

Abstract
Cardiac hypertrophy in response to multiple stimuli has important physiological and pathological significances. GATA4 serves as a nuclear integrator of several signalling pathways during cardiac hypertrophy. Sp1 and Sp3 are also reported to be involved in this process. However, the mechanism by which GATA4 acts as a mediator, integrating these ubiquitously expressed transcriptional factors, is poorly understood. We found that the expression of GATA4 and Sp1 was up-regulated in the myocardium of a pressure overload hypertrophy rat model, as well in phenylephrine-induced (PE-induced) hypertrophic growth of neonatal cardiomyocytes. GST pull-down assays demonstrated that GATA4 could interact with Sp1 in vitro. Therefore, we proposed that GATA4 cooperates with Sp1 in regulating ANF expression, as its reactivation is closely linked with hypertrophy. Further studies demonstrated that GATA4 could activate the ANF promoter synergistically with Sp1 through direct interaction. In contrast, Sp3 exhibited antagonistic function, and overexpression of Sp3 repressed the transcriptional synergy between Sp1 and GATA4. We also found that Sp1 alone could activate the ANF promoter in cardiomyocytes, whereas Sp3 exerted negative effects on ANF expression. Bioinformatics analysis revealed novel Sp-binding sites on the ANF promoter. The recruitment of GATA4 and Sp1 on the ANF promoter was enhanced during phenylephrine-mediated hypertrophy, whereas the recruitment of Sp3 was reduced. The phosphorylation of GATA4 by ERK1/2 kinase could enhance the affinity between GATA4 and Sp1. Thus, our findings revealed the critical interaction of GATA4 and Sp1 in modulating ANF expression, indicating their involvement in cardiac hypertrophy.
Keywords: GATA4, Sp1, Sp3, ANF expression, cardiac hypertrophy, transcription
Introduction
Hypertrophic signals change the transcriptional program and eventually result in cardiac hypertrophy. At the cellular level, cardiac hypertrophy is characterized by an increase in cell size and reactivation of foetal gene programs, as well as cytoskeletal reorganization. Large-scale expression and functional analyses have identified numerous transcription factors responsible for cardiac hypertrophy, including the general transcription factors AP1, NF-κB, NF-AT, c-myc and p300, and cardiac-specific transcription factors such as GATA4, SRF, MEF2C and NKX2.5 [1].
GATA4 has been recognized as a pivotal effector mediating cardiac gene transcription in response to hypertrophic stimuli. Targeted disruption of GATA4 showed that it is indispensable for terminal differentiation and survival of cardiac myocytes [2, 3]. Functional GATA-binding sites have been identified within the promoters of several cardiac genes, including atrial natriuretic factor (ANF), brain natriuretic peptide (BNP), α-myosin heavy chain (α-MHC) and β-MHC [4]. Overexpression of GATA4 alone led to cardiac hypertrophy both in cultured cardiomyocytes and transgenic mice, accompanied by up-regulated expression of ANF and other foetal genes [5, 6]. The forced expression of a dominant-negative GATA4 in cultured cardiomyocytes abrogated endothelin 1 (ET-1) or phenylephrine (PE)-induced increase in protein synthesis and hypertrophic gene expression.
GATA4 acts as a terminal integrator and effector in cardiac hypertrophy, on which converge divergent protein phosphorylation pathways, including p38, ERK1/2, PKA, PKC, AKT, JNK, CaM and Rho/ROCK signals [1, 7–13]. It is notable that GATA4 can function as a molecular bridge or scaffold linking multiple nuclear factors such as p300, MEF2C, SRF, NKX2.5, NF-AT and AP1 during cardiac hypertrophy [1, 13].
GATA families cooperate with Sp proteins, classically thought of as part of the basal transcriptional machinery, to mediate the expression of some tissue-specific genes [14]. The Sp family contains at least four members [15, 16], all having three zinc finger binding domains located at the C-terminal region for DNA binding and varying stretches of serine/threonine domains at the N-terminal region. Sp1 binds specifically to GC-rich sequences and is involved in the activation of promoters of various genes. Although Sp3 has the same binding specificity and ability as Sp1, its effect as an activator or repressor of transcription is mainly dependent on cell type, DNA-binding sites and its interactions with other nuclear factors. Sp3 often acts as a transcriptional repressor by competitively binding to the Sp1-bound GC box sequences and/or by directly functioning as a potent inhibitor [16]. Because Sp3 can inhibit Sp1-mediated transcription via competitive occupation, the ratio of Sp1/Sp3 affects the regulation of several genes [17, 18]. It has been reported that Sp1 can directly regulate a number of cardiac genes, such as ANF, connexin40, sarcoplasmic reticulum Ca2+ ATPase (SERCA), cardiac α-actin and cardiac troponin T (cTnT), etc. [19]. On the ANF promoter, loss of Sp1-binding sites resulted in an approximate 20-fold drop in reporter expression [20]. However, it is still unclear whether and how the Sp proteins cooperate with GATA4 functions during cardiac hypertrophy.
In the myocardium of a pressure overload hypertrophy rat model, we found that the expression of GATA4 and Sp1 was up-regulated, whereas Sp3 was down-regulated. We also observed the same expression changes in PE-induced hypertrophic growth of neonatal cardiomyocytes. These observations indicate the involvement of GATA4 and Sp1/Sp3. Further study demonstrated that Sp1 activated the ANF promoter in transiently transfected cardiomyocytes, whereas Sp3 repressed ANF promoter activity. Sp1 could be recruited to the ANF promoter, with PE treatment enhancing Sp1 recruitment in parallel with reduced Sp3 binding. GATA4 could cooperate with Sp1 to activate ANF transcription and there was a direct interaction between GATA4 and Sp1, but not Sp3. We found that the phosphorylation status of GATA4 at Ser105 site could affect its direct interaction with Sp1. Thus, our findings provide an important mechanism for GATA4 interacting with Sp1/Sp3 to participate in cardiac hypertrophy.
Materials and methods
Plasmid constructs
The plasmid constructs pN3-Sp1, pN3-Sp3 and pN3 vector were kind gifts from Dr. Suske G (Philipps-University, Marburg, Germany). pcDNA3-Sp1 (167–785aa), pcDNA3-Sp1 (167–565aa), pcDNA3-Sp1 (653–785aa), pcDNA3-Sp1 (680–785aa) and pcDNA3-Sp1 (708–785aa) were obtained from Dr. Sawaya BE (Center for Neurovirology and Cancer Biology, Temple University, Philadelphia, PA, USA). The dominant negative mutant of Sp1 (GST-Sp1, with an intact DNA-binding domain but without an activation domain) was a generous gift from Dr. Thiel G (University of Saarland Medical Center, Saarbrücken, Germany). Plasmids pCG-GATA4, ANF-luc and β-MHC were obtained from Dr. Nemer M (Clinical Research lnstitute of Montreal, Montreal, Canada). pFLAG-GATA4, GST-GATA4 N (1–214aa) and dHAND were gifts from Dr. Molkentin JD (Department of Pediatrics, University of Cincinnati, Cincinnati, OH, USA). pcDNA-MEF2C was obtained from Dr. Skerjanc IS (University of Western Ontario, London, Ontario, Canada). Constitutive-active ERK1 plasmid was a generous gift from Professor Ma DL (Center for Human Disease Genomics, Peking University, Beijing, China). pFLAG-GATA4 S105A and a mutant of -638 bp ANF-luc that lack the Sp1-binding sites, were constructed using the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) according to the manufacturer’s recommendation (PCR primers: see Table 1). Fragments of GATA4 were produced by PCR and cloned into pGEX-5X-1 vector (Promega, Madison, WI, USA) for GST-pull down assay (PCR primers: see Table 2).
