

Abstract
As activated microglia (MG) is an early sign that often precedes and triggers neuronal death, inhibition of microglial activation and reduction of subsequent neurotoxicity may offer therapeutic benefit. The present study demonstrates that rat primary cultured MG expressed Kir6.1 and SUR2 subunits of KATP channel, which was identical to that expressed in BV-2 microglial cell line. The classic KATP channel opener pinacidil and selective mitochondrial KATP (mito-KATP) channel opener diazoxide prevented rotenone-induc microglial activation and production of pro-inflammatory factors (tumour necrosis factor[TNF]-α and prostaglandin E2[PGE2]). And the effects of pinacidil and diazoxide were reversed by mito-KATP blocker 5-hydroxydecanoate (5-HD), indicating that mito-KATP channels participate in the regulation of microglial activation. Moreover, the underlying mechanisms involved the stabilization of mitocho drial membrane potential and inhibition of p38/c-Jun-N-terminal kinase (JNK) activation in microglia. Furthermore, the in vivo study confirmed that diazoxide exhibited neuroprotective effects against rotenone along with the inhibition of microglial activation and neuroinflammation. Thus, microglial mito-KATP channel might be a novel prospective target for the treatment of neuroinflammation-related degenerative disorders such as Parkinson's disease.
Keywords: ATP-sensitive potassium channel, diazoxide, microglia, neuroinflammation, neurodegenerative disease, pro-inflammatory factor
Introduction
Microglial activation has been demonstrated to be an early sign that often precedes and triggers neuronal death in neurodegenerative diseases, such as Parkinson's disease (PD) [1–3]. Excessive production of pro-inflammatory and neurotoxic factors from activated microglia (MG), such as tumour necrosis factor-α (TNF-α), prostaglandin E2 (PGE2), interleukin-1β (IL-1β) and reactive oxygen species (ROS), may trigger or exacerbate neuronal death [4–10]. Thus, seeking the endogenous target for controlling the extent of microglial activation and neuroinflammation may offer prospective clinical therapeutic benefit for inflammation-related neurodegenerative disorders.
A variety of K+ channels are expressed in MG and play an important role in regulating membrane potential and thereby microglial function [11]. ATP-sensitive potassium (KATP) channel is a special class of potassium channel, which links cell metabolic state to excitability. KATP channels consist of discrete pore-forming (Kir6.1/Kir6.2) and regulatory (SUR1/SUR2) subunits and are activated by a decrease in ATP/ADP ratio [12]. It is well documented that targeting KATP channels can provide neuroprotective effects for neurons and astrocytes against ischaemia, trauma and neuro-toxicants [13–16]. KATP channels in the brain do not belong to a homogenous group, and different combinations of possible KATP channel subunits give rise to functional KATP channels with a variety of biophysical, pharmacological and metabolic properties [12]. Kir6.2 forms the pore of most neuronal populations, while Kir6.1 is the principal pore-forming subunit of astrocyte and mitochondr-ial KATP (mito-KATP) channels in brain [12, 17]. Our previous study showed that Kir6.1/SUR2 KATP channels were expressed in BV-2 microglial cell line and involved in regulating the production of inflammatory factors [18]. Most recently, we reported that iptakalim, a novel drug with the properties of KATP channel opener, exhibited anti-neuroinflammatory effects [19]. Considering the differences between the property of BV-2 and MG, in the present study rat primary cultured MG were purified to investigate the subunit composition and function of KATP channels.
The aim of this study was to use the classic KATP channel opener pinacidil and selective mito-KATP channel opener diazoxide to investigate the regulatory effects of KATP channels on rotenone-induced MG activation and neuroinflammation.
Material and methods
Microglia-enriched cultures
Tissues from whole brains of postnatal (P1–P2) Sprague-Dawley (SD) rats were triturated and then cells were plated in Dulbecco's modified Eagle's medium (DMEM) containing 10% foetal calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin. After reaching a confluent monolayer of glial cells (10–14 d), MG were separated from astrocytes by shaking off for 5 hrs at 100 rpm in an orbit shaker at 37°C. Enriched MG were collected by centrifugation and reseeded in 24-well culture plates at a density of 105 cells/cm2. The enriched MG were >98% pure as determined by antibodies for OX-42 (a marker for MG, Serotec, USA) and glial fibrillary acidic protein (GFAP) (a marker for astrocyte, Sigma, USA).
Animals and treatment
Male SD rats aged 7 weeks were chosen for experiments. Rats (220–240 g) were kept five to a cage under standard laboratory conditions with free access to standard laboratory food and tap water, constant room temperature of 22 ± 1°C, 50–60% humidity and a natural day-night cycle. All experiments were carried out according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (publication No. 85–23, revised 1985) and the Guidelines for the Care and Use of Animals in Neuroscience Research by the Society for Neuroscience and approved by Institutional Animal Care and Use Committee of Nanjing Medical University(IACUC).
Dimethylsulfoxide (DMSO)/polyethylene glycol (PEG) (1/1) was used as vehicle for rotenone (Sigma, USA). Diazoxide (Sigma, USA) was dissolved in 100% DMSO as vehicle and diluted with sterile saline to a concentration of 1 mg/ml (0.2% DMSO) for administration to rats. Rats were randomly divided into three groups. Control rats (n= 16) received vehicle only. Rats of model group (n= 20) were subcutaneously injected with rotenone (2.5 mg/kg/day) for 4 weeks and daily dose of rotenone was administered t.i.d. at 08:00, 14:00 and 20:00, respectively. The injection volume was 0.1 ml/100 g body weight. Other rats were received intragas-tric administration with diazoxide (1.5 mg/kg/day, n= 16) for 3 days. Diazoxide was administrated directly into the rat stomach through oral cavity using a metal tube attached to a 2 ml syringe. On day 4, diazoxide was pre-treated 1 hr before the injection with rotenone (2.5 mg/kg/day, subcutaneously) on a daily basis for 4 weeks.
Rats of rotenone group underwent an obvious mortality (40%) after 4 week treatments. Twelve to 16 animals remained per group. After the last behaviour test, rats were killed and their brains were used for morphological (n= 6 per group) and biochemical (n= 4 per group) analyses.
Behavioural tests
The catalepsy test and rotarod test were performed for the assessment of the effects of drugs on rotenone-induced parkinsonian symptoms. All rats were tested for catalepsy and rotarod at five time points: before administration and 1, 2, 3 and 4 weeks after treatments. Only rats that completed all 4 weeks of the experiments were included in statistical analyses (n= 12 per group). For catalepsy test, rats were placed with both forepaws on bars 9 cm above and parallel from the base and were in a half-rearing position. Latency time of the removal of the paw was recorded. The basic requirement is for rotating roller (or rotarod), consisting of (i) a roller of the appropriate diameter (8 cm) for rats, (ii) a power source for turning the roller (control of rod speed) and (iii) four circular separators placed along the rod at suitable intervals to divided the roller into equal-sized compartments for simultaneous testing of four animals. For the rotarod test, rats were placed on the rod and sequentially tested at 5, 10, 15 r.p.m. for a maximum of 300 sec each speed, the length of time that each animal was able to stay on the rod at each rotarod speed was recorded.
