

Abstract
Current clinical protocols used for isolation and purification of mesenchymal stem cells (MSC) are based on long-term cultures starting with bone marrow (BM) mononuclear cells. Using a commercially available immunoselection kit for enrichment of MSC, we investigated whether culture of enriched BM-CD105+ cells could provide an adequate number of pure MSC in a short time for clinical use in the context of graft versus host disease and graft failure/rejection. We isolated a mean of 5.4 × 105 ± 0.9 × 105 CD105+ cells from 10 small volume (10–25 ml) BM samples achieving an enrichment >100-fold in MSC. Seeding 2 × 103 immunoselected cells/cm2 we were able to produce 2.5 × 108 ± 0.7 × 108 MSC from cultures with autologous serum enriched medium within 3 weeks. Neither haematopoietic nor endothelial cells were detectable even in the primary culture cell product. Expanded cells fulfilled both phenotypic and functional current criteria for MSC; they were CD29+, CD90+, CD73+, CD105+, CD45−; they suppressed allogeneic T-cell reaction in mixed lymphocyte cultures and retained in vitro differentiation potential. Moreover, comparative genomic hybridization analysis revealed chromosomal stability of the cultured MSC. Our data indicate that adequate numbers of pure MSC suitable for clinical applications can be generated within a short time using enriched BM-CD105+ cells.
Keywords: clinical scale expansion, mesenchymal stem cells, CD105+ cells
Introduction
The ability of mesenchymal stem cells (MSC) to differentiate along multiple pathways, together with their relatively easy isolation from BM and their extensive capacity for in vitro expansion, makes MSC potentially the most attractive population among stem cells for tissue engineering and cell therapy applicable on a variety of congenital and acquired diseases [1–3]. In addition to their role in regenerative therapies, MSC have been consistently shown to possess immunomodulatory properties, which may play a role in the maintenance of peripheral tolerance, the induction of transplantation-related tolerance and control autoimmunity [4, 5]. There are preliminary data supporting their clinical efficacy in controlling steroid resistant graft versus host disease (GVHD) and improving engraftment of donor cells in the allogeneic stem cell transplant setting. However, the beneficial role of MSC in GVHD prophylaxis is being questioned by small-scale randomized clinical trials [6, 7]. Further large-scale randomized studies are needed to clarify the benefits and hazards of MSC administration for the prevention and therapy of GVHD.
In all clinical protocols, MSC have been isolated from bone marrow mononuclear cells (BM-MNC) and amplified in culture media supplemented with foetal calf serum [8, 9], or human platelet lysate [10]. Such protocols, however, result in a heterogeneous initial population of adherent BM cells, of which a significant proportion represents adherent monocytic cells. Even when MSC have been harvested after three or more culture passages (>4weeks culture time), the proportion of macrophages identified represented 1–44% of stromal cells [11, 12]. Recent studies have shown that positive selection using several surface markers including stromal (STRO)-1, CD105, CD271 and stage specific embryonic antigen (SSEA)-4 makes it possible to obtain a homogenous MSC population without contaminating cultures with haematopoietic derived cells [13–16]. However, no such an enrichment method has yet been used in clinical scale ex vivo expansion of MSC. Here we describe the isolation and ex vivo expansion of MSC from BM-CD105+ cells in culture media enriched with autologous human serum for clinical application.