Table 1.
Primers used in site mutation assay
| Name | Primer sequences | |
|---|---|---|
| GATA4 | Top | 5′-CCGCCGCCCGTGGCCCCGCGCTTCTCT-3′ |
| S105A | Bottom | 5′-AGAGAAGCGCGGGGCCACGGGCGGCGG-3′ |
| mtsps1 | Top | 5′-GTGACAGAATGGTATTGGTTCCAGCTCT-3′ |
| Bottom | 5′-AGAGCTGGAACCAATACCATTCTGTCAC-3′ | |
| mtsps2 | Top | 5′-TGAGGGTCTGGGGGTCGACGGGTTACTGGAG-3′ |
| Bottom | 5′-CTCCAGTAACCCGTCGACCCCCAGACCCTCA-3′ | |
| mtsps3 | Top | 5′-TATTGCCTCTCCTAACGTTCTTATTTGGAG-3′ |
| Bottom | 5′-CTCCAAATAAGAACGTTAGGAGAGGCAATA-3′ | |
| mtsps4 | Top | 5′-GACATCTAGGGTTCTGCTGGGCTGTCTGG-3′ |
| Bottom | 5′-CCAGACAGCCCAGCAGAACCCTAGATGTC-3 |
Mutant sites are shown in bold.
Table 2.
Primers used in the construction of truncated GATA4 recombinants
| Name | Primer sequences | |
|---|---|---|
| GATA4-Nf | Forward | 5′-CTGAATTCATGTTCTCAGAAGGCAGAGAG-3′ |
| Reverse | 5′-ATCTCGAGCAGCCGGCGCTGAGGCTT-3′ | |
| GATA4-Cf | Forward | 5′-ATGAATTCTCTGCCTCCCGCCGGGTA-3′ |
| Reverse | 5′-GACTCGAGCTTCCGTTTTCTGGTTTG-3′ | |
| GATA4-C | Forward | 5′-CTGAATTCCCCAAGAATCTGAATAAA-3′ |
| Reverse | 5′-GACTCGAGTTACGCGGTGATTATGTC-3′ |
Restriction endonuclease sites are shown in bold.
Aortic stenosis model
Ascending aortic stenosis surgery was performed in male Sprague-Dawley rats (body weight, 50–55 g, obtained from the Medical Experimental Animal Center of Peking University, Beijing, China) as described previously [21]. Briefly, rats were anaesthetized with a ketamine–xylazine mixture (5:3, 1.32 mg/kg intraperitoneally). The thorax was opened and a silver clip (0.9-mm internal diameter) was placed on the ascending aorta. Sham-operated animals underwent an identical procedure but without the clip. To characterize the model, echocardiographic measurements were made using a Vevo 770 Imaging system (Visualsonics, Toronto, Canada) as described previously [22].
Cardiomyocytes culture
Neonatal rat ventricular myocytes (hereafter referred as cardiomyocytes) were prepared and cultured as previously described [23]. The cardiomyocytes were then treated with PE (Sigma Chemical, St. Louis, MO, USA) at a concentration of 10 μM for 24 hrs.
Luciferase assays
Cells were seeded at a density of 10 × 104 per well in a 24-well plate and transfected using Lipofectamine™2000 (Invitrogen, Carlsbad, CA, USA) when they reached 80% confluence. Luciferase assays were performed as previously described [24].
Immunocytochemistry
The cardiac myocytes were cultured in flask-style chambers on glass slides. Indirect immunofluorescent staining for α-actinin (A7811; Sigma Chemical) was performed using FITC-conjugated secondary antibody to visualize cellular morphology.
Western blot analysis
Western blots were performed as previously described [24]. Antibodies, including Sp1 (sc-59; Santa Cruz Biotechnology, Inc, Santa Cruz, CA, USA), Sp1 (sc-14027; Santa Cruz), Sp3 (sc-644; Santa Cruz), GATA4 (sc-1237; Santa Cruz), GATA4 (phospho S105) (ab5245; Abcam, Cambridge, UK), phospho-ERK1/2 (4376; Cell Signaling, Danvers, MA, USA), ERK1/2 (4695; Cell Signaling) and horseradish peroxidase-conjugated secondary from Santa Cruz Biotechnology were used.
Immunoprecipitation assay
Forty-eight hours after transfection, HeLa cells were harvested and lysed at 4°C in immunoprecipitation cell lysis buffer. Immunoprecipitations were performed using the anti-FLAG M2 affinity gel (A2220; Sigma Chemical) as previously described [24].
GST pull-down assays
GST-GATA4 fragment fusion proteins were expressed in BL21 Escherichia coli (E. coli) and purified according to standard protocols. HeLa cells transfected with 10 μg of different vectors (pN3-Sp1 or pN3-Sp3) were lysed and lysates were incubated overnight at 4°C with purified GST proteins. Sp1 and the deletion proteins were generated by in vitro translation in the presence of [35S] methionine using the TNT T7 SP6 Coupled Reticulocyte Lysate System (L5020; Sigma Chemical).
FLAG pull-down assays
HeLa cells were transfected with FLAG-GATA4 vector. Forty hours after transfection, cells were treated with 10 μM PD98059 (an ERK inhibitor, Promega, Madison, WI, USA) for 8 hrs in DMEM containing 10% FBS. At 48 hr post-transfection, cells were harvested and FLAG-GATA4 was immunoprecipitated by anti-FLAG antibody on protein G-agarose beads. The purified FLAG-GATA4 fusion protein was incubated with [35S] methionine-labelled Sp1 (167–785aa). Bound proteins were visualized by autoradiography as described previously [25]. To evaluate the effect of the S105 site of GATA4 for binding with Sp1 protein, HeLa cells were transfected with either the wild-type or S105A mutant of FLAG-GATA4 vector and subsequently treated as described earlier.
RT-PCR
RNA extraction was performed using Trizol Reagent (Invitrogen) based on the manufacturer’s instructions. Amplifications were performed in a T-gradient thermocycler (Biometra, Gottingen, Germany) with different primers and condition (Table 3).
Table 3.
Primers used in RT-PCR
| Name | Primer sequences | |
|---|---|---|
| ANF | Forward | 5′-GGCTCCTTCTCCTCACCAA-3′ |
| Reverse | 5′-CTCTGAGACGGGTTGACTTCC-3′ | |
| β-MHC | Forward | 5′-GAGTGGACGTTTATTGACTTCGG-3′ |
| Reverse | 5′-GCCTTTCTTTGCTTTGCCTTT-3′ | |
| GAPDH | Forward | 5′-GTCAGTGGTGGACCTGACCT-3′ |
| Reverse | 5′-AGGGGAGATTCAGTGTGGTG-3′ | |
| Sp1 | Forward | 5′-ACCTGGCGGTGATGGAAT-3′ |
| Reverse | 5′-GGTGGGTCTTGATATGCTTTG-3′ | |
| Sp3 | Forward | 5′-TACCGCTGCCCAACAAAT-3′ |
| Reverse | 5′-ACTCTTCCGATCAGGCTCTT-3′ | |
| GATA4 | Forward | 5′-AAGGTACCTGGACTTTGCCTGTTGGGG-3′ |
| Reverse | 5′-CTAAGCTTACTGCGGCTGAGCCTCGG-3′ |
Chromatin immunoprecipitation (ChIP) assay
ChIP experiments were performed according to the method described previously [24]. After cross-link reversal, precipitated DNA was analysed by PCR for fragments of the proximal ANF promoter with different primers and condition (Table 4).
