

Abstract
The role of obestatin, a 23-amino-acid peptide encoded by the ghrelin gene, on the control of the metabolism of pre-adipocyte and adipocytes as well as on adipogenesis was determined. For in vitro assays, pre-adipocyte and adipocyte 3T3-L1 cells were used to assess the obestatin effect on cell metabolism and adipogenesis based on the regulation of the key enzymatic nodes, Akt and AMPK and their downstream targets. For in vivo assays, white adipose tissue (WAT) was obtained from male rats under continuous subcutaneous infusion of obestatin. Obestatin activated Akt and its downstream targets, GSK3α/β, mTOR and S6K1, in 3T3-L1 adipocyte cells. Simultaneously, obestatin inactivated AMPK in this cell model. In keeping with this, ACC phosphorylation was also decreased. This fact was confirmed in vivo in white adipose tissue (omental, subcutaneous and gonadal) obtained from male rats under continuous sc infusion of obestatin (24 and 72 hrs). The relevance of obestatin as regulator of adipocyte metabolism was supported by AS160 phosphorylation, GLUT4 translocation and augment of glucose uptake in 3T3-L1 adipocyte cells. In contrast, obestatin failed to modify translocation of fatty acid transporters, FATP1, FATP4 and FAT/CD36, to plasma membrane. Obestatin treatment in combination with IBMX and DEX showed to regulate the expression of C/EBPα, C/EBPβ, C/EBPδ and PPARγ promoting adipogenesis. Remarkable, preproghrelin expression, and thus obestatin expression, increased during adipogenesis being sustained throughout terminal differentiation. Neutralization of endogenous obestatin secreted by 3T3-L1 cells by anti-obestatin antibody decreased adipocyte differentiation. Furthermore, knockdown experiments by preproghrelin siRNA supported that obestatin contributes to adipogenesis. In summary, obestatin promotes adipogenesis in an autocrine/paracrine manner, being a regulator of adipocyte metabolism. These data point to a putative role in the pathogenesis of metabolic syndrome.
Keywords: adipogenesis, adipocyte metabolism, Akt
Introduction
The program of adipogenesis is orchestrated by a sequential activation of transcription factors transducing information from intracellular and extracellular factors indicative of suitable conditions for differentiation [1–3]. Several factors have been associated with dynamic changes of adipose tissue [4]. Particular attention was recently given to the role of the gastric hormone, ghrelin, on its role in the regulation of adipocyte biology [5]. The net effect of prolonged ghrelin exposure is hyperphagia [6, 7]. At central level, ghrelin promote ubiquitous fat deposition independent of its orexigenic activity [6, 8, 9]. Peripheral ghrelin promotes the enlargement of adipocytes in specific abdominal white adipose tissue (WAT) deposits by enhancing lipid accumulation [10]. This is further supported by augment of adipogenesis [11, 12], intracellular lipid accumulation [13], triglyceride content [14] and reduction of lipolysis [10, 14, 15].
The ghrelin gene encodes a polypeptide called preproghrelin, which undergoes stepwise processing to produce ghrelin [16]. Preproghrelin undergoes additional proteolytic cleavage, generating a 23-amino-acid peptide named obestatin [17]. This peptide was originally isolated from stomach showing to be a circulating peptide whose secretion is pulsatile and displays an ultradian rhythmicity similar to ghrelin and growth hormone secretion [17]. It was originally reported to be the ligand for the orphan receptor GPR39, which belongs to the family of the ghrelin receptor GHS-R1a and the motilin receptor [17]. Despite the initial enthusiasm about the potential of this molecule as a physiological opponent of ghrelin, several observations related to this point have set its effectiveness into question [18–21]. Consequently, the state-of-knowledge on obestatin suffers from serious gaps, especially for the lack of reproducibility of its central activities. Keeping aside its controversial anorexigenic activity, there are data suggesting a relevant biological role, such as the mitogenic effect described in 3T3-L1 pre-adipocyte [21], human gastric carcinoma [22, 23] and pancreatic β cells [24]. Furthermore, obestatin induced c-fos expression in gastrointestinal and white adipose tissues through binding to GPR39 [21]. Of interest, GPR39 expression in white adipose tissues of rats was up-regulated during fasting whereas GPR39 levels were decreased in cultured mouse embryonic fibroblast cell lines during adipogenesis [25]. In human adipose tissue, decreased GPR39 expression was found in patients with obesity-associated type 2 diabetes mellitus [26]. These findings suggest a possible role for obestatin in adipocyte function. Thus, to shed light on the hypothetical role of obestatin in the biology and pathology of adipose tissue, we determined the effect of obestatin on the control of the metabolism of adipocyte and WAT as well as on adipogenesis focusing on the regulation of key enzymatic nodes for metabolism, Akt and AMPK [27, 28].
Methods
Materials
Rat/mouse obestatin was obtained from California Peptide Research (CA, USA). Mouse ghrelin was obtained from Global Peptides (CO, USA). Insulin was obtained from NovoNordisk (Bagsvaerd, Denmark). Anti-pAkt HM (S473), anti-pAMPKα(T172), anti-pGSK3α/β(S21/9), anti-pmTOR(S2448), anti-pS6K1(T389), anti-pACC(S79), anti-GLUT4 and anti-tubulin antibodies were from Cell Signaling Technology (MA, USA). Anti-pAS160(T642) antibody was obtained from Millipore (CA, USA). Preproghrelin siRNA, control siRNA, anti-GLUT1, anti-FAT/CD36, anti-FATP1, anti-FATP4, anti-C/EBPα, anti-C/EBPβ, anti-C/EBPδ, anti-PPARγ and anti-GHSR1a antibodies were obtained from Santa Cruz Biotechnology (CA, USA). GPR39 siRNAs were obtained from Applied Biosystems/Ambion (TX, USA). Anti-GPR39 antibody was obtained from Abcam (Cambridge, UK). For IHC, anti-obestatin antibody was from Alpha Diagnostic International (TX, USA). Anti-obestatin (obestatin neutralization assays) and anti-preproghrelin antibodies were obtained from Phoenix Pharmaceuticals (CA, USA). Secondary antibodies, enhanced chemiluminescence detection system and 2-[3H]deoxyglucose were from GE-Amersham (Buckinghamshire, UK). Alzet® osmotic minipumps (model 1003D) were purchased from Alzet Corporation (CA, USA). All other chemical reagents were from Sigma (MO, USA).
Animals
Adult male Sprague–Dawley rats (250 g; n = 40) were housed in 12-hr light/12-hr dark cycles with free access to standard rat chow diet and water. Alzet® minipumps (model 1003D) were sc implanted. The animals were assigned to one of four-matched experimental groups (n = 10/group): 1) 24 hr-minipump sc implanted group [containing obestatin (300 nmol/kg body weight/24 h)]; 2) 24 hr-minipumps sc control group (containing saline); 3) 72 hr-minipump sc implanted group [containing obestatin (300 nmol/kg body weight/24 hrs)] and 4) 72 hr-minipumps sc control group (containing saline). These minipumps hold 100 μl volume and deliver 1 μl/h. After 24 or 72 hrs, rats were euthanatized to obtain omental, subcutaneous and gonadal adipose tissues.
Cell culture and differentiation induction of 3T3-L1 pre-adipocytes
3T3-L1 pre-adipocytes were maintained in DMEM containing 10% foetal bovine serum (FBS), 100 U/ml penicillin and 100 U/ml streptomycin. For routine differentiation, confluent cells were treated with 0.5 mM isobutylmethylxantine (IBMX), 25 μM dexamethasone (DEX) and 861 nM (5 μg/ml) insulin for 3 days and maintained in DMEM containing 10% FBS, 100 U/ml penicillin, 100 U/ml streptomycin and supplemented with 172 nM (1 μg/ml) insulin for 10 days after the beginning of differentiation unless otherwise stated. Before each experiment 3T3-L1 pre-adipocytes or adipocyte cells were serum-starved 12 hrs in DMEM.
Western blot analysis
Tissue samples or serum-starved cells were stimulated with obestatin for the indicated time period at 37°C. The media was then aspirated and the cells were directly lysed in ice-cold RIPA buffer [Tris-HCl (pH 7.2), 50 mM; NaCl, 150 mM; EDTA, 1 mM; NP-40, 1% (v/v); Na-deoxycholate, 0.25% (w/v); protease inhibitor cocktail (Sigma); phosphatase inhibitor cocktail (Sigma)]. Lysates were clarified by centrifugation (14,000 × g for 15 min. at 4°C) and the protein concentration was quantified performed with the QuantiPro™ BCA assay kit (Sigma). For immunoblotting, equal amounts of protein were fractionated by SDS-PAGE and transferred onto nitrocellulose membranes. Immunoreactive bands were detected by enhanced chemiluminescence (ECL Plus Western blotting kit; GE-Amersham).