Materials and methods
BM was harvested from the posterior iliac crest of 10 normal donors for a related stem cell transplant, aged 10–30 years old, after informed consent, according to the Ethical Committee of the Aghia Sophia Children’s Hospital. Initially, we performed immunomagnetic isolation of BM-CD105+ cells using Milteny microbeads according to the manufacturer’s instructions (Miltenyi Biotech, Bergisch Gladbach, Germany). After the isolation, BM-CD105+ cells were suspended in DMEM (Stem Cell Technologies, Vancouver, BC, Canada) enriched with 10% autologous serum, and placed into a 75 cm2 flask (Corning Life Sciences, Corning, NY, USA) at a concentration of 2 × 103 cells/cm2. The flasks were incubated at 37°C in a humidified environment with 5% CO2. The medium was changed after 72 hrs and thereafter every 3–4 days. Fibroblastoid cells first became evident 2 to 4 days after inoculating culture flasks with CD105+ cells (Fig. 1C). By 9 to 12 days, a homogeneous population of adherent fibroblastoid cells was present (Fig. 1D). At this time-point cells were detached with 0.25% trypsin-ethylenediaminetetraacetic acid (Gibco BRL, Grand Island, NY, USA) for 5 min. at 37°C counted and subsequently re-plated at 4 × 103 cells/cm2 in 175 cm2 flasks until confluency. Cells derived from primary cultures of CD105+ cells were defined as Passage 0 (P0) and each cycle of reseeding of MSC after trypsinization was considered to be one additional passage. After a total time of 3 weeks, the cultures were discontinued. Surface marker expression of MSC derived from both primary (P0) and P1 cultures were analysed by flow cytometry. Cells were stained with monoclonal antibodies against human leukocyte antigen (HLA)-DR, CD14, CD29, CD34, CD45, CD31, CD73, CD105, CD44, CD73, and CD90. In order to investigate the multipotentiality of MSC, MSC derived both from P0 and P1 were directed towards the osteogenic, chondrogenic and adipogenic lineages. For osteogenic differentiation, cells were cultured in complete osteogenic differentiation medium (Stem Cell Technologies). Cells were fed twice a week. After a culture period ranged from 3 to 5 weeks, monolayer cultures were analysed morphologically for the formation of large osteogenic nodules. For adipogenic differentiation, cells were cultured for 2 weeks in complete adipogenic differentiation medium (Stem Cell Technologies). Adipogenic differentiation was confirmed by the formation of neutral lipid vacuoles. Chondrogenic differentiation was performed in high-density pellets containing 2.5 × 105 cells per pellet, cultured for 21 days in StemPro chondrogenesis differentiation medium (Invitrogen, Carlsbad, CA, USA). Total RNA was isolated from differentiated cells using RNeasy MINI Kit (Qiagen GmbH, Hilden, Germany). One microgram of each RNA sample was reverse transcribed with SuperScript III (Invitrogen) with use of Oligo(dT). The resulting cDNA was then used as a template for PCR amplification with Platinum Taq (Invitrogen); the following genes have been inquired: alkaline phosphatase (ALP, F: TCAGAAGCTCAACACCAACG; R: GTCAGGGACCTGGGCATT) [15] for osteogenesis, lipoprotein lipase (LPL, F: GAGATTTCTCTGTATGGCACC; R: CTGCAAATGAGACACTTTCTC) [17] for adipogenesis and collagen 2A1 (COL2A1, F: GGAAACTTTGCTGCCCAGATG; R: TCACCAGGTTCACCAGGATTGC) [17] for chondrogenesis. To estimate the immune inhibitory function of MSC, we used Ki-67-antigen staining in mixed lymphocyte cultures (MLC) in the presence of MSC as third party [18]. MLC were carried out with human peripheral blood mononuclear cells (hPBMC) from two HLA-DRB mismatched individuals used as effectors and stimulators. Stimulators and MSC were irradiated (30 Gy) before being cultured with effectors. Effectors and stimulators were seeded in triplicate at the concentration of 1 × 105 cells/500 μl/well in 24-well round-bottom plates (Nunc, Roskilde, Denmark) in the absence (control) or presence of decreasing number of third-party MSCs (ratio MSC/PBMC = 1, 0.5 and 0.1). After an incubation period of 96 hrs, intracellular expression of Ki-67 was determined by immunohistochemistry. An array-based comparative genomic hybridization (array-CGH) analysis was conducted in order to investigate the genomic stabilility of MSC. Genomic DNA was extracted from four MSC samples derived at the end of expansion culture using the Qiamp DNA Mini kit (Qiagen GmbH, Hilden, Germany). Agilent Human Genome CGH 4 × 44K microarrays with an average spatial resolution of 12 kb were used in the study (Agilent Technologies, Santa Clara, CA, USA). Genomic DNA from MSCs and pooled reference DNA of the same gender (Promega Corporation, Madison, WI, USA) were digested with AluI and RsaI (Promega Corporation) and labelled using Agilent Genomic DNA labelling Kit according to manufacturer’s instructions. MSCs and reference DNA were labelled with Cy3 and Cy5, respectively, and were co-hybridized to arrays for 40 hrs at 65°C in a rotating oven (Agilent Technologies) at 20 rpm. The arrays were then washed with Agilent wash buffer and scanned by using an Agilent Microarray Scanner. Data were extracted using Feature Extraction 9.1 software (Agilent Technologies) and analysed using CGH Analytics 3.4 software (Agilent Technologies). Genomic copy number changes were identified with the assistance of the Aberration Detection Method 1 algorithm with a threshold of 6. Centralization and fuzzy zero corrections were applied to remove putative variant intervals with small average log2 ratios. For the location of genes in the deleted/duplicated genomic segments the UCSC (http://genome.ucsc.edu/) and the Database of Genomic Variants (http://projects.tcag.ca/variation/) (human genome build 18) were used.