Table 4.
Primers used in ChIP assay
| Name | Primer sequences | |
|---|---|---|
| S1 | Forward | 5′-TGGAGCTGCTCAAGGCAAAG-3′ |
| Reverse | 5′-TCTGATGTTTGCTGTCTCGG-3′ | |
| S2 | Forward | 5′-AGGCGAGCGCCCAGGAAGAT-3′ |
| Reverse | 5′-CGGCGGCCAGGAGAAGATGC-3′ | |
| S3 | Forward | 5′-AGAGGTCCACCCACGAGGC-3′ |
| Reverse | 5′-TCTCCCTGGCAACGGGACCC-3′ | |
| S4 | Forward | 5′-GTGTAGCAGAATTCTTTAGA-3′ |
| Reverse | 5′-CAGGCAGCTGGGAGACAGCT-3′ | |
| GATA4 | Forward | 5′-AGGCGAGCGCCCAGGAAGAT-3′ |
| Reverse | 5′-CGGCGGCCAGGAGAAGATGC-3′ |
Electrophoretic mobility shift assays (EMSA)
The human recombinant protein Sp1 was purchased from Promega Company (E639A). Nuclear extracts were prepared from cardiomyocytes treated as described [24]. Oligonucleotides used in EMSA assay were labelled by [γ-32P] ATP. The sequences of the sense strand of these oligonucleotides are as follows:
ANF-GATA, 5′- gcttcgctggactgataactttaaaagg-3′;
wtANF-Sp-s1, 5′- GTGACAGAATGGGGAGGGTTCCAGCTCT -3′;
mtANF-Sp-s1, 5′- GTGACAGAATGGTATTGGTTCCAGCTCT -3′;
wtANF-Sp-s2, 5′- TGAGGGTCTGGGGGAGGGAGGGTTACTGGAG -3′;
mtANF-Sp-s2, 5′- TGAGGGTCTGGGGGTCGAAGGGTTACTGGAG -3′;
wtANF-Sp-s3, 5′- TATTGCCTCTCCTCCCGCCCTTATTTGGAG -3′;
mtANF-Sp-s3, 5′- TATTGCCTCTCCTAACGTTCTTATTTGGAG -3′;
wtANF-Sp-s4, 5′- GACATCTAGGGTGGGGGTGGGCTGTCTGG -3′;
mtANF-Sp-s4, 5′- GACATCTAGGGTTCTGCTGGGCTGTCTGG -3′.
The recombinant Sp1 or different nuclear proteins were mixed at room temperature prior to incubation with a radiolabelled probe. Protein-DNA complexes were resolved on a 6% native polyacrylamide gel and visualized by autoradiography.
Construction of Ad-GATA4
The AdEasy system (provided by Dr. He T, Howard Hughes Medical Institute, Baltimore, MD, USA) was used to construct recombinant adenovirus. Briefly, the pCG-GATA4 plasmid containing full-length GATA4 cDNA was digested with XbaI, and the target fragment was inserted into the pUC18 vector. The pUC18-GATA4 was digested with SalI and BamHI, resulting in a fragment containing GATA4 cDNA. pAdTrack-CMV inserted with the targeted fragment was linearized by digestion with PmeI, and subsequently co-transformed into E. coli. BJ5183 with pAdEasy-1. Recombinants were selected for kanamycin resistance and recombination was confirmed by digestion with PacI. Control virus AdEasy-β-gal is identical to Ad-GATA4 except that it contains a β-gal expression cassette rather than GATA4.
Statistical analysis
The data are expressed as means ± S.D. Comparisons between groups were analysed by Student’s t-test or ANOVA, and the Student–Newman–Kleuss method was used to estimate the level of significance. Differences were considered to be statistically significant at *P < 0.05.
Results
Involvement of GATA4, Sp1/Sp3 in cardiac hypertrophy
To elucidate whether Sp1 and Sp3 participate in cardiac hypertrophy, we created pressure-overload hypertrophy models induced by aortic stenosis [21] and PE-induced neonatal cardiomyocyte hypertrophy. About 7 weeks after aorta banding, the status of CHT (compensated hypertrophy) was identified by increased left ventricle wall thickness and normal contractile indices [26]. The expressions of GATA4 and Sp1 are up-regulated, whereas Sp3 is down-regulated in rats with pressure overload hypertrophy 7 weeks after aortic banding (Fig. 1A). PE is a α1-adrenoceptor agonist and is often used as a prohypertrophic stimulus. Cardiomyocytes stimulated by PE displayed an increase in cell size, total protein content and reorganization of myofibrils (Fig. 1B and C). Meanwhile, PE stimulation greatly increased the expression of foetal gene ANF and the expression of GATA4 and Sp1, whereas expression of Sp3 decreased (Fig. 1D).
Fig 1.
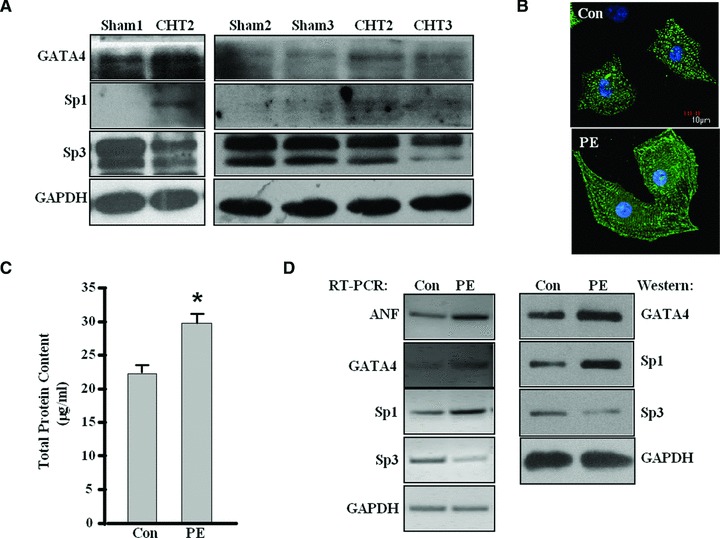
The expression of GATA4, Sp1 and Sp3 in rat cardiac hypertrophic tissues and PE-treated cardiomyocytes. (A) Cardiac tissues were collected from eight hypertrophy rats and four sham rats individually. Western blot analysis was performed on individual tissues to analyse the changes of GATA4, Sp1 and Sp3 at protein level in cardiac hypertrophic rat tissue compared with control rats. The protein level of GAPDH was used as an internal control. The figure shows a representative result with three CHT rats as well three control rats. (B) Neonatal rat cardiomyocytes were cultured with phenylephrine (PE, 10 μM for 24 h) and then stained with α-actinin antibody for immunofluorescence. The cardiomyocytes cultured without PE were used as a control (Con). The experiment was repeated at least three times, the figure shows a representative result. (C) The total protein content of cardiomyocytes treated as in (B) was determined using BCA protein assay kit. (Each bar represents mean ± S.D. from three independent experiments; *P < 0.05 versus control.) (D) RT-PCR and Western blot were performed with cardiomyocytes treated as in (B) to analyse the expression of Sp1, Sp3, GATA4 and ANF. GAPDH was used as internal controls. The experiment was repeated at least three times and similar results were obtained each time. The figure shows a representative result.
