

Abstract
The epidermal growth factor receptor (EGFR) and other members of the EGFR/ErbB receptor family of receptor tyrosine kinases (RTKs) are important regulators of proliferation, angiogenesis, migration, tumorigenesis and metastasis. Overexpression, mutations, deletions and production of autocrine ligands contribute to aberrant activation of the ErbB proteins. The signalling output from EGFR is complicated given that other ErbB proteins are often additionally expressed and activated in the same cell, resulting in formation of homo-and/or heterodimers. In particular, association of EGFR with ErbB2 prevents its down-regulation, underscoring the importance of the cellular background for EGFR effects. Signalling from ErbB proteins can either be terminated by dissociation of ligand resulting in dephosphorylation, or blunted by degradation of the receptors. Although proteasomal targeting of ErbB proteins has been described, lysosomal degradation upon ligand-induced endocytosis seems to play the major role in EGFR down-regulation. Preclinical and clinical data have demonstrated that EGFR is a central player in cancer, especially in carcinomas, some brain tumours and in non-small cell lung cancer. Such studies have further validated EGFR as an important molecular target in cancer treatment. This review focuses on mechanisms involved in ligand-induced EGFR activation and endocytic down-regulation. A better understanding of EGFR biology should allow development of more tumour-selective therapeutic approaches targeting EGFR-induced signalling.
Keywords: EGFR, growth factor, cancer, endocytosis, kinase activity, ubiquitination, clathrin
Introduction
Cellular growth, proliferation and migration are essential aspects of embryogenesis and tissue maintenance. When uncontrolled, such cellular processes can lead to cancer progression. Central in these processes are growth factor receptors, which convert information from the external milieu by interacting with exposed cellular proteins and soluble ligands. This activates signalling pathways, which trigger early phenotypic responses such as increased motility and can change gene expression. Signalling from growth factor receptors is normally tightly regulated at several levels, including desensitization and degradation of the receptors. A paradigmatic system of growth factors and receptors is the network of ErbB proteins and their interacting ligands [1, 2], largely because dysregulation of ErbB proteins is a key factor in progression of many human cancers [3–5]. The epidermal growth factor (EGF), which binds to EGFR/ErbB1, was one of the first growth factors to be described [6]. EGFR and the three other members of the EGFR family (ErbB2-4) are structurally similar type I transmembrane proteins, yet with noticeable differences in the ligand-binding domain for ErbB2 and the kinase domain for ErbB3. The ligand-binding extracellular domain in ErbB1, 3 and 4 undergoes a major conformational change during ligand binding, exposing a dimerization arm [7]. In contrast, the dimerization arm of ErbB2 is constitutively exposed in the absence of ligand, and no known soluble ligand binds to ErbB2. Nevertheless, ErbB2 is the preferred dimerization partner due to its conformation (for a review, see [4]). While the extracellular and the very C-terminal regions are divergent, the kinase domains share a high amount of homology, except for ErbB3 where it is inactive. At least seven ligands can bind to the extracellular part of EGFR including EGF and transforming growth factor alpha (TGFα) [1, 4, 8]. Ligand binding induces homo- and/or heterodimerization, thereby resulting in kinase activation and phosphorylation of tyrosines in the ErbB tail (see Fig. 1A). Although kinase-deficient, ErbB3 can be transphosphorylated upon heterodimerization [1]. Phosphorylation on tyrosine residues creates binding sites for src-homology 2 (SH2) and phosphotyrosine-binding (PTB) domain-containing proteins, serving as adaptors and effectors in signal transduction (Fig. 1A). Several important signal transduction pathways are initiated downstream of ErbB proteins, including activation of Ras/MAP kinase, phospholipase Cγ/protein kinase C, phos-phatidylinositol 3-kinase (PI3 K)/Akt, Jak/STAT and Src family kinases [9, 10]. This again activates gene transcription resulting in new proteins responsible for proliferation, migration, adhesion, differentiation and apoptosis [1, 8]. Also proteins involved in endocytic down-regulation can be recruited to phosphorylated ErbB proteins, but only activated EGFR seems to be efficiently endocytosed and sorted to lysosomes for degradation [1, 11] (see Fig. 1B). EGFR recruits adaptor and effector proteins involved in endocytosis, and the plasma membrane eventually pinches off to form EGFR-containing vesicles. These early vesicles rapidly fuse to generate EGFR-containing early endosomes. It was previously demonstrated that upon siRNA-induced depletion of the clathrin adaptor complex AP2, most clathrin-coated pits disappear [12]. Interestingly, we discovered that under such conditions, new coated pits are formed upon activation of the EGFR. These coated pits accommodate EGFR, but not the transferrin receptor [13]. EGFR-containing coated pits may therefore be distinct from coated pits carrying constitutively internalized cargo.
1.