RT-PCR
Total RNA was extracted from MG after stimulation using Trizol (Invitrogen Life technologies, USA). 2 μg of total RNA from primary cultured MG was reverse transcribed into single-stranded cDNA. PCR was performed on the equivalent cDNAs from each sample. Primers used are listed in Table 1. PCR cycles were as follows: Kir6.1 primers: 94°C, 4 min; 94°C, 30 sec; 57°C, 30 sec; 72°C, 30 sec; 72°C, 5 min (27 cycles). Kir6.2 primers: 94°C, 4 min; 94°C, 30 sec; 64°C, 30 sec; 72°C, 30 sec; 72°C, 5 min (33 cycles). SUR1 primers: 94°C, 4 min; 94°C, 30 sec; 64°C, 30 sec; 72°C, 30 sec; 72°C, 5 min (29 cycles). SUR2 primers: 94°C, 4 min; 94°C, 30 sec; 59°C, 30 sec; 72°C, 30 sec; 72°C, 5 min (35 cycles); TNF-α, cyclooxygenase-2 (COX-2) and GAPDH primers: 95°C, 5 min; 95°C, 30 sec; 63°C, 30 sec; 72°C, 30 sec; 72°C, 10 min (35 cycles). The PCR reactions were then visualized on a 1.8% agarose gel containing 0.06 μg/ml ethidium bromide and the resulting bands were confirmed using Molecular Image FX (BIO-RAD).
1.
Primers for RT-PCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Kir6.1 | 5′-GAGTGAACTGTCGCACCAGA-3′ | 5′-CGATCACCAGAACTCAGCAA-3′ |
| Kir6.2 | 5′-TCCAACAGCCCGCTCTAC-3′ | 5′-GATGGGGACAAAACGCTG-3′ |
| SUR1 | 5′-GGAGCAATCCAGACCAAGAT-3′ | 5′-AGCCAGCAGAATGATGACAG-3′ |
| SUR2 | 5′-ACCTGCTCCAGCACAAGAAT-3′ | 5′-TCTCTTCATCACAATGACCAGG-3′ |
| TNF-a | 5′-CAGACCCTCACACTCAGATCATCTT-3′ | 5′-CAGAGCAATGACTCCAAAGTAGACCT-3′ |
| COX-2 | 5′-TGATGACTGCCCAACTCCCATG-3′ | 5′-AATGTTGAAGGTGTCCGGCAGC-3′ |
| GAPDH | 5′-TGGTGCCAAAAGGGTCATCTCC-3′ | 5′-GCCAGCCCCAGCATCAAAGGTG-3′ |
Immunochemistry
Cells were fixed using 4% paraformaldehyde, followed by blocking with PBS containing 10% bovine serum albumin (BSA). After blocking, cells were incubated at 4°C overnight with the primary antibody (goat anti-kir6.1, kir6.2, SUR1and SUR2 at 1:100, Santa Cruz, USA). Cells were then incubated for 1 hr at room temperature with primary antibody for OX-42 (1:100). After washing, cells were exposed to goat antimouse tetramethyl rhodamine isothiocyanate (TRITC)-conjugated antibody at 1:100 and rabbit anti-goat fluorescein isothiocyanate (FITC)-conjugated antibody at 1:100. Stained cells were observed by scanning confocal microscopy. ED1 (a marker for activated microglia, Serotec, USA) immunofluorescence-staining was performed as above described at 1:100. And the total ED1 fluorescence intensities of every well were quantitated using image-analysis software (Simple PCI).
Rats were fixed by transcardial perfusion of 4% paraformaldehyde. Free-floating sections encompassing the entire midbrain were prepared by using a cryostat. For TH, ED1 and OX-42 immunostaining, tissue sections were incubated with primary antibodies overnight at 4°C. Primary antibodies used in this study were as follows: mouse anti-TH (1:4000, Sigma, USA), mouse anti-OX-42 (1:100), mouse anti-ED1 (1:100). Immunostaining was visualized by using 3, 3'-diaminobenzidine. Sections were then counterstained with hematoxylin. The total numbers of TH-pos-itive neurons and GFAP-positive cells in the substantia nigra were counted stereologically using the optical fractionator method [20].
Western blotting
Proteins in cell extracts were denatured with SDS sample buffer and separated by 10% SDS-PAGE. Proteins were transferred to nitrocellulose membranes using a Bio-Rad miniprotein-III wet transfer unit. The membranes were incubated with 5% BSA dissolved in Tris-buffered saline with Tween-20 (TBST) (pH 7.5, 10 mM Tris-HCl, 150 mM NaCl, and 0.1% Tween-20) at room temperature for 1 hr, washed three times and incubated with different antibodies against different subunits of KATP channel overnight at 4°C (goat anti-Kir6.1, Kir6.2, SUR1 and SUR2 at 1:100). The membranes were washed three times with TBST buffer and incubated with the secondary antibody (1:1000) for 1 hr followed by four washings. Signal detection was performed with an enhanced chemiluminescence kit.
For p38 and JNK western blotting, primary antibodies used in this study were as follows: rabbit anti-p38, anti-phospho-p38, anti-JNK and anti-phospho-JNK (1:1000, Cell Signal, USA).
Cell viability assay
Cell viability was measured by MTT method. The absorbance of each well was obtained using a Dynatech MR5000 plate counter at a test wavelength of 570 nm with a reference wavelength of 630 nm.
TNF-α and PGE2 assay
The amount of TNF-α in the culture medium was determined 24 hrs after treatment with a rat TNF-α enzyme-linked immunosorbent assay (ELISA) kit (BioSource International, USA). The content of TNF-α in rat brain tissue (BT) extracts was also determined by TNF-α ELISA kit. The production of PGE2 in microglial cultures was evaluated 24 hrs after treatment with a PGE2 radioimmumoassay (RIA) kit purchased from Blood Research Institute (Soochow University, China).
R0S assay
Intracellular production of ROS was measured by 2’, 7-dichloro-fluores-cein (DCFH) oxidation. DCFH-DA (sigma, USA) was dissolved in ethanol at 10 mM and was diluted 500-fold in DMEM to give DCFH-DA at 20 μM. Cells were exposed to DCFH-DA for 1 hr and then treated with DMEM containing corresponding concentration of drugs for 3 hrs. The fluorescence was visualized immediately at wavelengths of 485 nm for excitation and 530 nm for emission by a Nikcon Optical TE2000-S inverted fluorescence microscope. And the total green fluorescence intensities of every well were quantitated using image-analysis software (Simple PCI).