Western blot analysis of GLUT1, GLUT4, FAT/CD36, FATP1 and FATP4 in plasma membrane
Serum-starved cells were treated with obestatin (100 nM) or insulin (172 nM) for 60 min. and then harvested by scraping. After centrifugation, pellets were resuspended in homogenization buffer [HEPES (pH 7.4), 20 mM; sucrose, 255 mM; EDTA, 2 mM; protease inhibitor cocktail (Sigma); phosphatase inhibitor cocktail (Sigma)], homogenized with eight strokes, and then centrifuged at 1200 × g for 5 min. at 4°C to remove nuclei and unbroken cells. The supernatant was then centrifuged at 195,000 × g for 60 min. at 4°C to obtain total membranes. The final pellet was resuspended in homogenization buffer and used for western blot analysis.
Real time quantitative reverse transcription PCR (qRT-PCR)
For qRT-PCR, total RNA was isolated with Trizol (Invitrogen, CA, USA) and DNA-free kit (Applied Biosystems/Ambion, TX, USA) to generate first-strand cDNA synthesis performed with High-capacity cDNA Reverse Transcription kit (Applied Biosystems). qRT-PCR was performed using an ABI PRISM 7300 HT Sequence Detection System (Applied Biosystems). For the analysis of the preproghrelin gene, ACTB was used as housekeeping gene (TaqMan: Applied Biosystems). The fold change in gene expression was calculated using the 2−ΔΔCt relative quantitation method according to the manufacturer’s guidelines (Applied Biosystems).
Quantification and staining of lipids with Oil Red O
Treated cells were fixed for 1 hr with 4% buffered paraformaldehyde-PBS. Each dish was rinsed three times with Milli-Q water and then with 60% iso-propanol (5 min. at room temperature). Lipid droplets were stained for 10 min. at room temperature with a working solution of 60% Oil Red O. For quantification, cells were washed extensively with water to remove unbound dye, and iso-propanol was added to the stained culture plates and then analysed by spectrophotometry at 520 nm.
Immunohistochemistry
Cells were fixed on cover slips in 96% ethanol. White adipose tissue samples were fixed by immersion in 10% buffered formalin for 24 hrs, dehydrated and embedded in paraffin by a standard procedure. Sections, 5 μm thick, were mounted on Histobond Adhesion Microslides (Marienfeld, Lauda-Königshofen, Germany), dewaxed and rehydrated. Slides were consecutively incubated with: 1) anti-obestatin rabbit polyclonal antibody at a dilution of 1:100 in Dako ChemMate antibody diluent (Dako, Glostrup, Denmark); 2) EnVision peroxidase rabbit (Dako, CA, USA) used as the detection system and 3) 3,3′-diaminobenzidine-tetrahydrochloride (Dako Liquid DAB + Substrate-chromogen system). Cell and tissue sections were faintly counterstained with Harris’ haematoxylin. Immunohistochemistry controls were performed applying the primary antibody plus control antigen peptide (10 nmol/ml) to positive samples.
Small interfering RNA (siRNA) silencing of gene expression
Chemically synthesized double-stranded siRNA duplexes (Santa Cruz Biothecnology) were for preproghrelin: 3′-CAGAGAAAGGAAUCCAAGA-5′, 3′-CCUUCGAUGUUGGCAUCAA-5′ and 3′-CUCUCCUACCACUUUAAGA-5′. GPR39 targeted siRNAs (Ambion siRNA sequence numbers s89441, s89442, s89443) were selected from Silencer® Pre-designed sequences from Applied Biosystems. A non-silencing RNA duplex was used as a control for all siRNA experiments. 3T3-L1 cells were transfected with Lipofectamine 2000 (Invitrogen).
Immunofluorescence analysis of GLUT translocation
3T3-L1 cells were cultured on cover slips and differentiated into adipocytes. Serum-starved cells were treated with obestatin (100 nM) or insulin (172 nM) for 30 min. Intact cells were fixed with 4% buffered paraformaldehyde-PBS for 15 min., washed, permeabilized and blocked with PBT [1% Triton X-100, 1% Tween-20, 5% heat inactivated normal goat serum, 0.2% BSA in PBS] for 30 min., and then incubated with anti-Glut4 rabbit antibody diluted in PBT (1:100) overnight at 4°C. After three washes with PBS, cells were incubated with the secondary antibody (Alexa 594-conjugate goat anti-rabbit antibody) in PBT (1:500) for 45 min. at 37°C. Fluorescent images were captured with a Leica TCS SP 2 (LeicaMicrosystems, Heidelberg, Germany) confocal laser scanning microscope mounted on an Leica DM IRBE inverted microscope.
2-[3H]Deoxyglucose uptake (DOGU)
3T3-L1 adipocytes cells were cultured in six-well culture plates. Serum-starved cells (12 hrs) were washed with PBS buffer and incubated for 10 min. in PBS supplemented with 0.1% BSA. Then cells were either unstimulated or stimulated with obestatinghrelin or insulin at the indicated concentrations for 30 min. Glucose transport was determined by addition of 2-[3H]deoxyglucose (0.5 μCi/ml). The uptake was stopped after 5 min. by aspiration, and cells were washed three times with ice-cold PBS. Cells were lysed in 1% SDS, and deoxyglucose uptake was determined by scintillation counting in triplicate. Non-specific uptake was measured in the presence of 20 μM cytochalasin B (cyB).
Data analysis
Comparisons between groups were made by ANOVA. P < 0.05 was considered as statistically significant (*,#,##).
Results
Obestatin activates Akt phosphorylation and AMPK dephosphorylation in 3T3-L1 adipocyte cells
First, the dose dependence of Akt activation by obestatin was examined in 3T3-L1 adipocytes. Akt phosphorylation at C-terminal hydrophobic motif (S473) [HM(S473); pAkt(S473)] was observed using 10 nM obestatin, and the level of pAkt(S473) plateaued at 100–200 nM (data not shown). Next, we examined the kinetic of Akt activation in response to obestatin. Significant pAkt(S473) augment was observed 10 min. after addition of obestatin (100 nM), being sustained by 60 min. (Fig. 1A). Parallel to Akt activation, obestatin (100 nM) triggered the phosphorylation of a broad range of Akt downstream substrates: GSK3α/β (S21/9), AS160(T642), mTOR(S2448) and S6K1(T384) (Fig. 1B and C). Importantly, dephosphorylation of pAMPKα(T172) and its downstream substrate ACC(S79) was observed in response to obestatin (100 nM) (Fig. 1C).
Fig 1.
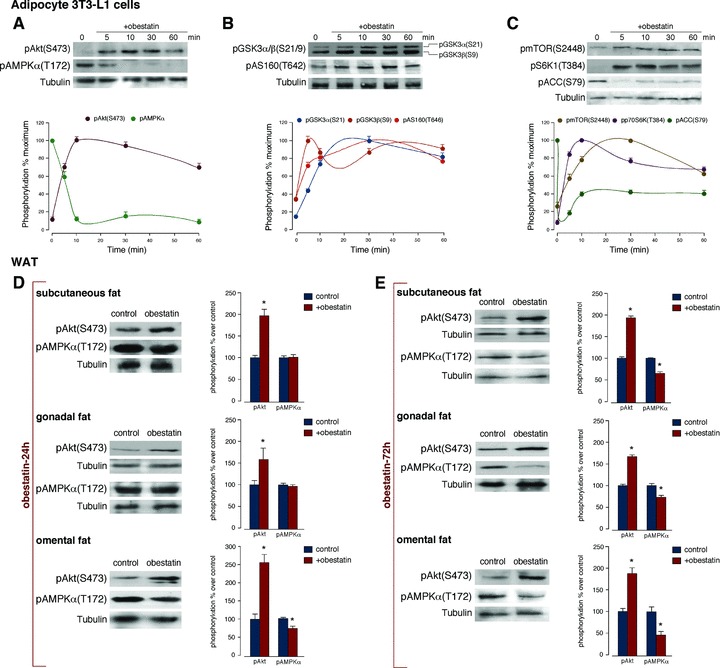
Obestatin activates Akt phosphorylation and AMPK dephosphorylation in 3T3-L1 adipocyte cells and WAT. Time-course of the effect of obestatin (100 nM) on: (A) pAkt(S473) and pAMPKα(T172); (B) pGSK3α/β(S21/9) and pAS160(T642) and (C) pmTOR(S2448), pS6K1(T384) and pACC(S79). Phosphorylation was expressed as a percentage of the maximal phosphorylation obtained for each residue (n = 3; mean±S.E.). Blots are representative of three independent experiments. (D) Effect of 24 hrs continuous sc infusion of obestatin (300 nmol/kg body weight/24 hrs; n = 10) on pAkt(S473) and pAMPKα(T172) from subcutaneous, gonadal and omental WAT obtained from male rats. (E) Effect of 72 hrs continuous sc infusion of obestatin (300 nmol/kg body weight/24 hrs; n = 10) on pAkt(S473) and pAMPKα(T172) from subcutaneous, gonadal and omental WAT obtained from male rats. Phosphorylation was expressed as a percentage of control obtained from 24 hrs or 72 hrs-minipump sc implanted saline group (n = 10; mean±S.E.). Asterisk (*) denotes P < 0.05 when comparing obestatin-treated group with untreated control group (saline).