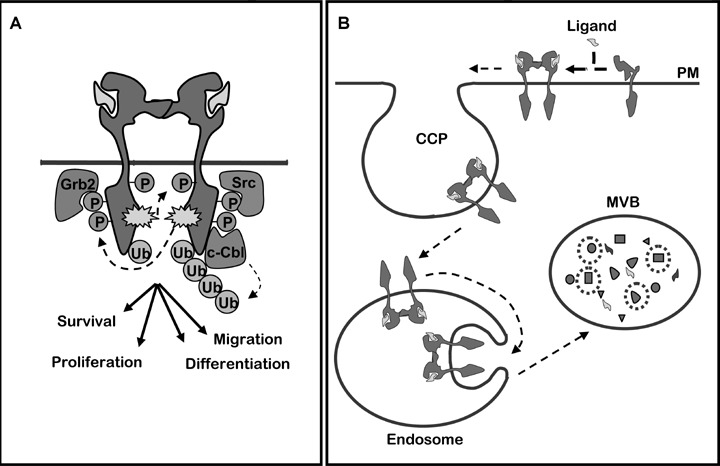
(A) EGFR dimerization upon ligand binding triggers auto-phosphorylation (P) and ubiquitination (Ub) of its cytoplasmic domain. This causes recruitment of effector proteins, like Grb2, Src and Cbl. (B) Intracellular route leading EGFR to down-regulation by lysosomal degradation. PM: plasma membrane, CCP: clathrin-coated pit, MVB: multivesicular body.
Dysregulation of EGFR is frequently observed in association with carcinogenesis. This can be the result of several unbalanced mechanisms controlling the quantitative and qualitative output from EGFR, caused by receptor overexpression, mutations, deletions and/or failure in down-regulation. EGFR can be constitutively activated in cells where the gene encoding EGFR is amplified or mutated [14], so that increased, tonic EGFR activity promotes neo-plastic transformation. First, in a number of tumours, both EGFR and its ligands are overexpressed. This scenario has been clearly demonstrated in the human epidermoid carcinoma cell line A431, where strong overexpression of EGFR and TGFa results in constitutive proliferative activity [14, 15]. Second, EGFR can be mutated, resulting in constitutively activated homo- and/or heterodimers. Somatic mutations have been identified in patients with advanced non-small lung cancer, who benefit from a dramatic clinical response to EGFR tyrosine kinase inhibitors [16]. Such EGFR mutations are often found in the first four exons of the kinase domain [16], directly dysregulating the kinase activity. Another example of EGFR mutant with constitutive signalling is EGFRvIII, which is characterized by a large extracellular deletion [3]. EGFRvIII is expressed in several cancer types, especially in glioblastomas and is sufficient to cause cell transformation [3, 17]. Although it was proposed that EGFRvIII undergoes a normal endocytic down-regulation [18], other groups reported insignificant degradation of EGFRvIII, due to impaired internalization combined with effective recycling of the small pool of internalized EGFR [19, 20]. It is important to recognize that heterodimers can be responsible for both increased and altered signalling, as well as decreased down-regulation of EGFR. A number of excellent reviews have summarized the role of EGFR in signal transduction and carcinogenesis. This review will mostly focus on how EGFR signalling can be terminated or altered by endocytic down-regulation and how EGFR down-regulation can be inhibited in cancer cells.
Endocytosis of EGFR
The requirements for efficient internalization of EGFR, which is a prerequisite for efficient down-regulation, are disputed. Recently, conflicting data on the necessity of EGFR kinase activity, clathrin-coated pits and ubiquitination have been published.
Kinase activity
EGFR normally occurs as monomer at the plasma membrane, prior to activation. Ligand binding triggers its dimerization, resulting in kinase activation, tyrosine phosphorylation and endocytosis. It has been published that EGFR kinase activity is required for recruitment of EGFR into clathrin-coated pits and subsequent endocytic down-regulation of EGF-bound EGFR [21–24]. Furthermore, it was reported that activation of EGFR induces the formation of clathrin-coated pits, by a mechanism requiring EGFR kinase activity [13]. It was recently proposed that EGFR dimerization is sufficient to induce endocytosis of kinase-dead EGFR [25].
However, EGFR kinase activity is required for tyrosine phosphorylation in the EGFR tail, and since binding of Grb2 to such phosphorylated tyrosines is key to clathrin-mediated, ligand-dependent endocytosis [26], there is reason to believe that EGFR kinase activity is indeed required for efficient ligand-dependent internalization. The fact that stress signals (UV irradiation and inflammatory cytokines) or drugs like cisplatin can induce endocytosis of the EGFR upon p38 MAP-kinase activation in a clathrin-dependent, but EGFR kinase-independent manner [27, 28], points to an alternative, ligand-independent mechanism whereby EGFR can be internalized under conditions of stress. This mechanism entails phosphorylation of the EGFR at multiple serines and threonines, as well as phosphorylation of the Rab5 effectors EEA1 and GDI [28]. It should be noted that stress signals do not appear to result in degradation of EGFR, but rather to temporarily sequester the EGFR inside the cell and eventually result in recycling of the EGFR to the plasma membrane [28]. Such internalization could enhance the cytotoxic effect of combining chemotherapy and EGFR-targeting drugs [28]. By temporarily preventing EGFR-mediated survival signalling, the cytotoxic effects of drugs like cisplatin could be enhanced.