Measurement of microglial mitochondrial membrane potential (ΔΨm)
Microglial Δψm was assessed with the fluorescent probe JC-1 (Molecular Probes, USA). At 490 nm, cells with depolarized mitochondria contained JC-1 predominantly in monomeric form and fluoresced green. Cells with polarized mitochondria predominantly contained JC-1 in aggregate form, and mitochondria fluoresced red-orange. MG were incubated with 5 μM of JC-1 for 10 min at 37°C, washed, and placed on a thermostatted stage at 37°C. Fluorescent images were visualized by a Nikon Optical TE2000-S inverted fluorescence microscope with excitation at 490 nm and emission at >520 nm. MG with polarized mitochondria were seen with distinct mitochondria fluorescing red-orange, and, in MG with depolarized mitochondria, the cell cytoplasm and mitochondria appeared green. Acquired signal was analysed with image-analysis software (Simple PCI). A minimum of six fields were selected and average intensity for each region was quantified. The ratio of J-aggregate to JC-1 monomer intensity for each region was calculated. A decrease in this ratio was interpreted as loss of ΔΨm, whereas an increase in the ratio was interpreted as gain in ΔΨm.
Statistical analysis
Values are means ± S.E.M. The significance of the difference between control and samples treated with various drugs was determined by one-way anova followed by the post-hoc Student-Newman-Keuls test using SPSS v10.0 for Windows (SPSS Inc., Chicago, IL). Differences were considered significant at P<0.05.
Results
Rat primary cultured microglia express KATP channel subunits Kir6.1 and SUR2 but not Kir6.2 and SUR1
RT-PCR was performed to detect mRNA level of Kir6.1, Kir6.2, SUR1 and SUR2. The results showed that in rat primary cultured MG, single-band amplification products of expected sizes for Kir6.1 (247 bp) and SUR2 (144 bp) subunits were observed, but no detectable bands at the predicted sizes of 167 bp for Kir6.2 or 248 bp for SUR1 (Fig. 1A). As positive controls, Kir6.2 and SUR1 subunits were observed in whole rat BT.
1.
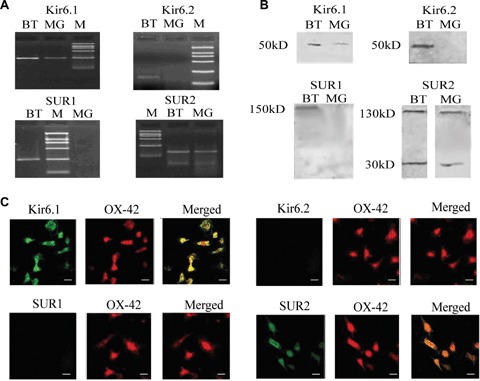
Primary cultured microglia expressed Kir6.1 and SUR2 subunits of KATP channels but not Kir6.2 and SUR1. (A) RT-PCR analysis of the Kir6.1 and SUR2 subunits in microglia. M, DNA marker; BT, whole brain tissue; MG, microglia. (B) Western blotting analysis of the Kir6.1 and SUR2 sub-units in microglia. (C) Immunolocalization of Kir6.1 and SUR2 subunits in microglia was examined using antibodies for Kir6.1 (green), SUR2 (green), colocalized with the microglial marker OX-42 (red). Scale bar: 25 μm.
Western blotting and double-immunolabelling experiments were performed to detect expression of subunit at protein level. The results of western blotting showed that in MG prominent bands at expected molecular weights of 50 kDa for Kir6.1, 130 kD and 30 kD for SUR2 were observed. However, no obvious bands at expected molecular weights of 50 kD for Kir6.2 and 150 kD for SUR1 were detected (Fig. 1B). As shown in Figure 1C, nearly all of microglial cells labelled with microglial marker OX-42 were Kir6.1 and SUR2 positive, but Kir6.2 and SUR1 were negative. These results indicate that KATP channel subunits, Kir6.1 and SUR2, are expressed in rat primary cultured MG.
Opening of mito-KATP channels inhibits rotenone-induced microglial activation and production of TNF-α, PGE2 and ROS
The effect of rotenone on the viability of MG was studied so as to determine its concentration used in following experiments. Notably, rotenone-induced microglial activation but failed to affect the microglial viability at 10 nM (Fig. 2A). Therefore, this concentration was chosen to investigate the effects of KATP channels on microglial activation.
2.
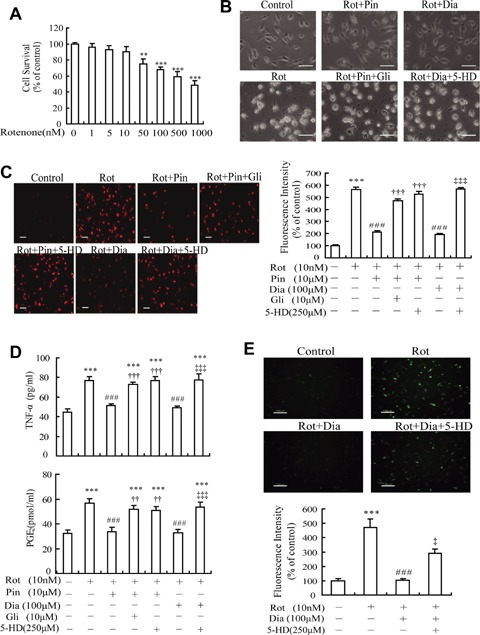
Effects of KATP channel openers on rotenone-induced microglial activation and production of pro-inflammatory and neurotoxic factors. (A) Rotenone induced a decrease in the viability of microglia in a concentration-dependent manner. (B) KATP channel openers attenuated rotenone-induced morphological change of microglia. Rot, rotenone; Pin, pinacidil; Dia, diazoxide; Gli, glibenclamide; 5-HD, 5-hydroxydecanoate. (C) KATP channel openers reduced fluorescence intensity of rotenone-induced ED1-positive microglia (red). (D) KATP channel openers inhibited rotenone-induced production of TNF-α and PGE2 from microglia. (E) KATP channel openers inhibited rotenone-induced production of intracellular reactive oxygen species (ROS) in microglia. Scale bar: 50 μm (B and C); 100 μm (E). **P<0.01, ***P <0.001 versus control group; ###P <0.001 versus Rot group; ††P <0.01, †††P <0.001 versus Rot + Pin group; ‡P <0.05, ‡‡‡P <0.001 versus Rot + Dia group. Data are presented as the mean ± S.E.M. of four independent experiments.
Then, we examined the effects of KATP channel openers on rotenone-induced microglial morphological changes. It is well documented that unstimulated MG are typically ramified and either bipolar or unipolar, indicative of a resting state, whereas activated microglial cells are altered, becoming round with enlarged and amoeboid cell bodies [21]. As shown in Fig. 2B, the morphological features considered as the resting state of MG were observed in control cells. Stimulated with 10 nM rotenone for 24 hrs, most MG were activated and consequently underwent dramatic morphological changes. Although KATP channel opener pinacidil (10 μM) or diazoxide (100 μM) alone failed to affect cell morphology of resting MG (data not shown), pre-incubation with 10 μM pinacidil for 20 min ameliorated rotenone-induced morphological alterations. Co-incubation of 10 μM glibenclamide, a classic KATP channel blocker, with pinacidil, abolished the effects of pinacidil. MG, which were pre-treated with 100 μM diazoxide (a selective mito-KATP channel opener) for 20 min before incubation with 10 nM rotenone for 24 hrs, were observed that the morphological alterations of rotenone-activated MG were also ameliorated. The effects of diazoxide were abolished by 250 μM 5-hydroxyde-canoate (5-HD), a selective mito-KATP channel blocker.