Obestatin activates Akt phosphorylation and AMPK dephosphorylation in WAT
For in vivo obestatin administration, osmotic minipumps were selected based on the short half-life of this peptide (∼22 min.) [29]. Male rats received a 24 hrs continuous sc infusion of obestatin (300 nmol/kg body weight/24 hrs) and dissected WATs were processed for Western blot analysis. Obestatin significantly activated Akt, measured as pAkt(S473), in subcutaneous, gonadal and omental WAT (Fig. 1D). pAMPKα(T172) was decreased by 20% to basal phosphorylation in omental WAT with no effect in subcutaneous and gonadal WAT (Fig. 1D). A 72 hrs continuous sc infusion of obestatin (300 nmol/kg body weight/24 hrs) increased pAkt(S473) and decreased pAMPKα(T172) in subcutaneous, gonadal and omental WAT (Fig. 1E). The divergence between 24 and 72 hrs might be explained by the stress generated by mini-pump implantation. Total Akt and AMPK levels were comparable in all instances (data not shown).
Obestatin increases GLUT4 levels in plasma membranes and glucose uptake in 3T3-L1 adipocyte cells
The amplitude of Akt activation was evaluated on the basis to the GLUT1 and GLUT4 translocation to plasma membrane in 3T3-L1 adipocyte cells. Acute treatment with obestatin (100 nM, 30 min.) resulted in an increase in GLUT4 translocation to the plasma membrane by ∼1.9-fold (Fig. 2A). Intriguingly, a modest GLUT1 redistribution was observed in response to 100 nM obestatin (30 min.) in 3T3-L1 adipocyte cells (∼1.3-fold; Fig. 2A). Insulin (172 nM, 30 min.) caused a higher increase of GLUT4 levels at the plasma membrane than obestatin in 3T3-L1 adipocyte cells (∼2.8-fold) with no significant effect on GLUT1 translocation (Fig. 2A). The effect of obestatin in the translocation of GLUT4 to the plasma membrane was confirmed by specific immunofluorescence staining (Fig. 2B). In quiescent 3T3-L1 adipocyte cells, the fluorescence was distributed throughout the cytoplasm. After exposure to obestatin (100 nM, 30 min.), the fluorescence almost completely disappeared from the cytoplasm to be redistributed in the plasma membrane.
Fig 2.
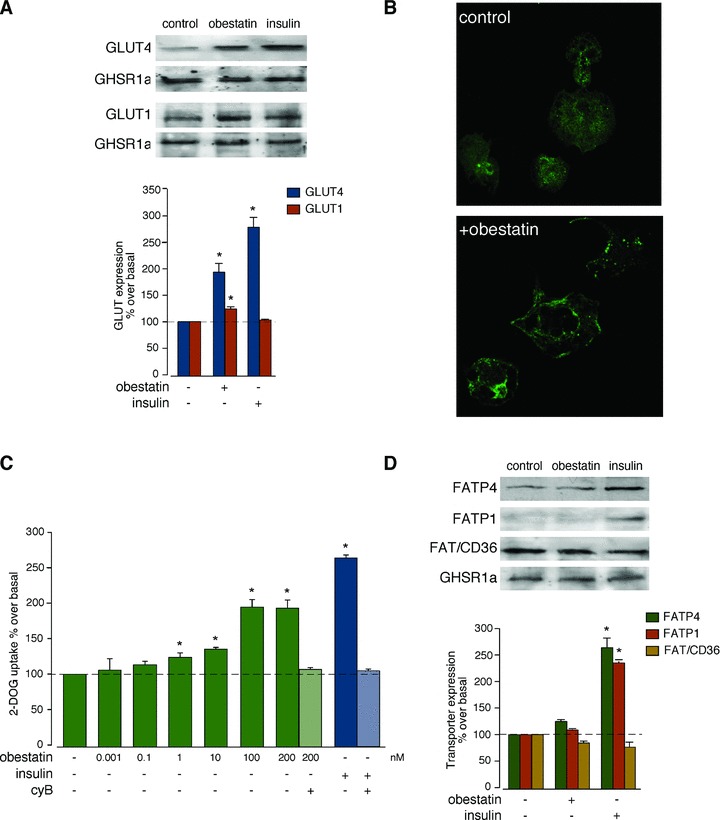
(A) Western blot analysis of GLUT4 and GLUT1 expression in membrane from 3T3-L1 adipocyte cells treated with obestatin (100 nM) or insulin (172 nM) for 30 min. GLUT expression was expressed as a percentage of basal expression obtained in control cells (n = 3; mean±S.E.). (B) Immunofluorescence analysis of obestatin-induced GLUT4 translocation in 3T3-L1 adipocyte cells. Serum-starved cells were stimulated with obestatin (100 nM, 30 min.) and the amount of GLUT4 was determined by labelling with anti-GLUT4 antibody in combination with Alexa 594-conjugate goat anti-rabbit antibody in permeabilized cells. (C) Obestatin dose dependently stimulated 2-[3H]deoxyglucose uptake in 3T3-L1 adipocyte cells (n = 3; mean±S.E.). Non-specific uptake was measured in the presence of 20 μM cytochalasin B (cyB). (D) Western blot analysis of FATP1, FATP4 and FAT/CD36 expression in membrane from 3T3-L1 adipocyte cells treated with 100 nM obestatin or with 172 nM insulin for 30 min. FATP1, FATP4 and FAT/CD36 expression were expressed as a percentage of basal expression obtained in control cells (n = 3; mean±S.E.). For (A, D) expression of GHSR1a was determined to ensure equal membrane protein loading. Blots are representative of three independent experiments. Asterisk (*) denotes P < 0.05 when comparing obestatin-treated group with untreated control group.
The effect of obestatin on DOGU was examined in 3T3-L1 adipocyte cells. Cells were incubated with different concentrations of obestatin (0.001–200 nM) for 30 min., resulting in a dose-dependent increase in DOGU with a maximal effect at 100 nM obestatin (∼2.0-fold; Fig. 2C). Insulin treatment (172 nM) caused an increase of DOGU by ∼2.8-fold in adipocyte cells (Fig. 2C).
Obestatin shows no effect on FATP1, FATP4 and FAT/CD36 translocation to plasma membrane in 3T3-L1 adipocyte cells
The levels of fatty acid (long chain) transport proteins 1 and 4 (FATP1 and FATP4) and fatty acid translocase (FAT/CD36) at the plasma membrane were examined by subcellular fractionation. As shown in Fig. 2D, obestatin (100 nM, 30 min.) did not modify FATP1, FATP4 and FAT/CD36 levels at the plasma membrane in 3T3-L1 adipocyte cells. Remarkably, FATP1 and FATP4 redistribution was significantly affected in response to insulin (172 nM, 30 min.) in ∼2.3- and 2.6-fold, respectively. In contrast to the FATP observation, FAT/CD36 redistribution was unaffected in response to insulin (172 nM, 30 min.; Fig. 2D).