Clathrin-coated pits
Whereas it is well established that EGFR is internalized in a clathrin-dependent manner [12, 29–32], it has also been suggested that ubiquitinated EGFR can be endocytosed by a clathrin-independent process. Sigismund and colleagues have proposed that EGFR is endocytosed from caveolae upon exposure to high concentrations of EGF [33], based on the observation that EGFR is frequently found in such plasma membrane microdomains under these conditions [34, 35]. However, despite the fact that EGFR function seems to be sensitive to the level of caveolin and/or membrane cholesterol [35, 36], no mobilization of caveolae could be found by FRAP analysis when high concentrations of EGF were added [29]. This strongly argues that even though EGFR can localize to caveolae, activated EGFR is not internalized from these invaginations, but from clathrin-coated pits. It has also been demonstrated that EGFR can activate small GTPases, like Cdc42 and Rac, and thereby be endocytosed into macropinosomes [37]. Furthermore, EGFR has been found to be effectively internalized via circular dorsal ruffles or ‘waves' [38]. However, induction of macropinocytosis and waves may be restricted to certain cell types or conditions, and there is now good agreement that functional clathrin-coated pits are the general portal for efficient EGFR endocytosis [29].
Ubiquitination
One of the EGFR tyrosine phosphorylation sites (pY1045) provides a direct docking site for the protooncoprotein and ubiquitin ligase, Cbl, which, upon EGFR activation, acts in concert with an E2 ubiquitin-loaded enzyme to covalently bind ubiquitin to lysine residues in the EGFR kinase domain as well as in the C-terminal tail [39–41]. In addition, Cbl can bind indirectly to the EGFR via Grb2 [40]. Whether the EGFR is mono- or poly-ubiquitinated has been disputed [42]. However, mass spectrometry analysis has clearly demonstrated both forms to occur and revealed that the ubiquitin chains on EGFR are mainly connected through lysine 63 [41]. While it is established (see below) that ubiquitination plays a central role in subcellular sorting and lysosomal degradation of the EGFR, it is still unclear whether ubiquitination is a signal required for initial steps of clathrin-dependent endocytosis. Mutations of EGFR impairing ubiquitination and thereby causing its stabilization have been described in cancer patients. Such EGFR mutants have further been demonstrated to promote growth and to protect against apoptosis [43]. When expressed in transfected cells, EGFR mutants that are inefficiently ubiquitinated are still efficiently endocytosed, arguing that ubiquitination is not required for EGFR internalization [41, 44, 45]. However, an EGFR mutant with impaired kinase activity was reported to depend on ubiquitination for internalization [45]. Thus, ubiquitination could potentially be required under conditions of inefficient activation of the EGFR kinase. Data comparing internalization of normal EGFR with EGFR constructs, endowed with normal kinase activity but abolished ubiquitination, are still warranted before concluding whether limited amounts of EGFR ubiquitination could be sufficient for clathrin-dependent EGFR endocytosis. However, it has so far not been possible to completely isolate effects on ubiquitination from effects on kinase activity [41, 45].
Eps15 and epsin, known to localize to clathrin-coated pits, are two proteins with ubiquitin binding capacity. Eps15 bears two ubiquitin-interaction motives (UIMs), interacts with AP2 and localizes to the edge of clathrin-coated pits [46–48]. Epsin proteins have two or three tandem UIMs [48] as well as clathrin- and AP2-interaction motives [49–53]. Epsin UIMs have been reported to efficiently interact with chains of four or more ubiquitins, preferentially those where lysine 63 in ubiquitin is isopeptide-linked to glycine (K63-linked polyubiquitin chains) [54]. Interestingly, while Eps15 localizes to the edge of clathrin-coated pits [46, 47], epsin was reported to localize along the entire curvature of these pits [47]. This could suggest that ubiquitinated EGFR is captured by Eps15 and subsequently handed off to epsin deeper in the coated pits, in order to be efficiently sequestered within clathrin-coated vesicles pinching off the plasma membrane (Fig. 2). However, the mechanisms involved in recruiting EGFR to clathrin-coated pits remain an open question.
2.
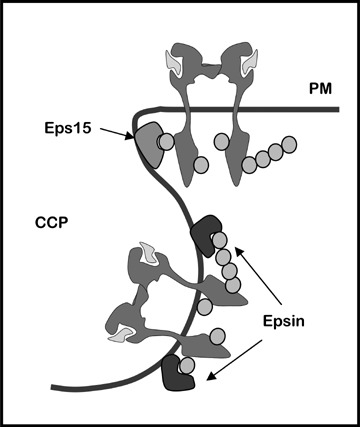
Tentative model of molecular interactions promoting regulated entry of EGFR into clathrin-coated pits (CCP). Initial recruitment of ubiquitinated EGFR to Eps15 at the rim of CCPs allows for its transfer to Epsin present in the entire CCP curvature. Green circles represent ubiquitin.
Effects of EGFR-ErbB2 heterodimerization on EGFR internalization
Whereas activated EGFR can in principle form heterodimers with all other ErbB proteins, ErbB2 is its preferred dimerization partner. Upon ErbB2 overexpression, activated EGFR is increasingly incorporated into EGFR-ErbB2 heterodimers [55]. Interestingly, amplification and overexpression of ErbB2 in breast cancer correlates with poor clinical outcome [56]. ErbB2 is normally trapped at the cell surface due to interaction with the molecular chaperone, heat-shock protein 90 (hsp90) and therefore escapes endocytosis [57, 58]. However, geldanamycin and related drugs of lesser toxicity, which specifically bind and inactivate the molecular chaperone Hsp90 [59, 60], disrupt the stabilizing interaction of Hsp90 with ErbB2 [61], resulting in degradation of ErbB2 as well as other Hsp90 target proteins (reviewed in [62, 63]). Remarkably, in addition to being itself endocytosis-deficient, ErbB2 also prevents internalization of EGFR upon heterodimerization [57, 58, 64]. This effect is ErbB2-concentration-dependent, and inhibition of EGFR endocytosis can be ascribed to blunted EGF-induced formation of clathrin-coated pits and recruitment of EGFR into such microdomains [58]. While EGF-induced formation of clathrin-coated pits was observed in cells when AP2 had been knocked down by siRNA and in serum-starved cells [13], EGF-induced formation was under similar conditions strongly inhibited when the cells overexpressed ErbB2 [58]. Mechanisms responsible for impaired EGF-induced coated pit formation in serum-starved cells are still unclear. While it was demonstrated that neither phosphorylation of tyrosines recruiting Grb2 and Cbl, nor ubiquitination of EGFR, was quantitatively affected upon overexpression of ErbB2, it cannot be excluded that ubiquitination was qualitatively altered [58].