Furthermore, cells were stained with ED1 antibody, a marker for microglial activation. As shown in representative confocal scanning laser microscopy micrographs of ED1, the intensity of red fluorescence was significantly increased when MG were incubated with rotenone for 24 hrs. The fluorescence intensity of MG pre-incubated with 10 μM pinacidil or 100 μM diazoxide was decreased by 62.5% and 65.6%, respectively, which was reversed by either non-selective KATP channel blocker glibenclamide (10 μM) or selective mito-KATP channel blocker 5-HD (250 μM) (Fig. 2C). These results suggest that the opening of microglial KATP channels, mainly mito-KATP channels, might prevent from rotenone-induced microglial activation.
It is well documented that excessive production of TNF-a, PGE2 and ROS from activated MG plays an important role in the process of neurodegenerative diseases [4], so we examined whether KATP channel openers affect rotenone-induced TNF-α, PGE2 and ROS production from MG. The results showed that neither 10 μM pinacidil nor 100 μM diazoxide alone affected TNF-a and PGE2 production from resting MG (data not shown). However the amount of TNF-α in the medium 24 hrs after the addition of rotenone was significantly reduced by pre-treatment with pinacidil (10 μM) or diazoxide (100 μM) down to 67.1% and 64.1% of rotenone-treated group, respectively. Pre-incubation of MG with glibenclamide (10 μM) or 5-HD (250 μM) for 20 min prior to the addition of pinacidil or diazoxide could eliminate the effects of pinacidil and diazoxide as shown in Fig. 2D. Similarly, pinacidil or diazoxide could also decrease the production of PGE2 from rotenone-treated MG, which was reversed by either glibenclamide or 5-HD (Fig. 2D). In addition, levels of intracellular ROS, the neu-rotoxic factors, were determined after stimulation with rotenone (10 nM) for 3 hrs. As shown in Fig. 2E, treatment with rotenone markedly elevated the production of ROS in MG, indicated by the increased green fluorescence intensity (up to 471% of control). A significant inhibition of the rotenone-stimulated ROS was observed in cultures pre-treated (20 min) with 100 μM diazoxide. And the effect of diazoxide was eliminated by 250 μM 5-HD. These results indicate that mito-KATP channels play an important role in the regulation of rotenone-induced production of inflammatory and neurotoxic factors from MG.
Opening of mito-KATP channels alleviates rotenone-induced mitochondrial membrane potential loss and p38/JNK phosphorylation in microglia
As mitochondrial membrane potential (Δψm) has been demonstrated to regulate microglial activation, molecular probe JC-1 was used to detect the effect of mito-KATP channel opener diazoxide on rotenone-induced microglial mitochondrial membrane potential change. Most cultured MG displayed a loss or collapse of ΔΨm 30 min after exposure to rotenone, indicated by fluorescence of JC-1 shifted from red-orange to greenish yellow. Pre-treatment of MG with 100 μM diazoxide prevented mitochondria from loss of Ψm. Furthermore, addition of 5-HD (250 μM) could reverse the preventive effect of diazoxide (Fig. 3A).
3.
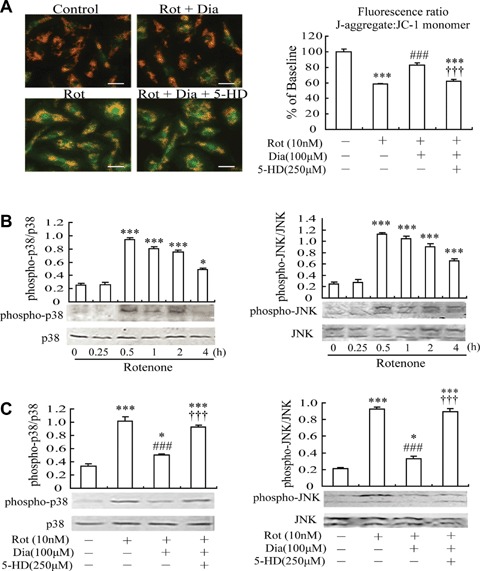
Effects of mito-KATP channel opener diazoxide on rotenone-induced mitochon-drial depolarization and p38/JNK phos-phorylation in microglia. (A) JC-1 fluorescence imaging of mitochondria. Quantification of mitochondrial membrane potential expressed as a ratio of J-aggregate to JC-1 monomer (red: green) fluorescence. Scale bar: 25 μm. (B) Rotenone induced p38/JNK phosphoryla-tion in microglia, indicative of p38/JNK activation. Microglia were treated with 10 nm rotenone for indicated times. (C) Pre-treatment with diazoxide suppressed rotenone-induced p38/JNK phosphoryla-tion in microglia. 30 min after treatment, microglia were harvested and phosphory-lated p38/JNK were analysed. Lower: Representative blots are shown. Upper: Densitometric analysis of the phosphory-lated forms of p38/JNK. *P <0.05, ***P <0.001 versus control group; ###P <0.001 versus Rot group; †††P <0.001 versus Rot + Dia group. Data are presented as the mean ± S.E.M. of four independent experiments.
Since p38 and JNK are the predominant signaling transduction pathways responsible for synthesis and production of pro-inflammatory factors (such as TNF-α and PGE2) from MG [22–25], we investigated whether mito-KATP opener diazoxide could affect rotenone-induced p38/JNK phosphorylation in MG, which might in turn regulate the production of pro-inflammatory factors. First, cells were treated with rotenone (10 nM) for different intervals of time to determine the involvement of p38/JNK in rotenone stimulation. Treatment with rotenone led to a rapid and transient phosphorylation of both p38 and JNK, indicative of p38 and JNK activation, with the peak levels of phospho-p38 and phospho-JNK occurring at 30 min (Fig. 3B). Phosphorylated p38 sustained up to 2 hrs after rotenone stimulation, but JNK phosphorylation was still apparent at 4 hrs. These data indicate that both p38 and JNK signalling pathway are activated in response to rotenone treatment in MG. Next, we detected whether mito-KATP opener diazoxide could regulate rotenone-induced p38/JNK phosphorylation. The following experiments with diazoxide were performed at the 30 min time point. As shown in Fig. 3C, pre-treatment with 100 μM diazoxide reduced rotenone-induced increase of phospho-p38 by over 50% and that of phospho-JNK by over 64%. And these effects were reversed by mito-KATP blocker 5HD (250 μM).
Mito-KATP channel opener diazoxide ameliorates rotenone-induced rat behavioural symptoms
Control rats were treated with vehicle, and death did not occur. Rotenone-treated rats (2.5 mg/kg/day for 28 days, subcutaneously) underwent significant weight loss (data not shown) and mortality (40%) during the treatment. However, rats with diazoxide (1.5 mg/kg/day, orally), which was pre-treated for 3 days and administered daily for 4 weeks with an injection of rotenone 1 hr later each time, had markedly increased weight and lower mortality (19%).