Fig 1.
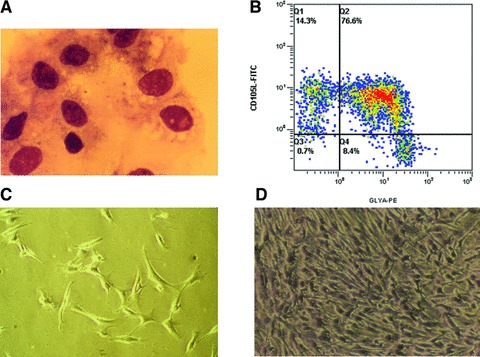
Characterization of freshly isolated BM-CD105+cells and cultured MSCs. (A) Morphology of immunomagnetically isolated BM-CD105+ cells after they were cytocentrifuged and counterstained with May-Grünwald solution. (B) Three colour flow cytometric analysis of sorted cells demonstrated two distinct CD105+ cell populations: CD105+Glycophorin-A+CD45− and CD105+Glycophorin-A−CD45−. (C–D) Fibroblastoid morphology of cells derived from CD105+ cells after 3 (C) and 10 days (D) in culture before first passage (primary cells).
Results
From BM samples of 10–25 ml (16 ml average) we were able to immunomagnetically isolate 5.4 × 105± 0.9 ± 105 cells. We detected two populations of CD105+CD45− cells (Fig. 1B): CD105+clycophorin+CD45− (erythroid cells) and CD105+clycophorin−CD45− cells (mesenchymal cells) comprising 78.6 ± 16.2% and 18.6 ± 4.8% of the total isolated CD105+ cells, respectively, whereas the proportion of the latter in the initial BM mononuclear cell population was 0.16% ± 0.12%. Light microscopic examination of cytospins prepared with freshly sorted CD105+ cells disclosed, apart from typical erythroblasts, median size agranular cells with heterochromatic nuclei and numerous projections of the cell membrane resembling reticular cells (Fig. 1A). Primary culture yielded 8.6 × 106 ± 1.2 × 106 MSC within a median time of 10 days. After replating at 4 × 103 cells/cm2, P0 MSC were further cultured for an additional period of 11 days, and resulted to a total production of 2.5 × 108 ± 0.7 × 108 MSC (Fig. 2). Flow cytometry revealed that P0 MSC did not express certain haematopoietic and endothelial markers such as CD45, CD31, CD14, CD34 and CD62L. On the other hand, cultured cells were found positive for CD105, CD90, CD73, CD44, CD29 and class I HLA (Fig. 3). No difference in marker expression was found for P0 or P1 MSC.
Fig 2.
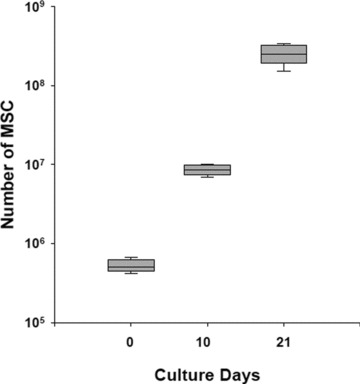
Expansion potential of MSC derived from selected BM-CD105+cells. Growth profile of CD105+ cells sorted on day 0 and plated at 2 × 103 cells/cm2 culture surface. On day 10, adherent cells were detached with thrypsin and replated at 4 × 103 cells/cm2 in culture for an additional period of 11 days. Boxes represent the interquartile range; lines inside boxes represent the median value; whiskers represent the lowest and highest observations, respectively.