Transcriptional synergy between GATA4 and Sp1
To reveal whether there was an interaction between Sp proteins and GATA4 in regulating hypertrophic gene expression, we first performed luciferase reporter assays. Co-transfection of GATA4 and Sp1 in HeLa cells increased ANF promoter activity 24-fold over the basal promoter activity alone (Fig. 2A). GATA4 co-transfected with increasing amount of Sp1 vector stimulated ANF promoter activity in a dose-dependent manner (Fig. 2B). No significant cooperation occurred between Sp1 and Nkx2.5, dHAND or MEF2C (Fig. 2A). In contrast, co-transfection of GATA4 with Sp3 produced an additive rather than a synergetic effect (Fig. 2A), although Sp3 and Sp1 share some homologies in both structure and function. Similar results were also obtained from transient transfection with β-MHC promoter (Fig. 2C). These results indicate a transcriptional synergy between GATA4 and Sp1, but not Sp3.
Fig 2.
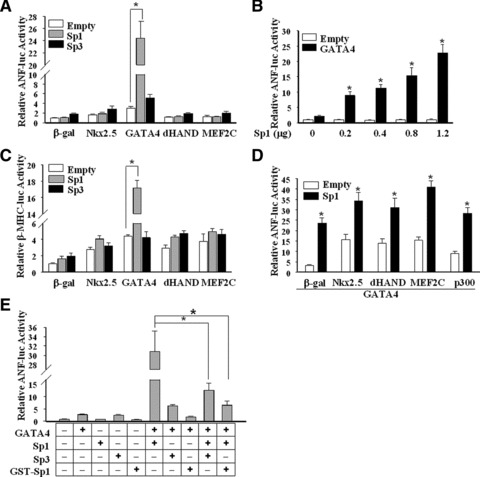
Transcriptional effects of Sp1/Sp3 and GATA4 on ANF promoter. Luciferase assays were performed in HeLa cells to evaluate the effects of Sp1 or Sp3 with other transcriptional factors on the ANF-638 bp promoter-luciferase reporter construct (ANF-luc) in (A), (B), (D) and (E), or β-MHC promoter-luciferase reporter construct (β-MHC-luc) in (C). GST-Sp1 is a dominant-negative mutant of Sp1, which contains an intact DNA-binding region without an activation domain on the N-terminus and thus competitively blocks putative Sp1 DNA-binding sites. The total DNA amounts for transfection were kept constant using pcDNA3 plasmid DNA. The activities of luciferase reporters (ANF-luc or β-MHC-luc) were set as 1.0. pRL was used to normalize the transfection efficiency. Each bar represents mean ± S.D. from three independent experiments, each sample in triplicate. Cells transfected with pcDNA3 plasmid DNA were used as controls (Empty) (*P < 0.05).
Previous reports found that Sp1 reduced the co-activation of serum response elements (SRF) and GATA4 or Nkx2.5 on the cardiac α-actin promoter [27]. However, in our system, Sp1 further increased the synergetic transcriptional activities between GATA4 and other cardiac-specific transactivators (Fig. 2D). Furthermore, overexpression of Sp3 or dominant-defective Sp1 (GST-Sp1) inhibited the co-activation of Sp1 and GATA4 (Fig. 2E). Increasing doses of Sp3 resulted in greater degrees of inhibition (data not shown), which indicates that Sp3 may function as a repressor in cardiac gene expression, possibly via competitive binding to GC boxes.
Interactions between GATA4 and Sp1 in vivo and in vitro
The functional synergy indicates an indirect or direct interaction between GATA4 and Sp1. Immunoprecipitation assays showed that Sp1, not Sp3, strongly interacts with GATA4 (Fig. 3A), consistent with results of the luciferase reporter assays. To further map the interaction between GATA4 and Sp1, GST pull-down assays were performed. As shown in Figure 3B, the C-terminal zinc finger of GATA4 is the major domain interacting with Sp1 rather than Sp3. Similarly, as shown in Figure 3C, truncated Sp1 fragments were capable of interacting strongly with GATA4 as long as they retain the zinc finger domain.
Fig 3.
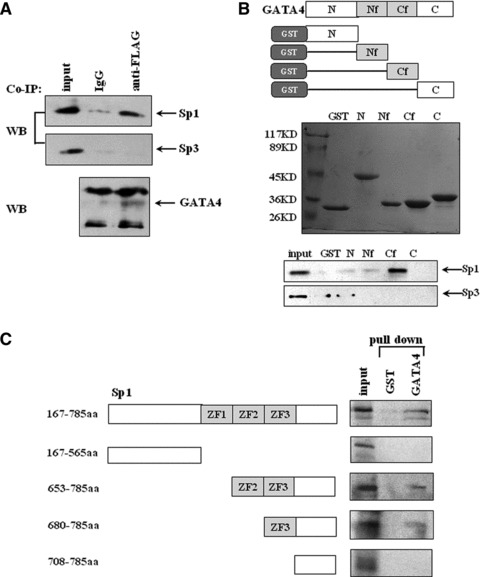
Physical interaction between Sp1/Sp3 and GATA4. (A) Co-immunoprecipitation was performed with cell lysates from HeLa cells transfected with FLAG-GATA4 in combination with Sp1 or Sp3 with anti-FLAG antibody or normal IgG as a control antibody, and then detected by anti-Sp1, Sp3 or GATA4 antibodies. (B) GST pull-down experiments were performed with cell lysates from HeLa cells transfected with Sp1 or Sp3 in combination with various fragments (as shown in the upper panel) of GATA4 fused to GST and then analysed by SDS-PAGE and Western blot. N, Nf, Cf and C stand for N terminal, N terminal zinc finger, C terminal zinc finger and C terminal domain, respectively. The middle panel shows GST-fusion proteins stained by Coomassie brilliant blue. The precipitated complexes were detected with Sp1 or Sp3 antibodies (the bottom panel). An equal amount of GST protein was used as a negative control. (C) GST pull-down was performed to analyse the interaction between in vitro translated 35S-labelled various fragments of Sp1 and GATA4 fused to GST. The labelled fragment bond with GATA4 was visualized by autoradiography. All the experiments were repeated at least three times.
Identification of Sp-binding sites on the proximal region of ANF
We further analysed this proximal region of the ANF gene and found four potential binding sites for Sp proteins, S1, S2, S3 and S4 (Fig. 4A). Using EMSA, we confirmed that four oligonucleotide probes, each containing one of the S1–S4 sites, could complex with purified Sp1 (Fig. 4B) or Sp3 (data not shown). The DNA-protein complexes could be competed by the non-labelled wild-type oligonucleotides but not by oligonucleotides with mutations of the Sp-binding sites. Thus, Sp proteins can bind to the four GC boxes located in the proximal region of the ANF gene.
Fig 4.