Using the catalepsy test, rotenone-treated rats showed prolonged latency time compared to that in the vehicle-treated control group. The latency time of rats pre-treated with diazoxide decreased by 91% (Fig. 4A). In the rotarod test, vehicle-treated animals learned the task quickly and, after a short training period, were able to remain on the rod at different speed (5–20 r.p.m.). They stepped voluntarily from the hand of the experimenter onto the rotarod, except at very high-rotation speeds. Almost all control animals remained on the rod for 300 sec at 5–20 r.p.m. The time-on-the-rod of rotenone-treated rats was significantly reduced with an obvious gradual decrease in time-on-the-rod as rod speed increased. Rats pre-treated with diazoxide had a longer length of time-on-the-rod compared with the rotenone-treated rats (Fig. 4B).
4.
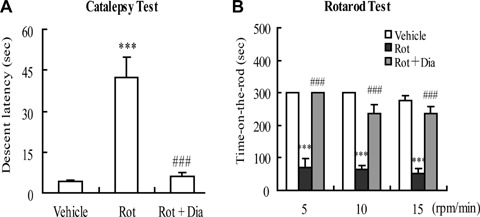
Effects of mito-KATP channel opener diazoxide on rotenone-induced rat behavioural symptoms. (A) Catalepsy test. (B) Rotarod test. ***P <0.001 versus control group; ###P <0.001 versus rotenone (2.5 mg/kg/day) group. Data are presented as the mean ± S.E.M., n= 12 per group.
Mito-KATP channel opener diazoxide alleviates rotenone-induced dopaminergic neuronal degeneration and inhibits microglial activation in rat substantia nigra
Since mito-KATP channel opener diazoxide can suppress rotenone-induced microglial activation and production of pro-inflammatory factors in vitro, diazoxide that can pass through the blood–brain barrier should theoretically protect dopaminergic neurons in rotenone-treated rats via inhibiting microglial activation. Thus, in vivo experiments were performed to investigate whether systematical administration with diazoxide exhibited neu-roprotection in rats accompanied with the suppression of microglial activation. As illustrated in Figures 5A and B, rotenone, 2.5 mg/kg/day for 28 days, caused dramatically reduction in the amount of substantia nigra pars compacta (SNpc) dopaminergic neurons compared with control, as evidenced by TH immunos-taining. Diazoxide (1.5 mg/kg/day), which was pre-treated for 3 days and administered daily for 4 weeks with an injection of rotenone 1 hr later each time, increased significantly the number of surviving SNpc TH-positive neurons.
5.
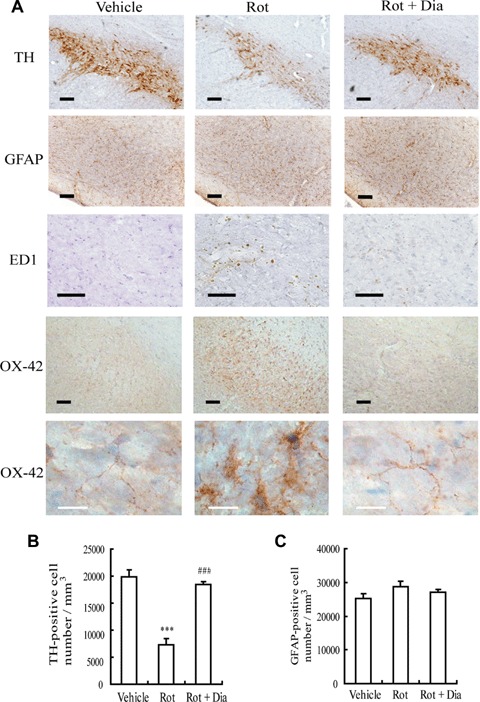
Effects of mito-KATP channel opener diazoxide on rotenone-induced dopaminer-gic neuronal death and microglial activation in rat substantia nigra. (A) Images of immunohistochemistry. TH-positive cells (brown) represent dopaminergic neurons. Anti-GFAP was employed for staining astro-cytes (brown). OX-42 and ED1 immunos-taining (brown) were carried out to study microglial activation. Scale bar: 100μm (black), 20 μm (white). (B) TH-positive cell numbers in substantia nigra. ***P<0.001 versus control group; ###P <0.001 versus rotenone (2.5 mg/kg/day) group. (C) GFAP-positive cell numbers in substantia nigra. Data are expressed as mean ± S.E.M., n = 6 per group.
Simultaneously, antibodies for OX-42 and ED1 were used to detect microglial activation and anti-GFAP was employed for staining astrocytes. Morphologically, robustly immunoreactive OX-42 and ED1-positive activated MG were observed in SNpc of rotenone-treated rats (Fig. 5A). And OX-42 and ED1 immunostaining of the rats pre-treated with diazoxide was similar to those seen in control rats. In SNpc of rotenone-treated rats, the cell body of most MG became larger, and the processes were poorly ramified, short and thick. The microglial appearance in SNpc of the rats pre-treated with diazoxide was similar to those observed in vehicle-treated rats, indicated by small cell body, ramified and thin processes (Fig. 5A). In addition, there was no significant difference in the morphology and numbers of GFAP-positive cells among rats of different treatments (Fig. 5A and C).
Mito-Katp channel opener diazoxide inhibits rotenone-induced production of pro-inflammatory factors in rat brain and peripheral blood
To further explore whether mito-KATP channel opener diazoxide could regulate MG-mediated neuroinflammation in vivo, the content of TNF-α in substantia nigra and peripheral blood serum was determined as well as the mRNA levels of TNF-α and COX-2 in striatum and substantia nigra. The results showed that chronic treatment with rotenone (2.5 mg/kg/day) significantly increased TNF-α content in rat substantia nigra (123 ± 13.9 pg/mg protein, P<0.001 versus control group) and peripheral blood (77 ± 4.4 pg/ml, P<0.001 versus control group). However, diazoxide (1.5 mg/kg/day) could attenuate the content of TNF-α either in substantia nigra or in peripheral blood compared with that of rotenone-treated rats (Fig. 6A). Consistently the results of RT-PCR showed that pre-treatment with diazoxide markedly down-regulated rotenone-induced elevation of TNF-α and COX-2 mRNA levels in rat substantia nigra and striatum (Fig. 6B).
6.
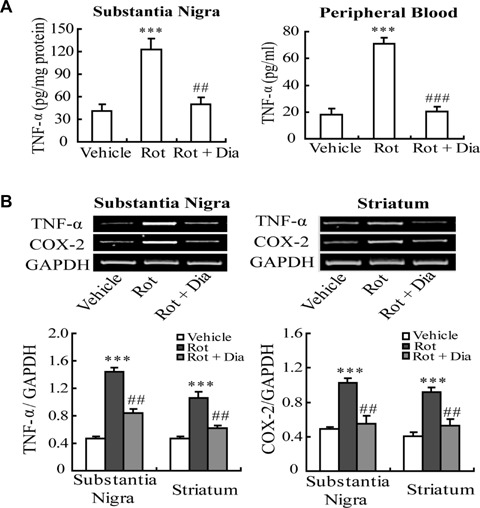
Effects of mito-KATP channel opener diazoxide on rotenone-induced production of pro-inflammatory factors in rat brain and peripheral blood. (A) Diazoxide decreased the content of TNF-α in subtantia nigra and peripheral blood compared with that of rotenone-treated rats. (B) Diazoxide down-regulated the mRNA levels of TNF-α and COX-2 in substantia nigra and striatum. ***P <0.001 versus vehicle group; ##P <0.01, ###P <0.001 versus Rot group. Data are expressed as mean ± S.E.M., n= 6 per group.