Fig 3.
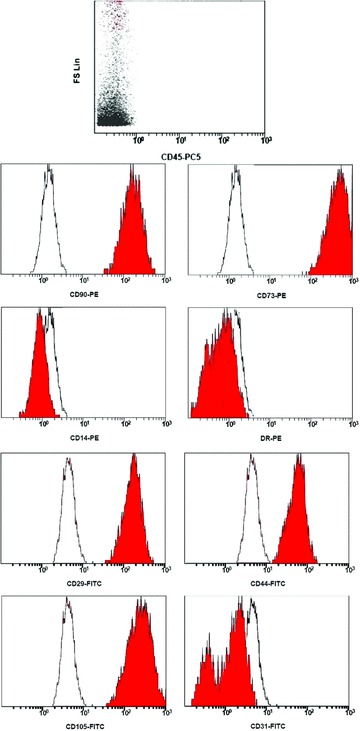
Immunophenotype of P0-MSCs. Flow cytometric plots showing the immunophenotype of cultured cells at the end of primary culture on day 10. Gray histograms show specific antibody staining profile and open histograms show isotype control IgG staining profile.
MSC from both primary and passage 1 (P0 and P1) cultures treated with osteogenic medium underwent a change in their morphology from spindle shaped to cuboidal, and formed large nodules after 18 days of induction. MSC cultured in adipocytic differentiation medium formed lipid vacuoles 14 days after induction. Chondrogenic, adipogenic and osteogenic differentiation were confirmed by COL2A1, LPL and ALP expression, respectively (Fig. 4).
Fig 4.
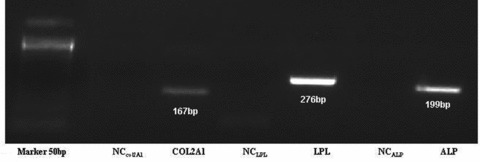
Chondrogenic, adipogenic and osteogenic differentiation of MSC. Chondrogenic, adipocytic and osteogenic differentiation was demonstrated by the expression of specific genes COL2A1, LPL and ALP, respectively.
We next assessed the ability of MSC to inhibit the alloreaction observed in MLC. As expected, MLC without MSC led to an increase in proliferation of effector lymphocytes quantified by the percentage of Ki-67+ cells. Both P0 and P1 MSC, when added to MLC at MSC/effector ratio of 1:1, efficiently reduced the percentage of Ki-67+ cells by approximately 85%. With decreasing MSC/effector ratios the proportion of proliferating effector cells was increased. However, even at low MSC to effector ratio of 0.1 there was an inhibition of proliferating cells of about 25–30% (Fig. 5).
Fig 5.
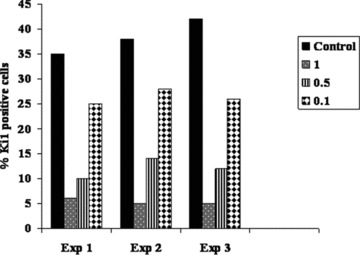
Inhibitory effect of MSC on lymphocyte proliferation in MLC. hPBMC from healthy donors were co-cultured in one-way MLC with irradiated allogeneic PBMC in the absence (control) or presence of decreasing number of unrelated MSC. At 96 hrs of culture, cells were stained for intracellular Ki-67 antigen. Ki-67 antigen positivity indicates proliferating responder cells. Bars represent the percentage of Ki-67+ cells in three independent experiments at 3 MSC to effector ratios of 1, 0.5 and 0.1.
We performed a whole-genome microarray-based CGH in order to look for chromosomal rearrangements with multiple loci involvement and for genome-wide aberrations in our MSC samples from normal donors. CGH-plotter software showed balanced profiles for all four MSC samples after culture expansion (Fig. 6). We detected several small (average size of 15 kb to 1.5 Mb) autosomal copy number variations, both deletions and duplications, which are also observed in normal individuals [19].