Obestatin promotes adipogenesis in vitro
To initiate the 3T3-L1 adipocyte differentiation process, IBMX, DEX and insulin are used. Among these three compounds, only insulin is capable of activating Akt system through IGF-1 receptor playing a pivotal role in adipogensis [30]. To ascertain the effect of obestatin on adipogenesis, we first analysed the effect of obestatin on Akt activation in 3T3-L1 pre-adipocytes. Consistent with the effect on 3T3-L1 adipocytes, obestatin (100 nM) induced significant Akt(S473) phosphorylation, which was parallel to AMPK dephosphorylation at T172 in 3T3-L1 pre-adipocyte cells (Fig. 3A). The effect of acute GPR39 deficiency was determined by means of siRNA in 3T3-L1 pre-adipocyte cells. Under these conditions, the constructs decreased GPR39 expression by 50±10% (Fig. 3B). In the presence of a non-targeting control siRNA, obestatin-activated Akt(S473) phosphorylation was identical to that observed without any transfection (data not shown). Silencing of GPR39 in pre-adipocyte cells decreased subsequent pAkt(S473) with respect to siRNA control (47±2% under treatment with obestatin (100 nM) for 5 min.; Fig. 3B). Next, we examined the role of obestatin as promoter of terminal adipocyte differentiation working with 3T3-L1 pre-adipocyte cells maintained in DMEM containing 10% FBS with obestatin (0.001–200 nM), ghrelin (0.001–200 nM) or insulin (172 nM) for 7 days after induction of differentiation under combination of 0.5 mM IBMX, 25 μM DEX and 861 nM insulin for 3 days. Oil red O staining revealed that the accumulation of lipid droplets was dose-dependent for obestatin or ghrelin treatments being maximal at 10 nM for both peptides (∼3.6-fold; Fig. 3C). Insulin (172 nM) treatment increased the formation of lipid droplets (∼7.8-fold; Fig. 3C). Cotreatment with obestatin or ghrelin (1 or 10 nM) and a half-dose of insulin (86 nM) further stimulated adipocyte differentiation compared to the treatments with obestatin or ghrelin alone (Fig. 3C).
Fig 3.
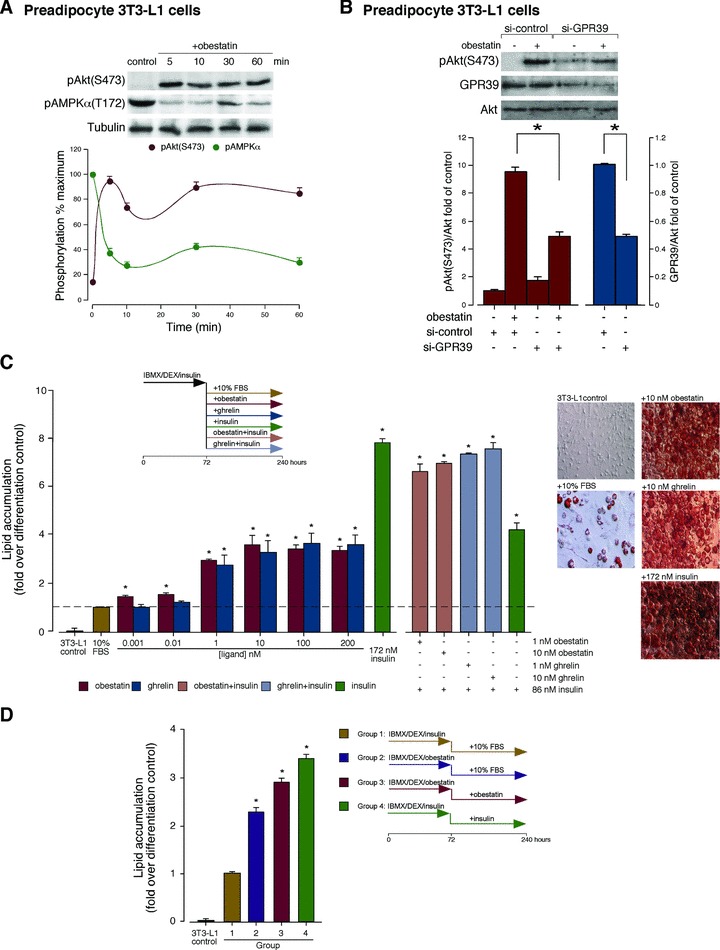
(A) Time-course of the effect of obestatin (100 nM) on pAkt(S473) and pAMPKα(T172) in 3T3-L1 pre-adipocyte cells. Phosphorylation was expressed as a percentage of the maximal phosphorylation obtained for each residue (n = 3; mean±S.E.). (B) Effect of siRNA depletion of GPR39 on obestatin-activated pAkt(S473) (100 nM, 5 min.) in 3T3-L1 pre-adipocyte cells. Expression of pAkt(S473) and GPR39 was expressed as fold of their levels in control siRNA-transfected cells (n = 3; mean±S.E.). (C) 3T3-L1 cells display an obestatin-dependent increase in adipogenesis. 3T3-L1 pre-adipocyte cells were maintained in DMEM containing 10% FBS with different concentrations of obestatin, ghrelin or insulin (172 nM) for 7 days after induction of differentiation by combination of 0.5 mM IBMX/25 μM DEX/861 nM insulin for 3 days. Lipid droplet accumulation was analysed by spectrophotometry at 520 nm by Oil red O staining. Results are expressed as a fold of lipid accumulation over differentiation control (cells maintained in DMEM/10% FBS/172 nM insulin for 7 days after induction of differentiation under treatment with 0.5 mM IBMX/25 μM DEX/861 nM insulin for 3 days; n = 3; mean±S.E.). Representative microscope fields of view (right) are shown at the same magnification. (D) Effect of obestatin as initiatr of adipogenesis. 3T3-L1 pre-adipocyte cells were maintained in DMEM containing 10% FBS with obestatin (392 nM) or insulin (172 nM) for 7 days after induction of differentiation by treatment with 0.5 mM IBMX, 25 μM DEX, 861 nM insulin or 1.96 μM obestatin for 3 days. Lipid droplet accumulation was analysed by spectrophotometry at 520 nm by Oil red O staining. Results are expressed as fold of lipid accumulation over differentiation control (cells maintained in DMEM/10% FBS/172 nM insulin for 7 days after induction of differentiation by treatment with 0.5 mM IBMX/25 μM DEX/861 nM insulin for 3 days; n = 3; mean±S.E.). Asterisk (*) denotes P < 0.05.
The capability of obestatin as initiator of adipocyte differentiation was explored by the combination of IBMX, DEX and obestatin (1.96 μM obestatin for 3 days and then maintained in DMEM containing 10% FBS with 392 nM obestatin for 7 days; Fig. 3D). Obestatin-stimulated adipocyte differentiation was detected at 10 nM, but maximal effects were observed at pharmacological levels for in vitro assays (≥1 μM). Obestatin treatment (1.96 μM) for 3 days (Group 2, Fig. 3D) induced more lipid droplets (∼1.6-fold) than insulin treatment (Group 1, Fig. 3D). The accumulation was higher in cells maintained in DMEM/10% FBS with obestatin (392 nM) for 7 days after induction of differentiation (Group 3, Fig. 3D), but fewer than that observed in full dose insulin-treated cells (172 nM) for 7 days (Group 4, Fig. 3D). Supporting this scenario, in obestatin-treated 3T3-L1 differentiating adipocytes, C/EBPβ protein expression markedly increased at 24 hrs for p32 and p35 isoforms, respectively (Fig. 4A). Furthermore, C/EBPγ protein expression peaked around 6 hrs after obestatin treatment (Fig. 4B). Consistently, obestatin treatment strongly increased the expression of PPARγ proteins peaking at 48 and 144 hrs of differentiation for PPARγ1 and PPARγ2 isoforms, respectively (Fig. 4C). In addition, this treatment also increased the expression of C/EBPα proteins peaking at 24 and 72 hrs for p30 and p42 isoforms, respectively (Fig. 4D).
Fig 4.
