to TP). MP was a Chicago Fellow of the University of Chicago and is a Natural Sciences and Engineering Research Council of Canada postdoctoral fellow.
Glossary
Abbreviations:
- ChIP-seq
crosslinking and immunoprecipitation followed by deep sequencing
- SNP
single nucleotide polymorphism
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/27361
References
- 1.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA, 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA, 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia-Naji N, Chen H, Rybin D, Liu CT, Bielak LF, Prokopenko I, et al. DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium. Multiple Tissue Human Expression Resource (MUTHER) Consortium A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44:659–69. doi: 10.1038/ng.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marth GT, Yu F, Indap AR, Garimella K, Gravel S, Leong WF, Tyler-Smith C, Bainbridge M, Blackwell T, Zheng-Bradley X, et al. 1000 Genomes Project The functional spectrum of low-frequency coding variation. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gravel S, Henn BM, Gutenkunst RN, Indap AR, Marth GT, Clark AG, Yu F, Gibbs RA, Bustamante CD, 1000 Genomes Project Demographic history and rare allele sharing among human populations. Proc Natl Acad Sci U S A. 2011;108:11983–8. doi: 10.1073/pnas.1019276108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conrad DF, Keebler JE, DePristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. 1000 Genomes Project Variation in genome-wide mutation rates within and between human families. Nat Genet. 2011;43:712–4. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, Eichler EE, 1000 Genomes Project Diversity of human copy number variation and multicopy genes. Science. 2010;330:641–6. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lappalainen T, Sammeth M, Friedländer MR, ’t Hoen PA, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. Geuvadis Consortium. Geuvadis Consortium Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–11. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khurana E, Fu Y, Colonna V, Mu XJ, Kang HM, Lappalainen T, Sboner A, Lochovsky L, Chen J, Harmanci A, et al. 1000 Genomes Project Consortium Integrative annotation of variants from 1092 humans: application to cancer genomics. Science. 2013;342:1235587. doi: 10.1126/science.1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan PP, Lowe TM. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37:D93–7. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodenbour JM, Pan T. Diversity of tRNA genes in eukaryotes. Nucleic Acids Res. 2006;34:6137–46. doi: 10.1093/nar/gkl725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soll D. Transfer RNA: An RNA for all seasons. in The RNA World (eds. Gesteland, R.F. & Atkins, J.F.) 157-184 (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1993). [Google Scholar]
- 13.Wek RC. eIF-2 kinases: regulators of general and gene-specific translation initiation. Trends Biochem Sci. 1994;19:491–6. doi: 10.1016/0968-0004(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 14.Mei Y, Yong J, Liu H, Shi Y, Meinkoth J, Dreyfuss G, Yang X. tRNA binds to cytochrome c and inhibits caspase activation. Mol Cell. 2010;37:668–78. doi: 10.1016/j.molcel.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rudinger-Thirion J, Lescure A, Paulus C, Frugier M. Misfolded human tRNA isodecoder binds and neutralizes a 3′ UTR-embedded Alu element. Proc Natl Acad Sci U S A. 2011;108:E794–802. doi: 10.1073/pnas.1103698108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parisien M, Wang X, Perdrizet G, 2nd, Lamphear C, Fierke CA, Maheshwari KC, Wilde MJ, Sosnick TR, Pan T. Discovering RNA-protein interactome by using chemical context profiling of the RNA-protein interface. Cell Rep. 2013;3:1703–13. doi: 10.1016/j.celrep.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kutter C, Brown GD, Gonçalves A, Wilson MD, Watt S, Brazma A, White RJ, Odom DT. Pol III binding in six mammals shows conservation among amino acid isotypes despite divergence among tRNA genes. Nat Genet. 2011;43:948–55. doi: 10.1038/ng.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hagenbüchle O, Larson D, Hall GI, Sprague KU. The primary transcription product of a silkworm alanine tRNA gene: identification of in vitro sites of initiation, termination and processing. Cell. 1979;18:1217–29. doi: 10.1016/0092-8674(79)90234-4. [DOI] [PubMed] [Google Scholar]