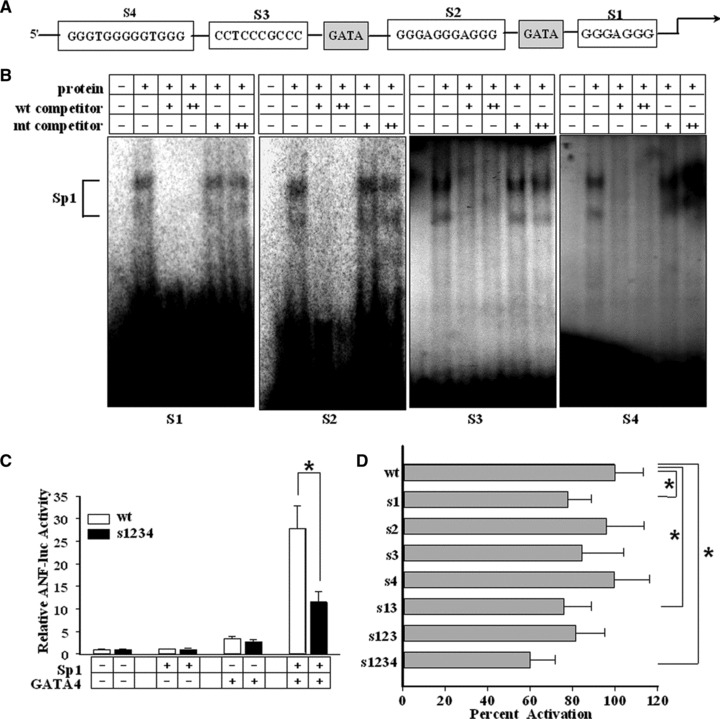
Sp1/Sp3 binding on ANF promoter and the effects on ANF promoter activity. (A) The schematic diagram shows the putative GC boxes (S1, S2, S3 and S4) for Sp1/Sp3 and consensus sequences for GATA4 (GATA) in the proximal region of the rat ANF promoter. (B) EMSA assays were performed with purified Sp1 protein to analyse its binding ability with 32P-labelled rat ANF S1, S2, S3 and S4 probes, respectively. The specific bands of Sp1 are indicated. (C, D) Luciferase assays were performed to evaluate the contributions of the Sp-binding sites to ANF promoter activity. HeLa cells (C) were co-transfected with GATA4 and Sp1 in combination with wild-type ANF luciferase-reporter construct (wt) or mutant ANF luciferase constructs (s1234) containing mutations in the four Sp-binding sites. (D) shows the results obtained in neonatal cardiomyocytes. The mutant ANF luciferase constructs include mutations in one or more Sp-binding sites (as indicated on the y-axis). (Each bar represents mean ± S.D. from three independent experiments; *P < 0.05.)
To investigate the contributions of the Sp-binding sites to total ANF promoter activity, luciferase assays were performed. As shown in Figure 4C, once all four Sp-binding sites on the ANF promoter were destroyed, the transcriptional cooperation between Sp1 and GATA4 was attenuated but not completely diminished.
We also performed luciferase assays in cardiomyocytes. As shown in Figure 4D, mutation of all four binding sites reduced promoter activity by approximately 40%. Individual mutation of S1 or S3 caused a slight drop in promoter activity whereas mutation of S2 or S4 sites seemed unable to affect the promoter activity. Thus, the individual roles of four sites may be complementary and redundant and they could act in a cooperative fashion for full proximal promoter activity.
Alteration of Sp1/Sp3 recruitment in response to PE stimulation
The expression changes of GATA4 and Sp1/Sp3 in the myocardium of rat cardiac hypertrophy model and PE-induced neonatal cardiomyocytes indicate their involvement in these processes. As Sp proteins can bind the ANF promoter directly, we performed ChIP assays to investigate their recruitment to the ANF promoter in response to PE stimulation. Four hours after PE stimulation, Sp1 recruitment onto the ANF promoter was dramatically augmented, whereas Sp3 binding was suppressed (Fig. 5A). The recruitment of GATA4 to the ANF promoter was also enhanced after PE stimulation (Fig. 5A). Forced GATA4 expression resulted in a significant increase in cardiomyocyte surface area and ANF expression (Fig. 5B), as well as in the DNA-binding activity of Sp1, but not Sp3, on the ANF promoter (Fig. 5C). However, Sp1 expression was not affected by GATA4 overexpression, indicating that the enhanced expression and recruitment of GATA4 under PE stimulation might further increase the recruitment of Sp1 on ANF promoter but not Sp1 expression.
Fig 5.
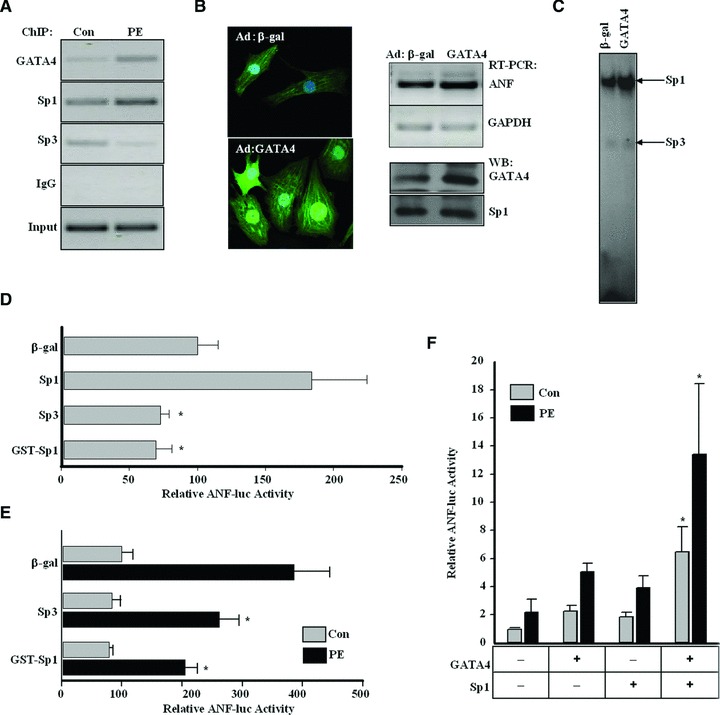
Alteration of Sp1/Sp3 recruitment on ANF promoter in response to PE stimulation. (A) ChIP assay was performed with cell lysates from neonatal cardiomyocytes of rats treated with phenylephrine (PE, 10 μM for 4 hrs) with anti-Sp1, Sp3 and GATA4 antibody or normal IgG as a control. Input DNA was used as a template to determine whether equal amounts of total DNA were used in each group. After cross-link reversal, precipitated DNA samples were used as templates for PCR. PCR amplified fragments cover the S1 site and GATA site on ANF promoter. (B) Neonatal cardiomyocytes were transduced either with β-gal or GATA4 recombinant adenovirus (Ad-β-gal or Ad-GATA4) at 100 multiplicity of infection. The morphological changes of infected cells were viewed under Laser Scanning Confocal Microscope (left panel), and the expressions of ANF, GATA4 and Sp1 in these infected cells were checked by RT-PCR and Western blotting (right panel). GAPDH was used as internal control. (C) EMSA assays were performed with nuclear extracts from neonatal cardiomyocytes treated as in B for the ability to bind with the 32P-labelled rat ANF S1 probe. (D) Luciferase assays were performed in neonatal cardiomyocytes transfected with ANF-638 bp promoter-luciferase reporter construct (ANF-luc) in combination with Sp1, Sp3 or a dominant-defective Sp1 (GST-Sp1) to evaluate their effects on ANF transcriptional activity. (Each bar represents mean ± S.D. from three independent experiments; *P < 0.05, versus control.) (E) Neonatal cardiomyocytes transfected as in (D) were further cultured with the addition of PE (10 μM) to evaluate their effects on ANF transcriptional activity during cardiomyocytes hypertrophy, and luciferase assay was then performed. (Each bar represents mean ± S.D. from three independent experiments; *P < 0.05, versus the control group treated with PE.) (F) Neonatal cardiomyocytes transfected with ANF-638 bp promoter-luciferase reporter construct (ANF-luc) in combination with Sp1 and GATA4 were further cultured with the addition of PE (10 μM) to evaluate their effects on ANF transcriptional activity, and luciferase assay was then performed. (Each bar represents mean ± S.D. from three independent experiments; *P < 0.05, versus control.)