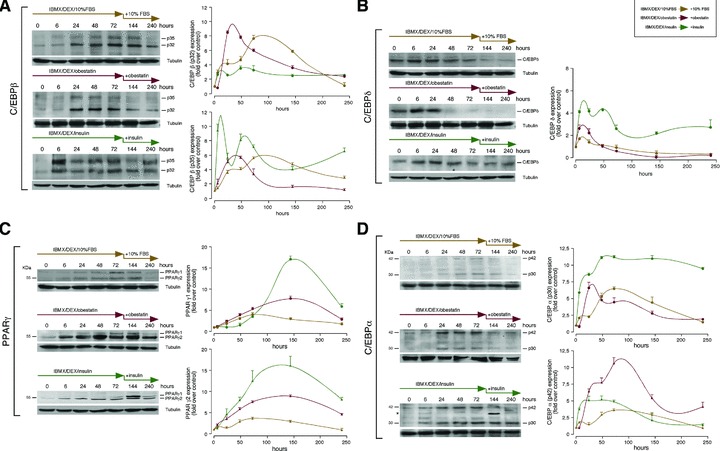
Immunoblots showing the expression pattern of: (A) C/EBPβ, (B) C/EBPδ, (C) PPARγ and (D) C/EBPα. 3T3-L1 pre-adipocyte cells were maintained in DMEM containing 10% FBS, obestatin (392 nM) or insulin (172 nM) for 7 days after induction of differentiation by treatment with 0.5 mM IBMX, 25 μM DEX, 10% FBS, 861 nM insulin or 1.96 μM obestatin for 3 days [in (D), asterisk (*) denotes unspecific band]. Data were expressed as percentage of control expression (n = 3; mean±S.E.). Blots are representative of three independent experiments.
Preproghrelin expression increases throughout adipogenesis
We first examine obestatin expression in 3T3-L1 pre-adipocyte and adipocyte cells at protein level, utilizing immunocytochemistry. 3T3-L1 adipocyte cells showed a stronger obestatin expression (Fig. 5B) compared to that observed in 3T3-L1 pre-adipocyte cells (Fig. 5A). No immunostaining was found with obestatin antibody pre-adsorption control in 3T3-L1 adipocyte cells (Fig. 5C). This result leads us to focus on preproghrelin expression along adipogenesis as source of obestatin. Preproghrelin expression was examined at mRNA and protein level, by RT-PCR and immunoblot, respectively, in 3T3-L1 pre-adipocyte cells. The cells were maintained in DMEM containing 10% FBS with insulin (172 nM) for 7 days after induction for 3 days (0.5 mM IBMX, 25 μM DEX and 861 nM insulin). The amount of preproghrelin mRNA showed a biphasic pattern of expression. A rapid augment was observed at 6 hrs (∼1.7-fold; Fig. 5D), to reach values bellow basal by 24 hrs after the induction of differentiation. Preproghrelin mRNA expression increased from 48 hrs, becoming maximal 72 hrs after induction to be sustained throughout terminal differentiation. As Fig. 5E shows, there is a rapid augment of preproghrelin protein expression at the time of differentiation into adipocytes (∼4.0-fold, 6 hrs after induction) reaching a maximum 24 hrs after induction of differentiation (∼5.8-fold) concomitant with the minimum of preproghrelin mRNA expression (Fig. 5D). Intriguingly, preproghrelin protein expression decreased 48 hrs after induction of differentiation to be sustained along terminal differentiation being higher than that in pre-adipocyte cells (∼2.4-fold at 240 hrs). Furthermore, GPR39 protein expression decreased in terminal differentiation compared to undifferentiated 3T3-L1 cells (∼40% reduction, 240 hrs after induction; Fig. 5F), showing two maximal expression levels concomitant with the maximum of preproghrelin expression (Fig. 5D). Consistent with the results from cultured cells, immunohistochemical analysis confirmed the expression of obestatin in subcutaneous, gonadal and omental WAT obtained from male ad libitum rats (Fig 5G–I, respectively).
Fig 5.
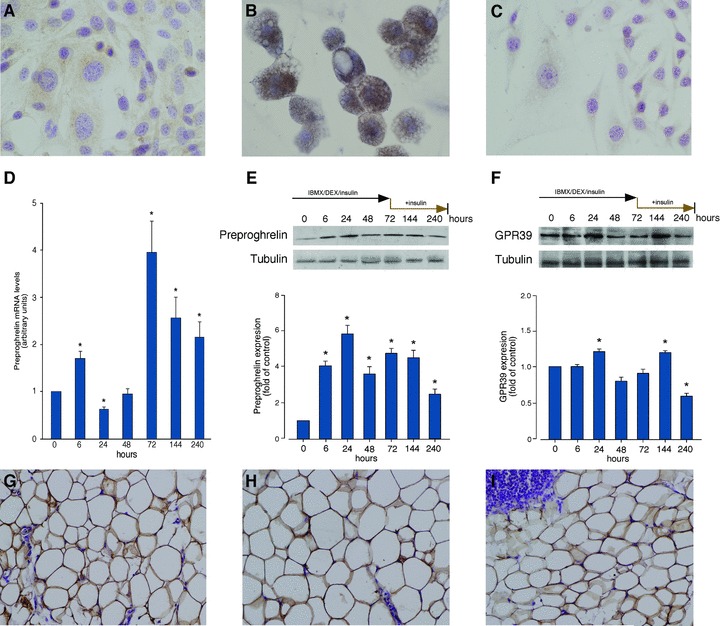
Immunocytochemical detection of obestatin in 3T3-L1 pre-adipocyte (A) and adipocyte cells (B) (objective magnification 40×). Obestatin immunostaining was mainly localized in the cytoplasm of 3T3-L1 adipocytes while it was faint in 3T3-L1pre-adipocyte cells. (C) Pre-adsorption control with mouse obestatin (10 nmol/ml) showed no positive immunostaining. (D) Preproghrelin mRNA levels in the course of adipogenesis. (E, F) Western blot analysis of preproghrelin (E) and GPR39 (F) expression in the course of adipogenesis. Preproghrelin expression at mRNA or protein levels, were examined in 3T3-L1 pre-adipocyte cells maintained in DMEM containing 10% FBS with insulin (172 nM) for 7 days after induction for 3 days (0.5 mM IBMX, 25 μM DEX and 861 nM insulin). Protein expression was expressed as a fold over control cells (n = 3; mean±S.E.). mRNA was quantified by RT-PCR and expressed as arbitrary units (n = 5; mean±S.E.). (G–I) Immunohistochemical detection of obestatin in subcutaneous (G), gonadal (H) and omental (I) WAT (objective magnification 20×). Asterisk (*) denotes P < 0.05 when comparing obestatin-treated group with untreated control group.
Autocrine/paracrine role of obestatin on adipogenesis
The autocrine/paracrine role of obestatin on adipogenesis was tested in vitro performed with a neutralizing obestatin antibody (anti-obestatin Ab). As shown in the Fig. 6A (left panel) neutralization of obestatin (100 nM) by preincubation with anti-obestatin Ab (1.3 μg/ml) reduced by ∼70±3% the level of pAkt(S473) in 3T3-L1 pre-adipocyte cells. By contrast, insulin-induced Akt activity was not affected by preincubation with anti-obestatin Ab. The autocrine/paracrine role on adipogenesis was then tested by combination of serum-free conditioned medium by 3T3-L1 adipocyte cells (CM, 24 and 48 hrs) with neutralizing obestatin antibody (5 μg/ml). 3T3-L1 pre-adipocyte cells were maintained in DMEM containing 0.1% BSA with obestatin (392 nM; Group 2), anti-obestatin Ab, (Group 3), obestatin (392 nM)+anti-obestatin Ab (Group 4), CM-24h (Group 5), CM-24h+anti-obestatin Ab (Group 6), CM-48h (Group 7) or CM-48h+anti-obestatin Ab (Group 8) for 7 days after induction of differentiation under treatment with 0.5 mM IBMX, 25 μM DEX and 861 nM insulin for 3 days. Oil Red O staining revealed that incubation with neutralizing obestatin Ab reduced by 35±3% accumulation of lipid droplets compared to 3T3-L1 pre-adipocyte cells maintained in DMEM/0.1% BSA/392 nM obestatin (Fig. 6A, Group 3 versus Group 2). Neutralization of obestatin (392 nM) by preincubation with anti-obestatin Ab reduced by 35±8% accumulation of lipid droplets compared to 3T3-L1 pre-adipocyte cells maintained in DMEM/0.1% BSA/392 nM obestatin (Group 4 versus Group 2; Fig. 6A). Serum-free conditioned medium, CM-24h or CM-48h, also activated accumulation of lipid droplets (∼2.0- and ∼2.5-fold, respectively; Groups 5 and 7 versus Group 1; Fig. 6A). Preincubation of the CM-24h or CM-48h with anti-obestatin Ab reduced accumulation of lipid droplets by 21±4% and 20±6%, respectively (Groups 6 versus Groups 5, and Group 8 versus Group 7, Fig. 6A). The effect of acute obestatin deficiency was determined by knockdown of preproghrelin by means of siRNA prior to induction of adipocyte differentiation. Under these conditions, the constructs decreased preproghrelin expression by 58±6% (Fig. 6B). In the presence of a non-targeting control siRNA, obestatin-activated adipocyte differentiation was identical to that observed without any transfection (data not shown). Silencing of preproghrelin in pre-adipocyte cells decreased subsequent adipocyte differentiation with respect to siRNA control [43±5% and 48±4% for 3T3-L1 pre-adipocyte cells under treatment with 0.5 mM IBMX, 25 μM DEX, and insulin (861 nM) for 3 days and then maintained in DMEM containing 10% FBS or insulin (172 nM) for 7 days, respectively (Fig. 6B)].