Since ErbB2 inhibits endocytic down-regulation of EGFR, EGFR-ErbB2 heterodimers accumulate at the plasma membrane. These heterodimers have increased ligand binding affinity [65] and represent potent oncogenic signalling units. Geldanamycin, which efficiently down-regulates such heterodimers in cultured cells, is unfortunately too toxic in vivo for clinical use, but new derivatives of geldanamycin have already been introduced in pre-clinical and clinical trials in cancers overexpressing ErbB2 [59]. An alternative approach has been developed, based on monoclonal antibodies. The humanized monoclonal antibody, pertuzumab, recognises the dimerization arm of ErbB2 and thus prevents its stable association with EGFR or ErbB3 [66]. By preventing the formation of EGFR-ErbB2 heterodimers, pertuzumab eventually causes accumulation of EGFR monomer at the cell surface. Moreover, since pertuzumab does not block EGF-induced EGFR activation [67], liganded EGFR monomers are induced to form endocytosis-competent EGFR homodimers. Altogether, these data predict that pertuzumab should result in altered signalling and down-regulation of EGFR in tumours overexpressing EGFR and ErbB2. As expected, pertuzumab has been demonstrated to counteract the growth-promoting and anti-apoptotic effects of both EGFR/ErbB2 and ErbB2/ErbB3 heterodimers, and in clinical studies to exhibit potent anti-tumour activity against ErbB2-expressing breast and prostate cancers and against lung cancers co-expressing ErbB2 and ErbB3.
Cellular and molecular requirements for lysosomal degradation of EGFR
Intracellular EGFR degradation depends on luminal sorting at multivesicular bodies
Upon translocation to clathrin-coated pits, activated EGFR is entrapped in clathrin-coated vesicles by a dynamin- and actin-dependent pinching off mechanism [68]. Uncoating of coated vesicles allows for homotypic fusion in a Rab5-dependent and EEA1-dependent manner, to form early sorting endosomes [69]. These endosomes initially have a molecular composition comparable to that of the plasma membrane, but become rapidly accessed by biosynthetic cargo from the trans-Golgi apparatus (TGN), including newly synthesized lysosomal hydrolases. Upon endosome maturation, internal vesicles are budding from the limiting membrane and accumulate in the lumen: they are the hallmark of multivesicular bodies (MVBs). Translocation of trans-membrane proteins from the limiting membrane to inner vesicles is a prerequisite for degradation to occur upon fusion with secondary lysosomes (for review see [70]).
Molecular requirements for EGFR sorting in multivesicular endosomes
Whereas the role of ubiquitination in EGFR internalization is still controversial, ubiquitination is clearly required for its intracellular sorting to lysosomes. Some reports have indicated that non-ubiquitinated proteins can be incorporated from the limiting membrane into budding internal vesicles of MVBs. However, recombinant addition of ubiquitin to transmembrane proteins normally localized to the plasma membrane, the Golgi apparatus or endosomes was shown to be sufficient to trigger their sorting at MVBs for eventual degradation in lysosomes [71–73]. Ubiquitinated EGFR is sorted into intraluminal vesicles of MVBs upon sequential interaction with four endosomal sorting complexes required for transport, referred to as ESCRT-0 to -III) (see Fig. 3). ESCRT-0, the first interacting partner of ubiquitinated EGFR, harbours both the Hepatocyte growth factor regulated tyrosine kinase substrate, Hrs, and the protein Signal-transducing adaptor molecule, STAM, each bearing an ubiquitin-binding motif (UIM). Likewise, the following partners ESCRT-I and II have ubiquitin-binding domains. In contrast, ESCRT-III complex has no constituent with a defined ubiquitin-interacting domain, and it is currently unclear how it recruits ubiquitinated transmembrane proteins [70].
3.
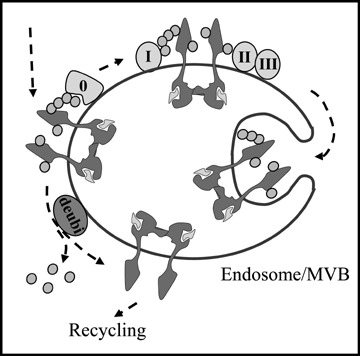
Molecular interactions regulating endosomal sorting of EGFR to the degradative pathway. At the limiting membrane of the late endosome/MVB, ubiquitinated EGFR interacts with ESCRT 0-III. This results into clustering to inward budding domains, eventually generating the inner vesicles of MVBs and degradation. Alternatively, EGFR can be deubiquitinated by de-ubiquiting enzymes (deubi) and recycled back to the plasma membrane. ESCRT complexes and ubiquitin are removed and recycled to the cytosolic pool before internal vesicles are formed.