Discussion
It is reported that several types of K+ channels are involved in microglial functions, such as proliferation, migration, release of IL-1 β and ROS [11]. We reported here that the KATP channel Kir6.1 and SUR2 subunits are expressed in rat primary cultured MG in the resting state, and that these KATP channels functionally modulate rotenone-induced microglial activation and subsequent production of TNF-α, PGE2 and ROS. Abundant studies have demonstrated that pro-inflammatory and neurotoxic factors produced from activated MG, such as TNF-α, PGE2 and ROS, are involved in mediating neuronal cell death [6, 8, 26, 27] and aggravating glutamate neurotoxicity by inhibiting glutamate uptake in astrocytes [9, 10]. Thus, opening microglial KATP channels may alleviate MG-induced toxic effects on neurons and astrocytes through inhibiting the production of pro-inflammatory and neurotoxic factors.
The present study showed that the inhibitory effects of KATP channel opener pinacidil and specific mito-KATP channel opener diazoxide on rotenone-induced microglial activation were reversed by specific mito-KATP channel blocker 5-HD. These results suggest that mito-KATP channels may play a pivotal role in regulating microglial activation and subsequent production of pro-inflammatory factors. It has been demonstrated that exposure to rotenone results in mito-chondrial depolarization [28, 29]. The present study demonstrated that pre-treatment with diazoxide inhibited rotenone-induced mito-chondrial membrane depolarization. And the inhibitory action of diazoxide was abolished by 5-HD, indicating that opening of mito-KATP channels is responsible for the observed maintenance of polarized states of mitochondrial membrane potentials during rotenone stimulation. It is strongly supported by the reports that activation of mito-KATP channel reduced anoxic injury [30] and glutamate excito-toxicity [31]via restoring mitochondrial membrane potential. Thus, opening of mito-KATP channel may maintain microglial normal function and attenuate microglial activation by preventing rotenone-induced mitochondrial membrane potential loss. However, the detailed mechanisms by which opening of mito-KATP channels inhibits microglial activation need to be further investigated.
Mitogen-activated protein kinases (MAPKs) are downstream signal proteins of mito-KATP channel [32–34]. A cardioselective KATP channel opener KR-31378 protected H9c2 cells against chemical hypoxia-induced cell death by blocking the activation of p38 and JNK [35]. The present study showed that opening of mito-KATP channels by diazoxide inhibited rotenone-induced phosphorylation of p38 and JNK in MG. Since MAPKs play a key regulatory role in COX-2 expression, PGE2 and TNF-α production [22, 24, 25], our results imply that opening of mito-KATP channels might modulate PGE2 and TNF-α production through MAPK pathway. Recently, ROS generated in the mitochondrial respiratory chain is considered as an important mediator of signal transduction in ischaemic pre-conditioning [36], which activates p38/JNK through the activation of upstream signalling MKK. Mito-KATP channel opening increases production of protective ROS during pre-conditioning and decreases the levels of ROS produced during reperfusion [37]. So we propose that opening of mito-KATP channels may inhibit the rotenone-induced production of PGE2 and TNF-α from MG through suppression of mitochondria-derived ROS and downstream MAPKs phosphorylation.
It is well documented that microglial activation and subsequent production of TNF-α, PGE2 and ROS participate in rotenone-induced dopaminergic neuronal degeneration [2–4]. Since KATP channel openers improved rotenone-elicited motor and neurochemical alterations [16, 38], the rotenone-induced Parkinsonism rat model was used to investigate whether the neuroprotective effects of KATP channel openers implicated the inhibition of microglial activation and subsequent neuroinflammation in substantia nigra. The activated state of microglial cells is indicated by the increased density of OX-42-posi-tive cells and amoeboid morphological alteration. As expected, our in vivo results demonstrated that mito-KATP channel opener diazoxide alleviated rotenone-induced dopaminergic neuronal degeneration along with the inhibition of microglial activation and subsequent neuroinflammation in rat substantia nigra. On the other hand, the minimal astrocytosis in substantia nigra after rotenone treatment was consistent with a previous study that reactive astrocytosis was not prominent in cortex or nigra and limited around the lesion in stratium of chronic rotenone-treated rats [39]. Systematical administration with selective mito-KATP channel opener diazoxide also attenuated rotenone-induced production of pro-inflammatory factors in brain and peripheral blood, further confirming that opening of mito-KATP channels may exert indirect neuroprotection through the effect of anti-neuroinflammation. These findings suggest that mito-KATP channel may be an important molecular target for treating inflammation-related neurodegenerative disorders such as PD.
Increasing evidence has shown that selective activation of mito-KATP channels elicits protective effects against ischaemia, degeneration or metabolic challenges [13, 31, 40–43]. However, little is known about the subunit composition or functional regulation of the mito-KATP channels. Our previous study showed that novel KATP channel opener iptakalim inhibited microglial activation [19] and could open Kir6.1/SUR2-composed KATP channels [44], which is same as the composition of mito-KATP channels [45–47]. The results in the present study demonstrate that microglial KATP channels are formed by Kir6.1 and SUR2 subunits and selective mito-KATP channel opener diazoxide inhibited microglial activation. Thus, it is reasonable that detectable Kir6.1 and SUR2 subunits constitute functional mito-KATP channels in MG, which appear to play a critical role in regulating microglial activation.
In conclusion, we demonstrated that Kir6.1 and SUR2 subunits of KATP channels were expressed in rat primary cultured MG and opening microglial mito-KATP channels inhibited microglial activation and subsequent production of pro-inflammatory factors. The underlying mechanisms involved the regulation of mitochondrial membrane potential and p38/JNK activation in MG. Since the in vivostudy further showed that opening mito-KATP channels exerted neuroprotective effects along with the inhibition of neuroinflamma-tion, it is prospective that targeting microglial mito-KATP channels may be a promising therapeutic strategy for treating neuroinflam-mation-related neurodegenerative diseases such as PD.
Acknowledgments
This work was partly supported by a grant from the National Natural Science Foundation of China (No. 30625038 No.30600758 and No. 30572172), a grant from the Key Project of Natural Science Foundation of Jiangsu Educational Department (No. 05KJA31014 and No. 06KJA31029), and a Specialized Research Fund for the Doctoral Program of Higher Education of China (No. 20040312004).