Fig 6.
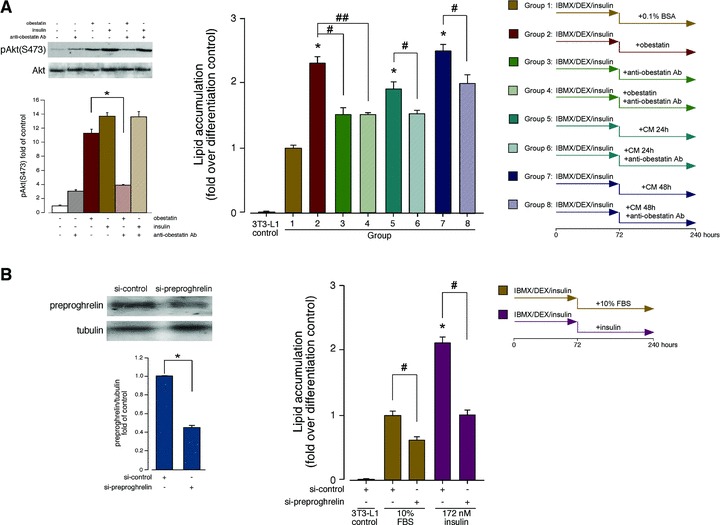
(A) Autocrine/paracrine role of obestatin on adipogenesis. (Right panel) 3T3-L1 pre-adipocyte cells were maintained in DMEM containing 0.1% BSA with obestatin (392 nM; Group 2), anti-obestatin antibody (5 μg/ml; anti-obestatin Ab; Group 3), obestatin (392 nM)+anti-obestatin Ab (5 μg/ml; Group 4), CM-24h (Group 5), CM-24h+anti-obestatin Ab (5 μg/ml; Group 6), CM-48h (Group 7) or CM-48h+anti-obestatin Ab (5 μg/ml; Group 8) for 7 days after induction of differentiation by treatment with 0.5 mM IBMX, 25 μM DEX and 861 nM insulin for 3 days. Lipid droplet accumulation was analysed by spectrophotometry (520 nm) by Oil red O staining. Results are expressed as fold of lipid accumulation over differentiation control (Group 1; cells maintained in DMEM/0.1% BSA for 7 days after induction of differentiation by treatment with 0.5 mM IBMX/25 μM DEX/861 nM insulin for 3 days; mean±S.E. of three independent experiments). Asterisk (*) denotes P < 0.05 when comparing treated Groups with control Group (Group1); daggers (#, ##) denote P < 0.05 when comparing treated Groups with antibody-treated Groups. (Left panel) Anti-obestatin antibody specificity. Effect of anti-obestatin antibody (1.3 μg/ml), obestatin (100 nM), insulin (100 nM), anti-obestatin antibody+obestatin and anti-obestatin antibody +insulin on pAkt(S473) in 3T3-L1 pre-adipocytes. Phosphorylation was expressed as fold of control (n = 3; mean±S.E.). Blot is representative of three independent experiments. Asterisk (*) denotes P < 0.05 when comparing obestatin-treated group with anti-obestatin antibody+obestatin. (B) Effect of siRNA depletion of preproghrelin on adipogenesis. 3T3-L1 cells transfected with preproghrelin siRNA prior to induction of adipocyte differentiation (DMEM/10% FBS with insulin (172 nM) for 7 days after induction for 3 days under 0.5 mM IBMX, 25 μM DEX and 861 nM insulin). Equal amounts of protein in each sample were used to assess the expression of preproghrelin (left panel) by western blotting. Expression of preproghrelin was expressed as fold of the level of preproghrelin in control siRNA-transfected cells (mean±S.E.). Lipid droplet accumulation was analysed by spectrophotometry at 520 nm by Oil red O staining (right panel). Results are expressed as fold of lipid accumulation over differentiation control. Asterisk (*) denotes P < 0.05 when comparing treated control siRNA group with control siRNA group; dagger (#) denotes P < 0.05 when comparing preproghrelin siRNA group with control siRNA group.
Discussion
In this study, we provide the first evidence that obestatin regulates adipocyte metabolism and adipogenesis. Preproghrelin expression, and thus obestatin, increased during adipogenesis being sustained throughout terminal differentiation of 3T3-L1 cells. In vitro 3T3-L1 cells secreted obestatin whose effects on adipocyte differentiation were neutralized by an obestatin antibody. Preproghrelin knockdown experiments revealed that obestatin contributes to adipogenesis, a fact that is supported by the effect of exogenous obestatin on the expression of C/EBPα, C/EBPβ, C/EBPδ and PPARγ and, consequently, lipid accumulation. In 3T3-L1 adipocyte cells, obestatin activated Akt phosphorylation and its downstream targets, GSK3α/β, mTOR, S6K1 and inhibited AMPK activity. This fact was confirmed in vivo in omental, subcutaneous and gonadal WAT obtained from male rats under continuous sc infusion of obestatin. The relevance of obestatin as regulator of pre-adipocyte/ adipocyte metabolism was also supported by AS160 phosphorylation, GLUT4 translocation and augment of glucose uptake in 3T3-L1 adipocyte cells. By contrast, obestatin failed to modify translocation of fatty acid transporters, FATP4 and FAT/CD36, to plasma membrane.
Exposure of 3T3-L1 cells, a well-characterized model for studying the differentiation of white adipocytes, to obestatin regulated Akt activity. This study also demonstrated that chronic in vivo obestatin administration in male rats enhanced Akt activity in omental, subcutaneous and gonadal fat. It is well known that Akt regulates mammalian cell cycle progression and cell survival [27]. Furthermore, there is strong evidence, including results obtained from genetic mouse models, supporting the concept that Akt is a key node for regulation of a multitude of metabolic events such as glucose uptake, glycogen synthesis, glyconeogenesis, lipid metabolism, protein synthesis and differentiation processes as adipogenesis [27, 30–34]. One of the physiological functions of Akt is to stimulate glucose uptake by GLUT4 trafficking through the Rab GAP (GTPase-acting protein) known as AS160 [35–38]. It was described five putative Akt phosphorylating sites on AS160 of which T642 appears to be critical for mediating the GLUT4 translocation and glucose uptake in response to insulin [36]. Our results showed that obestatin increased AS160 phosphorylation on T642, consistent with Akt mediating obestatin-induced phosphorylation. AS160 keeps a GAP domain that maintains its target Rab(s) in their inactive GDP-state. AS160 phosphorylation suppresses its GAP activity, shifting the equilibrium of its targets Rab(s) to an active GTP form, enabling it to mediate GLUT4 trafficking [39]. Indeed, obestatin increased GLUT4 membrane translocation and glucose transport in 3T3-L1 adipocyte cells, although in smaller extension to that of insulin. Inside the cell, glucose is converted to glucose 6-phosphate that can be stored by conversion to glycogen or catabolized by glycolysis, both processes under the control of Akt signalling [27]. In this sense, obestatin inactivated GSK3α/β, an Akt substrate, through phosphorylation in 3T3-L1 adipocyte cells. This inactivation leads to a decrease in the phosphorylation of glycogen synthase (GS) resulting in its activation, thereby stimulating glycogen synthesis [40]. Apart from GS, translation initiation factor 2B (eIF2B) can be also activated in a similar way leading to an increase in the glycogen synthesis and the protein synthesis [40]. Furthermore, inactivation of GSK3 has been shown to avoid degradation of the sterol regulatory element-binding proteins (SREBPs), which are transcription factors that trigger the expression of genes involved in cholesterol and fatty acid biosynthesis [41]. Consequently, Akt-directed signaling coordinates the obestatin-evoked glucose uptake and storage as glycogen in adipose tissue by regulation of AS160 and GSK3. In addition to glucose uptake, obestatin showed to regulate mTORC1/S6K1 signaling, a critical element integrating cellular metabolism with growth factor signaling [42]. mTORC1 controls many aspects of cellular metabolism including fat metabolism [42–44]. In particular, mTORC1/S6K1 plays an important role in adipogenesis and, thus, in lipid accumulation [43]. Loss of mTORC1/S6K1 activity correlates with a decrease in fat accumulation, suggesting that the mTOR pathway is required for fat accumulation [45, 46]. Furthermore, activation of mTORC1 signaling is a critical step in adipocyte differentiation [46]. The capacity of obestatin to modulate Akt activity in pre-adipocyte and adipocyte cells is not an isolated fact since this kinase is also activated by this peptide in pancreatic®-cell lines, human islets [24] and gastric cell lines [23]. In gastric cell lines, a signaling pathway involving a β-arrestin 1 scaffolding complex and EGFR to activate Akt signaling is proposed [23]. It will clearly be of interest to establish whether this also occurs in adipose cells.