Whether proteins should be mono- or poly-ubiquitinated for efficient endosomal sorting to occur is presently unclear. While it has been generally accepted that mono-ubiquitination is sufficient for proteins to interact with UIM-containing proteins on the limiting membrane of endosomes [70], this view has recently been questioned. Indeed, whereas both monomeric CD4 chimeras with K63-linked polyubiquitin chains or tetramers of mono-ubiquitinated CD4 chimeras were rapidly targeted to the lysosome, lysosomal delivery of single CD4 mono-ubiquitin chimeras exposing K48-linked ubiquitin chains was delayed, and lysosomal delivery of monoubiquitinated CD4 chimeras was undetectable [74]. The requirement for poly- and multi-ubiquitination suggests that polyvalent interactions are required to overcome low-affinity binding of ubiquitin to the UIMs of ubiquitin-binding adaptors like Hrs and STAM. The different sorting efficiency mediated by K48- and K63-linked ubiquitin chains could potentially be explained by different specificity of deubiquitinating enzymes, which are also differently recruited to ESCRT complexes [74]. Moreover, Hrs was found to preferentially interact with K63-ubiquitin chains and negligibly with mono-ubiquitin [74]. This is interesting in view of the recently published proteomics studies demonstrating that EGFR occurs in both mono- and poly-ubiquitinated forms and that the polyubiquitin chains are mostly K63-linked [41].
EGFR mutants, frequently found in human cancer, can escape efficient ubiquitination and thereby avoid lysosomal degradation [75]. Like natural ErbB2, these EGFR mutants have recently been reported to bind Hsp90. The Hsp90 inhibitor geldanamycin, known to induce down-regulation of ErbB2 (see above) has importantly been demonstrated to also promote down-regulation of poorly ubiquitinated EGFR mutants [43, 75]. This potentially opens new treatment avenues for the numerous cancers bearing such EGFR mutations.
Acknowledgments
We are grateful to Thoralf Christoffersen, Pierre Courtoy and Espen Stang for critical reading of this manuscript.
References
- 1.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–37. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 2.Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–67. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuan CT, Wikstrand CJ, Bigner DD. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr Relat Cancer. 2001;8:83–96. doi: 10.1677/erc.0.0080083. [DOI] [PubMed] [Google Scholar]
- 4.Yarden Y. The EGFR family and its ligands in human cancer: signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37:S3–8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 5.Harari PM. Epidermal growth factor receptor inhibition strategies in oncology. Endocr Relat Cancer. 2004;11:689–708. doi: 10.1677/erc.1.00600. [DOI] [PubMed] [Google Scholar]
- 6.Cohen S, Carpenter G. Human epidermal growth factor: isolation and chemical and biological properties. Proc Natl Acad Sci USA. 1975;72:1317–21. doi: 10.1073/pnas.72.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell. 2003;11:507–17. doi: 10.1016/s1097-2765(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 8.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–54. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 9.Raymond E, Faivre S, Armand JP. Epidermal growth factor receptor tyrosine kinase as a target for anticancer therapy. Drugs. 2000;60:S15–23. doi: 10.2165/00003495-200060001-00002. [DOI] [PubMed] [Google Scholar]
- 10.Hirata A, Ogawa S, Kometani T, Kuwano T, Naito S, Kuwano M, Ono M. ZD1839 (Iressa) induces antiangiogenic effects through inhibition of epidermal growth factor receptor tyrosine kinase. Cancer Res. 2002;62:2554–60. [PubMed] [Google Scholar]
- 11.Baulida J, Kraus MH, Alimandi M, Di Fiore PP, Carpenter G. All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J Biol Chem. 1996;271:5251–7. doi: 10.1074/jbc.271.9.5251. [DOI] [PubMed] [Google Scholar]
- 12.Motley A, Bright NA, Seaman MN, Robinson MS. Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol. 2003;162:909–18. doi: 10.1083/jcb.200305145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johannessen LE, Pedersen NM, Pedersen KW, Madshus IH, Stang E. Activation of the epidermal growth factor (EGF) receptor induces formation of EGF receptor- and Grb2-containing clathrin-coated pits. Mol Cell Biol. 2006;26:389–401. doi: 10.1128/MCB.26.2.389-401.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gullick WJ. Prevalence of aberrant expression of the epidermal growth factor receptor in human cancers. Br Med Bull. 1991;47:87–98. doi: 10.1093/oxfordjournals.bmb.a072464. [DOI] [PubMed] [Google Scholar]
- 15.Reiss M, Stash EB, Vellucci VF, Zhou ZL. Activation of the autocrine transforming growth factor alpha pathway in human squamous carcinoma cells. Cancer Res. 1991;51:6254–62. [PubMed] [Google Scholar]