References
- 1.Castano A, Herrera AJ, Cano J, Machado A. Lipopolysaccharide intrani-gral injection induces inflammatory reaction and damage in nigrostriatal dopamin-ergic system. J Neurochem. 1998;70:1584–92. doi: 10.1046/j.1471-4159.1998.70041584.x. [DOI] [PubMed] [Google Scholar]
- 2.Gao HM, Hong JS, Zhang W, Liu B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2002;22:782–90. doi: 10.1523/JNEUROSCI.22-03-00782.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Minghetti L, Ajmone-Cat MA, De Berardinis MA, De Simone R. Microglial activation in chronic neurodegenerative dis-eases: roles of apoptotic neurons and chronic stimulation. Brain Res Rev. 2005;48:251–6. doi: 10.1016/j.brainresrev.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 4.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 5.Gao HM, Liu B, Zhang W, Hong JS. Novel anti-inflammatory therapy for Parkinson's disease. Trends Pharmacol Sci. 2003;24:395–401. doi: 10.1016/S0165-6147(03)00176-7. [DOI] [PubMed] [Google Scholar]
- 6.Ahmad AS, Saleem S, Ahmad M, Dore S. Prostaglandin EP1 receptor contributes to excitotoxicity and focal ischemic brain damage. Toxicol Sci. 2006;89:265–70. doi: 10.1093/toxsci/kfj022. [DOI] [PubMed] [Google Scholar]
- 7.Bernardino L, Xapelli S, Silva AP, Jakobsen B, Poulsen FR, Oliveira CR, Vezzani A, Malva JO, Zimmer J. Modulator effects of interleukin-1beta and tumor necrosis factor-alpha on AMPA-induced excitotoxicity in mouse organ-otypic hippocampal slice cultures. J Neurosci. 2005;25:6734–44. doi: 10.1523/JNEUROSCI.1510-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–9. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- 9.Korn T, Magnus T, Jung S. Autoantigen specific T cells inhibit glutamate uptake in astrocytes by decreasing expression of astrocytic glutamate transporter GLAST: a mechanism mediated by tumor necrosis factor-alpha. FASEB J. 2005;19:1878–80. doi: 10.1096/fj.05-3748fje. [DOI] [PubMed] [Google Scholar]
- 10.Zou JY, Crews FT. TNF alpha potentiates glutamate neurotoxicity by inhibiting gluta-mate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res. 2005;1034:11–24. doi: 10.1016/j.brainres.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 11.Eder C. Regulation of microglial behavior by ion channel activity. J Neurosci Res. 2005;81:314–21. doi: 10.1002/jnr.20476. [DOI] [PubMed] [Google Scholar]
- 12.Liss B, Roeper J. Molecular physiology of neuronal K-ATP channels. Mol Membr Biol. 2001;18:117–27. [PubMed] [Google Scholar]
- 13.Busija DW, Lacza Z, Rajapakse N, Shimizu K, Kis B, Bari F, Domoki F, Horiquchi T. Targeting mitochondrial ATP-sensitive potassium channels–a novel approach to neuroprotection. Brain Res Rev. 2004;46:282–94. doi: 10.1016/j.brainresrev.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Hu LF, Wang S, Shi XR, Yao HH, Sun YH, Ding JH, Liu SY, Hu G. ATP-sensitive potassium channel opener iptakalim protected against the cytotoxicity of MPP+ on SH-SY5Y cells by decreasing extracellular glutamate level. J Neurochem. 2005;94:1570–9. doi: 10.1111/j.1471-4159.2005.03306.x. [DOI] [PubMed] [Google Scholar]
- 15.Yamada K, Inagaki N. Neuroprotection by KATP channels. J Mol Cell Cardiol. 2005;38:945–9. doi: 10.1016/j.yjmcc.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 16.Yang Y, Liu X, Long Y, Wang F, Ding JH, Liu SY, Sun YH, Yao HH, Wang H, Wu J, Hu G. Activation of mitochondrial ATP-sensitive potassium channels improves rotenone-related motor and neurochemical alterations in rats. Int J Neuropsychopharmacol. 2006;9:51–61. doi: 10.1017/S1461145705005547. [DOI] [PubMed] [Google Scholar]
- 17.Liss B, Haeckel O, Wildmann J, Miki T, Seino S, Roeper J. K-ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nat Neurosci. 2005;12:1742–51. doi: 10.1038/nn1570. [DOI] [PubMed] [Google Scholar]
- 18.Liu X, Wu JY, Zhou F, Sun XL, Yao HH, Yang Y, Ding JH, Hu G. The regulation of rotenone-induced inflammatory factor production by ATP-sensitive potassium channel expressed in BV-2 cells. Neurosci Lett. 2006;394:131–5. doi: 10.1016/j.neulet.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 19.Zhou F, Wu JY, Sun XL, Yao HH, Ding JH, Hu G. Iptakalim alleviates rotenone-induced degeneration of dopaminergic neurons through inhibiting microglia-mediated neuroinflammation. Neuro-psy-chopharmacol. 2007;32:2570–80. doi: 10.1038/sj.npp.1301381. [DOI] [PubMed] [Google Scholar]
- 20.West MJ. New stereological methods for counting neurons. Neurobiol Aging. 1993;14:275–85. doi: 10.1016/0197-4580(93)90112-o. [DOI] [PubMed] [Google Scholar]
- 21.Zhang W, Qin L, Wang T, Wei SJ, Gao HM, Liu J, Wilson B, Liu B, Zhang W, Kim HC, Hong JS. 3-hydroxymorphinan is neurotrophic to dopaminergic neurons and is also neuroprotective against LPS-induced neurotoxicity. FASEB J. 2005;19:395–7. doi: 10.1096/fj.04-1586fje. [DOI] [PubMed] [Google Scholar]
- 22.Akundi RS, Candelario-Jalil E, Hess S, Hull M, Lieb K, Gebicke-Haerter PJ, Fiebich BL. Signal transduction pathways regulating cyclooxygenase-2 in lipopolysaccharide-activated primary rat microglia. Glia. 2005;51:199–208. doi: 10.1002/glia.20198. [DOI] [PubMed] [Google Scholar]
- 23.Ciallella JR, Saporito M, Lund S, Leist M, Hasseldam H, McGann N, Smith CS, Bozyczko-Coyne D, Flood DG. CEP-11004, an inhibitor of the SAPK/JNK pathway, reduces TNF-alpha release from lipopolysaccharide-treated cells and mice. Eur J Pharmacol. 2005;515:179–87. doi: 10.1016/j.ejphar.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 24.Lund S, Porzgen P, Mortensen AL, Hasseldam H, Bozyczko-Coyne D, Morath S, Hartung T, Bianchi M, Ghezzi P, Bsibsi M, Dijkstra S, Leist M. Inhibition of microglial inflammation by the MLK inhibitor CEP-1347. J Neurochem. 2005;92:1439–51. doi: 10.1111/j.1471-4159.2005.03014.x. [DOI] [PubMed] [Google Scholar]
- 25.Waetzig V, Czeloth K, Hidding U, Mielke K, Kanzow M, Brecht S, Goetz M, Lucius R, Herdegen T, Hanisch UK. c-Jun N-ter-minal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia. 2005;50:235–46. doi: 10.1002/glia.20173. [DOI] [PubMed] [Google Scholar]