Fig 6.
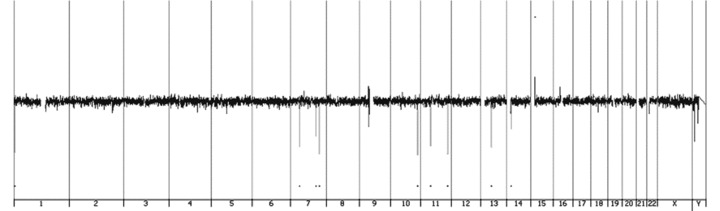
Whole-genome array CGH profile of MSC. The X-axis represents the 22 autosomes, the X and Y chromosomes, and the Y-axis shows the log2 of the fluorescence intensity ratio of all spots of the chromosome. Every coloured vertical peak represents a commonly found copy number variations; duplications are represented above the baseline with log2 ratio >0.25 and deletions below the baseline with log2 ratio <–0.25.
Discussion
Our aim was to establish a culture system where sufficient numbers of pure MSC could be generated within reasonable time for ‘emergency type’ therapeutic strategies in the context of allogeneic stem cell transplant, like treatment of steroid-resistant GVHD and graft failure/rejection. It is important to note that MSC contaminated with a low percentage of CD45+ cells and MSC of late passage do not exhibit in vivo significant anti-GVHD effect, as recently suggested [20], indicating that early passage and significant immunodepletion are required for MSC-based cell therapy of GVHD. Therefore, we had to develop an in vitro approach which would strictly fulfil two requirements: (1) The generation of a 2 × 106/per kg adult patient weight, that is the upper cell dose used for an adult patient, within a maximum of 3 weeks, capable to suppress the alloreaction in vitro and (2) the absence of any contamination of MSC with haematopoietic or/and endothelial cells. Based on our previous experience with chimerism studies of MSC derived from immunoselected CD105+ BM cells [21] and the reduced seeding density approach established by Prockop’s group [22], we combined these methods aiming to obtain adequate numbers of MSC with immunosuppressive properties within 3 weeks. Starting with CD105+ cells isolated from small volume BM samples, we achieved MSC propagation which corresponded to a cell dose of >2 × 106/kg for an adult patient within 3 weeks. More importantly, even MSC from primary culture could be given to patients safely, because they were completely devoid of haematopoietic cells. We also investigated the maintenance of genomic stability using a whole-genome microarray based CGH at the end of the expansion procedure. Although we analysed MSC after a high in vitro proliferation rate, they did not show genomic alterations. By comparing our results with published data from clinical studies, similar numbers of MSC were obtained after culturing BM-MNC isolated from >40 ml BM samples for 4–6 weeks [23, 24]. According to our own experience, when cultures started from BM-MNC population, a larger volume of BM is needed (>30 mL) and the median time of expansion is much longer, almost 35 days to obtain adequate numbers for MSC infusion. Two recent, independent studies, however, described the successful production of a large MSC number (>2 × 108) within fewer than 4 weeks derived from small BM aspiration samples [25, 26]. Both research groups were based on the fact that reduced MSC seeding density results in marked increase of MSC proliferation. Nevertheless, the fact that the cultures were initiated with unmanipulated BM [25] or BM-MNC [26] and MSC for clinical use were derived from primary culture or P1, respectively, makes the contamination with haematopoietic cells a matter of concern. At the time of writing this report, Poloni et al. [27] were able to produce 1 × 109 MSC using BM-CD271+ cells immunoselected from 5 ml BM in 30 days. However, they did not mention whether the produced cells possessed in vitro immunomodulatory abilities. In our hands, expansion cultures of CD271+ cells using foetal bovine serum (FBS) or human serum did not produce MSC solely, but also a percentage of neurosphere-like structures indicating a spontaneous differentiation to neural cell lineage. That was the reason for choosing the CD105+ cells.