- 16.Janne PA, Johnson BE. Effect of epidermal growth factor receptor tyrosine kinase domain mutations on the outcome of patients with non-small cell lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2006;12:S4416–20. doi: 10.1158/1078-0432.CCR-06-0555. [DOI] [PubMed] [Google Scholar]
- 17.Pedersen MW, Pedersen N, Damstrup L, Villingshøj M, Sønder SU, Rieneck K, Bovin LF, Spang-Thomsen, Poulsen HS. Analysis of the epidermal growth factor receptor specific transcriptome: effect of receptor expression level and an activating mutation. J Cell Biochem. 2005;96:412–27. doi: 10.1002/jcb.20554. [DOI] [PubMed] [Google Scholar]
- 18.Davies GC, Ryan PE, Rahman L, Zajac-Kaye M, Lipkowitz S. EGFRvIII undergoes activation-dependent downregulation mediated by the Cbl proteins. Oncogene. 2006;25:6497–509. doi: 10.1038/sj.onc.1209662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han W, Zhang T, Yu H, Foulke JG, Tang CK. Hypophosphorylation of residue Y1045 leads to defective downregulation of EGFRvIII. Cancer Biol Ther. 2006;5:1361–8. doi: 10.4161/cbt.5.10.3226. [DOI] [PubMed] [Google Scholar]
- 20.Grandal MV, Zandi R, Pedersen MW, Willumsen BM, Van Deurs B, Poulsen HS. EGFRvIII escapes down-regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis. 2007;28:1408–17. doi: 10.1093/carcin/bgm058. [DOI] [PubMed] [Google Scholar]
- 21.Felder S, LaVin J, Ullrich A, Schlessinger J. Kinetics of binding, endocytosis, and recycling of EGF receptor mutants. J Cell Biol. 1992;117:203–12. doi: 10.1083/jcb.117.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamaze C, Schmid SL. Recruitment of epidermal growth factor receptors into coated pits requires their activated tyrosine kinase. J Cell Biol. 1995;129:47–54. doi: 10.1083/jcb.129.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiley HS, Herbst JJ, Walsh BJ, Lauffenburger DA, Rosenfeld MG, Gill GN. The role of tyrosine kinase activity in endocytosis, compartmentation, and down-regulation of the epidermal growth factor receptor. J Biol Chem. 1991;66:11083–94. [PubMed] [Google Scholar]
- 24.Sorkina T, Huang F, Beguinot L, Sorkin A. Effect of tyrosine kinase inhibitors on clathrin-coated pit recruitment and internalization of epidermal growth factor receptor. J Biol Chem. 2002;277:27433–41. doi: 10.1074/jbc.M201595200. [DOI] [PubMed] [Google Scholar]
- 25.Wang Q, Villeneuve G, Wang Z. Control of epidermal growth factor receptor endocytosis by receptor dimerization, rather than receptor kinase activation. EMBO Rep. 2005;6:942–8. doi: 10.1038/sj.embor.7400491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang X, Huang F, Marusyk A, Sorkin A. Grb2 regulates internalization of EGF receptors through clathrin-coated pits. Mol Biol Cell. 2003;14:858–70. doi: 10.1091/mbc.E02-08-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vergarajauregui S, San Miguel A, Puertollano R. Activation of p38 mitogen-activated protein kinase promotes epidermal growth factor receptor internalization. Traffic. 2006;7:686–98. doi: 10.1111/j.1600-0854.2006.00420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zwang Y, Yarden Y. p38 MAP kinase mediates stress-induced internalization of EGFR: implications for cancer chemotherapy. EMBO J. 2006;25:4195–206. doi: 10.1038/sj.emboj.7601297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kazazic M, Roepstorff K, Johannessen LE, Pedersen NM, Van Deurs B, Stang E, Madshus IH. EGF-induced activation of the EGF receptor does not trigger mobilization of caveolae. Traffic. 2006;7:1518–27. doi: 10.1111/j.1600-0854.2006.00487.x. [DOI] [PubMed] [Google Scholar]
- 30.Huang F, Khvorova A, Marshall W, Sorkin A. Analysis of clathrin-mediated endocyto-sis of epidermal growth factor receptor by RNA interference. J Biol Chem. 2004;279:16657–61. doi: 10.1074/jbc.C400046200. [DOI] [PubMed] [Google Scholar]
- 31.Carpentier JL, Gorden P, Anderson RG, Goldstein JL, Brown MS, Cohen S, Orci L. Co-localization of 125I-epidermal growth factor and ferritin-low density lipoprotein in coated pits: a quantitative electron microscopic study in normal and mutant human fibroblasts. J Cell Biol. 1982;95:73–7. doi: 10.1083/jcb.95.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanover JA, Willingham MC, Pastan I. Kinetics of transit of transferrin and epidermal growth factor through clathrin-coated membranes. Cell. 1984;39:283–93. doi: 10.1016/0092-8674(84)90006-0. [DOI] [PubMed] [Google Scholar]
- 33.Sigismund S, Woelk T, Puri C, Maspero E, Tacchetti C, Transidico P, Di Fiore PP, Polo S. Clathrin-independent endocytosis of ubiquitinated cargos. Proc Natl Acad Sci USA. 2005;102:2760–5. doi: 10.1073/pnas.0409817102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mineo C, Gill GN, Anderson RG. Regulated migration of epidermal growth factor receptor from caveolae. J Biol Chem. 1999;274:30636–43. doi: 10.1074/jbc.274.43.30636. [DOI] [PubMed] [Google Scholar]