Consistent with the binding of Sp1 onto its consensus sites, Sp1 activated ANF promoter in cardiomyocytes while Sp3 or GST-Sp1 (the dominant negative mutant) exhibited a repression effect on reporter activity (Fig. 5D). Because Sp3 could inhibit ANF promoter activity in cardiomyocytes, we investigated whether Sp3 could inhibit ANF expression in cardiac hypertrophy. As shown in Figure 5E, in the presence of PE, the transcriptional activity of ANF promoter was greatly increased, which was partly attenuated by co-transfection with Sp3 or GST-Sp1. Meanwhile, GATA4 and Sp1 synergistically activated ANF promoter in cardiomyocytes with or without PE stimulation (Fig. 5F).
Reinforced recruitment of Sp1 by phosphorylated GATA4
It has been reported that phosphorylation of GATA4 at Ser105 by ERK1/2 kinase could enhance the transcriptional activity and DNA-binding affinity of GATA4 [10]. As shown in Figure 6A, the DNA-binding activity of GATA4 was attenuated after cardiomyocytes were treated with PD98059 (ERK1/2 inhibitor) or when GATA4 Ser105 was mutated. We speculated that phosphorylation triggered conformational changes in GATA4, leading to higher affinity with basal transcriptional apparatus such as Sp1. We thus measured the effect of phosphorylation of GATA4 on recruitment of Sp1. As shown in Figure 6B, when phosphorylation of GATA4 was interfered with by the application of PD98059 or Ser105 mutation, the level of Sp1 immunoprecipitated by GATA4 was reduced, suggesting that phosphorylation of GATA4 plays an important role in recruiting Sp1. GATA4 S105A mutant also exhibited lower efficiency than wild type in cooperation with Sp1 to activate the ANF promoter, especially when the ERK1 pathway was activated (Fig. 6C). Consistent with these findings, we also observed that in vivo phosphorylation of ERK1/2 and GATA4 S105 were enhanced (Fig. 6D) and the level of p-GATA4 S105 immunoprecipitated by Sp1 was also increased in response to PE stimulation in cardiomyocytes (Fig. 6E). Thus, our data established a mechanism by which GATA4 phosphorylated by ERK1/2 kinases can recruit Sp1 and convey hypertrophic response to Sp1.
Fig 6.
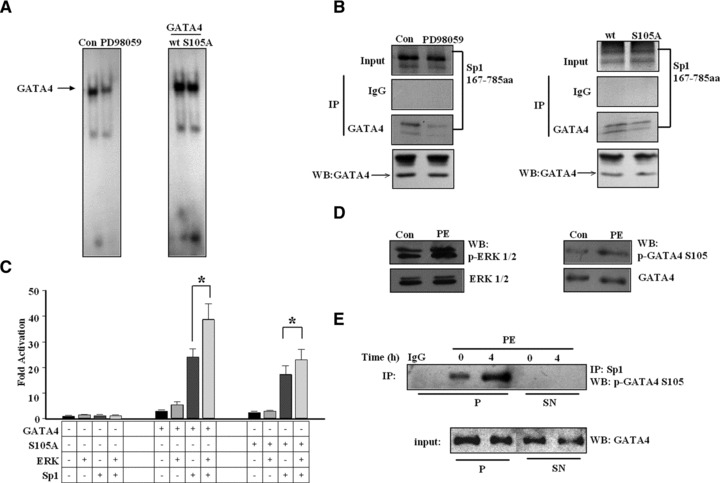
The influence of phosphorylation status of GATA4 on Sp1 recruitment and the synergetic cooperation between GATA4 and Sp1. (A) EMSA assays were performed with nuclear extracts from neonatal cardiomyocytes treated with 10 μM PD98059 or HeLa cells transfected with a mutant GATA4 (S105A) for the ability to bind the 32P-labelled rat ANF GATA probe. (B) Immunoprecipitation assays were performed with 35S-labelled Sp1 protein and nuclear extracts from HeLa cells treated with either 10 μM PD98059, or transfected with wild-type GATA4 (wt) or a mutant GATA4 (S105A), both with FLAG tag. The detection was performed with anti-FLAG antibody or normal IgG as a control antibody, and then visualized by autoradiography. (C) Luciferase assays were performed in HeLa cells transfected with ANF-638 bp promoter-luciferase reporter construct (ANF-luc) in combination with Sp1, a constitutively activated ERK, wild-type GATA4 or a mutant GATA4 (S105A) as indicated. (Each bar represents mean ± S.D. from triplicate experiments; *P < 0.05.) (D) Western blot were performed with cardiomyocytes treated with 10 μM PE for 4 hrs to analyse the phosphorylation of ERK1/2 and GATA4 S105. Total ERK1/2 and GATA4 were used as internal controls respectively. The experiment was repeated at least three times; the figure shows a representative result. (E) Cardiac myocytes were exposed to 10 μM PE for the times indicated. Cell lysates were immunoprecipitated with anti-Sp1 antibody or normal IgG as a control. The immunoprecipitates were separated into pellet and supernatant fractions. Supernatant fractions were subjected to the second immunoprecipitation with anti-Sp1 antibody. After immunoprecipitation, the phosphorylation of GATA4 was demonstrated by immunoblot analysis with anti-GATA4 (phospho S105) antibody. Total GATA4 was used as input. Similar results were obtained from three independent experiments. P: pellet; Sn: supernatant.
Discussion
The data presented in this study demonstrated the manner and effects of the cooperative interaction between GATA4 and Sp1 in PE-induced cardiomyocyte hypertrophy. PE stimulation enhanced the expression and DNA-binding ability of GATA4 and Sp1. Sp1 alone activated ANF promoter in cardiomyocytes and novel Sp1-binding sites were revealed in the ANF promoter. GATA4 could cooperate with Sp1 to enhance ANF transcription. A direct interaction was observed between GATA4 and Sp1, which was affected by the phosphorylation status of the Ser105 site on GATA4.
It has been reported that GATA4 could play important roles in the process of cardiac hypertrophy [1, 6]. It can act as a molecular bridge to form a complex with other cofactors. Sp1 was also reported to regulate a number of cardiac genes. Whether GATA4 could cooperate with Sp proteins, especially in cardiac hypertrophy, was unclear. Some reports indicated a physical and functional interaction between Sp1 and GATA family members. GATA5 could interact with numerous nuclear factors including Sp1 [14]. Sp1 could have direct interaction with GATA1 erythroid specific promoter [28]. Therefore, we proposed that GATA4 may interact with Sp proteins. In this study, we choose a widely used marker gene, ANF, as a target gene to evaluate the interactions between Sp proteins and GATA4. Not to our surprise, the results show that GATA4 could regulate ANF expression synergistically with Sp1 not only in HeLa cells (Fig. 2 and 6C) but also in rat neonatal cardiomyocytes (Fig. 5F). The association between hypertrophy and the expression level of transcription factors demonstrated in cardiac tissue (Fig. 1A) was also present in cardiomyocytes as shown in Figure 1D and 5E. Several GATA-binding sites have been reported on the ANF promoter [4–6]. Bioinformatics analysis revealed four Sp-binding sites on the proximal region of the ANF promoter, providing a structural basis for the interaction. Both GATA4 and Sp1 have direct interactions with the ANF promoter. However, their effects on activating the ANF promoter were not very strong by themselves (Fig. 2E). Their synergistic effect could be due to protein–protein interaction between GATA4 and Sp1, which was shown by the data from Co-IP and GST pull-down experiments (Fig. 3). It is worthy to note that the binding of Sp1 on the ANF promoter is also important for its synergistic effects with GATA4. Once Sp-binding sites on the ANF promoter were deleted, the synergistic effects were inhibited (Fig. 4C). Further investigation demonstrated that overexpression of GATA4 could enhance the recruitment of Sp1 on the ANF promoter (Fig. 5C), but not Sp1 expression, indicating that the relationship among Sp1/Sp3, GATA4 and ANF is very complicated.