Several criteria are required for clinical application of MSC in human beings. It is of major importance to use a rapid method to produce adequate numbers of uncontaminated MSC with proven immunosuppressive potential. Especially, in the clinical setting of resistant GVHD or graft failure, alternative treatments are required urgently, timing is crucial and shortening the culture period even by a few days can prove beneficial. The use of purified CD105+ BM cells makes the method more expensive, but in terms of cost-effectiveness, the benefit from the time saved can prove valuable. We have shown that our method of deriving MSC from enriched CD105+-BM cells fulfils the criteria for a clinically applicable cell therapy protocol.
Acknowledgments
K.G. is supported by a scholarship from the Alexander S. Onassis Public Benefit Foundation.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Alhadlaq A, Mao JJ. Mesenchymal stem cells: isolation and therapeutics. Stem Cells Dev. 2004;13:436–48. doi: 10.1089/scd.2004.13.436. [DOI] [PubMed] [Google Scholar]
- 2.Deans RJ, Moseley AB. Mesenchymal stem cells: biology and potential clinical uses. Exp Hematol. 2000;28:875–84. doi: 10.1016/s0301-472x(00)00482-3. [DOI] [PubMed] [Google Scholar]
- 3.Torrente Y, Polli E. Mesenchymal stem cell transplantation for neurodegenerative diseases. Cell Transplant. 2008;17:1103–13. doi: 10.3727/096368908787236576. [DOI] [PubMed] [Google Scholar]
- 4.Keyser KA, Beagles KE, Kiem HP. Comparison of mesenchymal stem cells from different tissues to suppress T-cell activation. Cell Transplant. 2007;16:555–62. doi: 10.3727/000000007783464939. [DOI] [PubMed] [Google Scholar]
- 5.Ucceli A, Moretta L, Pistoia V. Immunoregulatory function of mesenchymal stem cells. Eur J Immunol. 2006;36:2566–73. doi: 10.1002/eji.200636416. [DOI] [PubMed] [Google Scholar]
- 6.Lazarus HM, Koc ON, Devine SM, et al. Cotransplantation of HLA-identical sibling culture-expanded mesenchymal stem cells and hematopoietic stem cells in hematologic malignancy patients. Biol Blood Marrow Transplant. 2005;11:389–98. doi: 10.1016/j.bbmt.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Ning H, Yang F, Jiang M, et al. The correlation between cotransplantation of mesenchymal stem cells and higher recurrence rate in hematologic malignancy patients: outcome of a pilot clinical study. Leukemia. 2008;22:593–9. doi: 10.1038/sj.leu.2405090. [DOI] [PubMed] [Google Scholar]
- 8.Koç O, Gerson SL, Cooper BW, et al. Rapid hematopoietic recovery after coinfusion of autologous-blood stem cells in advanced breast cancer patients receiving high-dose chemotherapy. J Clin Oncol. 2000;18:307–16. doi: 10.1200/JCO.2000.18.2.307. [DOI] [PubMed] [Google Scholar]
- 9.Le Blanc K, Frassoni F, Ball L, et al. Developmental Committee of the European Group for Blood and Marrow Transplantation. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008;371:1579–86. doi: 10.1016/S0140-6736(08)60690-X. [DOI] [PubMed] [Google Scholar]
- 10.Von Bonin M, Stoelzel F, Goedecke A, et al. Treatment of refractory acute GVHD with third-party MSC expanded in platelet lysate-containing medium. Bone Marrow Transplant. 2009;43:245–51. doi: 10.1038/bmt.2008.316. [DOI] [PubMed] [Google Scholar]
- 11.Awaya N, Rupert K, Bryant E, et al. Failure of adult marrow-derived stem cells to generate marrow stroma after successful hematopoietic stem cell transplantation. Exp Hematol. 2002;30:937–42. doi: 10.1016/s0301-472x(02)00821-4. [DOI] [PubMed] [Google Scholar]
- 12.Ismail M, Gibson FM, Marks K, et al. Combined immunocytochemistry and FISH: an improved method to study engraftment of accessory bone marrow stromal cells. Clin Lab Haematol. 2002;24:239–335. doi: 10.1046/j.1365-2257.2002.00463.x. [DOI] [PubMed] [Google Scholar]