- 35.Ringerike T, Blystad FD, Levy FO, Madshus IH, Stang E. Cholesterol is important in control of EGF receptor kinase activity but EGF receptors are not concentrated in caveolae. J Cell Sci. 2002;115:1331–40. doi: 10.1242/jcs.115.6.1331. [DOI] [PubMed] [Google Scholar]
- 36.Park WY, Cho KA, Park JS, Kim DI, Park SC. Attenuation of EGF signaling in senescent cells by caveolin. Ann N Y Acad Sci. 2001;928:79–84. doi: 10.1111/j.1749-6632.2001.tb05638.x. [DOI] [PubMed] [Google Scholar]
- 37.Kerr MC, Lindsay MR, Luetterforst R, Hamilton N, Simpson F, Parton RG, Gleeson PA, Teasdale RD. Visualisation of macropinosome maturation by the recruitment of sorting nexins. J Cell Sci. 2006;119:3967–80. doi: 10.1242/jcs.03167. [DOI] [PubMed] [Google Scholar]
- 38.Orth JD, McNiven MA. Get off my back! Rapid receptor internalization through circular dorsal ruffles. Cancer Res. 2006;66:11094–6. doi: 10.1158/0008-5472.CAN-06-3397. [DOI] [PubMed] [Google Scholar]
- 39.Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol. 2003;5:461–6. doi: 10.1038/ncb983. [DOI] [PubMed] [Google Scholar]
- 40.Waterman H, Katz M, Rubin C, Shtiegman K, Lavi S, Elson A, Jovin T, Yarden Y. A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J. 2002;21:303–13. doi: 10.1093/emboj/21.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang F, Kirkpatrick D, Jiang X, Gygi S, Sorkin A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol Cell. 2006;21:737–48. doi: 10.1016/j.molcel.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 42.Galcheva-Gargova Z, Theroux SJ, Davis RJ. The epidermal growth factor receptor is covalently linked to ubiquitin. Oncogene. 1995;11:2649–55. [PubMed] [Google Scholar]
- 43.Yang S, Qu S, Perez-Tores M, Sawai A, Rosen N, Solit DB, Arteaga CL. Association with HSP90 inhibits Cbl-medi-ated down-regulation of mutant epidermal growth factor receptors. Cancer Res. 2006;66:6990–7. doi: 10.1158/0008-5472.CAN-06-1042. [DOI] [PubMed] [Google Scholar]
- 44.Jiang X, Sorkin A. Epidermal growth factor receptor internalization through clathrin-coated pits requires Cbl RING finger and proline-rich domains but not receptor polyubiquitylation. Traffic. 2003;4:529–43. doi: 10.1034/j.1600-0854.2003.t01-1-00109.x. [DOI] [PubMed] [Google Scholar]
- 45.Huang F, Goh LK, Sorkin A. EGF receptor ubiquitination is not necessary for its internalization. Proc Natl Acad Sci USA. 2007;104:16904–9. doi: 10.1073/pnas.0707416104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tebar F, Sorkina T, Sorkin A, Ericsson M, Kirchhausen T. Eps15 is a component of clathrin-coated pits and vesicles and is located at the rim of coated pits. J Biol Chem. 1996;271:28727–30. doi: 10.1074/jbc.271.46.28727. [DOI] [PubMed] [Google Scholar]
- 47.Stang E, Blystad FD, Kazazic M, Bertelsen V, Brodahl T, Raiborg C, Stenmark H, Madshus IH. Cbl-dependent ubiquitination is required for progression of EGF receptors into clathrin-coated pits. Mol Biol Cell. 2004;15:3591–604. doi: 10.1091/mbc.E04-01-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Polo S, Confalonieri S, Salcini AE, Di Fiore PP. EH and UIM: endocytosis and more. Sci STKE. 2003;213:1–17. doi: 10.1126/stke.2132003re17. [DOI] [PubMed] [Google Scholar]
- 49.Owen DJ, Vallis Y, Noble ME, Hunter JB, Dafforn TR, Evans PR, McMahon HT. A structural explanation for the binding of multiple ligands by the alpha-adaptin appendage domain. Cell. 1999;97:805–15. doi: 10.1016/s0092-8674(00)80791-6. [DOI] [PubMed] [Google Scholar]
- 50.Traub LM, Downs MA, Westrich JL, Fremont DH. Crystal structure of the alpha appendage of AP-2 reveals a recruitment platform for clathrin-coat assembly. Proc Natl Acad Sci USA. 1999;96:8907–12. doi: 10.1073/pnas.96.16.8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hussain NK, Yamabhai M, Ramjaun AR, Guy AM, Baranes D, O’Bryan JP, Der CJ, Kay BK, McPherson PS. Splice variants of intersectin are components of the endocytic machinery in neurons and non-neuronal cells. J Biol Chem. 1999;274:15671–7. doi: 10.1074/jbc.274.22.15671. [DOI] [PubMed] [Google Scholar]
- 52.Rosenthal JA, Chen H, Slepnev VI, Pellegrini L, Salcini AE, Di Fiore PP, De Camilli P. The epsins define a family of proteins that interact with components of the clathrin coat and contain a new protein module. J Biol Chem. 1999;274:33959–65. doi: 10.1074/jbc.274.48.33959. [DOI] [PubMed] [Google Scholar]
- 53.Drake MT, Traub LM. Interaction of two structurally distinct sequence types with the clathrin terminal domain beta-propeller. J Biol Chem. 2001;276:28700–9. doi: 10.1074/jbc.M104226200. [DOI] [PubMed] [Google Scholar]