Interestingly, PE stimulation up-regulated the expression of GATA4 and Sp1, enhancing their binding on the ANF promoter, as well as facilitating the interaction between GATA4 and Sp1. It has been reported that several pathways are involved in cardiac hypertrophy. Our data demonstrated that the inhibition of ERK1/2 pathway weakened its DNA-binding activity and reduced its cooperation with Sp1 on ANF promoter. However, both effects were not completely diminished when a mutation at S105 of GATA4 was induced, indicating that there might be other mechanisms or different phosphorylation sites involved in this process. It has been reported that S105 of GATA4 could also be phosphorylated by p38, and S261 of GATA4 could be phosphorylated by PKA pathway in gonadal cells [29]. A further study could be carried out to exam whether S261 of GATA4 is also involved in cardiac hypertrophy.
Compared with Sp1, Sp3 inhibits inducible expression of cardiac genes by competing with Sp1 for GC boxes and breaking the cooperation of Sp1 and GATA4. Sp3 is a bifunctional transcriptional regulator that plays active or repressive roles depending on the cell type and its interactions with other nuclear factors. Sp3 represses Sp1-mediated transcriptional activation of the cTnT gene in embryonic cardiomyocytes by a mechanism of competitive binding/inhibition [30]. Although Sp3 and Sp1 recognize GC boxes with similar specificity, Sp3 prefers to bind multiple GC boxes, forming more stable complexes than monomeric Sp3-DNA complexes or multimeric Sp1-DNA complexes. As a consequence, Sp3 can efficiently compete with Sp1 for promoter occupancy, thereby blocking the synergistic transactivation function of Sp1 [31]. Interestingly, the proximal region of the ANF promoter harbours multiple GC boxes, which is the preferential binding region of Sp3. In our report, transient expression assays showed that Sp3 functioned as a competitive inhibitor of Sp1-mediated transactivation of the ANF reporter gene in cardiomyocytes. Furthermore, after exposure of cardiomyocytes to PE, the occupation of Sp3 on ANF was down-regulated. Therefore, Sp3 more favourably occupies the adjacent GC boxes on the ANF promoter in the absence of hypertrophic stimuli. GATA4 and Sp1 were simultaneously recruited onto the ANF promoter in response to PE treatment, implicating a functional link. Further investigation demonstrated a physical interaction and function cooperation between Sp1 and GATA4. No significant interaction of Sp3 and GATA4 was detected. Therefore, our results further point out that overexpressed Sp3 may competitively occupy the GC boxes, excluding Sp1’s access to the promoter and leading to the disassembly of active complex of Sp1 and GATA4. In contrast, the default Sp3 interaction with GATA4 leads to its inability to induce expression of ANF.
Sp proteins are usually considered constitutively expressed transcriptional factors. The expression levels are normally kept constant. However, upon PE stimulation, the expression of Sp1 was up-regulated, whereas Sp3 expression was down-regulated. This may be due to the reactivation of foetal genes in response to hypertrophic stimuli. It is also consistent with a previous report that Sp1 expression was increased in hypertrophied cardiac chambers, whereas Sp3 expression was decreased [30]. Thus, the recruitment of Sp1 on ANF promoter underwent a biphasic increase in response to hypertrophic stimuli. At the early stage of induced hypertrophy, Sp1 was recruited onto the ANF promoter by activated GATA4; at the late stage, Sp1 was up-regulated in protein abundance. Both alterations should contribute to an increased ratio of Sp1/Sp3, overriding the inhibitory effect of Sp3 occupation on the ANF promoter.
Taken together, we revealed that GATA4 and Sp1/Sp3 are dynamically modified at the promoter region of ANF, indicating their synergistic involvement in cardiac hypertrophy. This mechanism, by which the ratio of Sp1/Sp3 determines the expression of cardiac genes in cooperation with GATA4, may also function in expression regulation of other genes characterized by similar promoter structure to the ANF gene that contain multiple GATA and GC boxes. It should be noted that transcription factor regulation is not the only mechanism in cardiac hypertrophy regulation. The involvement of microRNA in this process has attracted much attention recently. MicroRNAs 199a, 133a and 208a have been reported to be regulators of cardiac hypertrophy [32–34]. The regulation network has been suggested between cardiac specific transcriptional factors and microRNA. For example, miR-1/miR-133 and miR-762 precursors are directly regulated by MEF2c and GATA-4, respectively [35, 36], whereas miR-1 also negatively regulated the expression of MEF2a and GATA4 [37]. Therefore, the regulation of cardiac hypertrophy must be complicated. More detailed mechanisms of Sp1 and GATA4 regulation in cardiac hypertrophy remain to be discovered.
Acknowledgments
This work was supported by the National Natural Sciences Foundation of China (30871253, 90919022) and the 111 Project of China (B07001). We thank Dr. Jason Wong, University of Cambridge, UK, for his kind help in the preparation of this manuscript.
Conflicts of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Akazawa H, Komuro I. Roles of cardiac transcription factors in cardiac hypertrophy. Circ Res. 2003;92:1079–88. doi: 10.1161/01.RES.0000072977.86706.23. [DOI] [PubMed] [Google Scholar]
- 2.Molkentin JD, Lin Q, Duncan SA, et al. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11:1061–72. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- 3.Kuo CT, Morrisey EE, Anandappa R, et al. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997;11:1048–60. doi: 10.1101/gad.11.8.1048. [DOI] [PubMed] [Google Scholar]
- 4.Liang Q, Molkentin JD. Divergent signaling pathways converge on GATA4 to regulate cardiac hypertrophic gene expression. J Mol Cell Cardiol. 2002;34:611–6. doi: 10.1006/jmcc.2002.2011. [DOI] [PubMed] [Google Scholar]
- 5.Charron F, Tsimiklis G, Arcand M, et al. Tissue-specific GATA factors are transcriptional effectors of the small GTPase RhoA. Genes Dev. 2001;15:2702–19. doi: 10.1101/gad.915701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang Q, De Windt LJ, Witt SA, et al. The transcription factors GATA4 and GATA6 regulate cardiomyocyte hypertrophy in vitro and in vivo. J Biol Chem. 2001;276:30245–53. doi: 10.1074/jbc.M102174200. [DOI] [PubMed] [Google Scholar]
- 7.Chandrasekar B, Mummidi S, Claycomb WC, et al. Interleukin-18 is a pro-hypertrophic cytokine that acts through a phosphatidylinositol 3-kinase-phosphoinositide-dependent kinase-1-Akt-GATA4 signaling pathway in cardiomyocytes. J Biol Chem. 2005;280:4553–67. doi: 10.1074/jbc.M411787200. [DOI] [PubMed] [Google Scholar]