- 13.Simmons PJ, Torok-Storb B. Identification of stromal cell precursors in human bone marrow by a novel monoclonal antibody, STRO-1. Blood. 1991;78:55–62. [PubMed] [Google Scholar]
- 14.Barry FP, Boynton RE, Haynesworth S, et al. The monoclonal antibody SH-2, raised against human mesenchymal stem cells, recognizes an epitope on endoglin (CD105) Biochem Biophys Res Commun. 1999;265:134–9. doi: 10.1006/bbrc.1999.1620. [DOI] [PubMed] [Google Scholar]
- 15.Quirici N, Soligo D, Bossolasco P, et al. Isolation of bone marrow mesenchymal stem cells by anti-nerve growth factor receptor antibodies. Exp Hematol. 2002;30:783–91. doi: 10.1016/s0301-472x(02)00812-3. [DOI] [PubMed] [Google Scholar]
- 16.Gang EJ, Bosnakovski D, Figueiredo CA, et al. SSEA-4 identifies mesenchymal stem cells from bone marrow. Blood. 2007;109:1743–51. doi: 10.1182/blood-2005-11-010504. [DOI] [PubMed] [Google Scholar]
- 17.Nesti LJ, Jackson WM, Shanti RM, et al. Differentiation potential of multipotent progenitor cells derived from war-traumatized muscle tissue. J Bone Joint Surg Am. 2008;90:2390–8. doi: 10.2106/JBJS.H.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang L, Lange C, Engel M, et al. Sensitive balance of suppressing and activating effects of MSC on T cell proliferation. Transplantation. 2006;82:1370–3. doi: 10.1097/01.tp.0000232450.62408.f9. [DOI] [PubMed] [Google Scholar]
- 19.Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polchert D, Sobinsky S, Douglas GW, et al. IFN-γ activation of mesenchymal stem cells for treatment and prevention of graft versus host disease. Eur J Immunol. 2008;38:1745–55. doi: 10.1002/eji.200738129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goussetis E, Spiropoulos A, Theodosaki M, et al. Culture of bone marrow CD105+ cells allows rapid selection of pure BM-stromal cells for chimerism studies in patients undergoing allogeneic bone marrow transplantation. Bone Marrow Transplant. 2005;36:557–9. doi: 10.1038/sj.bmt.1705083. [DOI] [PubMed] [Google Scholar]
- 22.Colter DC, Class R, DiGirolamo CM, et al. Rapid expansion of recycling stem cells in cultures of plastic-adherent cells from human bone marrow. Proc Natl Acad Sci USA. 2000;97:3213–8. doi: 10.1073/pnas.070034097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ning H, Yang F, Jiang M, et al. The correlation between cotransplantation of mesenchymal stem cells and higher recurrence rate in hematologic malignancy patients: outcome of a pilot clinical study. Leukemia. 2008;22:593–9. doi: 10.1038/sj.leu.2405090. [DOI] [PubMed] [Google Scholar]
- 24.McMillan ML, Blazar BR, DeFor TE, et al. Transplantation of ex vivo culture-expanded parental haploidentical mesenchymal stem cells to promote engraftment in pediatric recipients of unrelated donor umbilical cord blood: results of a phase I-II clinical trial. Bone Marrow Transplant. 2009;43:245–51. doi: 10.1038/bmt.2008.348. [DOI] [PubMed] [Google Scholar]
- 25.Schallmoser K, Rohde E, Reinisch A, et al. Rapid large-scale expansion of functional mesenchymal stem cells from unmanipulated bone marrow without animal serum. Tissue Eng. 2008;14:185–95. doi: 10.1089/ten.tec.2008.0060. [DOI] [PubMed] [Google Scholar]
- 26.Bartmann C, Rohde E, Schallmoser K, et al. Two steps to functional mesenchymal stromal cells for clinical application. Transfusion. 2007;47:1426–35. doi: 10.1111/j.1537-2995.2007.01219.x. [DOI] [PubMed] [Google Scholar]
- 27.Poloni A, Maurizi G, Rosini V, et al. Selection of CD271+ cells and human AB serum allows a large expansion of mesenchymal stromal cells from human bone marrow. Cytotherapy. 2009;11:153–62. doi: 10.1080/14653240802582125. [DOI] [PubMed] [Google Scholar]