- 54.Hawryluk MJ, Keyel PA, Mishra SK, Watkins SC, Heuser JE, Traub LM. Epsin 1 is a polyubiquitin-selective clathrin-associated sorting protein. Traffic. 2006;7:262–81. doi: 10.1111/j.1600-0854.2006.00383.x. [DOI] [PubMed] [Google Scholar]
- 55.Yarden Y. Biology of HER2 and its importance in breast cancer. Oncology. 2001;61:1–13. doi: 10.1159/000055396. [DOI] [PubMed] [Google Scholar]
- 56.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 57.Hommelgaard AM, Lerdrup M, Van Deurs B. Association with membrane protrusions makes ErbB2 an internalization-resistant receptor. Mol Biol Cell. 2004;4:1557–67. doi: 10.1091/mbc.E03-08-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haslekas C, Breen K, Pedersen KW, Johannessen LE, Stang E, Madshus IH. The inhibitory effect of ErbB2 on epidermal growth factor-induced formation of clathrin-coated pits correlates with retention of epidermal growth factor receptor-ErbB2 oligomeric complexes at the plasma membrane. Mol Biol Cell. 2005;16:5832–42. doi: 10.1091/mbc.E05-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arslan MA, Kutuk O, Basaga H. Protein kinases as drug targets in cancer. Curr Cancer Drug Targets. 2006;6:623–34. doi: 10.2174/156800906778742479. [DOI] [PubMed] [Google Scholar]
- 60.Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem. 1999;42:260–6. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 61.Xu W, Mimnaugh E, Rosser MF, Nicchitta C, Marcu M, Yarden Y, Neckers L. Sensitivity of mature Erbb2 to gel-danamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J Biol Chem. 2001;276:3702–8. doi: 10.1074/jbc.M006864200. [DOI] [PubMed] [Google Scholar]
- 62.Neckers L, Ivy SP. Heat shock protein 90. Curr Opin Oncol. 2003;15:419–24. doi: 10.1097/00001622-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 63.Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3:213–7. doi: 10.1016/s1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 64.Wang Z, Zhang L, Yeung TK, Chen X. Endocytosis deficiency of epidermal growth factor (EGF) receptor-ErbB2 het-erodimers in response to EGF stimulation. Mol Biol Cell. 1999;10:1621–36. doi: 10.1091/mbc.10.5.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilkinson JC, Beechem JM, Staros JV. A stopped-flow fluorescence anisotropy method for measuring hormone binding and dissociation kinetics with cell-surface receptors in living cells. J Recept Signal Transduct Res. 2002;22:357–71. doi: 10.1081/rrs-120014607. [DOI] [PubMed] [Google Scholar]
- 66.Franklin MC, Carey KD, Vajdos FF, Leahy DJ, De Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5:317–28. doi: 10.1016/s1535-6108(04)00083-2. [DOI] [PubMed] [Google Scholar]
- 67.Sakai K, Yokote H, Murakami-Murofushi K, Tamura T, Saijo N, Nishio K. Pertuzumab, a novel HER dimerization inhibitor, inhibits the growth of human lung cancer cells mediated by the HER3 signaling pathway. Cancer Sci. 2007;98:1498–503. doi: 10.1111/j.1349-7006.2007.00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dawson JC, Legg JA, Machesky LM. Bar domain proteins: a role in tubulation, scission and actin assembly in clathrin-medi-ated endocytosis. Trends Cell Biol. 2006;16:493–8. doi: 10.1016/j.tcb.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 69.Simonsen A, Lippe R, Christoforidis S, Gaullier JM, Brech A, Callaghan J, Toh BH, Murphy C, Zerial M, Stenmark H. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 1998;394:494–8. doi: 10.1038/28879. [DOI] [PubMed] [Google Scholar]
- 70.Raiborg C, Rusten TE, Stenmark H. Protein sorting into multivesicular endosomes. Curr Opin Cell Biol. 2003;15:446–55. doi: 10.1016/s0955-0674(03)00080-2. [DOI] [PubMed] [Google Scholar]
- 71.Urbanowski JL, Piper RC. Ubiquitin sorts proteins into the intralumenal degradative compartment of the late-endo-some/vacuole. Traffic. 2001;2:622–30. doi: 10.1034/j.1600-0854.2001.20905.x. [DOI] [PubMed] [Google Scholar]
- 72.Reggiori F, Pelham HR. Sorting of proteins into multivesicular bodies: ubiquitin-dependent and -independent targeting. EMBO J. 2001;20:5176–86. doi: 10.1093/emboj/20.18.5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Raiborg C, Bache KG, Gillooly DJ, Madshus IH, Stang E, Stenmark H. Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat Cell Biol. 2002;4:394–8. doi: 10.1038/ncb791. [DOI] [PubMed] [Google Scholar]
- 74.Barriere H, Nemes C, Du K, Lukacs GL. Plasticity of polyubiquitin recognition as lysosomal targeting signals by the endosomal sorting machinery. Mol Biol Cell. 2007;18:3952–65. doi: 10.1091/mbc.E07-07-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Padron D, Sato M, Shay JW, Gazdar AF, Minna JD, Roth MG. Epidermal growth factor receptors with tyrosine kinase domain mutations exhibit reduced Cbl association, poor ubiquitylation, and down-regulation but are efficiently internalized. Cancer Res. 2007;67:7695–702. doi: 10.1158/0008-5472.CAN-07-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]