Parallel to Akt activation, AMPK inactivation was triggered by obestatin in 3T3-L1 pre-adipocyte and adipocyte cells. Similarly, 72 hrs continuous sc infusion of obestatin in vivo resulted in decreased AMPK activity in WAT similar to that observed in vitro. Thus, a decline in AMPK phosphorylation would be expected to lead to a reduction in ACC phosphorylation, and thus an increase in the activity of this enzyme. Accordingly, we found that ACC phosphorylation was also decreased by obestatin in this study. Based on it, increased ACC activity consequently leads to increased malonyl Co-A levels, which mediate an inhibitory effect on cholinephosphotransferase 1 (CPT-1), preventing fatty acid transport into mitochondria and fatty acid oxidation, promoting fatty acid synthesis [47]. Thus, Akt activation and AMPK inactivation seem to be inversely correlated during obestatin stimulation. This fact was confirmed in vivo in omental, subcutaneous and gonadal WAT obtained from male rats under continuous sc infusion of obestatin. This enzymatic activity favours fat deposition with the consequent inhibition of breakdown plus burning stored fat and reduction of body weight. Furthermore, obestatin works in a rapid, hormone-like manner, being its effect insulin independent.
This study demonstrates a novel role for obestatin as an adipogenic molecule. Based on the promoter role exerted by obestatin on insulin-induced adipogenesis and the fact that obestatin expression increased in adipocytes compared to pre-adipocyte cells, we postulated that obestatin might exert an autocrine/paracrine effect on the differentiation process. The fact that preproghrelin expression increased in the course of differentiation process being sustained throughout terminal differentiation of 3T3-L1 cells supports our hypothesis. Furthermore, preproghrelin knockdown experiments revealed its contribution to adipogenesis, although it would involve both ghrelin and obestatin. The implication of obestatin seems to be confirmed by neutralization of obestatin with specific antibody in 3T3-L1 cells that reduced their adipogenic potential. This is further supported by the effect of exogenous obestatin on the expression of master regulators of adipocyte fate, C/EBPα, C/EBPβ, C/EBPδ and PPARγ [48]. We can hypothesize that ghrelin and obestatin mutually contribute to adipogenesis as pro-adipogenic factors promoting lipogenesis. Indeed, it has been shown that ghrelin stimulates lipid accumulation in vitro [13, 49] and in vivo [5, 10]. Thus, the original concept that these two peptides derived from the same gene show opposite actions is not completely correct. Interestingly, ghrelin and obestatin knockdown by preproghrelin siRNA revealed a key contribution of both peptides to insulin-induced terminal adipogenesis suggesting that the insulin effect might be mediated by expression and secretion of both peptides on the late stage of adipogenesis.
The expression of GPR39 in 3T3-L1 cells and its expression changes during adipogenesis suggests its implication in adipocyte differentiation. This fits well with the expression pattern of GPR39 in mouse embryonic fibroblast cells during adipocyte differentiation [25]. This hypothesis seems to be consistent with the up-regulation of preproghrelin expression and, in consequence, with the increase of the biosynthesis and secretion of obestatin by adipocytes, which exerts a paracrine control on pre-adipocyte via a GPR39-dependent mechanism.
In summary, in this report we describe a novel role of obestatin in the autocrine/paracrine regulation of adipogenesis. The adipogenic role of obestatin is supported by: 1) the activation of Akt signaling, 2) the regulation of adipocyte metabolism and, 3) the inhibition of adipogenesis by disruption of preproghrelin or netralization of obestatin. Intriguingly, circulating obestatin and ghrelin levels do not increase in obese subjects, a fact shared with ghrelin [50–52]. Therefore, neither obestatin nor ghrelin appear to be determinant in maintaining obesity. Nonetheless this fact does not rule out a function for obestatin in the development of obesity. In this sense, it is remarkable that the magnification of WAT is related to the development of the metabolic disorders, and the identification of factors that participate to this phenomenon would provide new aspects for the understanding of adipogenesis as well as the pathogenesis of obesity and associated comorbidities.
Acknowledgments
We thank to Cecilia Castelao-Taboada, Carlos Seoane-Mosteiro, Andrea Salgado-Suárez, and Laura Parente-Noya for their technical assistance in the development of this study. Dr. P.V. Lear is greatly acknowledged for helpful discussions in immunocytochemistry assays. This work was supported by grants from Instituto de Salud Carlos III (Ministerio de Ciencia e Innovación, Spain; PI070908, PS09/02202, PS09/02075), Ministerio de Ciencia e Innovación (SAF2010–20451) and Xunta de Galicia (PGIDIT07PXIB208112PR, INCITE09918374PR). The work of JP Camina and Y Pazos are funded by the Instituto de Salud Carlos III and SERGAS through a research-staff stabilization contract. LM Seoane and M Pardo are funded by the Instituto de Salud Carlos III and SERGAS through a research-staff contract Miguel Servet. AB Crujeiras is funded by the Instituto de Salud Carlos III and SERGAS through a research-staff contract Sara Borrell. U Gurriarán-Rodriguez and A Roca-Rivada are funded by Xunta de Galicia through a research-staff contract Isabel Barreto and Lucas Labrada, respectively.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131:242–56. doi: 10.1016/j.cell.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 2.Park KW, Halperin DS, Tontonoz P. Before they were fat: adipocyte progenitors. Cell Metab. 2007;8:454–7. doi: 10.1016/j.cmet.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 4.Wang P, Mariman E, Renes J, et al. The secretory function of adipocytes in the physiology of white adipose tissue. J Cell Physiol. 2008;216:3–13. doi: 10.1002/jcp.21386. [DOI] [PubMed] [Google Scholar]
- 5.Wells T. Ghrelin—defender of fat. Prog Lipid Res. 2009;48:257–74. doi: 10.1016/j.plipres.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Tschöp M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000;407:908–13. doi: 10.1038/35038090. [DOI] [PubMed] [Google Scholar]
- 7.Wren AM, Small CJ, Abbott CR, et al. Ghrelin causes hyperphagia and obesity in rats. Diabetes. 2001;50:2540–7. doi: 10.2337/diabetes.50.11.2540. [DOI] [PubMed] [Google Scholar]
- 8.Tschöp M, Flora DB, Mayer JP, et al. Hypophysectomy prevents ghrelin-induced adiposity and increases gastric ghrelin secretion in rats. Obes Res. 2002;10:991–9. doi: 10.1038/oby.2002.135. [DOI] [PubMed] [Google Scholar]
- 9.Theander-Carrillo C, Wiedmer P, Cettour-Rose P, et al. Ghrelin action in the brain controls adipocyte metabolism. J Clin Invest. 2006;116:1983–93. doi: 10.1172/JCI25811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies JS, Kotokorpi P, Eccles SR, et al. Ghrelin induces abdominal obesity via GHS-R-dependent lipid retention. Mol Endocrinol. 2009;23:914–24. doi: 10.1210/me.2008-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi K, Roh SG, Hong YH, et al. The role of ghrelin and growth hormone secretagogues receptor on rat adipogenesis. Endocrinology. 2003;144:754–9. doi: 10.1210/en.2002-220783. [DOI] [PubMed] [Google Scholar]
- 12.Thompson NM, Gill DA, Davies R, et al. Ghrelin and des-octanoyl ghrelin promote adipogenesis directly in vivo by a mechanism independent of the type 1a growth hormone secretagogue receptor. Endocrinology. 2004;145:234–42. doi: 10.1210/en.2003-0899. [DOI] [PubMed] [Google Scholar]
- 13.Rodríguez A, Gómez-Ambrosi J, Catalán V, et al. Acylated and desacyl ghrelin stimulate lipid accumulation in human visceral adipocytes. Int J Obes (Lond) 2009;33:541–52. doi: 10.1038/ijo.2009.40. [DOI] [PubMed] [Google Scholar]