- 19.Ouyang C, Martinez MJ, Young LS, Sprague KU. TATA-Binding protein-TATA interaction is a key determinant of differential transcription of silkworm constitutive and silk gland-specific tRNA(Ala) genes. Mol Cell Biol. 2000;20:1329–43. doi: 10.1128/MCB.20.4.1329-1343.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmeing TM, Voorhees RM, Kelley AC, Ramakrishnan V. How mutations in tRNA distant from the anticodon affect the fidelity of decoding. Nat Struct Mol Biol. 2011;18:432–6. doi: 10.1038/nsmb.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geslain R, Pan T. Functional analysis of human tRNA isodecoders. J Mol Biol. 2010;396:821–31. doi: 10.1016/j.jmb.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geslain R, Pan T. tRNA: Vast reservoir of RNA molecules with unexpected regulatory function. Proc Natl Acad Sci U S A. 2011;108:16489–90. doi: 10.1073/pnas.1113715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clarke L, Zheng-Bradley X, Smith R, Kulesha E, Xiao C, Toneva I, Vaughan B, Preuss D, Leinonen R, Shumway M, et al. 1000 Genomes Project Consortium The 1000 Genomes Project: data management and community access. Nat Methods. 2012;9:459–62. doi: 10.1038/nmeth.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33:W686-9. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giegé R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26:5017–35. doi: 10.1093/nar/26.22.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stombaugh J, Zirbel CL, Westhof E, Leontis NB. Frequency and isostericity of RNA base pairs. Nucleic Acids Res. 2009;37:2294–312. doi: 10.1093/nar/gkp011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sponer J, Mokdad A, Sponer JE, Spacková N, Leszczynski J, Leontis NB. Unique tertiary and neighbor interactions determine conservation patterns of Cis Watson-Crick A/G base-pairs. J Mol Biol. 2003;330:967–78. doi: 10.1016/S0022-2836(03)00667-3. [DOI] [PubMed] [Google Scholar]
- 29.Siegfried NA, O’Hare B, Bevilacqua PC. Driving forces for nucleic acid pK(a) shifting in an A(+).C wobble: effects of helix position, temperature, and ionic strength. Biochemistry. 2010;49:3225–36. doi: 10.1021/bi901920g. [DOI] [PubMed] [Google Scholar]
- 30.Ardell DH. Computational analysis of tRNA identity. FEBS Lett. 2010;584:325–33. doi: 10.1016/j.febslet.2009.11.084. [DOI] [PubMed] [Google Scholar]
- 31.Burkard ME, Turner DH. NMR structures of r(GCAGGCGUGC)2 and determinants of stability for single guanosine-guanosine base pairs. Biochemistry. 2000;39:11748–62. doi: 10.1021/bi000720i. [DOI] [PubMed] [Google Scholar]
- 32.Pan B, Mitra SN, Sundaralingam M. Crystal structure of an RNA 16-mer duplex R(GCAGAGUUAAAUCUGC)2 with nonadjacent G(syn).A+(anti) mispairs. Biochemistry. 1999;38:2826–31. doi: 10.1021/bi982122y. [DOI] [PubMed] [Google Scholar]
- 33.Wolfson AD, Uhlenbeck OC. Modulation of tRNAAla identity by inorganic pyrophosphatase. Proc Natl Acad Sci U S A. 2002;99:5965–70. doi: 10.1073/pnas.092152799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Köhrer C, Sullivan EL, RajBhandary UL. Complete set of orthogonal 21st aminoacyl-tRNA synthetase-amber, ochre and opal suppressor tRNA pairs: concomitant suppression of three different termination codons in an mRNA in mammalian cells. Nucleic Acids Res. 2004;32:6200–11. doi: 10.1093/nar/gkh959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dittmar KA, Goodenbour JM, Pan T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006;2:e221. doi: 10.1371/journal.pgen.0020221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 37.Dale T, Uhlenbeck OC. Amino acid specificity in translation. Trends Biochem Sci. 2005;30:659–65. doi: 10.1016/j.tibs.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Wohlgemuth I, Pohl C, Mittelstaet J, Konevega AL, Rodnina MV. Evolutionary optimization of speed and accuracy of decoding on the ribosome. Philos Trans R Soc Lond B Biol Sci. 2011;366:2979–86. doi: 10.1098/rstb.2011.0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ledoux S, Olejniczak M, Uhlenbeck OC. A sequence element that tunes Escherichia coli tRNA(Ala)(GGC) to ensure accurate decoding. Nat Struct Mol Biol. 2009;16:359–64. doi: 10.1038/nsmb.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varshavsky A. The N-end rule and regulation of apoptosis. Nat Cell Biol. 2003;5:373–6. doi: 10.1038/ncb0503-373. [DOI] [PubMed] [Google Scholar]
- 41.Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol. 2005;59:407–50. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- 42.Ivanov P, Emara MM, Villen J, Gygi SP, Anderson P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol Cell. 2011;43:613–23. doi: 10.1016/j.molcel.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamasaki S, Ivanov P, Hu GF, Anderson P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J Cell Biol. 2009;185:35–42. doi: 10.1083/jcb.200811106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pavon-Eternod M, Gomes S, Rosner MR, Pan T. Overexpression of initiator methionine tRNA leads to global reprogramming of tRNA expression and increased proliferation in human epithelial cells. RNA. 2013;19:461–6. doi: 10.1261/rna.037507.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gaj T, Gersbach CA, Barbas CF., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ledoux S, Uhlenbeck OC. [3′-32P]-labeling tRNA with nucleotidyltransferase for assaying aminoacylation and peptide bond formation. Methods. 2008;44:74–80. doi: 10.1016/j.ymeth.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.