- 8.Liang Q, Wiese RJ, Bueno OF, et al. The transcription factor GATA4 is activated by extracellular signal-regulated kinase 1- and 2-mediated phosphorylation of serine 105 in cardiomyocytes. Mol Cell Biol. 2001;21:7460–9. doi: 10.1128/MCB.21.21.7460-7469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morisco C, Seta K, Hardt SE, et al. Glycogen synthase kinase 3 beta regulates GATA4 in cardiac myocytes. J Biol Chem. 2001;276:28586–97. doi: 10.1074/jbc.M103166200. [DOI] [PubMed] [Google Scholar]
- 10.Tenhunen O, Sarman B, Kerkela R, et al. Mitogen-activated protein kinases p38 and ERK 1/2 mediate the wall stress-induced activation of GATA-4 binding in adult heart. J Biol Chem. 2004;279:24852–60. doi: 10.1074/jbc.M314317200. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Paradis P, Aries A, et al. Convergence of protein kinase C and JAK-STAT signaling on transcription factor GATA-4. Mol Cell Biol. 2005;25:9829–44. doi: 10.1128/MCB.25.22.9829-9844.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yanazume T, Hasegawa K, Wada H, et al. Rho/ROCK pathway contributes to the activation of extracellular signal-regulated kinase/GATA-4 during myocardial cell hypertrophy. J Biol Chem. 2002;277:8618–25. doi: 10.1074/jbc.M107924200. [DOI] [PubMed] [Google Scholar]
- 13.Morimoto T, Hasegawa K, Wada H, et al. Calcineurin-GATA4 pathway is involved in beta-adrenergic agonist-responsive endothelin-1 transcription in cardiac myocytes. J Biol Chem. 2001;276:34983–9. doi: 10.1074/jbc.M005498200. [DOI] [PubMed] [Google Scholar]
- 14.Kiela PR, LeSueur J, Collins JF, et al. Transcriptional regulation of the rat NHE3 gene. Functional interactions between GATA-5 and Sp family transcription factors. J Biol Chem. 2003;278:5659–68. doi: 10.1074/jbc.M209473200. [DOI] [PubMed] [Google Scholar]
- 15.Suske G. The Sp-family of transcription factors. Gene. 1999;238:291–300. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- 16.Li L, He S, Sun JM, et al. Gene regulation by Sp1 and Sp3. Biochem Cell Biol. 2004;82:460–71. doi: 10.1139/o04-045. [DOI] [PubMed] [Google Scholar]
- 17.Wooten LG, Ogretmen B. Sp1/Sp3-dependent regulation of human telomerase reverse transcriptase promoter activity by the bioactive sphingolipid ceramide. J Biol Chem. 2005;280:28867–76. doi: 10.1074/jbc.M413444200. [DOI] [PubMed] [Google Scholar]
- 18.Fandos C, Sanchez-Feutrie M, Santalucia T, et al. GLUT1 glucose transporter gene transcription is repressed by Sp3. J Mol Biol. 1999;294:103–19. doi: 10.1006/jmbi.1999.3216. [DOI] [PubMed] [Google Scholar]
- 19.Biesiada E, Hamamori Y, Kedes L, et al. Myogenic basic helix-loop-helix proteins and Sp1 interact as components of a multiprotein transcriptional complex required for activity of the human cardiac alpha-actin promoter. Mol Cell Biol. 1999;19:2577–84. doi: 10.1128/mcb.19.4.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McDonough PM, Hanford DS, Sprenkle AB, et al. Collaborative roles for c-Jun N-terminal kinase, c-Jun, serum response factor, and Sp1 in calcium-regulated myocardial gene expression. J Biol Chem. 1997;272:24046–53. doi: 10.1074/jbc.272.38.24046. [DOI] [PubMed] [Google Scholar]
- 21.Feldman AM, Weinberg EO, Ray PE, et al. Selective changes in cardiac gene expression during compensated hypertrophy and the transition to cardiac decompensation in rats with chronic aortic banding. Circ Res. 1993;73:184–92. doi: 10.1161/01.res.73.1.184. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Xu N, Feng X, et al. Targeted disruption of Smad4 in cardiomyocytes results in cardiac hypertrophy and heart failure. Circ Res. 2005;97:821–8. doi: 10.1161/01.RES.0000185833.42544.06. [DOI] [PubMed] [Google Scholar]
- 23.Palm-Leis A, Singh US, Herbelin BS, et al. Mitogen-activated protein kinases and mitogen-activated protein kinase phosphatases mediate the inhibitory effects of all-trans retinoic acid on the hypertrophic growth of cardiomyocytes. J Biol Chem. 2004;279:54905–17. doi: 10.1074/jbc.M407383200. [DOI] [PubMed] [Google Scholar]
- 24.Liu Z, Li T, Liu Y, et al. WNT signaling promotes Nkx2.5 expression and early cardiomyogenesis via downregulation of Hdac1. Biochim Biophys Acta. 2009;1793:300–11. doi: 10.1016/j.bbamcr.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 25.Gupta P, Huq MD, Khan SA, et al. Regulation of co-repressive activity of and HDAC recruitment to RIP140 by site-specific phosphorylation. Mol Cell Proteomics. 2005;4:1776–84. doi: 10.1074/mcp.M500236-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Xu M, Zhou P, Xu S, et al. Intermolecular failure of L-type Ca2+ channel and ryanodine receptor signaling in hypertrophy. PLoS Biol. 2007;5:0203–11. doi: 10.1371/journal.pbio.0050021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sepulveda JL, Vlahopoulos S, Iyer D, et al. Combinatorial expression of GATA4, Nkx2–5, and serum response factor directs early cardiac gene activity. J Biol Chem. 2002;277:25775–82. doi: 10.1074/jbc.M203122200. [DOI] [PubMed] [Google Scholar]
- 28.Hou CH, Huang J, He QY, et al. Involvement of Sp1/Sp3 in the activation of the GATA-1 erythroid promoter in K562 cells. Cell Res. 2008;18:302–10. doi: 10.1038/cr.2008.10. [DOI] [PubMed] [Google Scholar]
- 29.Tremblay JJ, Viger RS. Transcription factor GATA-4 is activated by phosphorylation of serine 261 via the cAMP/protein kinase A signaling pathway in gonadal cells. J Biol Chem. 2003;278:22128–35. doi: 10.1074/jbc.M213149200. [DOI] [PubMed] [Google Scholar]
- 30.Zakie A, Fineman JR, He Y. Sp3 inhibits Sp1-mediated activation of the cardiac troponin T promoter and is downregulated during pathological cardiac hypertrophy in vivo. Am J Physiol Heart Circ Physiol. 2006;291:H600–11. doi: 10.1152/ajpheart.01305.2005. [DOI] [PubMed] [Google Scholar]
- 31.Fluck CE, Miller WL. GATA-4 and GATA-6 modulate tissue-specific transcription of the human gene for P450c17 by direct interaction with Sp1. Mol Endocrinol. 2004;18:1144–57. doi: 10.1210/me.2003-0342. [DOI] [PubMed] [Google Scholar]
- 32.Song XW, Li Q, Lin L, et al. MicroRNAs are dynamically regulated in hypertrophic hearts, and miR-199a is essential for the maintenance of cell size in cardiomyocytes. J Cell Physiol. 2010;225:437–43. doi: 10.1002/jcp.22217. [DOI] [PubMed] [Google Scholar]
- 33.Matkovich SJ, Wang W, Tu Y, et al. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res. 2010;106:166–75. doi: 10.1161/CIRCRESAHA.109.202176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Callis TE, Pandya K, Seok HY, et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest. 2009;119:2772–86. doi: 10.1172/JCI36154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cordes KR, Srivastava D. MicroRNA regulation of cardiovascular development. Circ Res. 2009;104:724–32. doi: 10.1161/CIRCRESAHA.108.192872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oliveira E, Tang Y, Qian K, et al. Different microRNA in mouse heart, cardiac stem cells and cardiac stem cells with GATA4, indicate different sets of microRNA involved in stem cell differentiation by gene repression. Circulation. 2008;118:S491. [Google Scholar]
- 37.Ikeda S, He A, Kong SW, et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol. 2009;29:2193–204. doi: 10.1128/MCB.01222-08. [DOI] [PMC free article] [PubMed] [Google Scholar]