- 26.Hemmer K, Fransen L, Vanderstichele H, Vanmechelen E, Heuschling P. An in vitro model for the study of microglia-induced neurodegeneration: involvement of nitric oxide and tumor necrosis factor-alpha. Neurochem Int. 2001;38:557–65. doi: 10.1016/s0197-0186(00)00119-4. [DOI] [PubMed] [Google Scholar]
- 27.Tomas-Camardiel M, Rite I, Herrera AJ, De Pablos RM, Cano J, Machado A, Venero JL. Minocycline reduces the lipopolysaccharide-induced inflammatory reaction, peroxynitrite-mediated nitration of proteins, disruption of the blood-brain barrier, and damage in the nigral dopamin-ergic system. Neurobiol Dis. 2004;16:190–201. doi: 10.1016/j.nbd.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 28.Moon Y, Lee KH, Park JH, Geum D, Kim K. Mitochondrial membrane depolarization and the selective death of dopaminergic neurons by rotenone: protective effect of coenzyme Q10. J Neurochem. 2005;93:1199–208. doi: 10.1111/j.1471-4159.2005.03112.x. [DOI] [PubMed] [Google Scholar]
- 29.Radad K, Rausch WD, Gille G. Rotenone induces cell death in primary dopaminer-gic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem Int. 2006;49:379–86. doi: 10.1016/j.neuint.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Xu M, Wang Y, Ayub A, Ashraf M. Mitochondrial K(ATP) channel activation reduces anoxic injury by restoring mito-chondrial membrane potential. Am J Physiol Heart Circ Physiol. 2001;281:H1295–303. doi: 10.1152/ajpheart.2001.281.3.H1295. [DOI] [PubMed] [Google Scholar]
- 31.Yamauchi T, Kashii S, Yasuyoshi H, Zhang S, Honda Y, Akaike A. Mitochondrial ATP-sensitive potassium channel: a novel site for neuroprotection. Invest Ophthalmol Vis Sci. 2003;44:2750–6. doi: 10.1167/iovs.02-0815. [DOI] [PubMed] [Google Scholar]
- 32.Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Abe Y. Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension. 2005;45:438–44. doi: 10.1161/01.HYP.0000157169.27818.ae. [DOI] [PubMed] [Google Scholar]
- 33.Lin YF, Raab-Graham K, Jan YN, Jan LY. NO stimulation of ATP-sensitive potassium channels: Involvement of Ras/mitogen-activated protein kinase pathway and contribution to neuroprotection. Proc Natl Acad Sci USA. 2004;101:7799–804. doi: 10.1073/pnas.0402496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Samavati L, Monick MM, Sanlioglu S, Buettner GR, Oberley LW, Hunninghake GW. Mitochondrial K(ATP) channel openers activate the ERK kinase by an oxidant-dependent mechanism. Am J Physiol Cell Physiol. 2002;283:C273–81. doi: 10.1152/ajpcell.00514.2001. [DOI] [PubMed] [Google Scholar]
- 35.Jung YS, Lee DH, Lim H, Yi KY, Yoo SE, Kim E. KR-31378 protects cardiac H9c2 cells from chemical hypoxia-induced cell death via inhibition of JNK/p38 MAPK activation. Jpn J Physiol. 2004;54:575–83. doi: 10.2170/jjphysiol.54.575. [DOI] [PubMed] [Google Scholar]
- 36.Otani H. Reactive oxygen species as mediators of signal transduction in ischemic preconditioning. Antioxid Redox Signal. 2004;6:449–69. doi: 10.1089/152308604322899521. [DOI] [PubMed] [Google Scholar]
- 37.O'Rourke B. Evidence for mitochondrial K+channels and their role in cardioprotection. Circ Res. 2004;94:420–32. doi: 10.1161/01.RES.0000117583.66950.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Y, Liu X, Long Y, Wang F, Ding JH, Liu SY, Sun YH, Yao HH, Wang H, Wu J, Hu G. Systematic administration of iptakalim, an ATP-sensitive potassium channel opener, prevents rotenone-induced motor and neurochemical alterations in rats. J Neurosci Res. 2005;80:442–9. doi: 10.1002/jnr.20467. [DOI] [PubMed] [Google Scholar]
- 39.Sherer TB, Betarbet R, Kim JH, Greenamyre JT. Selective microglial activation in the rat rotenone model of Parkinson's disease. Neurosci Lett. 2003;341:87–90. doi: 10.1016/s0304-3940(03)00172-1. [DOI] [PubMed] [Google Scholar]
- 40.Farkas E, Annahazi A, Institoris A, Mihaly A, Luiten PG, Bari F. Diazoxide and dimethyl sulphoxide alleviate experimental cerebral hypoperfusion-induced white matter injury in the rat brain. Neurosci Lett. 2005;373:195–9. doi: 10.1016/j.neulet.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 41.Liu D, Lu C, Wan R, Auyeung WW, Mattson MP. Activation of mitochondrial ATP-dependent potassium channels protects neurons against ischemia-induced death by a mechanism involving suppression of Bax translocation and cytochrome c release. J Cereb Blood Flow Metab. 2002;22:431–43. doi: 10.1097/00004647-200204000-00007. [DOI] [PubMed] [Google Scholar]
- 42.Teshima Y, Akao M, Li RA, Chong TH, Baumgartner WA, Johnston MV, Marban E. Mitochondrial ATP-sensitive potassium channel activation protects cerebellar gran-ule neurons from apoptosis induced by oxidative stress. Stroke. 2003;34:1796–802. doi: 10.1161/01.STR.0000077017.60947.AE. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Takashi E, Xu M, Ayub A, Ashraf M. Downregulation of protein kinase C inhibits activation of mitochondrial K(ATP) channels by diazoxide. Circulation. 2001;104:85–90. doi: 10.1161/01.cir.104.1.85. [DOI] [PubMed] [Google Scholar]
- 44.Hu J, Hu G, Wang H, Lukas RJ, Wu J. Iptakalim as a human nicotinic acetyl-choline receptor antagonist. J Pharmacol Exper Therap. 2006;316:914–25. doi: 10.1124/jpet.105.094987. [DOI] [PubMed] [Google Scholar]
- 45.Bajgar R, Seetharaman S, Kowaltowski AJ, Garlid KD, Paucek P. Identification and properties of a novel intracellular (mito-chondrial) ATP-sensitive potassium channel in brain. J Biol Chem. 2001;276:33369–74. doi: 10.1074/jbc.M103320200. [DOI] [PubMed] [Google Scholar]
- 46.Lacza Z, Snipes JA, Kis B, Szabo C, Grover G, Busija DW. Investigation of the subunit composition and the pharmacology of the mitochondrial ATP-dependent K+ channel in the brain. Brain Res. 2003;994:27–36. doi: 10.1016/j.brainres.2003.09.046. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki M, Kotake K, Fujikura K, Inagaki N, Suzuki T, Gonoi T, Seino S, Takata K. Kir6.1: a possible subunit of ATP-sensi-tive K+ channels in mitochondria. Biochem Biophys Res Commun. 1997;241:693–7. doi: 10.1006/bbrc.1997.7891. [DOI] [PubMed] [Google Scholar]