- 14.Tsubone T, Masaki T, Katsuragi I, et al. Ghrelin regulates adiposity in white adipose tissue and UCP1 mRNA expression in brown adipose tissue in mice. Regul Pept. 2005;130:97–103. doi: 10.1016/j.regpep.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 15.Muccioli G, Pons N, Ghè C, et al. Ghrelin and des-acyl ghrelin both inhibit isoproterenol-induced lipolysis in rat adipocytes via a non-type 1a growth hormone secretagogue receptor. Eur J Pharmacol. 2004;498:27–35. doi: 10.1016/j.ejphar.2004.07.066. [DOI] [PubMed] [Google Scholar]
- 16.Zhu X, Cao Y, Voogd K, et al. On the processing of proghrelin to ghrelin. J Biol Chem. 2006;281:38867–70. doi: 10.1074/jbc.M607955200. [DOI] [PubMed] [Google Scholar]
- 17.Zhang JV, Ren PG, Avsian-Kretchmer O, et al. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake. Science. 2005;310:996–9. doi: 10.1126/science.1117255. [DOI] [PubMed] [Google Scholar]
- 18.Seoane LM, Al-Massadi O, Pazos Y, et al. Central obestatin administration does not modify either spontaneous or ghrelin-induced food intake in rats. J Endocrinol Invest. 2006;29:RC13–5. doi: 10.1007/BF03344174. [DOI] [PubMed] [Google Scholar]
- 19.Chartrel N, Alvear-Perez R, Leprince J, et al. Comment on “Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake”. Science. 2007;315:766. doi: 10.1126/science.1135047. [DOI] [PubMed] [Google Scholar]
- 20.Nogueiras R, Pfluger P, Tovar S, et al. Effects of obestatin on energy balance and growth hormone secretion in rodents. Endocrinology. 2007;148:21–6. doi: 10.1210/en.2006-0915. [DOI] [PubMed] [Google Scholar]
- 21.Zhang JV, Jahr H, Luo CW, et al. Obestatin induction of early-response gene expression in gastrointestinal and adipose tissues and the mediatory role of G protein-coupled receptor, GPR39. Mol Endocrinol. 2008;22:1464–75. doi: 10.1210/me.2007-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pazos Y, Alvarez CJ, Camiña JP, et al. Stimulation of extracellular signal-regulated kinases and proliferation in the human gastric cancer cells KATO-III by obestatin. Growth Factors. 2007;25:373–81. doi: 10.1080/08977190801889313. [DOI] [PubMed] [Google Scholar]
- 23.Alvarez CJ, Lodeiro M, Theodoropoulou M, et al. Obestatin stimulates Akt signalling in gastric cancer cells through beta-arrestin-mediated epidermal growth factor receptor transactivation. Endocr Relat Cancer. 2009;16:599–611. doi: 10.1677/ERC-08-0192. [DOI] [PubMed] [Google Scholar]
- 24.Granata R, Settanni F, Gallo D, et al. Obestatin promotes survival of pancreatic beta-cells and human islets and induces expression of genes involved in the regulation of beta-cell mass and function. Diabetes. 2008;57:967–79. doi: 10.2337/db07-1104. [DOI] [PubMed] [Google Scholar]
- 25.Egerod KL, Holst B, Petersen PS, et al. GPR39 splice variants versus antisense gene LYPD1: expression and regulation in gastrointestinal tract, endocrine pancreas, liver, and white adipose tissue. Mol Endocrinol. 2007;21:1685–8. doi: 10.1210/me.2007-0055. [DOI] [PubMed] [Google Scholar]
- 26.Catalán V, Gómez-Ambrosi J, Rotellar F, et al. The obestatin receptor (GPR39) is expressed in human adipose tissue and is down-regulated in obesity-associated type 2 diabetes mellitus. Clin Endocrinol (Oxf) 2007;66:598–601. doi: 10.1111/j.1365-2265.2007.02777.x. [DOI] [PubMed] [Google Scholar]
- 27.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89:1025–78. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 29.Zizzari P, Longchamps R, Epelbaum J, et al. Obestatin partially affects ghrelin stimulation of food intake and growth hormone secretion in rodents. Endocrinology. 2007;148:1648–53. doi: 10.1210/en.2006-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu J, Liao K. Protein kinase B/AKT 1 plays a pivotal role in insulin-like growth factor-1 receptor signaling induced 3T3-L1 adipocyte differentiation. J Biol Chem. 2004;279:35914–22. doi: 10.1074/jbc.M402297200. [DOI] [PubMed] [Google Scholar]
- 31.Chen WS, Xu PZ, Gottlob K, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–8. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho H, Thorvaldsen JL, Chu Q, et al. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–52. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 33.Garofalo RS, Orena SJ, Rafidi K, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berggreen C, Gormand A, Omar B, et al. Protein kinase B activity is required for the effects of insulin on lipid metabolism in adipocytes. Am J Physiol Endocrinol Metab. 2009;296:E635–46. doi: 10.1152/ajpendo.90596.2008. [DOI] [PubMed] [Google Scholar]
- 35.Kane S, Sano H, Liu SC, et al. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem. 2002;277:22115–8. doi: 10.1074/jbc.C200198200. [DOI] [PubMed] [Google Scholar]
- 36.Sano H, Kane S, Sano E, et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 37.Eguez L, Lee A, Chavez JA, et al. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2005;2:263–72. doi: 10.1016/j.cmet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 38.Larance M, Ramm G, Stöckli J, et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005;280:37803–13. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- 39.Zaid H, Antonescu CN, Randhawa VK, et al. Insulin action on glucose transporters through molecular switches, tracks and tethers. Biochem J. 2008;413:201–15. doi: 10.1042/BJ20080723. [DOI] [PubMed] [Google Scholar]
- 40.Rayasam GV, Tulasi VK, Sodhi R, et al. Glycogen synthase kinase 3: more than a namesake. Br J Pharmacol. 2009;156:885–98. doi: 10.1111/j.1476-5381.2008.00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sundqvist A, Bengoechea-Alonso MT, Ye X, et al. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7) Cell Metab. 2005;1:379–91. doi: 10.1016/j.cmet.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 42.Um SH, D’Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006;3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 43.Kim JE, Chen J. Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes. 2004;53:2748–56. doi: 10.2337/diabetes.53.11.2748. [DOI] [PubMed] [Google Scholar]
- 44.Dann SG, Selvaraj A, Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med. 2007;13:252–9. doi: 10.1016/j.molmed.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 45.Um SH, Frigerio F, Watanabe M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–5. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 46.Zhang HH, Huang J, Düvel K, et al. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS One. 2009;4:e6189. doi: 10.1371/journal.pone.0006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Munday MR. Regulation of mammalian acetyl-CoA carboxylase. Biochem Soc Trans. 2002;30:1059–64. doi: 10.1042/bst0301059. [DOI] [PubMed] [Google Scholar]
- 48.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7:885–96. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 49.Kim MS, Yoon CY, Jang PG, et al. The mitogenic and antiapoptotic actions of ghrelin in 3T3-L1 adipocytes. Mol Endocrinol. 2004;18:2291–301. doi: 10.1210/me.2003-0459. [DOI] [PubMed] [Google Scholar]
- 50.Vicennati V, Genghini S, De Iasio R, et al. Circulating obestatin levels and the ghrelin/obestatin ratio in obese women. Eur J Endocrinol. 2007;157:295–301. doi: 10.1530/EJE-07-0059. [DOI] [PubMed] [Google Scholar]
- 51.Huda MS, Durham BH, Wong SP, et al. Plasma obestatin levels are lower in obese and post-gastrectomy subjects, but do not change in response to a meal. Int J Obes. 2008;32:129–35. doi: 10.1038/sj.ijo.0803694. [DOI] [PubMed] [Google Scholar]
- 52.Zamrazilová H, Hainer V, Sedlácková D, et al. Plasma obestatin levels in normal weight, obese and anorectic women. Physiol Res. 2008;57:S49–55. doi: 10.33549/physiolres.931489. [DOI] [PubMed] [Google Scholar]